

Abstract
The UDP glucuronosyltransferase (UGT) 1A gene cluster encodes nine UGT1A family members via splicing of individual first exons to common exons 2 through 5. Each of these nine UGT1As can also undergo alternative splicing at their 3′ ends by using an alternate exon 5, resulting in 27 different UGT1A mRNA species with each UGT1A gene encoding three different combinations of 5A and 5B UGT1A exons. To examine the importance of UGT1A exon 5 splice variants on overall UGT1A activity, a nested quantitative polymerase chain reaction assay was developed to accurately assess the combined expression of exon 5 splice variants (termed v2/v3) versus the expression of wild-type (termed v1) for each specific UGT1A. v1 expression was 16-, 17-, 57- and 29-fold higher than that observed for the levels of v2/v3 for UGTs 1A1, 1A4, 1A6, and 1A9, respectively, in normal human liver specimens. In a series of 58 normal human liver specimens, the expression of both UGT1A1 v1 and v2/v3 mRNAs was positively correlated with raloxifene glucuronidation activity in corresponding microsomes prepared from the same specimens (p < 0.0001, r2 = 0.720; p = 0.0002, r2 = 0.241, respectively), with expression of both variants lower in individuals homozygous for the UGT1A1*28 allele (42% for v1, p = 0.041; 53% for v2/v3, p = 0.0075). The expression of UGT1A1 v2/v3 was 1.6-fold higher than v1 (p = 0.03) in HepG2 cells, and short interfering RNA knockdown of HepG2 v2/v3 increased raloxifene glucuronidation activity by 83%. Together, these data suggest that hepatic UGT1A v2/v3 mRNA species are minor form variants in human livers from most individuals.
Introduction
UDP-glucuronosyltransferases (UGTs) play an important role in the metabolism and excretion of endogenous and xenobiotic compounds including drugs, carcinogens, and chemotherapeutic agents. UGTs mediate the conjugation of glucuronic acid to their substrates, thereby increasing the polarity and facilitating excretion of the conjugate in the urine, bile, and feces (Dutton, 1980). Based on sequence homology, the UGTs are divided into two major subfamilies. The nine UGT1A family members are encoded by a single locus on chromosome 2q37, where each family member has a unique exon 1 and shares exons 2 to 5 (Gong et al., 2001). The unique first exon confers substrate specificity for the enzyme, whereas the common region contains the UDP glucuronic acid (UDPGA) cofactor binding domain and other essential motifs (Girard et al., 2007).
An alternative exon 5 in the common region of the UGT1A gene cluster leads to the expression of 18 additional mRNA species from this locus (Lévesque et al., 2007). UGT1A mRNAs that incorporate only exon 5a are known as variant 1 (v1) and encode the active isoform 1 (i1) proteins (Girard et al., 2007). When the alternative exon 5b is incorporated as the last exon [variant 2 (v2)] or when both exons 5a and 5b are incorporated together [variant 3 (v3)], the resulting protein is catalytically inactive (Girard et al., 2007). The v2 and v3 mRNAs each have the same open reading frame and thus encode the same protein [isoform 2 (i2)] (Girard et al., 2007). In vitro cotransfection studies demonstrated a dominant negative effect of i2 proteins on i1 glucuronidation activity (Benoit-Biancamano et al., 2009; Bellemare et al., 2010a,b,c). In addition, concomitant expression of UGT1A1 i1 and i2 proteins in HEK293 cells leads to significantly decreased activity against a wide range of substrates (Bellemare et al., 2010a). These studies suggested that the function of the i2 proteins may be to negatively modulate the catalytic activity of the UGT1A i1 proteins. However, neither expression of the UGT1A v2 and v3 splice variants nor UGT1A i2-mediated regulation of UGT1A i1 activity have been explored in human tissues.
We hypothesized that interindividual variation in the relative abundance of v1 and v2/v3 mRNA expression affects glucuronidation capacity in human liver. The goal of the present study was to assess the importance of the exon 5 splice variants on overall UGT1A activity by quantifying mRNA expression of the v1 versus v2/v3 UGT1A splice variants in individual human liver specimens and determine whether there are correlations with glucuronidation activity in those same specimens.
Materials and Methods
Chemicals and Materials.
UDPGA, dl-2-lysophosphatidyl choline palmital C16:0, and Escherichia coli β-glucuronidase were purchased from Sigma (St. Louis, MO). Pfu DNA polymerase was purchased from Agilent Technologies (Santa Clara, CA), and the 1kb-plus DNA ladder, reverse transcription (RT) kits, cell culture reagents, and siRNA reagents were purchased from Invitrogen (Carlsbad, CA). High-performance liquid chromatography (HPLC)-grade solvents were purchased from Thermo Fisher Scientific (Waltham, MA). Suppliers of other reagents included QIAGEN (Valencia, CA) for extraction kits, Agilent Technologies for Pfu polymerase, Applied Biosystems (Foster City, CA) for gene expression assays, and Thermo Fisher Scientific for bicinchoninic acid reagents.
Tissue Collection.
Liver specimens were collected at the H. Lee Moffitt Cancer Center (Tampa, FL) from 58 individuals undergoing surgery for excision of hepatocellular carcinoma and were confirmed to be histologically normal as described previously (Fang and Lazarus, 2004). Tissue microsomes were prepared from 58 normal liver specimens through differential centrifugation, total protein concentrations were measured with the bicinchoninic acid assay (Thermo Fisher Scientific), and samples were stored (10–20 mg protein/ml) at −70°C in 100-μl aliquots (Coughtrie et al., 1987). All protocols involving the collection and analysis of tissue specimens were approved by the Institutional Review Board at Penn State University and were in accordance with assurances filed with and approved by the United States Department of Health and Human Services. All samples were isolated and quick-frozen at −70°C within 2 h postsurgery.
RNA Collection and cDNA Synthesis.
RNA was extracted from all liver tissue specimens by the Tissue Procurement Facility at the H. Lee Moffitt Cancer Center, and demographic data were provided for all subjects. Cell line RNA was extracted by using the RNeasy Mini Kit (QIAGEN) according to the manufacturer's protocol. Samples were subjected to DNase I digestion during extraction to prevent genomic DNA contamination. RNA concentrations were determined by using a Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific), and RNA purity was assessed by absorbance ratios A260/A280 (>1.9) and A260/A230 (>1.8). RNA integrity was determined by using an Agilent 2100 Bioanalyzer with Agilent RNA 6000 Nano chips (Agilent Technologies, Santa Clara, CA). Two samples with RNA integrity number values of 7.9 or less were excluded from further analysis because of degradation. Reverse transcription was performed by using the Superscript First Strand cDNA synthesis kit (Invitrogen) with either oligo(dT) or random heximer primers and 1 μg of starting RNA per sample. A negative control without RNA and a negative control without enzyme were analyzed in parallel.
First-Step PCR.
For the initial amplification of the UGT1A mRNA species, the four exon 1-specific sense primers used were as follows: UGT1A1, 5′-catgctgggaagatactgttga-3′ (+75 to +97); UGT1A4, 5′-cttctgctgagatggccaga-3′ (−11 to +9); UGT1A6, 5′-tttggggcatggttgtaggt-3′ (+59 to +78); and UGT1A9, 5′-ccactggttcaccatgaggt-3′ (+108 to +127) (locations provided are relative to the ATG translation start site for each UGT). The two exon 5-specific antisense primers were v1, 5′-cgcatgatgttctccttgtaactt-3′ (−300 to −276) and v2/v3, 5′-tgggaagtcagtcatcagtcct-3′ (+74 to + 96) (both relative to the TGA translation stop codon for the v1 and v2/v3 variants, respectively). Because UGT1A v2 and v3 mRNAs encode the same inactive protein (Girard et al., 2007), the reverse primer was designed to target a shared region of the exon 5b 3′ untranslated region (UTR) so that both cDNAs could be amplified simultaneously. The reverse primer specific for UGT1A v1 mRNA was designed such that the primer overlapped the exon 4/5a junction, which is the only region completely unique to variant 1. The UGT1A exon 1 sense primers were designed so that the amplification product from the first PCR was specific to the UGT1A enzyme of interest and would contain the binding site for the primers and probes of the subsequent real-time PCR. Differences in amplification efficiency caused by primer characteristics were lessened by using a primer design strategy that minimized differences in GC content (42–55%), melting temperature (54.7–57.4°C), primer length (20–25 bp), and hairpin formation (ΔG > −2 kcal/mol). The specificity of each primer set was verified by using a 40-cycle PCR amplification, followed by visualization and extraction of the amplification product and subsequent sequencing in the forward and reverse directions to confirm its identity. To optimize the first-step PCR, PCR amplification was carried out for 10, 15, or 20 cycles by using Pfu Turbo DNA polymerase (Agilent Technologies) and a 25-ng RNA equivalent of cDNA. PCR conditions were as follows: 95°C for 2 min, then 10, 15, or 20 cycles of 95°C for 20 s, 60°C for 30 s, and 72°C for 2 min, and finally 1 cycle of 72°C for 10 min.
Real-Time Quantitative PCR.
The target sequence and GC content of the different UGT1A amplicons are highly homologous, making it easier to achieve similar amplification efficiency for these cDNA species than for heterogenous DNA sequences. After the first-step PCR amplification, cDNA samples were diluted 100-fold and subjected to a Taqman exon 1-specific real-time PCR (Applied Biosystems). Because proper control selection is imperative for accurate normalization via the 2−ΔΔCt method (Freeman et al., 1999; Livak and Schmittgen, 2001; Vandesompele et al., 2002; VanGuilder et al., 2008), all UGT1A genes were normalized to expression of the MT-ATP6 gene, which was determined to be the most stably expressed gene among a panel of 32 putative control genes in a subset of liver samples (data not shown). In brief, 32 putative control genes were analyzed in six liver specimens by using Taqman Express Endogenous Control Plates (Applied Biosystems). MT-ATP6 was determined to be the most stably expressed control gene among the panel, with the lowest S.D. in amplification cycle number between the six liver tissues examined. Quadruplicate real-time PCRs were performed for each cDNA sample by using a 10-μl final reaction volume containing 5 μl of 2× Taqman Universal PCR Master Mix, 4.5 μl of diluted cDNA, and 0.5 μl of gene expression assay. Reactions were performed in 384-well plates by using the ABI 7900 HT Sequence Detection System (Applied Biosystems) under the following conditions: 1 cycle at 50°C for 2 min, 1 cycle at 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min.
Relative quantification (RQ) of expression was calculated by using the ΔΔCt method. In brief, ΔCt was calculated as the Ct value of the target gene minus the Ct of the control gene (MT-ATP6). The ΔΔCt was then calculated as the ΔCt of the sample minus the ΔCt of a calibrator sample, in this case the lowest-expressing liver sample. RQ was determined with the formula (1 + Ex)−ΔΔCt where Ex is the amplification efficiency of the given qPCR gene expression assay. Amplification efficiencies were calculated from UGT1A variant standard curves with the formula Ex = [10(−1/slope) − 1]. Standard curves for the eight UGT1A variants were generated as follows: pooled cDNA from eight livers was amplified for 20 cycles by using primer sets specific to each of the eight full-length UGT1A cDNA species as described above. Resulting amplification products were subjected to a serial 10-fold dilution. Serially diluted UGT1A amplification products were then amplified in quadruplicate for 40 cycles by qPCR, and the average Ct value for each dilution was plotted against the log of the DNA concentration.
Cell Line Generation.
As positive controls for UGT1A variant expression, total RNA from cell lines individually overexpressing UGTs 1A1 v1, 1A4 v1, 1A6 v1, and 1A9 v1, as well as 1A1 v2, 1A4 v2, 1A6 v2, and 1A9 v2, were used to control for any differences in primer pair efficiency that may confound the quantification of the exon 1 and exon 5 splice variants. Generation of the HEK293 cell line overexpressing UGT1A v1 variants has been described previously (Coffman et al., 1995; Ethell et al., 2001; Olson et al., 2009). UGTs 1A1 v2-, 1A4 v2-, 1A6 v2-, and 1A9 v2-overexpressing cell lines were generated by stable transfection by using the Lipofectamine reagent (Invitrogen) according to the manufacturer's protocol. The primers used for cloning the UGT1A v2 variants were as follows: UGT1A1 sense, 5′-ccatggctgtggagtccc-3′ (−2 to +16); UGT1A4 sense, 5′-tggcttctgctgagatggccag-3′ (−14 to +9); UGT1A6 sense, 5′-ggagccctgtgatttggagagtg-3′ (−63 to −40); and UGT1A9 sense, 5′-cagttctctgatggcttgca-3′ (−10 to +10) (all locations listed are relative to the respective ATG translation start site). The antisense primer used for cloning the v2 variants is the same as listed above for the nested PCR. In brief, pcDNA3.1/V5-His-TOPO/UGT1A v2 constructs were transfected into HEK293 cells grown to 80% confluence at 37° and 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 4.5 mM glucose, 10 mM HEPES, 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. At 24 h post-transfection, cells were passaged and grown in geneticin (700 μg/ml medium) for the selection of geneticin-resistant cells, with selection medium changed every 3 to 4 days. For validation of the nested PCR method, RNA (1 μg) isolated from the eight UGT1A v1 and v2/v3 cell lines was reverse-transcribed, and the resulting cDNA was incubated with primers that amplified a product encompassing the entire UGT1A1 open reading frame and portions of the 5′ and 3′ UTRs. After this initial amplification, the resulting cDNA products were diluted 1:100 and subjected to a second PCR amplification by using exon 1-specific real-time PCR assays as described above.
To generate a cell line coexpressing UGT1A1_i1 and UGT1A1_i2 protein, UGT1A1 v2 was cloned into the pcDNA 6.2/V5/GW/D-TOPO vector by using the cloning primers described above, which amplify the entire coding region of the UGT1A1_i2 protein. This vector was transfected into UGT1A1_i1_-overexpressing cells grown to 80% confluence at 37° and 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 4.5 mM glucose, 10 mM HEPES, 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 700 μg/ml geneticin. At 24 h post-transfection, cells were passaged and grown in blasticidin for the selection of blasticidin-resistant cells, with selection medium changed every 3 to 4 days.
siRNA Knockdown of UGT1A v2/v3 Variants in HepG2 Cells.
Knockdown of the UGT1A v2/v3 species in HepG2 cells was accomplished by using Lipofectamine 2000 reagent and 10 nM of the v2/v3-specific siRNA (5′-cagcagucaggaagacagaugugaa-3′). Control cells were transfected with scrambled (SCR) siRNA (5′-uucguaaccugucuuuccgucacug-3′). siRNA was designed by using the Invitrogen Block-iT siRNA design module. Because adherent HepG2 cells are notoriously difficult to transfect, cells were subjected to neofection, which enables a higher percentage of cells to be exposed to the transfection reagent, which can be a problem with adherent HepG2 cells that tend to layer on top of each other. In brief, cells were first trypsinized and collected. siRNA and Lipofectamine 2000 reagent were then complexed in Invitrogen Optimem reduced serum media according to the manufacturer's protocol. Two milliliters of the resulting siRNA/Lipofectamine complexes were added to 150-mm culture dishes. Finally, 6 × 106 HepG2 cells were added to each plate so that the confluence of the cells would be ∼30% after 24 h. Cells were allowed to grow for 96 h before isolation of RNA and homogenate.
Glucuronidation Assays.
Glucuronidation activities of HLM and HepG2 cell homogenates were performed after an initial incubation of 500 μg of HepG2 cell homogenate or 15 μg of HLM with alamethicin (50 μg/mg protein) for 15 min in an ice bath. Glucuronidation reactions were then performed in a final reaction volume of 50 μl at 37°C in 50 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 4 mM UDPGA, and tested substrate. Raloxifene (1–128 μM) was used for kinetic analysis in HLMs from six individual subjects. For glucuronidation rate analysis, 2 μM raloxifene was used for HLM, and 50 μM raloxifene was used for HepG2 cell homogenates. For UGT1A1_i1/UGT1A1_i2 coexpression experiments, 200 μg of protein homogenate from cell lines overexpressing either UGT1A1_i1 or UGT1A1 i1+i2 protein were used for kinetic rate determination against the substrate raloxifene (1–200 μM). Reactions were terminated by the addition of 50 μl of cold acetonitrile on ice. Mixtures were centrifuged for 10 min at 4°C at 16,100g, and the supernatants were collected.
Raloxifene glucuronidation was analyzed by using a Waters (Milford, MA) ACQUITY ultra-pressure liquid chromatography (UPLC)/tandem mass spectrometry system with a 1.7-μ ACQUITY UPLC BEH C18 analytical column (2.1 mm × 50 mm; Waters) in series with a 0.2-μm Waters assay frit filter (2.1 mm). The gradient elution conditions, using a flow rate of 0.3 ml/min, were as follows: starting with 5% acetonitrile and 95% buffer A (5 mM ammonium acetate, pH 5.0) for 1 min, a subsequent linear gradient to 100% acetonitrile over 5 min was performed and then maintained at 100% acetonitrile for 2 min. Raloxifene-glucuronides (ral-6-Gluc and ral-4′-Gluc) were confirmed by their stability in 1 M NaOH but sensitivity to the treatment of β-glucuronidase. In addition, confirmation of raloxifene glucuronide formation was performed by loading up to 5 μl of incubation product onto an UPLC identical to that described above in tandem with a Waters TQD triple quadrupole MS system. By using a positive mode, the parent compound [M + H]+ peak and their corresponding glucuronide [M-Gluc + H]+ peaks were characterized.
The ral-6-Gluc and ral-4′-Gluc formed could not be distinguished by tandem mass spectrometry because they have the same molecular weight and mass spectrum. By using standard compounds and matching their migration time in chromatograms, two glucuronide isomers were identified. Standard stock solutions of raloxifene, ral-6-Gluc, ral-4′-Gluc, and their internal standards were prepared in dimethyl sulfoxide. Combined internal standards solution containing raloxifene-d4, ral-6-Gluc-d4, and ral-4′-Gluc-d4 was prepared at concentrations of 20, 20, and 5 μg/ml, respectively. The combined standard solution was then serially diluted by dimethyl sulfoxide to make standard working solutions from 24 ng/ml to 25 μg/ml for raloxifene and 195 ng/ml to 100 μg/ml for ral-6-Gluc and ral-4′-Gluc. All solutions were kept at −20°C before use.
Quantification of raloxifene, ral-6-Gluc, and ral-4′-Gluc was performed by using multiple reaction monitoring of the transitions of m/z 474.2 → 112.2 for raloxifene, m/z 650.5 → 474.3 for ral-6-Gluc and ral-4′-Gluc, m/z 478.2 → 116.2 for raloxifene-d4, and m/z 654.5 → 478.3 for ral-6-Gluc-d4 and ral-4′-Gluc-d4. Standard curves were constructed by plotting the ratio of analyte peak area to corresponding internal standard peak area versus analyte concentration (concentration range described above). Concentrations of raloxifene and its metabolites were determined by measuring the analyte/internal standard peak area ratio and then calculating the concentration from the standard curves. All data were quantified by MassLynx NT 4.1 software with the QuanLynx program (Waters).
The analysis of bilirubin glucuronidation in the 58 HLM samples analyzed in the present study had been determined and described previously (Fang and Lazarus, 2004). In brief, glucuronidation of bilirubin was assayed in a 100-μl reaction volume by using 0.25 mg of HLM protein incubated for 1 h at 37°C in 50 mM sodium citrate, pH 7.4, containing 0.7 mM bilirubin, 4 mM UDPGA, 10 mM MgCl2, and dl-2-lysophosphatidyl choline palmital C16:0 (10 μg/100 μg of protein). Reactions were terminated by the addition of an equal volume (100 μl) of 2% ascorbic acid in ethanol.
Bilirubin glucuronides were analyzed by HPLC using a Waters binary pump (model 1525) HPLC system equipped with a dual λ absorbance detector operated at 450 nm, an automatic injector (model WISP 710B; Waters), and a β-RAM radioactive flow detector (IN/US Systems, Tampa, FL). Samples were injected onto a Waters μBondapak (3.9 × 300 mm) 5 μC18 column. HPLC separations were performed by using solvent A (0.01% trifluoroacetic acid in acetonitrile) and solvent B (0.01% trifluoroacetic acid in water) and the following linear gradient conditions: 0 to 20 min, 10 to 90% solvent A and 20 to 25 min, 90 to 100% solvent A. The HPLC flow rate was 1 ml/min, and the scintillation fluid flow rate was 4 ml/min. The column was routinely washed with 100% solvent A for 15 min and then equilibrated after every HPLC run with 10% solvent A for at least 20 min. Absorbance (450 nm) and 14C-labeled peaks corresponding to glucuronide conjugates of bilirubin were tentatively identified by relative retention times, and then confirmed by sensitivity to E. coli β-glucuronidase treatment (1000 units; overnight incubation at 37°C), using HPLC as described above.
Statistical Analyses.
Pearson correlation of qPCR data with glucuronidation of raloxifene in HLM was done by using Prism version 5.00 (GraphPad Software Inc., San Diego, CA) with significance at p value < 0.05. The Student's t test (two-sided) was used to compare raloxifene glucuronidation rates in siRNA-treated versus untreated HepG2 cells.
Results
Quantification of UGT1A v1 and v2/v3 Splice Variant mRNA Levels.
Previous studies examining the relative expression of UGT1A mRNAs before the identification of UGT1A exon 5b used real-time PCR assays containing exon 1-specific antisense primers (Zheng et al., 2002; Nakamura et al., 2008; Izukawa et al., 2009; Ohno and Nakajin, 2009). Therefore, quantification values reported in those studies encompassed the entire population of v1, v2, and v3 mRNAs for each UGT1A enzyme. To develop a quantitative assay that could distinguish between the v1 and v2/v3 mRNA variants for each UGT1A enzyme, a nested real-time PCR technique was designed for quantification purposes, because the distance between the unique first exons of each of the UGT1A enzymes and the alternate fifth exons is >1 kilobase, a distance suboptimal for traditional qPCR techniques. Because for each individual UGT1A the v2 and v3 variant mRNA species encode the same nonfunctional protein, no attempt was made to distinguish between v2 and v3 transcripts in the present study. This nested real-time PCR approach (Fig. 1) consisted of a RT reaction using olig(dT) as primer and relied on a first-step PCR that was carried out for 20 cycles and used primers specific for eight UGT1A mRNA species (UGT1A1 v1, UGT1A1 v2/v3, UGT1A4 v1, UGT1A4 v2/v3, UGT1A6 v1, UGT1A6 v2/v3, UGT1A9 v1, and UGT1A9 v2/v3). UGTs 1A5, 1A8, and 1A10 were not included because they are expressed at very low or undetectable levels in human liver, and UGT1A3 was excluded because all primer pairs that were developed for this gene resulted in cross-amplification of UGT1A4 (results not shown). A nested real-time assay was developed for the extra-hepatic UGT1A7 (Beaulieu et al., 1998; Zheng et al., 2002; Izukawa et al., 2009) as a negative control for these experiments. The second-step PCR was a real-time PCR assay that was performed by using UGT1A exon 1-specific primers and probes (Fig. 1). To demonstrate the specificity of the chosen primer pairs, pooled cDNA from eight human liver samples were PCR-amplified for 40 cycles. As shown in Fig. 2, each primer pair resulted in a single PCR product of the appropriate size, and subsequent DNA sequencing confirmed that each PCR product corresponded to the intended UGT1A isoform (results not shown). The same primer pairs were used to amplify the pooled liver cDNA for 20 cycles, and none of the UGT1A cDNA species could be visualized by gel electrophoresis (results not shown), indicating that the PCR assay had not yet reached the plateau phase, a requirement for the initial reaction of the nested PCR technique.
Fig. 1.

Schematic of UGT1A expression analysis. The first-step PCR amplification involved a 20-cycle reaction to distinguish between the different exon 5 splice variants. Because UGT1A v1 variants encode the active i1 proteins, whereas UGT1A v2 and v3 variants encode the same inactive i2 proteins, UGT1A v2 and v3 were grouped together and designated UGT1A v2/v3 mRNA as described previously (Girard et al., 2007). The arrows represent the approximate location of the primers, with the same antisense primer used for amplification of both UGT1A v2 and v3 mRNA. UGT1A exon 1 is representative of the nine different UGT1A exon 1 species that can be alternatively spliced to the common UGT1A exons 2 to 5. The second-step real-time PCR assay was then performed by using UGT1A exon 1-specific real-time PCR primers and probes. FAM, approximate location of fluorescent 6-carboxyfluorescein probe.
Fig. 2.
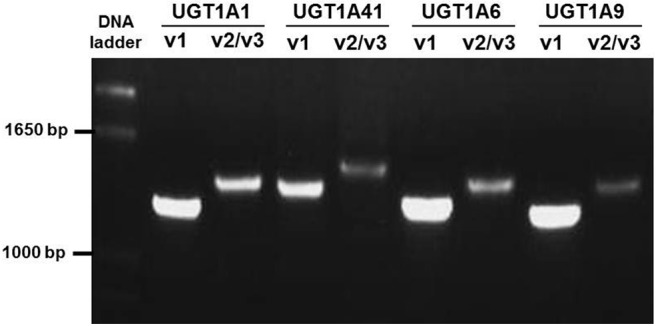
UGT1A sequence-confirmed amplification products from human liver cDNA. Pooled cDNA was reverse-transcribed from total RNA from eight human liver specimens as described under Materials and Methods. Sense primers were chosen to exploit sequence differences between the highly homologous UGT1A exon 1 regions. UGT1A isoforms were PCR-amplified for 40 cycles. Expected band sizes were: UGT1A1_v1, 1250 bp; UGT1A1_v2/v3, 1349 bp; UGT1A4_v1, 1338 bp; UGT1A4_v2/v3, 1437 bp; UGT1A6_v1, 1264 bp; UGT1A6_v2/v3, 1363 bp; UGT1A9_v1, 1209 bp; and UGT1A9_v2/v3, 1308 bp.
To assess the efficiency of the different primer sets used during the first-step PCR amplification, total RNA from HEK293 cell lines that overexpressed each of the eight UGT1A variants was used. After reverse transcription, the nested PCR technique was used to amplify and quantify UGT1A mRNA from each of these cell lines. The first-step PCR amplification was carried out for 10, 15, or 20 cycles by using the respective primer pairs for v1 or v2/v3, and each reaction was subsequently amplified in a second-step reaction by using the real-time PCR assay described above and under Materials and Methods. As shown in Fig. 3A, the mean qPCR Ct values were linear for all UGT1A mRNAs for the range of 10 to 20 cycles used in the first-step amplification reaction. The maximum efficiency difference between any of the eight primer pairs used in the first reaction was 5.1%.
Fig. 3.

Validation of nested PCR methodology. A, total RNA from UGTs 1A1, 1A4, 1A6, and 1A9 v1- and v2/v3-overexpressing cell lines was reverse-transcribed, and the resulting cDNA was PCR-amplified for 10, 15, or 20 cycles before amplification by real-time PCR. Amplification efficiency calculated from the slope of the curves is shown for the amplification of both v1 and v2/v3 for each UGT. B, pooled cDNA from eight liver samples was PCR-amplified for 20 cycles by using primers specific for each of the eight full-length UGT1A variants, and the resulting amplification products were serially diluted and analyzed by real-time PCR. Average Ct values were calculated by taking the arithmetic mean of four replicate data points. Data are reported as mean ± S.D. ●, v1; □, v2/v3.
In addition to controlling for differences in primer efficiency for the first-step PCR, it was necessary to control for differences in amplification efficiency of the predesigned Applied Biosystems qPCR assays used for the second-step PCR amplification. qPCR efficiency was calculated by generating UGT1A v1 and v2/v3 standard curves as described under Materials and Methods. The calculated qPCR efficiency values were later used to adjust the UGT1A splice variant RQ values in individual liver specimens. The qPCR efficiency values for the different predesigned Applied Biosystems assays used for the second-step PCR amplification for the analysis of UGT1A enzyme expression ranged from 86 to 104% by using HEK293 cDNA (Fig. 3B).
Relative Expression Levels of UGT1A v1 and UGT1A v2/v3 mRNA in Human Liver.
The validated nested PCR technique described above was used to determine the relative expression levels of UGT1A mRNA variants in liver samples from 10 individuals. The mean RQ values for the UGT1A v1 species were: UGT1A1, 1233 ± 154; UGT1A4, 645 ± 103; UGT1A6, 228 ± 27; and UGT1A9, 626 ± 79. The mean RQ values for UGT1A v2/v3 mRNA species were: UGT1A1, 93 ± 19; UGT1A4, 43 ± 5.7; UGT1A6, 6.1 ± 1.4; and UGT1A9, 29 ± 5.3 (Fig. 4A). The relative abundance of v1 versus v2/v3, expressed as the log of the ratio of v1:v2/v3 for each of the four different UGT1As analyzed, is shown in Fig. 4B. Although some interindividual differences in the relative abundance of v1 versus v2/v3 mRNAs were observed, v2/v3 mRNA levels did not approach the levels of v1 mRNA for any of the UGT1A species examined. On average, all of the UGT1A v2/v3 mRNA species were expressed at levels that were at least 16-fold lower than that of the corresponding UGT1A v1 mRNA species in the 10 liver specimens examined, with mean v1:v2/v3 mRNA ratios of 16 ± 1.7, 17 ± 2.6, 57 ± 10, and 29 ± 5.2 for UGTs 1A1, 1A4, 1A6, and 1A9, respectively. No expression of either the UGT1A7 v1 or v2/v3 variants was observed in any of the human liver specimens analyzed in this study.
Fig. 4.

Relative mRNA expression levels of UGT1A variants in 10 liver RNA samples. UGT1A v1 and v2/v3 mRNA expression levels were assessed in liver cDNA from 10 individuals by using the two-step quantitative PCR methodology. A, the log of the relative expression levels for the v1 and v2/v3 variants for UGTs 1A1, 1A4, 1A6, and 1A9 is shown. B, the relative abundance is expressed as the log of the ratio of v1:v2/v3 for UGTs 1A1, 1A4, 1A6, and 1A9. Boxes, 25th to 75th percentile; whiskers, minimum-maximum; ●, outlier (>1.5 interquartile range outside of lowest quartile).
Because the v1 and v2/v3 transcripts differ at the 3′ end, the strength of the polyadenylation site was a potential source of variability of RT reaction efficiency. To address potential differences in RT priming efficiency by using different RT primers, pooled liver RNA from 10 samples was reverse-transcribed by using random hexamer primers, and the nested PCR technique was used to quantify the hepatic UGT1A v1 and v2/v3 cDNAs. The relative abundance ratio of v1:v2/v3 using random hexamers was as follows: 26-fold higher for UGT1A1, 19-fold higher for UGT1A4, 45-fold higher for UGT1A6, and 34-fold higher for UGT1A9. These values are very similar to the average values reported above with oligo(dT) as the RT primer.
Expression of UGT1A1 v1 and v2/v3 mRNA and Correlation with Glucuronidation Activity in Human Liver.
The relative expression of UGT1A v1 and v2/v3 was determined in 58 human liver RNA samples. There was a significant correlation between the expression of UGT1A1 v1 mRNA and UGT1A1 v2/v3 mRNA in these samples (Fig. 5; p<0.0001; r2 = 0.4213), suggesting that the rate of incorporation of the UGT1A1 alternate exon 5b is lower than exon 5a in human liver tissue. The average relative expression ratio of UGT1A1 v1 versus UGT1A1 v2/v3 mRNA was determined to be 17 ± 1.5 (mean ± S.E.) in the 58 samples. In all of the liver specimens examined, expression of UGT1A1 v1 was found to be at least 3-fold higher than the expression of the v2/v3 variants, with two samples exhibiting a difference of less than 5-fold and nine samples exhibiting a difference of less than 10-fold (range: 3- to 60-fold). The interindividual variability in the UGT1A1 v1:v2/v3 ratio was 20-fold, with the middle 90% of samples displaying a v1:v2/v3 ratio between 7- and 35-fold.
Fig. 5.
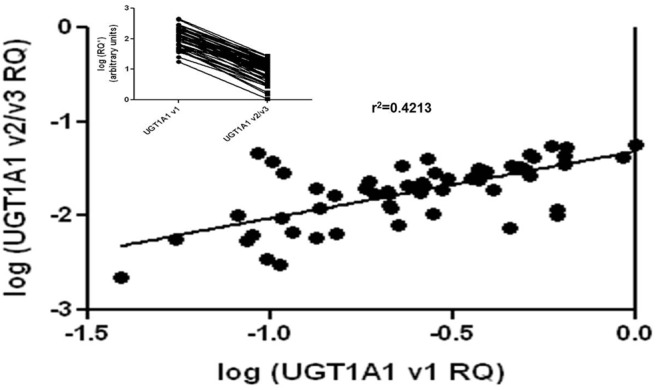
Relative expression levels of UGT1A1 v1 and UGT1A1 v2/v3 mRNAs in 58 human liver specimens. The nested qPCR developed in this study was used to determine the relative expression of UGT1A1 v1 and UGT1A1 v2/v3 in total RNA isolated from the liver specimens described under Materials and Methods. Shown is the correlation between the expression of the v1 and v2/v3 variants in the 58 individual liver specimens. Inset, lines connect the UGT1A1 v1 RQ value of an individual specimen with the corresponding UGT1A1 v2/v3 RQ value from the same specimen.
Raloxifene is glucuronidated extensively by family 1A UGTs, with its major glucuronide metabolite, raloxifene-6-O-glucuronide, primarily formed by UGT1A1 (Trdan Lusin et al., 2011). In the present study, UGT1A1 v1 expression was significantly (p<0.0001; r2 = 0.7195) correlated with raloxifene-6-O-glucuronide formation in HLMs (Fig. 6A). A correlation was also observed for raloxifene-6-O-glucuronide formation and UGT1A1 v2/v3 expression (p = 0.0002; r2 = 0.241), whereas a minimal correlation was observed with the ratio of v1:v2/v3 expression (p = 0.0114; r2 = 0.117). Similar results were observed when using another UGT1A1-specific substrate, bilirubin, in these experiments (Fig. 6B).
Fig. 6.

Correlation of UGT1A1 mRNA expression with HLM glucuronidation activity. A and B, UGT1A1 v1 and UGT1A1 v2/v3 mRNA levels were assayed in 58 human liver specimens and correlated with glucuronidation activity against raloxifene (A) and bilirubin (B) in HLMs derived from the same specimens. C and D, correlations of UGT1A1 v1 (C) and UGT1A1 v2/v3 (D) expression with raloxifene-6-O-glucuronide formation are shown after stratification by UGT1A1 TATAA box genotype. RQ values were calculated for v1 and v2/v3 mRNA separately by the ΔΔCt method.
The UGT1A1*28 allelic variant contains an additional (TA) dinucleotide repeat in the TATAA box of the UGT1A1 promoter [(TA)6 > (TA)7] that has been linked to decreased expression of the UGT1A1 gene and decreased overall UGT1A1 activity (Bosma et al., 1995b; Beutler et al., 1998; Burchell and Hume, 1999). After stratifying the HLMs examined in the present study by the UGT1A1 TATAA box genotype, significant correlations were observed between raloxifene-6-O-glucuronide formation and UGT1A1 v1 expression for HLMs from subjects who exhibited either the (*1/*1), (*1/*28), or (*28/*28) genotypes (Fig. 6C). Although a significant correlation was observed between raloxifene-6-O-glucuronide formation and UGT1A1 v2/v3 expression for HLMs from UGT1A1 (*1/*1) subjects (p = 0.02; r2 = 0.2512), no significant correlation was observed for HLMs from subjects who exhibited the UGT1A1 (*1/*28) or (*28/*28) genotypes (p > 0.05; Fig. 6D). The same trends were observed when using bilirubin as substrate (results not shown). As observed in previous studies (Bosma et al., 1995a,b; Monaghan et al., 1996; Beutler et al., 1998; Guillemette et al., 2000, 2001; Bosma, 2003; Fang and Lazarus, 2004), the expression of both UGT1A1 v1 and v2/v3 mRNAs was found to be significantly lower in livers from subjects with the UGT1A1 (*28/*28) genotype (42% lower for v1, p = 0.0413; 53% lower for v2/v3, p = 0.0075) compared with livers from UGT1A1 (*1/*1) subjects.
Effect of UGT1A v2/v3 Knockdown on Raloxifene Glucurondation Activities in Cell Lines.
Coexpression of UGT1A1 v1 and v2/v3 had previously been shown to reduce glucuronidation activity in vitro (Lévesque et al., 2007; Bellemare et al., 2010a,b,c; Benoit-Biancamano et al., 2009). In the present study, UGT1A1_i1 and UGT1A1_i2 were coexpressed in HEK293 cells to study the effect of the i2 protein on raloxifene-6-O-glucuronide formation. Cells coexpressing the UGT1A1_i1 and UGT1A1_i2 proteins displayed a 3.6-fold decrease in the Vmax of raloxifene-6-O-glucuronide formation compared with the Vmax of untreated UGT1A1_i1-overexpressing cells (Fig. 7A). This pattern was partially reversed (60%) when a v2/v3-specific siRNA was used. Treatment with SCR siRNA alone did not have an effect on the expression of UGT1A1 v1 or v2/v3 in any of the cell lines (data not shown).
Fig. 7.

Effect of siRNA treatment of UGT1A_v2 mRNA on raloxifene glucuronidation activity in cell lines. A, representative kinetic curves for raloxifene-6-O-glucuronide formation are shown for HEK293 cells overexpressing UGT1A1 v1 alone or in combination with the UGT1A1 v2 variant. Cells coexpressing UGT1A1 v1 and UGT1A1 v2 were treated with either 10 nM SCR or anti-UGT1A1 v2/v3-specific siRNA for 96 h before conducting glucuronidation assays. B, the two-step quantitative method described under Materials and Methods was used to assess the relative endogenous expression of UGT1A1 v1 and UGT1A1 v2/v3 variants in the HepG2 cell line. C, the relative quantification of UGT1A1 v2/v3 mRNA after knockdown with 10 nM SCR or anti-v2/v3-specific siRNA in the HepG2 cell line is shown. Inset, a representative electrophoresis gel of the RT-PCR of UGT1A1 in HepG2 cells treated with SCR or anti-v2/v3-specific siRNA after 37 cycles of amplification. D, effect of UGT1A1 v2/v3 knockdown on raloxifene-6-O-glucuronide formation in HepG2 cells treated with SCR or anti-v2/v3-specific siRNA.
To examine the effects of high levels of endogenous expression of v2/v3 on UGT1A activity, the relative expression of UGT1A v1 versus v2/v3 were examined in the HepG2 cell line. Unlike that observed in human liver tissue, this cancer-derived cell line expressed UGT1A1 v2/v3 mRNA at a level that was 1.6-fold higher than UGT1A1 v1 mRNA (Fig. 7B; p = 0.030), suggesting that this cell line could serve as a model for high relative endogenous expression of the UGT1A v2/v3 splice variants. siRNA knockdown of UGT1A v2/v3 splice variants resulted in a >95% reduction in UGT1A1 v2/v3 mRNA levels in HepG2 cells (Fig. 7C), resulting in a v1:v2/v3 ratio of 13.6, which was similar to that observed for human liver tissue. This decrease in expression corresponded with a significant (p = 0.003) 82.5% increase in raloxifene-6-O-glucuronide formation in anti-v2/v3 siRNA-treated HepG2 cells versus SCR-treated controls (Fig. 7D).
Discussion
This is the first quantitative study of the UGT1A gene locus to simultaneously distinguish between the 5′ splice variants, which determine the individual UGT1A species, and the 3′ splice variants, which result from incorporation of different fifth exons. Previous quantitative expression studies of UGT1A enzymes used exon 1-specific primers that could not distinguish between the three exon 5 splice variants. Therefore, quantification of UGT1A v1 transcripts would be confounded by the presence of the UGT1A v2 and v3 mRNA variants. Although the nested PCR methodology outlined in this study results in a small loss of accuracy in favor of increased sensitivity and specificity, control experiments demonstrated that the maximal efficiency difference between the primers used for the 20-cycle amplification was 5.3%. After 20 cycles, this would result in a maximum quantitation error of 61%, much lower than the >10-fold difference in relative abundance between the v1 and v2/v3 variants.
The mean hepatic expression of UGT1A v1 variants was as follows: UGT1A1 > UGT1A4 > UGT1A9 > UGT1A6. This order of expression and the high relative abundance of UGT1A1 relative to other UGT1A species matches closely with previous reports (Nakamura et al., 2008; Izukawa et al., 2009; Ohno and Nakajin, 2009). The mean hepatic expression of the UGT1A v2/v3 splice variants was also highest for UGT1A1. These variants were found to be expressed at an average of 2.2-fold higher than UGT1A4 v2/v3, 3.2-fold higher than UGT1A9 v2/v3, and 15-fold higher than UGT1A6 v2/v3. In all cases, UGT1A v1 mRNAs were found to be expressed, on average, at least 16-fold higher than their corresponding v2/v3 mRNAs. There was, however, some interindividual variability in the v1:v2/v3 ratio between specimens, with two of the 58 liver specimens examined exhibiting less than a 5-fold difference in the levels of the v1:v2/v3 relative abundance ratio for UGT1A1. This suggests that a small subset of individuals may have higher levels of exon 5b inclusion. Because UGT1A i2 proteins have been shown to negatively modulate the activity of i1 proteins through protein-protein interactions(Bellemare et al., 2010b), with negative regulation of i1 protein activity shown to occur in the presence of as little as 5-fold less i2 protein (Girard et al., 2007; Bellemare et al., 2010b), this is consistent with a potential effect of i2 UGT1A proteins on hepatic glucuronidation activity in some individuals.
To better assess the potential correlation between hepatic glucuronidation activity and the relative abundance of hepatic UGT1A v1 and/or v2/3 mRNA, expression of UGT1A1 variants was examined and correlated with hepatic glucuronidation activity. Significant positive correlations were observed between UGT1A1 v1 mRNA levels and raloxifene-6-O-glucuronide formation or bilirubin glucuronidation in the present study. A modest positive correlation was also observed for UGT1A1 v2/v3. However, if increased v2/v3 expression was affecting hepatic glucuronidation activities, a negative correlation would be expected. These data are consistent with a lack of an effect by v2/v3 UGT1A1 mRNA and its corresponding i2 protein on overall hepatic UGT1A1 glucuronidation activity in most individuals.
The i2 protein has been previously shown to be expressed in human carcinoma tissue and cell lines, and siRNA knockdown of the v2/v3 variant resulted in increased activity against a range of substrates in colon cancer cell lines (Bellemare et al., 2010c, 2011). In the current study, it was observed that the human hepatocellular carcinoma cell line HepG2 exhibited relatively high endogenous expression of exon 5b-containing variants for UGT1A1, with v2/v3 variant expression reaching levels that were slightly greater than that observed for UGT1A1 v1. siRNA-induced silencing of v2/v3 variants in HepG2 cells resulted in a 95% decrease in v2/v3 expression and a corresponding >80% increase in HepG2 activity against raloxifene. In addition, in cells coexpressing UGT1A1_i1 and UGT1A1_i2 the addition of anti-v2/v3-specific siRNA increased raloxifene glucuronidation. Together, these data support the apparent lack of an effect by endogenous UGT1A v2/v3 variants on hepatic glucuronidation activity in individuals with low levels of hepatic UGT1A1 v2/v3 expression.
The expression of UGT1A1 mRNA has been shown to correlate with UGT1A1_i1 protein expression in human liver (Izukawa et al., 2009), and it is known that reduced transcription of UGT1A1 by the *28 allele leads to differences in activity against substrates such as bilirubin and irinotecan (Bosma et al., 1995a; Monaghan et al., 1996; Iyer et al., 1999; Bosma, 2003; Hoskins et al., 2007). However, previous studies have not differentiated between v1 and v2/v3 mRNA species. The common TATA box promoter polymorphism associated with the UGT1A1*28 allele is known to reduce transcription of the UGT1A1 gene and decrease the metabolism of bilirubin and other substrates of UGT1A1 such as irinotecan (Ando et al., 1998; Iyer et al., 1999, 2002). When the HLMs examined in the present study were stratified by UGT1A1 genotype a significant correlation between hepatic UGT1A1 v1 mRNA levels and HLM raloxifene-6-O-glucuronide formation was observed for all three UGT1A1 TATAA box genotypes identified. These results were confirmed when using a second UGT1A1-specific substrate, bilirubin. The 42% decrease in hepatic UGT1A1 v1 mRNA expression observed in individuals homozygous for the UGT1A1*28 allele compared with individuals with one or zero copies (p = 0.0413; data not shown) is comparable with previous reports investigating the effect of this genotype on UGT1A1 transcription (Beutler et al., 1998; Guillemette et al., 2001). Levels of UGT1A1 v2/v3 were found to be 53% lower in individuals homozygous for the UGT1A1*28 allele compared with individuals with zero copies of this allele (p = 0.0075; data not shown). These data suggest that the TATAA box polymorphism associated with the UGT1A1*28 allele coordinately regulates the transcription of both the wild-type v1 and variant v2/v3 mRNAs in human liver.
The overall low relative expression of v2/v3 variants in human liver is not surprising considering that the v2/v3-specific UGT1A exon 5b 3′ UTR contains a sequence element that is associated with low expression. The exon 5b 3′ UTR evolved from a primate-specific Alu transposable element, sharing 78% sequence homology with the Alu consensus sequence (Price et al., 2004). It is estimated that 4% of human genes contain exonized transposable elements (Nekrutenko and Li, 2001; Sorek et al., 2002; Kim et al., 2008), most commonly Alu elements. Alu elements comprise ∼11% of the genome, with more than 10,000 Alus inserted into the 3′ UTR of genes (Chen et al., 2009). In general, transcripts with intronic, Alu-derived 3′ UTR sequences are minor form variants (Kim et al., 2004; Roy-Engel et al., 2005; Smalheiser and Torvik, 2006; Chen et al., 2008), consistent with our findings in the present study. In addition, the low relative abundance of v2/v3 mRNA species in human liver is consistent with the observation of low i2 protein levels in individual liver specimens observed previously (Lévesque et al., 2007). In the current study, v2/v3 mRNA levels were found to be, on average, between 2 and 7% of the levels of v1 mRNAs for the different hepatic UGT1A enzymes, agreeing well with the observation in the 2007 study that i2 protein levels were <10% of the i1 protein levels in all four of the individual liver specimens analyzed (Lévesque et al., 2007).
Given that the liver is the primary metabolic organ with a near constant need for high expression and activity of UGT enzymes, there may be little need for UGT1A i2-associated negative regulation of UGT1A i1 protein in liver, but this does not rule out an extra-hepatic, tissue-specific regulatory role for i2 proteins. In a panel of 10 tissues and five cell lines, more than 8000 alternatively spliced terminal exons were detected, nearly half of which were found to be expressed in a tissue-specific manner (Wang et al., 2008), a pattern observed previously for human Alu-derived exons (Amit et al., 2007; Mersch et al., 2007; Lin et al., 2008). For example, only the i2 proteins are expressed in the kidney where they may have their own tissue-specific function (Bellemare et al., 2011). In addition, the kidney and esophagus express only the v2/v3 variants of certain UGT1A species, suggesting a “switch-like” regulation mechanism favoring the expression of the nonfunctional variants (Girard et al., 2007).
In summary, this study indicates that, although there is a positive correlation between hepatic UGT1A v1 and v2/v3 expression for all hepatic UGTs tested, UGT1A v2/v3 mRNAs represent a minor form of the total UGT1A transcript profile in humans. The positive correlations observed between the hepatic expression of UGT1A1 v1 and UGT1A1 v2/v3 mRNA with glucuronidation activity suggests that v2/v3 expression may serve as a marker for v1 expression. There was, however, a small subset of human liver specimens examined in this study that exhibited UGT1A1 v2/v3 expression levels that were within 5-fold the levels observed for UGT1A1 v1, suggesting that v2/v3 UGT1A expression and its corresponding i2 proteins may be exerting a functional effect on hepatic glucuronidation activity in some individuals. To better understand how this complex gene locus is expressed and regulated throughout the body, future quantitative studies are required to determine organ locations where the relative expression of UGT1A v2/v3 mRNA is high and leads to functional regulation of UGT activity.
Acknowledgments
We thank the staff at the Functional Genomics Core Facility at the Penn State University College of Medicine for real-time PCR services; the staff at the Chandlee Lab at Penn State University Park for DNA sequencing services; and Diane McCloskey for editorial assistance.
These studies were supported in part by the National Institutes of Health National Institute of Dental and Craniofacial Research [Grant R01-DE13158] (to P.L.); and the Pennsylvania Department of Health's Health Research Formula Funding Programs [Grant 4100038714] (to P.L.).
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- UGT
- UDP glucuronosyltransferase
- UDPGA
- UDP glucuronic acid
- v1
- variant 1
- v2
- variant 2
- v3
- variant 3
- i1
- isoform 1
- i2
- isoform 2
- RT
- reverse transcription
- PCR
- polymerase chain reaction
- qPCR
- quantitative PCR
- RQ
- relative quantification
- UPLC
- ultra-performance liquid chromatography
- HPLC
- high-performance liquid chromatography
- UTR
- untranslated region
- HEK
- human embryonic kidney
- bp
- base pairs
- siRNA
- short interfering RNA
- SCR
- scrambled
- HLM
- human liver microsome
- ral-Gluc
- raloxifene-glucuronide.
Authorship Contributions
Participated in research design: Jones, Sun, Freeman, and Lazarus.
Conducted experiments: Jones and Sun.
Performed data analysis: Jones, Sun, Freeman, and Lazarus.
Wrote or contributed to the writing of the manuscript: Jones, Sun, Freeman, and Lazarus.
References
- Amit M, Sela N, Keren H, Melamed Z, Muler I, Shomron N, Izraeli S, Ast G. (2007) Biased exonization of transposed elements in duplicated genes: A lesson from the TIF-IA gene. BMC Mol Biol 8:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- tk;2 Ando Y, Saka H, Asai G, Sugiura S, Shimokata K, Kamataki T. (1998) UGT1A1 genotypes and glucuronidation of SN-38, the active metabolite of irinotecan. Ann Oncol 9:845–847 [DOI] [PubMed] [Google Scholar]
- Beaulieu M, Lévesque E, Hum DW, Bélanger A. (1998) Isolation and characterization of a human orphan UDP-glucuronosyltransferase, UGT2B11. Biochem Biophys Res Commun 248:44–50 [DOI] [PubMed] [Google Scholar]
- Bellemare J, Rouleau M, Girard H, Harvey M, Guillemette C. (2010a) Alternatively spliced products of the UGT1A gene interact with the enzymatically active proteins to inhibit glucuronosyltransferase activity in vitro. Drug Metab Dispos 38:1785–1789 [DOI] [PubMed] [Google Scholar]
- Bellemare J, Rouleau M, Harvey M, Guillemette C. (2010b) Modulation of the human glucuronosyltransferase UGT1A pathway by splice isoform polypeptides is mediated through protein-protein interactions. J Biol Chem 285:3600–3607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellemare J, Rouleau M, Harvey M, Popa I, Pelletier G, Têtu B, Guillemette C. (2011) Immunohistochemical expression of conjugating UGT1A-derived isoforms in normal and tumoral drug-metabolizing tissues in humans. J Pathol 223:425–435 [DOI] [PubMed] [Google Scholar]
- Bellemare J, Rouleau M, Harvey M, Têtu B, Guillemette C. (2010c) Alternative-splicing forms of the major phase II conjugating UGT1A gene negatively regulate glucuronidation in human carcinoma cell lines. Pharmacogenomics J 10:431–441 [DOI] [PubMed] [Google Scholar]
- Benoit-Biancamano MO, Connelly J, Villeneuve L, Caron P, Guillemette C. (2009) Deferiprone glucuronidation by human tissues and recombinant UDP glucuronosyltransferase 1A6: an in vitro investigation of genetic and splice variants. Drug Metab Dispos 37:322–329 [DOI] [PubMed] [Google Scholar]
- Beutler E, Gelbart T, Demina A. (1998) Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc Natl Acad Sci U S A 95:8170–8174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosma P, Chowdhury JR, Jansen PH. (1995a) Genetic inheritance of Gilbert's syndrome. Lancet 346:314–315 [DOI] [PubMed] [Google Scholar]
- Bosma PJ. (2003) Inherited disorders of bilirubin metabolism. J Hepatol 38:107–117 [DOI] [PubMed] [Google Scholar]
- Bosma PJ, Chowdhury JR, Bakker C, Gantla S, de Boer A, Oostra BA, Lindhout D, Tytgat GN, Jansen PL, Oude Elferink RP. (1995b) The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert's syndrome. N Engl J Med 333:1171–1175 [DOI] [PubMed] [Google Scholar]
- Burchell B, Hume R. (1999) Molecular genetic basis of Gilbert's syndrome. J Gastroenterol Hepatol 14:960–966 [DOI] [PubMed] [Google Scholar]
- Chen C, Ara T, Gautheret D. (2009) Using Alu elements as polyadenylation sites: a case of retroposon exaptation. Mol Biol Evol 26:327–334 [DOI] [PubMed] [Google Scholar]
- Chen LL, DeCerbo JN, Carmichael GG. (2008) Alu element-mediated gene silencing. EMBO J 27:1694–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman BL, Green MD, King CD, Tephly TR. (1995) Cloning and stable expression of a cDNA encoding a rat liver UDP-glucuronosyltransferase (UDP-glucuronosyltransferase 1.1) that catalyzes the glucuronidation of opioids and bilirubin. Mol Pharmacol 47:1101–1105 [PubMed] [Google Scholar]
- Coughtrie MW, Burchell B, Bend JR. (1987) Purification and properties of rat kidney UDP-glucuronosyltransferase. Biochem Pharmacol 36:245–251 [DOI] [PubMed] [Google Scholar]
- Dutton GF. (1980). Glucuronidation of Drugs and Other Compounds. CRC Press, Boca Raton, FL [Google Scholar]
- Ethell BT, Beaumont K, Rance DJ, Burchell B. (2001) Use of cloned and expressed human UDP-glucuronosyltransferases for the assessment of human drug conjugation and identification of potential drug interactions. Drug Metab Dispos 29:48–53 [PubMed] [Google Scholar]
- Fang JL, Lazarus P. (2004) Correlation between the UDP-glucuronosyltransferase (UGT1A1) TATAA box polymorphism and carcinogen detoxification phenotype: significantly decreased glucuronidating activity against benzo(a)pyrene-7,8-dihydrodiol(−) in liver microsomes from subjects with the UGT1A1*28 variant. Cancer Epidemiol Biomarkers Prev 13:102–109 [DOI] [PubMed] [Google Scholar]
- Freeman WM, Walker SJ, Vrana KE. (1999) Quantitative RT-PCR: pitfalls and potential. Biotechniques 26:112–122, 124–125 [DOI] [PubMed] [Google Scholar]
- Girard H, Lévesque E, Bellemare J, Journault K, Caillier B, Guillemette C. (2007) Genetic diversity at the UGT1 locus is amplified by a novel 3′ alternative splicing mechanism leading to nine additional UGT1A proteins that act as regulators of glucuronidation activity. Pharmacogenet Genomics 17:1077–1089 [DOI] [PubMed] [Google Scholar]
- Gong QH, Cho JW, Huang T, Potter C, Gholami N, Basu NK, Kubota S, Carvalho S, Pennington MW, Owens IS, et al. (2001) Thirteen UDPglucuronosyltransferase genes are encoded at the human UGT1 gene complex locus. Pharmacogenetics 11:357–368 [DOI] [PubMed] [Google Scholar]
- Guillemette C, De Vivo I, Hankinson SE, Haiman CA, Spiegelman D, Housman DE, Hunter DJ. (2001) Association of genetic polymorphisms in UGT1A1 with breast cancer and plasma hormone levels. Cancer Epidemiol Biomarkers Prev 10:711–714 [PubMed] [Google Scholar]
- Guillemette C, Millikan RC, Newman B, Housman DE. (2000) Genetic polymorphisms in uridine diphospho-glucuronosyltransferase 1A1 and association with breast cancer among African Americans. Cancer Res 60:950–956 [PubMed] [Google Scholar]
- Hoskins JM, Goldberg RM, Qu P, Ibrahim JG, McLeod HL. (2007) UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J Natl Cancer Inst 99:1290–1295 [DOI] [PubMed] [Google Scholar]
- Iyer L, Das S, Janisch L, Wen M, Ramírez J, Karrison T, Fleming GF, Vokes EE, Schilsky RL, Ratain MJ. (2002) UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2:43–47 [DOI] [PubMed] [Google Scholar]
- Iyer L, Hall D, Das S, Mortell MA, Ramírez J, Kim S, Di Rienzo A, Ratain MJ. (1999) Phenotype-genotype correlation of in vitro SN-38 (active metabolite of irinotecan) and bilirubin glucuronidation in human liver tissue with UGT1A1 promoter polymorphism. Clin Pharmacol Ther 65:576–582 [DOI] [PubMed] [Google Scholar]
- Izukawa T, Nakajima M, Fujiwara R, Yamanaka H, Fukami T, Takamiya M, Aoki Y, Ikushiro S, Sakaki T, Yokoi T. (2009) Quantitative analysis of UDP-glucuronosyltransferase (UGT) 1A and UGT2B expression levels in human livers. Drug Metab Dispos 37:1759–1768 [DOI] [PubMed] [Google Scholar]
- Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A. (2004) Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res 14:1719–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Goren A, Ast G. (2008) Alternative splicing: current perspectives. BioEssays 30:38–47 [DOI] [PubMed] [Google Scholar]
- Lévesque E, Girard H, Journault K, Lépine J, Guillemette C. (2007) Regulation of the UGT1A1 bilirubin-conjugating pathway: role of a new splicing event at the UGT1A locus. Hepatology 45:128–138 [DOI] [PubMed] [Google Scholar]
- Lin L, Shen S, Tye A, Cai JJ, Jiang P, Davidson BL, Xing YS. (2008). Diverse splicing patterns of exonized Alu elements in human tissues. PLoS Genet 4:e1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- Mersch B, Sela N, Ast G, Suhai S, Hotz-Wagenblatt A. (2007) SERpredict: detection of tissue- or tumor-specific isoforms generated through exonization of transposable elements. BMC Genet 8:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan G, Ryan M, Seddon R, Hume R, Burchell B. (1996) Genetic variation in bilirubin UPD-glucuronosyltransferase gene promoter and Gilbert's syndrome. Lancet 347:578–581 [DOI] [PubMed] [Google Scholar]
- Nakamura A, Nakajima M, Yamanaka H, Fujiwara R, Yokoi T. (2008) Expression of UGT1A and UGT2B mRNA in human normal tissues and various cell lines. Drug Metab Dispos 36:1461–1464 [DOI] [PubMed] [Google Scholar]
- Nekrutenko A, Li WH. (2001) Transposable elements are found in a large number of human protein-coding genes. Trends Genet 17:619–621 [DOI] [PubMed] [Google Scholar]
- Ohno S, Nakajin S. (2009) Determination of mRNA expression of human UDP-glucuronosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug Metab Dispos 37:32–40 [DOI] [PubMed] [Google Scholar]
- Olson KC, Dellinger RW, Zhong Q, Sun D, Amin S, Spratt TE, Lazarus P. (2009) Functional characterization of low-prevalence missense polymorphisms in the UDP-glucuronosyltransferase 1A9 gene. Drug Metab Dispos 37:1999–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Eskin E, Pevzner PA. (2004) Whole-genome analysis of Alu repeat elements reveals complex evolutionary history. Genome Res 14:2245–2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy-Engel AM, El-Sawy M, Farooq L, Odom GL, Perepelitsa-Belancio V, Bruch H, Oyeniran OO, Deininger PL. (2005) Human retroelements may introduce intragenic polyadenylation signals. Cytogenet Genome Res 110:365–371 [DOI] [PubMed] [Google Scholar]
- Smalheiser NR, Torvik VI. (2006) Alu elements within human mRNAs are probable microRNA targets. Trends Genet 22:532–536 [DOI] [PubMed] [Google Scholar]
- Sorek R, Ast G, Graur D. (2002) Alu-containing exons are alternatively spliced. Genome Res 12:1060–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trdan Lusin T, Trontelj J, Mrhar A. (2011) Raloxifene glucuronidation in human intestine, kidney, and liver microsomes and in human liver microsomes genotyped for the UGT1A1*28 polymorphism. Drug Metab Dispos 39:2347–2354 [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:RESEARCH0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanGuilder HD, Vrana KE, Freeman WM. (2008) Twenty-five years of quantitative PCR for gene expression analysis. Biotechniques 44:619–626 [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. (2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456:470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Fang JL, Lazarus P. (2002) Glucuronidation: an important mechanism for detoxification of benzo[a]pyrene metabolites in aerodigestive tract tissues. Drug Metab Dispos 30:397–403 [DOI] [PubMed] [Google Scholar]