

Abstract
Polycyclic aromatic hydrocarbons (PAHs) in cigarette smoke are among the most likely causes of lung cancer. PAHs require metabolic activation to initiate the carcinogenic process. Phenanthrene (Phe), a noncarcinogenic PAH, was used as a surrogate of benzo[α]pyrene and related PAHs to study the metabolic activation of PAHs in smokers. A dose of 10 μg of deuterated Phe ([D10]Phe) was administered to 25 healthy smokers in a crossover design, either as an oral solution or by smoking cigarettes containing [D10]Phe. Phe was deuterated to avoid interference from environmental Phe. Intensive blood and urine sampling was performed to quantitate the formation of deuterated r-1,t-2,3,c-4-tetrahydroxy-1,2,3,4-tetrahydrophenanthrene ([D10]PheT), a biomarker of the diol epoxide metabolic activation pathway. In both the oral and smoking arms approximately 6% of the dose was metabolically converted to diol epoxides, with a large intersubject variability in the formation of [D10]PheT observed. Two diagnostic plots were developed to identify subjects with large systemic exposure and significant lung contribution to metabolic activation. The combination of the two plots led to the identification of subjects with substantial local exposure. These subjects produced, in one single pass of [D10]Phe through the lung, a [D10]PheT exposure equivalent to the systemic exposure of a typical subject and may be an indicator of lung cancer susceptibility. Polymorphisms in PAH-metabolizing genes of the 25 subjects were also investigated. The integration of phenotyping and genotyping results indicated that GSTM1-null subjects produced approximately 2-fold more [D10]PheT than did GSTM1-positive subjects.
Introduction
According to the International Agency for Research on Cancer (2004), tobacco smoking is associated with 90% of the lung cancer deaths in the United States. Polycyclic aromatic hydrocarbons (PAHs) are among the strongest carcinogens in cigarette smoke and are considered to be a major etiological factor in lung cancer (Pelkonen and Nebert, 1982; Hecht, 2003). PAHs are very lipophilic and could theoretically accumulate in tissues and reach toxic concentrations (Lang and Pelkonen, 1999). Thanks to evolution, humans have developed the capacity to detoxify foreign chemicals, including carcinogens, by converting them to more water-soluble metabolites, which may enhance their elimination from the body. Enzymes involved in the detoxification of PAHs include cytochromes P450 1A1 (CYP1A1) and 1B1 (CYP1B1), glutathione transferases (GSTs), UDP-glucuronosyltransferases, and sulfotransferases (Shimada, 2006; Hecht, 2010). However, at the cost of solving the acute problem (lethal accumulation of xenobiotics), a small portion of PAHs is transformed to electrophilic intermediates. These intermediates do not cause lethal hazards in the short term, but may initiate carcinogenesis in the long term by attacking nucleophilic macromolecules of the cell, causing gene mutations that may eventually lead to the development of tumors (Hecht, 1999). The conversion of PAHs to active intermediates is called metabolic activation or bioactivation. Enzymes involved in metabolic activation include CYP1A1, CYP1A2, and CYP1B1, as well as epoxide hydrolase (Shimada, 2006).
Fewer than 20% of lifelong smokers develop lung cancer, indicating that some individuals are more susceptible than others (International Agency for Research on Cancer, 1986; Peto et al., 2000). How to identify susceptible subjects is still not clear. Numerous epidemiological studies have been conducted to investigate the relationship of cancer susceptibility to genetic polymorphisms in PAH-metabolizing enzymes (Kawajiri et al., 1990; Nakachi et al., 1991; Tefre et al., 1991; Shields et al., 1993; Okada et al., 1994; Xu et al., 1996; Ishibe et al., 1997; Persson et al., 1999; Bartsch et al., 2000; Williams, 2001; Kiyohara et al., 2002; Shi et al., 2008). However, the results of those studies were mixed and sometimes even contradictory, perhaps because of the complexity of PAH metabolism and the multiple factors involved. For example, these studies have not considered gene-environment interactions and tissue-specific metabolic enzyme expression. Subjects with a higher expression of activation enzymes and lower expression of detoxification enzymes would theoretically have larger systemic exposure to active intermediates than other subjects. In addition, if the pulmonary enzymes of these subjects played a major role in activation, then lung exposure would be substantial, which could lead to an increased susceptibility to lung cancer. Because of the important role of metabolic activation of PAHs in lung carcinogenesis, the present study focused on identification of subjects with large systemic and local (lung) exposure and presumably high lung cancer risk.
Benzo[α]pyrene (BaP) is found in cigarette smoke and is a prototypic and widely studied compound for the investigation of carcinogenesis by PAHs (Straif et al., 2005; Uno et al., 2006; Jiang et al., 2007). A major bioactivation pathway of BaP (Fig. 1) is its conversion to r-7,t-8,9,c-10-tetrahydroxy-7,8,9,10-tetrahydrobenzo(α)pyrene via the formation of the “bay region diol epoxide,” BaP-7,8-diol-9,10-epoxide (BPDE), which reacts readily with DNA and is mutagenic and carcinogenic (International Agency for Research on Cancer, 2010). BPDE is thought to be one of the electrophilic reactive intermediates responsible for BaP mutagenesis and carcinogenesis (Conney, 1982; Cooper et al., 1983; Geacintov et al., 1997; Szeliga and Dipple, 1998; Hecht, 1999), and related bay region diol epoxides are considered to be major ultimate carcinogens of a number of other PAHs (International Agency for Research on Cancer, 2010). Because most PAHs, such as BaP, are carcinogenic and cannot be administered to humans, there have not been any detailed pharmacokinetic (PK) studies of PAHs in humans. To address this issue, a novel biomarker approach has been proposed: the use of phenanthrene (Phe) as a surrogate of BaP (Hecht et al., 2003). Phe is a noncarcinogenic PAH ubiquitous in the environment that can be safely administered to human subjects because all humans are exposed to Phe. The conversion of Phe to r-1,t-2,3,c-4-tetrahydroxy-1,2,3,4-tetrahydrophenanthrene (PheT) mimics the formation of the diol epoxide metabolic activation of BaP, including the intermediates formed and enzymes involved (Fig. 1) (International Agency for Research on Cancer, 1983; Carmella et al., 2004), although there are some differences (Hecht et al., 2010).
Fig. 1.

Metabolic activation pathways of BaP and Phe (modified from Carmella et al. (2004). BaPT, r-7,t-8,9,c-10-tetrahydroxy-7,8,9,10-tetrahydrobenzo(α)pyrene.
In the present study, deuterated Phe ([D10]Phe) was administered to 25 subjects, and the metabolism of [D10]Phe to deuterated PheT ([D10]PheT) was characterized to evaluate each individual's capacity to carry out the diol epoxide metabolism pathway. We used [D10]Phe to avoid interference from ubiquitous exposure to environmental Phe. An intermediate formed during the activation of [D10]Phe was deuterated anti-1,2-dihydroxy-3,4-epoxy-1,2,3,4-tetrahydrophenanthrene ([D10]PheDE), a biomarker of the diol epoxide pathway leading to BPDE from BaP (Fig. 1). The primary objective of our study was to quantify the amount of [D10]PheDE formed in the metabolism of [D10]Phe. [D10]PheDE cannot be quantified directly because it reacts rapidly with H2O, producing [D10]PheT, which was measured in this study (Carmella et al., 2004).
Preliminary reports from a subset of subjects demonstrated the rapid formation of diol epoxides and potential immediate negative health consequences of smoking (Zhong et al., 2011a) and examined the metabolism of [D10]Phe administered to 16 smokers either in a cigarette or orally as a biomarker of the activation pathway (Zhong et al., 2011b). With the completion of the clinical trial, a comprehensive analysis of the bioactivation of Phe is now reported.
Materials and Methods
Clinical Study Design.
The study was approved by the U.S. Food and Drug Administration and the University of Minnesota Institutional Review Board. Details on the recruitment of subjects and dosing protocols were reported previously (Zhong et al., 2011a,b). Subjects were recruited by using advertisements on the radio, television, or metropolitan and campus newspapers. Volunteers interested in the study called the University of Minnesota Transdisciplinary Tobacco Use Research Center and were informed about the study. The preliminary screening was performed over the phone to select subjects meeting the following specific inclusion criteria: the smoking of at least 10 cigarettes daily for the past year and good physical and mental health. Female subjects who were pregnant or nursing were excluded. Eligible subjects were further invited to the clinic site for an orientation visit to fill out a detailed questionnaire regarding their smoking and medical history. Pregnancy tests were also conducted. Subject recruitment incentives were used, and an average of $500 was paid to each subject for the completion of the study.
A total of 25 eligible subjects, eight men, were recruited between April 2008 and September 2010. The subjects were between 23 and 54 years with a mean age (± S.D.) of 36.2 ± 10.4 years. Their weights ranged from 61 to 113 kg with a mean weight (± S.D.) of 87.4 ± 15.4 kg. Twelve subjects were white, eight were African-American, and five reported being multiracial.
A dose of 10 μg (53.2 nmol) [D10]Phe was administered to 25 subjects, either as an oral solution or by smoking cigarettes to which [D10]Phe had been added. [D10]Phe (98%; containing 2% nondeuterated Phe) was purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA), and then repurified in the University of Minnesota Molecular and Cellular Therapeutics GMP facility. The study had a randomized, open-label, single-dose, crossover design. The order of administration was randomized, and each dose was separated by a washout period of at least 1 week. For the oral dose, each subject was given 10 μg of [D10]Phe (5 ml of 20% ethanol-80% water solution). The dosing bottle was rinsed twice with water to ensure accurate dosing. For the inhalation dose, subjects followed a standard smoking protocol designed to deliver 10 μg of [D10]Phe, as monitored by a smoking topography device (Plowshare Technologies, Baltimore, MD) (Zhong et al., 2011a). The administration of [D10]Phe by smoking was performed in a specially ventilated room at the Transdisciplinary Tobacco Use Research Center. Subjects underwent an adaptation trial before smoking the cigarettes containing [D10]Phe. Specific instructions about the puff volume (55 ml), puff duration (2 s), and puff interval (30 s) were given to the subjects. Subjects smoked the cigarettes through the smoking topography device, which recorded the puff volume, puff duration, and puff number. A Marlboro cigarette (Philip Morris, Richmond, VA) was used in the practice session, after which subjects were then allowed to smoke the cigarette containing [D10]Phe. The smoking process was also observed by the clinician to ensure good compliance. The smoking session usually lasted approximately 4 to 5 min.
Blood samples of 10 ml each were taken before dosing and 15, 30, 45, 60, 90, 120, 150, 240, 360, 540, 720, or 1440 min after completion of administration. Blood samples were centrifuged to obtain plasma, which was stored at −20°C until analysis. Urine samples were obtained predosing and at the following postdosing intervals: 0 to 30, 30 to 60, 60 to 120, 120 to 360, 360 to 720, 720 to 1440, and 1440 to 2880 min. The volume of each urine collection was measured, and an aliquot of 50 ml was stored at −20°C until analysis. Gas chromatography-mass spectrometry was used to analyze the plasma and urine samples as described previously (Zhong et al., 2011a). It should be noted that because approximately 90% of the PheT in human urine exists as sulfate and glucuronide conjugates (Hecht et al., 2003), plasma and urine samples were incubated with β-glucuronidase and arylsulfatase before quantitation. Hence the reported level of [D10]PheT in both plasma and urine is the sum of the free [D10]PheT and its conjugates.
Pharmacokinetic Analysis.
Noncompartmental analysis was performed with the use of Phoenix WinNonlin (version 6.1; Pharsight, Mountain View, CA) to calculate the area under the [D10]PheT plasma concentration-time curve (AUC(PheT)) and the elimination half-life (t1/2) of [D10]PheT.
The clearance of [D10]PheT (CL7; Fig. 2) was estimated from the slope of the urinary excretion rate versus midpoint plasma concentration curve:
where Cpmid was the plasma concentration of [D10]PheT at the midpoint of the urine collection interval. The urinary excretion rate (ΔX/Δt) was determined from the amount of [D10]PheT excreted in urine at each interval (ΔX) divided by the length of the collection interval (Δt). Urine collections at 720 to 1440 and 1440 to 2880 min were not included in the estimate of CL7 because very low concentrations were present in urine in those intervals, and each of those intervals was >1 half-life of [D10]PheT (Gibaldi and Perrier, 1982).
Fig. 2.

The PK view of metabolic activation of [D10]Phe after administration (detailed description in Appendix).
The total amount of [D10]PheDE formed during activation (Aact) was estimated by two methods (eqs. 2 and 3; see Appendix for detailed derivation of the equations). The first method was using the product of AUC(PheT) and CL7:
The second method was to calculate the amount of [D10]PheT collected in urine up to 48 h (Xu(PheT),t = 48):
The percentage of dose activated (f) was calculated as:
 |
where D is the administered dose of [D10]Phe (10 μg or 53.2 nmol).
Aact,plasma and Aact,urine are two indicators of systemic exposure to [D10]PheT and thus formed the basis of diagnostic plot I (Fig. 3, top).
Fig. 3.

Diagnostic plots I (top) and II (bottom).
Differences in Aact between smoking and oral dosing were calculated as:
where Aact,smk and Aact,oral are Aact after smoking and oral dosing, respectively. Aact(lung) is a measure of lung contribution to the formation of [D10]PheT. A positive value of Aact(lung) indicates that the lung contributed more to the formation of [D10]PheT than liver, and hence played a major role in metabolic activation. In addition, the larger the value of Aact(lung), the larger local (lung) exposure to [D10]PheT.
The relative bioavailability of [D10]PheT after oral dosing compared with the smoking administration was calculated from the ratio of oral/smoking AUC(PheT) values (FAUC) and the ratio of oral/smoking Xu(PheT),t = 48 values (FXu). FAUC and FXu were used as two indicators of lung contribution to activation and formed the basis for diagnostic plot II (Fig. 3, bottom). The combination of plots I and II allowed the identification of subjects with substantial local exposure.
According to eq. A23 in Appendix, the amount of [D10]PheDE formed after oral dosing and smoking could be calculated as
where fa, fact, and fm are the fraction of the dose absorbed, the fraction of the dose converted to [D10]PheDE during first-pass activation, and the fraction of [D10]Phe converted to [D10]PheDE in the systemic circulation, respectively. Because changes in the route of administration would affect only fa and fact, but fm would remain the same, eq. 6 could be rewritten as eqs. 6.1 and 6.2 for the oral and smoking arm, respectively.
fa(oral) and fa(smk) are the fraction of [D10]Phe dose absorbed after oral dosing and smoking, respectively. fact(oral) and fact(smk) are the fraction of [D10]Phe converted to active intermediates during first-pass metabolism after oral dosing and smoking, respectively.
Genotyping.
Twelve polymorphisms of metabolizing enzymes of Phe were determined by the BioMedical Genomics Center at the University of Minnesota. Genotyping was performed by using the iPLEX Gold method (Sequenom, San Diego, CA). Similar methods have been reported previously (Hecht et al., 2006). In brief, the method is based on the primer-extension reaction that generates allele-specific products with distinct masses detected by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. The method started with polymerase chain reaction amplification followed by shrimp alkaline phosphatase treatment to remove unincorporated dNTPs. Single-base extension (SBE) was carried out by the addition of SBE primers, iPLEX enzymes, and buffers. SBE products were measured with the MassARRAY system (Sequenom), and mass spectra were analyzed with TYPER software (Sequenom). iPLEX reagents and protocols for multiplex polymerase chain reaction, SBE, and generation of mass spectra were based on the manufacturer's instructions.
The polymorphisms of CYP1A1 and CYP1B1 investigated in the study were CYP1A1MspI, CYP1A1I462V, CYP1B1R48G, CYP1B119S, CYP1B1L432V, and CYP1B1N453S. In addition, EPHX1Y113H and EPHX1H139R, two polymorphisms of microsomal epoxide hydrolase 1 (EPHX1), were also measured. The polymorphisms of the detoxification enzymes GST P1 (GSTP1), T1 (GSTT1), and M1 (GSTM1) measured in the study were GSTP1A114V, GSTP1I105V, GSTT1 null, and GSTM1 null.
Statistical Analysis.
A paired t test was used to compare t1/2, CL7, and AUC(PheT) between the oral and smoking arm. The measurement of relative bioavailability by two methods (FAUC versus FXu) was compared by using a paired t test after the logarithmic transformation of the original data. Two-way analysis of variance was used to investigate the influence of data source (plasma versus urinary data) and route of administration (oral versus smoking) on the estimates of systemic exposure to [D10]PheT. One-way analysis of variance was used to identify polymorphisms that might have effects on an individual's capacity to activate PAHs. A p value <0.05 was considered to be significant.
Results
Table 1 reports the half-life, clearance (CL7), and AUC(PheT) of [D10]PheT after oral dosing and smoking administration of [D10]Phe. No significant difference was observed in t1/2, CL7, or AUC(PheT) between the oral and smoking arms of the study, consistent with the results reported previously for 16 subjects (Zhong et al., 2011b).
TABLE 1.
Half-life, clearance and AUC of [D10]PheT after oral dosing and smoking
Paired t test was performed to compare the effects of routes of administration. Results were not significant, p > 0.05.
| ID |
t1/2 |
CL7 |
AUC(PheT) |
|||
|---|---|---|---|---|---|---|
| Oral | Smoking | Oral | Smoking | Oral | Smoking | |
| h | l/h | nmol · h/l | ||||
| 1 | 7.2 | 10.9 | 7.8 | 13.2 | 1.07 | 1.43 |
| 2 | 17.7 | 8.1 | 3.1 | 7.3 | 0.13 | 0.11 |
| 3 | 12.6 | 11.4 | 5.8 | 6.4 | 0.97 | 1.06 |
| 4 | 11.0 | 6.7 | 4.9 | 6.4 | 0.37 | 0.38 |
| 5 | 3.9 | 4.8 | 5.5 | 4.8 | 0.24 | 0.48 |
| 6 | 6.3 | 7.5 | 4.3 | 7.3 | 1.10 | 1.49 |
| 7 | 12.6 | 8.1 | 4.5 | 13.0 | 0.97 | 0.13 |
| 8 | 5.5 | 5.9 | 5.4 | 4.6 | 0.46 | 0.51 |
| 9 | 6.4 | 9.8 | 3.2 | 1.3 | 1.04 | 0.81 |
| 10 | 6.9 | 7.5 | 5.1 | 7.1 | 0.55 | 0.48 |
| 11 | 4.9 | 4.5 | 7.5 | 3.1 | 0.85 | 0.70 |
| 12 | 4.4 | 6.4 | 3.2 | 3.6 | 0.43 | 0.48 |
| 13 | 7.9 | 9.9 | 6.0 | 7.0 | 0.58 | 0.76 |
| 14 | 7.4 | 7.1 | 4.0 | 4.5 | 1.70 | 1.11 |
| 15 | 8.8 | 6.0 | 4.6 | 4.0 | 0.94 | 0.53 |
| 16 | 5.1 | 5.2 | 4.4 | 4.5 | 0.93 | 0.87 |
| 17 | 9.3 | 7.5 | 1.9 | 1.9 | 1.98 | 1.27 |
| 18 | 6.1 | 11.0 | 2.2 | 2.2 | 0.80 | 0.65 |
| 19 | 5.4 | N.A. | 1.8 | N.A. | 0.90 | N.A. |
| 20 | 5.2 | 6.2 | 3.7 | 4.1 | 1.04 | 1.02 |
| 21 | 7.4 | 9.8 | 3.2 | 2.9 | 1.45 | 0.69 |
| 22 | 5.8 | 8.9 | 2.8 | 4.7 | 0.82 | 1.53 |
| 23 | 7.0 | 6.7 | 3.0 | 5.1 | 2.08 | 1.45 |
| 24 | 4.8 | 7.3 | 5.3 | 3.0 | 1.29 | 1.57 |
| 25 | 6.3 | 6.9 | 2.4 | 2.9 | 0.65 | 0.79 |
| Mean | 7.4 | 7.7 | 4.2 | 5.2 | 0.93 | 0.85 |
| S.D. | 3.2 | 2.0 | 1.6 | 3.0 | 0.49 | 0.44 |
N.A., not available because the plasma concentration of [D10]PheT of subject 19 after smoking was below the limit of quantitation.
Aact is an estimate of the systemic exposure to [D10]PheT and was calculated by two methods: use of plasma data (Aact, plasma) and use of urinary data (Aact,urine). As shown in Table 2, Aact,plasma and Aact,urine were 4.23 ± 3.94 and 3.06 ± 1.91 nmol, respectively, in the smoking arm. The percentage of the [D10]Phe dose activated was calculated as 7.96 ± 7.41 and 5.75 ± 3.60 based on plasma and urinary data, respectively. Similar results were obtained from the oral arm. Route of administration (smoking versus oral) or source of data (plasma versus urine) did not have a significant impact on the estimate of Aact.
TABLE 2.
Aact of oral and smoking arm calculated by two methods
Two-way analysis of variance was performed to evaluate the influence of route of administration and data source (plasma, urine), which was found to be not significant, p > 0.05.
| ID | Oral Arm |
Smoking Arm |
||||||
|---|---|---|---|---|---|---|---|---|
| Aact,plasma | Aact,urine | Aact,plasma | Aact,urine | |||||
| nmol | % of dose | nmol | % of dose | nmol | % of dose | nmol | % of dose | |
| 1 | 8.25 | 15.51 | 5.62 | 10.56 | 18.81 | 35.36 | 8.00 | 15.04 |
| 2 | 0.41 | 0.77 | 0.28 | 0.53 | 0.79 | 1.48 | 0.43 | 0.81 |
| 3 | 5.57 | 10.47 | 3.37 | 6.33 | 6.74 | 12.67 | 3.45 | 6.48 |
| 4 | 1.79 | 3.36 | 1.33 | 2.50 | 2.41 | 4.53 | 2.06 | 3.87 |
| 5 | 1.32 | 2.48 | 1.49 | 2.80 | 2.30 | 4.32 | 2.27 | 4.27 |
| 6 | 4.62 | 8.68 | 6.37 | 11.97 | 10.77 | 20.24 | 7.03 | 13.21 |
| 7 | 4.36 | 8.20 | 3.29 | 6.18 | 1.71 | 3.21 | 1.08 | 2.03 |
| 8 | 2.45 | 4.61 | 1.89 | 3.55 | 2.30 | 4.32 | 2.46 | 4.62 |
| 9 | 3.24 | 6.09 | 5.29 | 9.94 | 1.00 | 1.88 | 3.76 | 7.07 |
| 10 | 2.83 | 5.32 | 2.28 | 4.29 | 3.44 | 6.47 | 2.22 | 4.17 |
| 11 | 6.31 | 11.86 | 6.11 | 11.48 | 2.14 | 4.02 | 3.22 | 6.05 |
| 12 | 1.36 | 2.56 | 1.72 | 3.23 | 1.73 | 3.25 | 1.58 | 2.97 |
| 13 | 3.43 | 6.45 | 2.45 | 4.61 | 5.26 | 9.89 | 2.96 | 5.56 |
| 14 | 6.75 | 12.69 | 5.85 | 11.00 | 4.91 | 9.23 | 4.40 | 8.27 |
| 15 | 4.3 | 8.08 | 4.45 | 8.36 | 2.10 | 3.95 | 3.29 | 6.18 |
| 16 | 4.08 | 7.67 | 4.20 | 7.89 | 3.92 | 7.37 | 4.13 | 7.76 |
| 17 | 3.72 | 6.99 | 3.89 | 7.31 | 2.34 | 4.40 | 2.33 | 4.38 |
| 18 | 1.74 | 3.27 | 1.68 | 3.16 | 1.39 | 2.61 | 0.77 | 1.45 |
| 19 | 1.59 | 2.99 | 1.31 | 2.46 | N.A. | N.A. | 0.26 | 0.49 |
| 20 | 3.83 | 7.20 | 3.43 | 6.45 | 4.11 | 7.73 | 2.86 | 5.38 |
| 21 | 4.6 | 8.65 | 2.66 | 5.00 | 1.96 | 3.68 | 1.42 | 2.67 |
| 22 | 2.29 | 4.30 | 2.33 | 4.38 | 7.14 | 13.42 | 4.29 | 8.06 |
| 23 | 6.24 | 11.73 | 5.95 | 11.18 | 7.37 | 13.85 | 5.26 | 9.89 |
| 24 | 6.77 | 12.73 | 5.63 | 10.58 | 4.68 | 8.80 | 5.13 | 9.64 |
| 25 | 1.57 | 2.95 | 1.07 | 2.01 | 2.31 | 4.34 | 1.87 | 3.52 |
| Mean | 3.74 | 7.02 | 3.36 | 6.31 | 4.23 | 7.96 | 3.06 | 5.75 |
| S.D. | 2.06 | 3.87 | 1.86 | 3.50 | 3.94 | 7.41 | 1.91 | 3.60 |
N.A., not available because plasma concentration of [D10]PheT after smoking was below the limit of quantitation.
Although at the group level no difference was observed in the means of Aact, a large intersubject variability (>20-fold) in Aact was observed in both the smoking and oral arms (Table 2). One purpose of this study was to identify people with a potentially increased susceptibility to carcinogenesis by PAH because of their ability to carry out the bay region diol epoxide pathway. Because the route of administration of [D10]Phe in the smoking arm mimicked the uptake of carcinogens by cigarette smokers, plasma and urinary data of the smoking arm were used to identify subjects with a large systemic exposure to [D10]PheT, as shown in diagnostic plot I (Fig. 3, top). Six subjects (1, 6, 14, 22, 23, and 24) whose estimates of systemic exposure (Aact) were in the top 30% of the population as measured by plasma data (≥4.68 nmol by eq. 2) and urine data (≥4.29 nmol by eq. 3) fell into zone A (large systemic exposure zone). Likewise, five subjects (2, 7, 12, 18, and 21) whose estimates of systemic exposure were in the lowest 30% of the population as measured by plasma data (≤1.96 nmol by eq. 2) and urine data (≤1.58 nmol by eq. 3), fell into zone B (low systemic exposure zone).
The relative bioavailability of [D10]PheT after oral dosing compared with the smoking administration was calculated from the ratio of oral/smoking AUC(PheT) values (FAUC) and the ratio of oral/smoking Xu(PheT),t = 48 values (FXu). FAUC and FXu were used as two indicators of lung contribution to metabolic activation. As shown in Fig. 4, after the administration of [D10]Phe by smoking, the parent molecule [D10]Phe passes through the lung before it reaches the systemic circulation. After oral dosing the parent molecule passes through the liver before it reaches the systemic circulation. Therefore, if FAUC and FXu are less than 1, the lung has a greater contribution to the formation of [D10]PheT. As shown in Table 3, the relative bioavailability measured by FAUC and FXu was 1.35 ± 1.33 and 1.35 ± 0.96, respectively. No significant difference was observed in relative bioavailability measured by the two methods. At the group level no significant difference was observed in the disposition of parent molecule by lung and liver. However, a large intersubject variability was observed (Table 3), illustrating a more than 9-fold range in both FAUC and FXu. Diagnostic plot II was then developed to identify subjects with significant lung contribution to metabolic activation. Nine subjects (1, 3, 4, 5, 6, 8, 13, 22, and 25) with both FAUC and FXu values of less than 1 were identified in zone C (Fig. 3, bottom).
Fig. 4.

Comparison of metabolic activation of [D10]Phe in the oral and smoking arm.
TABLE 3.
Relative bioavailability by two methods (FAUC and FXu)
Ninety percent confidence interval (90% CI) is of geometric means. Paired t test was performed to compare two methods of calculating relative bioavailability, and results were not significant, p > 0.05.
| ID | Relative Bioavailability |
|
|---|---|---|
| FAUC | FXu | |
| 1 | 0.75 | 0.70 |
| 2 | 1.20 | 0.66 |
| 3 | 0.92 | 0.98 |
| 4 | 0.98 | 0.65 |
| 5 | 0.50 | 0.66 |
| 6 | 0.73 | 0.91 |
| 7 | 7.32 | 3.05 |
| 8 | 0.91 | 0.77 |
| 9 | 1.29 | 1.41 |
| 10 | 1.14 | 1.03 |
| 11 | 1.21 | 1.90 |
| 12 | 0.88 | 1.09 |
| 13 | 0.76 | 0.83 |
| 14 | 1.53 | 1.33 |
| 15 | 1.80 | 1.36 |
| 16 | 1.06 | 1.02 |
| 17 | 1.56 | 1.67 |
| 18 | 1.22 | 2.19 |
| 19 | N.A. | 5.01 |
| 20 | 1.02 | 1.20 |
| 21 | 2.11 | 1.87 |
| 22 | 0.53 | 0.54 |
| 23 | 1.43 | 1.13 |
| 24 | 0.82 | 1.10 |
| 25 | 0.83 | 0.57 |
| Mean | 1.35 | 1.35 |
| SD | 1.33 | 0.96 |
| 90% CI | 0.93 −1.35 | 0.95 −1.38 |
N.A., not available because the plasma concentration of [D10]PheT after smoking was below the limit of quantitation.
The combination of zones A (diagnostic plot I) and C (diagnostic plot II) led to the identification of three subjects (1, 6, and 22) with both a large systemic exposure and a significant lung contribution to activation. These three subjects formed much more [D10]PheT after smoking than oral dosing (Table 4). In addition, Aact(lung) of these three subjects was between 0.66 and 10.56 nmol. This amount of [D10]PheT indicated substantial local exposure, especially considering that the total exposure to [D10]PheT in a typical subject after smoking ranged from 3.06 to 4.23 nmol.
TABLE 4.
Significant lung contribution to metabolic activation in subjects 1, 6, and 22
Data of a typical subject were based on the group mean (n = 25).
| ID |
Aact,plasma |
Aact(lung)a |
Aact,urine |
Aact(lung)b | ||
|---|---|---|---|---|---|---|
| Smoking | Oral | Smoking | Oral | |||
| nmol | ||||||
| 1 | 18.81 | 8.25 | 10.56 | 8.00 | 5.62 | 2.38 |
| 6 | 10.77 | 4.62 | 6.15 | 7.03 | 6.37 | 0.66 |
| 22 | 7.14 | 2.29 | 4.85 | 4.29 | 2.33 | 1.96 |
| A typical subject | 4.23 | 3.74 | 3.06 | 3.36 | ||
The difference of Aact,plasma between smoking and oral dosing.
The difference of Aact,urine between smoking and oral dosing
Table 5 shows the effects of metabolic enzyme polymorphisms on an individual's capacity to activate PAHs as measured by estimates of Aact (eqs. 2 and 3) of both the oral and smoking arms. Among 12 polymorphisms tested only the GSTM1 polymorphism was associated with a difference in systemic exposure. Figure 5, a (plasma data) and b (urine data), shows the maximum, minimum, and median of Aact in GSTM1-negative and -positive subjects after smoking. A more than 2-fold difference was observed in Aact between GSTM1-negative subjects (Aact,plasma: 5.87 ± 4.85; Aact,,urine: 4.16 ± 2.00 nmol; n = 12) and GSTM1-positive subjects (Aact,plasma: 2.60 ± 1.76; Aact,,urine: 2.05 ± 1.16 nmol; n = 13) after smoking (p < 0.05). To further confirm the effects of GSTM1 on the metabolic activation, estimates of Aact from the oral arm were investigated (Fig. 5, c and d), and similar results were observed.
TABLE 5.
Effects of polymorphisms on an individual's capacity to activate PAHs
| Gene Polymorphism | Occurrence |
p valuesa |
|||||
|---|---|---|---|---|---|---|---|
| Oral |
Smoking |
||||||
| Normal | Heterozygotes | Homozygotes | Aact,plasma | Aact,urine | Aact,plasma | Aact,urine | |
| % | |||||||
| CYP1A1 MspI (rs4646903) | 64 | 36 | 0 | N.S. | N.S. | N.S. | N.S. |
| CYP1A1 I462V (rs1048943) | 88 | 12 | 0 | N.S. | N.S. | N.S. | N.S. |
| CYP1B1 R48G (rs10012) | 32 | 60 | 8 | N.S. | N.S. | N.S. | N.S. |
| CYP1B1 A119S (rs1056827) | 8 | 92 | 0 | N.S. | N.S. | N.S. | N.S. |
| CYP1B1 L432V (rs1056836) | 20 | 52 | 28 | N.S. | N.S. | N.S. | N.S. |
| CYP1B1 N453S (rs1800440) | 84 | 16 | 0 | N.S. | N.S. | N.S. | N.S. |
| GSTP1 I105V (rs1695) | 36 | 52 | 12 | N.S. | N.S. | N.S. | N.S. |
| GSTP1 A114V (rs1138272) | 84 | 16 | 0 | N.S. | N.S. | N.S. | N.S. |
| EPHX1 Y113H (rs1051740) | 32 | 68 | 0 | N.S. | N.S. | N.S. | N.S. |
| EPHX1 H139R (rs2234922) | 64 | 28 | 8 | N.S. | N.S. | N.S. | N.S. |
| GSTT1 | 12 (presence) | 88 (null) | N.S. | N.S. | N.S. | N.S. | |
| GSTM1 | 52 (presence) | 48 (null) | * | * | * | ** | |
N.S., not significant p > 0.05.
, p < 0.05;
, p < 0.01.
One-way analysis of variance was performed to determine the influence of genotype on Aact.
Fig. 5.

Comparison of GSTM1-negative (−) and -positive (+) subjects after smoking (a and b) and oral dosing (c and d). Dotted lines indicate median. *, p < 0.05; **, p < 0.01.
Figure 6 integrates diagnostic plot I and GSTM1 genotype information and reveals an interesting correlation between genotyping and phenotyping results. The occurrence of the GSTM1-negative genotype was 100% (n = 6) in zone A compared with 48% in the group as a whole (n = 25). In contrast, the occurrence of the GSTM1-negative genotype was only 20% (n = 5) in zone B. High occurrence of the GSTM1-negative genotype in subjects with a large systemic exposure to [D10]PheT, and low occurrence of the GSTM1-negative genotype in subjects with a low systemic exposure to [D10]PheT, clearly suggests a correlation between GSTM1 genotype and an individual's capacity to activate PAHs. It is of interest to note that the three subjects (1, 6, and 22) with both high systemic exposure and significant lung contribution to activation all were GSTM1-negative and African American.
Fig. 6.

Correlation between genotyping and phenotyping (▵, GSTM1-positive; ●, GSTM1-negative).
Discussion
Because lung cancer treatment is not particularly effective (5-year survival rate <16%), prevention is an important alternative, especially considering that tobacco smoking accounts for 90% of cases of the disease in the United States (International Agency for Research on Cancer, 2004; Spiro and Silvestri, 2005). The outcome of smoking cessation, a major prevention strategy, depends on the intensity of intervention. The rate of successful smoking cessation at 1 year is 3 to 5% when smokers simply try to stop, 7 to 16% when behavioral intervention is provided, and up to 24% when smokers receive both pharmacological treatment and behavioral support (Laniado-Laborín, 2010). Therefore, the successful identification of susceptible individuals could lead to increasing the intensity of intervention for these individuals. This would potentially improve the outcome of smoking cessation interventions. The current study aimed to quantitate an individual's capacity to metabolically activate PAHs (one group of carcinogens involved in lung cancer) and identify subjects with extensive activation and presumably higher lung cancer risk.
The large intersubject variability (>20-fold) in the capacity to activate [D10]Phe was consistent with the large intersubject variability in the activation of PAHs reported in the literature (Cohen et al., 1979) and further justified the use of a PK approach to identify highly susceptible individuals. In our PK study of 25 subjects, the fraction of [D10]Phe converted to [D10]PheT ranged from 0.49 to 15.04% (Aact,urine) in the smoking arm. Even though only a relatively small fraction of a PAH, as represented by [D10]Phe, is metabolically activated in humans, it is believed to be critical in the initiation of carcinogenesis (Gelboin, 1980; Conney, 1982; Cooper et al., 1983; Dipple et al., 1984; Geacintov et al., 1997; Szeliga and Dipple, 1998). Metabolites not quantified in this study include phenols and dihydrodiols as well as unidentified material.
The quantitation of an individual's capacity to activate PAHs was the primary objective of the current study. The unique PK approach used here quantitated both systemic and local exposure to [D10]PheT. There are no other published studies on PAH PK in humans to our knowledge. Although the measurement of systemic exposure is important in the identification of susceptible subjects, local (lung) exposure is a physiologically more relevant measurement for two reasons: 1) it captures the magnitude of exposure to carcinogenic intermediates at the specific site where carcinogenesis occurs; and 2) [D10]Phe was used as a surrogate of BaP and other related PAHs, which generally have their strongest carcinogenic effects at the site of application, although there are exceptions (International Agency for Research on Cancer, 1983, 2010; Hecht, 2003). For locally acting carcinogens, lung exposure is much more relevant to tobacco smoke carcinogenesis of lung cancer than systemic exposure. Despite its crucial role in carcinogenesis by PAHs, local exposure is usually very difficult to measure in clinical trials. In the present study the integration of systemic exposure and relative bioavailability revealed important information regarding local exposure. The concept is that subjects with both large systemic exposure (Fig. 3, top, zone A of plot I) and significant lung contribution to activation (Fig. 3, bottom, zone C of plot II) had substantial local (lung) exposure. As shown in Fig. 4, for a given individual, the difference in systemic exposure after oral dosing and smoking is caused by the different pathways that [D10]Phe takes before reaching the systemic circulation, i.e., one single pass of parent molecule through the liver or lung. In other words, if more [D10]Phe were activated after smoking than oral dosing (FAUC < 1 or FXu < 1), then this subject's lung contributed more to the activation than did liver. Therefore, subjects with relative bioavailability less than 1 and subjects with large systemic exposure were of interest.
If relative bioavailability is less than 1, then by definition Aact(oral) < Aact(smk). According to eqs. 6.1 and 6.2, there are three scenarios when Aact(oral) < Aact(smk): 1) fa(oral) < fa(smk) and fact(oral) < fact(smk); 2) fa(oral) > fa(smk) and fact(oral) < fact(smk); and 3) fa(oral) < fa(smk) and fact(oral) > fact(smk).
In the first case, because fa(oral) < fa(smk), the lung allowed a larger fraction of [D10]Phe to enter systemic circulation than did the liver, indicating a larger systemic exposure after smoking than oral dosing. In addition, because fact(oral) < fact(smk), more active intermediates would be produced in lung than liver, indicating substantial local exposure. Therefore, case 1 was large systemic exposure, large local exposure. Likewise, cases 2 and 3 would be considered low systemic exposure, large local exposure and large systemic exposure, low local exposure, respectively. Although in case 3 lung may receive lower exposure to the active intermediates than does the liver, given the substantial amount of active intermediates in the circulation, the attack of electrophilic intermediates against lung DNA would still probably be substantial. Therefore, for all the cases related to a relative bioavailability of less than 1, potentially negative health consequences caused by substantial local exposure may be likely. As such, diagnostic plot II was developed to identify subjects with a significant lung contribution to metabolic activation.
As shown in plot I (Fig. 3, top), systemic exposure was calculated by two methods (eqs. 2 and 3). Likewise, in plot II (Fig. 3, bottom) the relative bioavailability was estimated by two methods (FAUC and FXu) to confirm the identification of subjects with significant lung contribution to activation. The combination of plots I and II led to the identification of three subjects. In these three subjects one single pass through lung produced the same level of [D10]PheT as total exposure in a typical subject. This suggests that these subjects may have higher susceptibility than the rest of the study group.
There was a statistically significant difference in systemic exposure to [D10]PheT between GSTM1-negative and -positive subjects. Because GSTM1 is a detoxification enzyme that catalyzes the conjugation of electrophilic intermediates with glutathione, the deletion of the GSTM1 gene reduces the efficiency of the detoxification pathways. As a result, more [D10]Phe may go through the activation pathway, and more end product of the activation pathway was observed in GSTM1-negative subjects. Furthermore, approximately a 2-fold difference in activation caused by GSTM1 might lead to clinically different outcomes considering the important role of PAHs in lung carcinogenesis and decades of uptake of PAHs from cigarette smoking. It has been reported that GSTM1-negative subjects have higher PAH-DNA adducts in the lung than GSTM1-positive subjects (Rojas et al., 1998, 2000). It is worth noting that the three subjects identified by diagnostic plots I and II all were GSTM1-negative. In addition, all three subjects were African American. The development of a population PK model is in progress that will estimate the population mean and intersubject variability of the efficiency of the activation pathway. Age, gender, weight, race, renal function, genotype, and smoking history will be incorporated in the population PK analysis to examine their influence on lung cancer susceptibility.
In other studies, we have found that conjugation of Phe diol epoxides by glutathione is a relatively minor pathway, at least based on the amount of the appropriate N-acetylcysteine conjugate found in urine (Hecht et al., 2008). The amounts seem to be too low to account for the decreased levels of [D10]PheT in the GSTM1-competent versus GSTM1-null individuals observed here. It is possible that glutathione conjugation occurs earlier in the pathway that produces [D10]PheT, for example, by reaction with Phe-1,2-epoxide (Fig. 1).
Although no effects of other polymorphisms on systemic exposure were observed, it is likely that multiple genes are involved in the metabolic activation of PAHs (Shimada, 2006; Hecht, 2010). The low frequency of minor alleles and relatively small number of subjects in the present study may explain the failure to detect an association of these polymorphisms with our measures of exposure.
One limitation of the present study was the lack of a PK profile of the parent compound [D10]Phe caused by its extremely low concentrations in plasma (< 25 pg/ml) and urine (< 5 pg/ml) after the 10-μg dose of [D10]Phe (Zhong et al., 2011b). Animal PK studies have been reported using a dose of 4.5 to 10 mg/kg, which was at least 30,000- fold higher than the dose used in this study (Chu et al., 1992; Schober et al., 2010). The half-life and oral clearance of phenanthrene in a mouse study with an oral dose of 4.5 mg/kg were reported to be 0.32 h and 2.18 liter/h · kg, respectively. Another study in rat and guinea pig indicated that more than 90% of the parent compound was converted to metabolites after oral administration of 10 mg/kg 14C-labeled Phe. The distribution of Phe to tissues was not well characterized, but Phe is probably extensively distributed to tissues because of its lipophilicity. Collectively, the low dose of [D10]Phe in human study, extensive metabolism, and rapid distribution are likely to contribute to the low concentration of [D10]Phe in plasma and urine.
In summary, in the present study the metabolic activation of Phe in humans has been investigated. Plasma and urinary data of the smoking arm indicated a more than 20-fold difference in an individual's capacity to activate PAHs and formed the basis of diagnostic plot I to identify subjects with large systemic exposure. The relative bioavailability between oral dosing and smoking also showed a large intersubject variability (> 9-fold), and formed the basis of diagnostic plot II to identify subjects with significant lung contribution to activation. The combination of plots I and II led to the identification of subjects with substantial lung exposure. This approach may have significant potential in the prediction of lung cancer risk. The integration of phenotyping and genotyping results indicated that GSTM1 played an important role in the detoxification of Phe.
Acknowledgments
We thank Dr. Andrea Yoder, Claire Brookmeyer, and Dr. Peter Villalta for consulting on gas chromatography-mass spectrometry.
Appendix
In the PK approach to identify susceptible subjects, one primary objective was to quantitate the [D10]PheT formed and use it as a surrogate for the amount of “active” metabolite [D10]PheDE formed during activation (Aact). This is an indicator of systemic exposure. As shown in Fig. 1, [D10]PheDE is the surrogate for the active intermediate BPDE, which binds to DNA and initiates carcinogenesis. However, direct measurement of [D10]PheDE was very difficult because of the poor stability of this intermediate. Our investigation turned to [D10]PheT, the end product of activation pathway. Based on our understanding of the metabolic activation of Phe as shown in Fig. 2, Aact could be estimated by quantitating [D10]PheT in plasma or urine.
The absorption and disposition of [D10]Phe after extravascular administration (oral or smoking) could be summarized in four steps (Fig. 2). Step 1, after delivery to the site of absorption, [D10]Phe was transferred to systemic circulation intact (absorption), in the form of [D10]PheT (first-pass activation) and other metabolites; the clearances of these three pathways of step 1 were CL1, CL2, and CL3, respectively. Step 2, after entering the systemic circulation, a small fraction of [D10]Phe was converted to active metabolite [D10]PheDE (activation), and the majority of [D10]Phe was transformed to other metabolites; the clearances of these two pathways of step 2 were CL5 and CL4, respectively. Step 3, [D10]PheDE was converted to [D10]PheT (hydrolysis) and the clearance of this process was CL6. Step 4, [D10]PheT was further converted to sulfate or glucuronide conjugates followed by renal excretion. The clearance of the process that eliminates [D10]PheT from the body was CL7.
Several assumptions were made in steps 3 and 4. The first assumption is that in step 3 all [D10]PheDE was converted to [D10]PheT. The second assumption is that in step 4 all of the [D10]PheT formed was excreted in urine in 48 h.
As such, the following differential equations were derived:
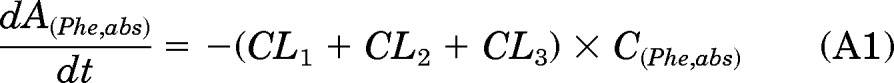 |
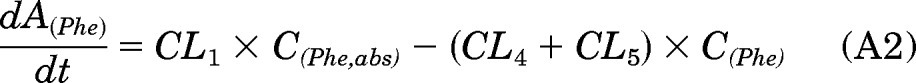 |
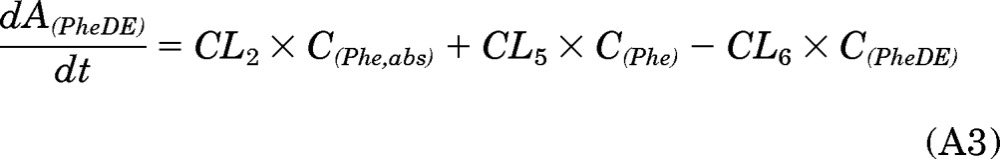 |
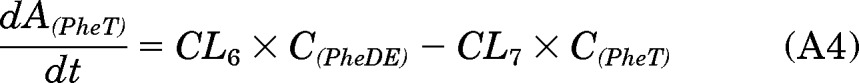 |
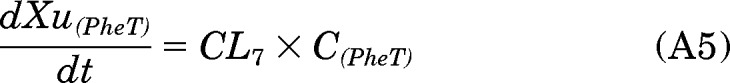 |
Equation A1 could also be written as:
The integration of eq. A6 with regard to time from t = 0 to t = ∞ provides the following expression:
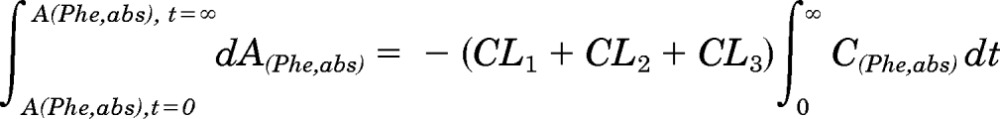 |
A(Phe,abs) integrated between t = 0 and t = ∞ is equal to the dose of [D10]Phe (D).
Therefore, D = (CL1 + CL2 + CL3) × AUC(Phe,abs).
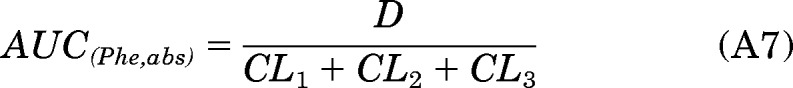 |
Likewise, the integration of eqs. A2, A3, A4, and A5 from t = 0 to t = ∞ provides the following expressions:
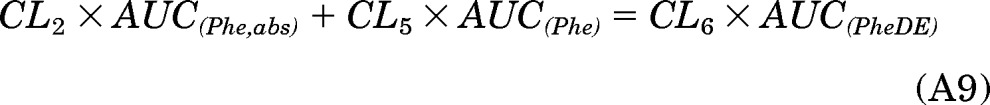 |
It is assumed that Xu(PheT),t = ∞ = Xu(PheT),t =48; that is, almost all of the [D10]PheT formed is excreted into the urine in 48 h.
The integration of eq. A3 from t = 0 to t = T (0 < T < ∞) provides the following expression:
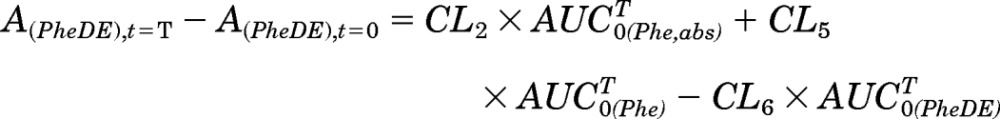 |
Because A(PheDE),t=0 equals 0, then
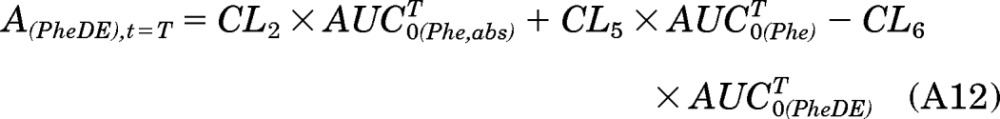 |
Likewise, the integration of eqs. A4 and A5 from t = 0 to t = T could result in eqs. A13 and A14:
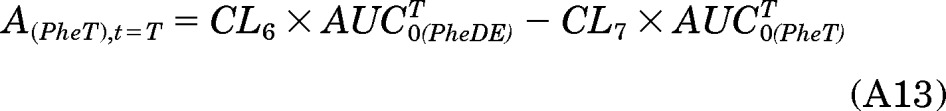 |
At time T, the [D10]PheDE that has been produced from activation can exist in three different forms: [D10]PheDE in the body, [D10]PheT in the body, and [D10]PheT in urine. Therefore, the amount of [D10]PheDE formed at time T is the sum of A(PheDE),t=T, A(PheT),t=T, and Xu(PheT), t=T.
Combining eqs. A12, A13, and A14:
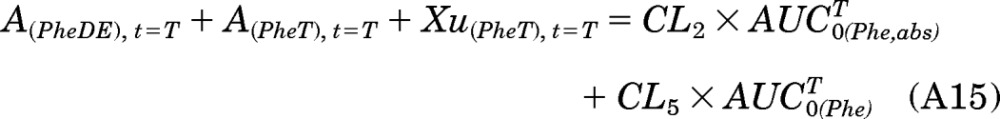 |
When time T approaches infinity, eq. A15 can be written as:
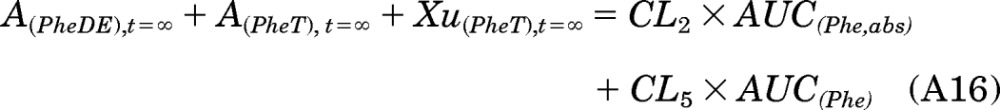 |
By definition, the left side of eq. A16 is equal to Aact:
Combining eqs. A16 and A17 to obtain eq. A18:
Substituting eqs. A7 and A8 into A18 to obtain eq. A19:
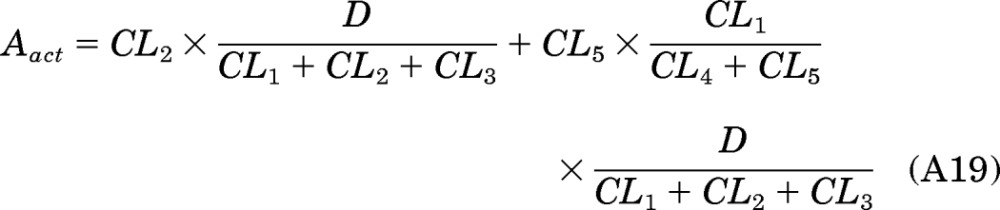 |
If fa, fact, and fm are defined as the following:
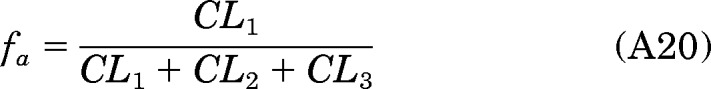 |
 |
 |
then
where fa, fact, and fm are the fraction of the dose absorbed, the fraction of the dose converted to [D10]PheDE during first-pass activation, and the fraction of [D10]Phe converted to [D10]PheDE in the systemic circulation, respectively.
Because A(PheDE),t = ∞ and A(PheT),t = ∞ both equal 0, eq. A17 could be reduced to:
In addition, it is assumed that all the [D10]PheT formed was excreted in urine in 48 h. Therefore,
The combination of eqs. A24 and A11 leads to eq. A26:
This study was supported by the National Institutes of Health National Cancer Institute [Grant CA-92025].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- PAH
- polycyclic aromatic hydrocarbon
- BaP
- benzo[α]pyrene
- Phe
- phenanthrene
- PheT
- r-1,t-2,3,c-4-tetrahydroxy-1,2,3,4-tetrahydrophenanthrene
- [D10]Phe
- deuterated Phe
- [D10]PheT
- deuterated PheT
- [D10]PheDE
- deuterated anti-1,2-dihydroxy-3,4-epoxy-1,2,3,4-tetrahydrophenanthrene
- BPDE
- benzo[α]pyrene-7,8-diol-9,10-epoxide
- GST
- glutathione transferase
- GSTM1
- GST mu 1
- PK
- pharmacokinetic
- AUC
- area under the curve
- CL
- clearance
- SBE
- single-base extension
- EPHX1
- epoxide hydrolase 1
- Aact
- total amount of [D10]PheDE formed during metabolic activation
- A(Phe,abs)
- amount of [D10]PheDE at the site of absorption
- A(Phe,abs),t = 0
- amount of [D10]PheDE at the site of absorption at time 0
- A(Phe,abs),t = T
- amount of [D10]Phe at the site of absorption at time T
- A(PheDE)
- amount of [D10]Phe in the body
- A(PhDE),t = 0
- amount of [D10]PheDE in the body at time 0
- A(PhDE),t = T
- amount of [D10]PheDE in the body at time T
- A(PhDE),t = ∞
- amount of [D10]PheDE in the body at time infinity
- A(PheT)
- amount of [D10]PheT in the body
- A(PheT),t= 0
- amount of [D10]PheT in the body at time 0
- A(PheT),t= T
- amount of [D10]PheT in the body at time T
- A(PheT),t= ∞
- amount of [D10]PheT in the body at time infinity
- AUC(Phe,abs)
- area under the C(Phe,abs)-time curve from time 0 to infinity
- AUC0T(Phe,abs)
- area under the C(Phe,abs)-time curve from time 0 to T
- AUC(Phe)
- area under the C(Phe)-time curve from time 0 to infinity
- AUC0T(Phe)
- area under the C(Phe)-time curve from time 0 to T
- AUC(PheDE)
- area under the C-(PheDE)-time curve from time 0 to infinity
- AUC0T(PheDE)
- area under the C(PheDE)-time curve from time 0 to T
- AUC(PheT)
- area under the C(PheT)-time curve from time 0 to infinity
- AUC0T(PheT)
- area under the C(PheT)-time curve from time 0 to T
- C(Phe,abs)
- concentration of [D10]Phe at the site of absorption
- C(Phe)
- plasma concentration of [D10]Phe
- C(PheDE)
- plasma concentration of [D10]PheDE
- C(PheT)
- plasma concentration of [D10]PheT
- fa
- the fraction of dose absorbed
- fact
- the fraction of dose converted to [D10]PheDE during first-pass activation
- fm
- the fraction of [D10]Phe converted to [D10]PheDE in the systemic circulation
- Xu(PheT)
- cumulative amount of [D10]PheT collected in the urine
- Xu(PheT),t = T
- amount of [D10]PheT collected in the urine at time T
- Xu(PheT),t = 48
- amount of [D10]PheT collected in the urine at 48 h
- Xu(PheT),t = ∞
- amount of [D10]PheT collected in the urine at time infinity.
Authorship Contributions
Participated in research design: Hatsukami, Hecht, and Zimmerman.
Conducted experiments: Wang, Zhong, Hochalter, Rauch, Oliver, and Jensen.
Contributed new reagents or analytic tools: Carmella and Upadhyaya.
Performed data analysis: Wang and Zimmerman.
Wrote or contributed to the writing of the manuscript: Wang and Zimmerman.
References
- Bartsch H, Nair U, Risch A, Rojas M, Wikman H, Alexandrov K. (2000) Genetic polymorphism of CYP genes, alone or in combination, as a risk modifier of tobacco-related cancers. Cancer Epidemiol Biomarkers Prev 9:3–28 [PubMed] [Google Scholar]
- Carmella SG, Chen M, Yagi H, Jerina DM, Hecht SS. (2004) Analysis of phenanthrols in human urine by gas chromatography-mass spectrometry: potential use in carcinogen metabolite phenotyping. Cancer Epidemiol Biomarkers Prev 13:2167–2174 [PubMed] [Google Scholar]
- Chu I, Ng KM, Benoit FM, Moir D. (1992) Comparative metabolism of phenanthrene in the rat and guinea pig. J Environ Sci Health B 27:729–749 [DOI] [PubMed] [Google Scholar]
- Cohen GM, Mehta R, Meredith-Brown M. (1979) Large interindividual variations in metabolism of benzo(α)pyrene by peripheral lung tissue from lung cancer patients. Int J Cancer 24:129–133 [DOI] [PubMed] [Google Scholar]
- Conney AH. (1982) Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons: G. H. A. Clowes Memorial Lecture. Cancer Res 42:4875–4917 [PubMed] [Google Scholar]
- Cooper CS, Grover PL, Sims P. (1983) The metabolism and activation of benzo[a]pyrene. Prog Drug Metab 7:295–396 [Google Scholar]
- Dipple A, Moschel RC, Bigger CAH. (1984) Polynuclear aromatic hydrocarbons, in Chemical Carcinogens, 2nd ed, ACS Monograph 182 (Searle CE. ed) pp 41–163, American Chemical Society, Washington, DC [Google Scholar]
- Geacintov NE, Cosman M, Hingerty BE, Amin S, Broyde S, Patel DJ. (1997) NMR solution structures of stereoisomeric covalent polycyclic aromatic carcinogen-DNA adducts: principles, patterns, and diversity. Chem Res Toxicol 10:111–146 [DOI] [PubMed] [Google Scholar]
- Gelboin HV. (1980) Benzo[α]pyrene metabolism, activation and carcinogenesis: role and regulation of mixed-function oxidases and related enzymes. Physiol Rev 60:1107–1166 [DOI] [PubMed] [Google Scholar]
- Gibaldi M, Perrier D. (1982) Pharmacokinetics, 2nd ed Informa Healthcare, New York [Google Scholar]
- Hecht SS. (1999) Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst 91:1194–1210 [DOI] [PubMed] [Google Scholar]
- Hecht SS. (2003) Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat Rev Cancer 3:733–744 [DOI] [PubMed] [Google Scholar]
- Hecht SS. (2010) Tobacco smoke carcinogens and lung cancer, in Chemical Carcinogenesis (Penning TM. ed) pp 53–74, Springer Verlag, New York [Google Scholar]
- Hecht SS, Carmella SG, Villalta PW, Hochalter JB. (2010) Analysis of phenanthrene and benzo[a]pyrene tetraol enantiomers in human urine: relevance to the bay region diol epoxide hypothesis of benzo[a]pyrene carcinogenesis and to biomarker studies. Chem Res Toxicol 23:900–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht SS, Carmella SG, Yoder A, Chen M, Li ZZ, Le C, Dayton R, Jensen J, Hatsukami DK. (2006) Comparison of polymorphisms in genes involved in polycyclic aromatic hydrocarbon metabolism with urinary phenanthrene metabolite ratios in smokers. Cancer Epidemiol Biomarkers Prev 15:1805–1811 [DOI] [PubMed] [Google Scholar]
- Hecht SS, Chen M, Yagi H, Jerina DM, Carmella SG. (2003) r-1,t-2,3,c-4-Tetrahydroxy-1,2,3,4-tetrahydrophenanthrene in human urine: a potential biomarker for assessing polycyclic aromatic hydrocarbon metabolic activation. Cancer Epidemiol Biomarkers Prev 12:1501–1508 [PubMed] [Google Scholar]
- Hecht SS, Villalta PW, Hochalter JB. (2008) Analysis of phenanthrene diol epoxide mercapturic acid detoxification products in human urine: relevance to molecular epidemiology studies of glutathione-S-transferase polymorphisms. Carcinogenesis 29:937–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Agency for Research on Cancer (1983) Chemical, environmental, and experimental Data, in IARC Monographs on the Evaluation of the Carcinog Risk of Chemicals to Humans, pp 419–430 International Agency for Research on Cancer, Lyon, France [Google Scholar]
- International Agency for Research on Cancer (1986) Tobacco smoking, in IARC Monographs on the Evaluation of the Carcinog Risk of Chemicals to Humans, pp 127–135 International Agency for Research on Cancer, Lyon, France [Google Scholar]
- International Agency for Research on Cancer (2004) Tobacco smoke and involuntary smoking, in IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, pp 36–40 International Agency for Research on Cancer, Lyon, France: [PMC free article] [PubMed] [Google Scholar]
- International Agency for Research on Cancer (2010) Some nonheterocyclic polycyclic aromatic hydrocarbons and some related exposures, in IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, pp 35–818 International Agency for Research on Cancer, Lyon, France [Google Scholar]
- Ishibe N, Wiencke JK, Zuo ZF, McMillan A, Spitz M, Kelsey KT. (1997) Susceptibility to lung cancer in light smokers associated with CYP1A1 polymorphisms in Mexican- and African-Americans. Cancer Epidemiol Biomarkers Prev 6:1075–1080 [PubMed] [Google Scholar]
- Jiang H, Gelhaus SL, Mangal D, Harvey RG, Blair IA, Penning TM. (2007) Metabolism of benzo[a]pyrene in human bronchoalveolar H358 cells using liquid chromatography-mass spectrometry. Chem Res Toxicol 20:1331–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawajiri K, Nakachi K, Imai K, Yoshii A, Shinoda N, Watanabe J. (1990) Identification of genetically high risk individuals to lung cancer by DNA polymorphisms of the cytochrome P450IA1 gene. FEBS Lett 263:131–133 [DOI] [PubMed] [Google Scholar]
- Kiyohara C, Otsu A, Shirakawa T, Fukuda S, Hopkin JM. (2002) Genetic polymorphisms and lung cancer susceptibility: a review. Lung Cancer 37:241–256 [DOI] [PubMed] [Google Scholar]
- Lang M, Pelkonen O. (1999) Metabolism of xenobiotics and chemical carcinogenesis, in Metabolic Polymorphisms and Susceptibility to Cancer (Vineis P, Malats N, Lang M, d'Errico A, Caporaso N, Cuzick J, Boffeta P. eds) pp 13–14 International Agency for Research on Cancer, Lyon, FR [Google Scholar]
- Laniado-Laborín R. (2010) Smoking cessation intervention: an evidence-based approach. Postgrad Med 122:74–82 [DOI] [PubMed] [Google Scholar]
- Nakachi K, Imai K, Hayashi S, Watanabe J, Kawajiri K. (1991) Genetic susceptibility to squamous cell carcinoma of the lung in relation to cigarette smoking dose. Cancer Res 51:5177–5180 [PubMed] [Google Scholar]
- Okada T, Kawashima K, Fukushi S, Minakuchi T, Nishimura S. (1994) Association between a cytochrome P450 CYPIA1 genotype and incidence of lung cancer. Pharmacogenetics 4:333–340 [DOI] [PubMed] [Google Scholar]
- Pelkonen O, Nebert DW. (1982) Metabolism of polycyclic aromatic hydrocarbons: etiologic role in carcinogenesis. Pharmacol Rev 34:189–222 [PubMed] [Google Scholar]
- Persson I, Johansson I, Lou YC, Yue QY, Duan LS, Bertilsson L, Ingelman-Sundberg M. (1999) Genetic polymorphism of xenobiotic metabolizing enzymes among Chinese lung cancer patients. Int J Cancer 81:325–329 [DOI] [PubMed] [Google Scholar]
- Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. (2000) Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ 321:323–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas M, Alexandrov K, Cascorbi I, Brockmöller J, Likhachev A, Pozharisski K, Bouvier G, Auburtin G, Mayer L, Kopp-Schneider A, et al. (1998) High benzo[a]pyrene diol-epoxide DNA adduct levels in lung and blood cells from individuals with combined CYP1A1 MspI/MspI-GSTM1*0/*0 genotypes. Pharmacogenetics 8:109–118 [PubMed] [Google Scholar]
- Rojas M, Cascorbi I, Alexandrov K, Kriek E, Auburtin G, Mayer L, Kopp-Schneider A, Roots I, Bartsch H. (2000) Modulation of benzo[a]pyrene diolepoxide-DNA adduct levels in human white blood cells by CYP1A1, GSTM1, and GSTT1 polymorphism. Carcinogenesis 21:35–41 [DOI] [PubMed] [Google Scholar]
- Schober W, Pusch G, Oeder S, Reindl H, Behrendt H, Buters JT. (2010) Metabolic activation of phenanthrene by human and mouse cytochromes P450 and pharmacokinetics in CYP1A2 knockout mice. Chem Biol Interact 183:57–66 [DOI] [PubMed] [Google Scholar]
- Shi X, Zhou S, Wang Z, Zhou Z, Wang Z. (2008) CYP1A1 and GSTM1 polymorphisms and lung cancer risk in Chinese populations: a meta-analysis. Lung Cancer 59:155–163 [DOI] [PubMed] [Google Scholar]
- Shields PG, Caporaso NE, Falk RT, Sugimura H, Trivers GE, Trump BF, Hoover RN, Weston A, Harris CC. (1993) Lung cancer, race, and a CYP1A1 genetic polymorphism. Cancer Epidemiol Biomarkers Prev 2:481–485 [PubMed] [Google Scholar]
- Shimada T. (2006) Xenobiotic-metabolizing enzymes involved in activation and detoxification of carcinogenic polycyclic aromatic hydrocarbons. Drug Metab Pharmacokinet 21:257–276 [DOI] [PubMed] [Google Scholar]
- Spiro SG, Silvestri GA. (2005) One hundred years of lung cancer. Am J Respir Crit Care Med 172:523–529 [DOI] [PubMed] [Google Scholar]
- Straif K, Baan R, Grosse Y, Secretan B, El Ghissassi F, Cogliano V, and WHO International Agency for Research on Cancer Monograph Working Group (2005) Carcinogenicity of polycyclic aromatic hydrocarbons. Lancet Oncol 6:931–932 [DOI] [PubMed] [Google Scholar]
- Szeliga J, Dipple A. (1998) DNA adduct formation by polycyclic aromatic hydrocarbon dihydrodiol epoxides. Chem Res Toxicol 11:1–11 [DOI] [PubMed] [Google Scholar]
- Tefre T, Ryberg D, Haugen A, Nebert DW, Skaug V, Brøgger A, Børresen AL. (1991) Human CYP1A1 (cytochrome P1 450) gene: lack of association between the MspI restriction fragment length polymorphism and incidence of lung cancer in a Norwegian population. Pharmacogenetics 1:20–25 [DOI] [PubMed] [Google Scholar]
- Uno S, Dalton TP, Dragin N, Curran CP, Derkenne S, Miller ML, Shertzer HG, Gonzalez FJ, Nebert DW. (2006) Oral benzo[a]pyrene in Cyp1 knockout mouse lines: CYP1A1 important in detoxication, CYP1B1 metabolism required for immune damage independent of total-body burden and clearance rate. Mol Pharmacol 69:1103–1114 [DOI] [PubMed] [Google Scholar]
- Williams JA. (2001) Single nucleotide polymorphisms, metabolic activation and environmental carcinogenesis: why molecular epidemiologists should think about enzyme expression. Carcinogenesis 22:209–214 [DOI] [PubMed] [Google Scholar]
- Xu X, Kelsey KT, Wiencke JK, Wain JC, Christiani DC. (1996) Cytochrome P450 CYP1A1 MspI polymorphism and lung cancer susceptibility. Cancer Epidemiol Biomarkers Prev 5:687–692 [PubMed] [Google Scholar]
- Zhong Y, Carmella SG, Upadhyaya P, Hochalter JB, Rauch D, Oliver A, Jensen J, Hatsukami D, Wang J, Zimmerman C, et al. (2011a) Immediate consequences of cigarette smoking: rapid formation of polycyclic aromatic hydrocarbon diol epoxides. Chem Res Toxicol 24:246–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Wang J, Carmella SG, Hochalter JB, Rauch D, Oliver A, Jensen J, Hatsukami DK, Upadhyaya P, Zimmerman C, et al. (2011b) Metabolism of [D10]phenanthrene to tetraols in smokers for potential lung cancer susceptibility assessment: comparison of oral and inhalation routes of administration. J Pharmacol Exp Ther 338:353–361 [DOI] [PMC free article] [PubMed] [Google Scholar]