

Abstract
Purpose
We tested the hypothesis that the combination of tremelimumab and interferon alfa-2b acting via different and possibly synergistic mechanisms would overcome tumor immune tolerance and lead to significant and durable clinical responses.
Patients and Methods
We conducted a phase II study in which patients were administered tremelimumab 15 mg/kg/course (three cycles [one cycle = 4 weeks]) intravenously every 12 weeks. High-dose interferon alfa-2b (HDI) was administered concurrently, including intravenous induction at 20 MU/m2/d for 5 d/wk for 4 weeks followed by maintenance at 10 MU/m2/d subcutaneously three times a week for 8 weeks per course. From course 2 onward, HDI maintenance was administered subcutaneously.
Results
Thirty-seven patients with American Joint Committee on Cancer stage IV (9M1a, 6M1b, and 22M1c) were enrolled. Two patients had previously treated brain metastases. Grades 3 and 4 toxicities included neutropenia (six patients; 17%), diarrhea/colitis (four patients; 11%), liver enzyme increase (four patients; 11%), rash (four patients; 11%), fatigue (15 patients; 40%), and anxiety/depression (five patients; 14%). Response data were available for 35 patients. The best objective response rate (RR; Response Evaluation Criteria in Solid Tumors) by intention to treat was 24% (90% CI, 13% to 36%; four complete responses [CRs] and five partial responses [PRs] that lasted 6, 6, > 12, > 14, > 18, 20, > 28, 30, and > 37 months, respectively). Fourteen patients (38%) had stable disease (SD) that lasted 1.5 to 21 months. The median progression-free survival was 6.4 months (95% CI, 3.3 to 12.1 months). The median overall survival (OS) was 21 months (95% CI, 9.5 to not reached). There was a weak association between therapy-induced autoimmunity and clinical benefits (CR/PR/SD; P = .0059), baseline C-reactive protein (CRP) less than or equal to 2.7× the upper limit of normal and clinical benefits (P = .0494) and improved probability of survival (P = .0032), and baseline lymphocyte count of at least 1,000/μL and response (CR/PR; P = .0183) and clinical benefits (CR/PR/SD; P = .0255). Biomarker associations were not significant after adjustment for multiple comparisons.
Conclusion
HDI can be administered combined with tremelimumab with acceptable toxicity and promising durable antitumor efficacy that warrant further testing in a randomized trial.
INTRODUCTION
Robust advances in our understanding of melanoma molecular biology and host immunity have opened the field of melanoma therapy onto new immunotherapeutic approaches that unlock the immune response, including cytotoxic T-cell lymphocyte-4 (CTLA-4) blockade, and molecularly targeted agents, including BRAF kinase inhibitors that have shown a significant impact on the clinical outcome.1–3 Although clinical benefits from these agents are unprecedented, they appear to be limited in duration and/or confined to subgroups of patients.
In advanced melanoma, the quality of the host immune response has been shown to be compromised, with a strong bias toward melanoma antigen-specific T helper type 2–type polarization,4 that yields a microenvironment that facilitates the progression of disease (PD).5 Strategies for overcoming tumor-induced immune suppression that build on the success of interferon alfa (IFN-α) and its immunomodulatory qualities as demonstrated in the adjuvant setting6 through the downregulation of the CTLA-4 suppressive regulatory elements are desirable.7 High-dose IFN-α (HDI) has been shown to play a critical role in the interruption of tumor immune tolerance by both improving tumor immunogenicity and increasing dendritic-cell (DC) activation and survival.7,8 IFN-α upregulates major histocompatibility complex antigen processing and co-stimulatory molecules, which leads to more efficient antigen presentation that may elicit previously low-affinity autoreactive T cells.7–9 Moreover, in their immature state, IFN-treated DCs induce a polarized T helper type 1 (Th1) cytokine microenvironment.10 Similarly, IFNs polarize lymphocytes toward the proinflammatory Th1 phenotype.11–13 This significant impact of type 1 IFNs in the cytotoxic T-cell compartment induces potent anti–tumor cell–mediated cytotoxicity,14 and promotes natural-killer cell–mediated proliferation and cytotoxicity.15 The IFN-induced Th1 bias can be detected in the circulating blood of patients with melanoma as upregulated proinflammatory cytokine response (Th1 polarization) as we have previously shown in the context of the adjuvant E1694 trial.16 In addition, locally produced type 1 IFNs induce the expression of integrins and chemokine receptors and the recruitment of natural-killer cells and macrophages that lead to Th1 rather than T helper type 2 lymphocyte traffic to the tumor site.17 This effect has been demonstrated clinically in which responding patients had significantly greater increases in intratumor CD11c+ DCs and CD3+ T cells in a neoadjuvant melanoma study of HDI.18
This potent antitumor impact of IFN-α can be suppressed by tumor tolerogenic mechanisms, which explains the limited clinical activity of IFN-α as a monotherapy in metastatic melanoma. Combination with the CTLA-4 blockade may alter this balance and downregulate CTLA-4-mediated counter regulatory mechanisms and possibly also release inhibitory influences on activated CD4 and CD8 effector cells. CTLA-4 is a key element in immune tolerance and a central negative regulator of T cell-mediated antitumor immune responses, and preclinical studies suggested that CTLA-4 serves as a natural braking mechanism for T-cell activation.19–22 The inhibitory signal produced by CTLA-4 is, therefore, blocked by anti–CTLA-4 antibodies (tremelimumab or ipilimumab), and T-cell activation is enhanced.23–27 Tremelimumab has been demonstrated to have a significant immune modulating role in which it unlocks the immune response by disrupting CTLA-4, enhances proinflammatory T-cell cytokine production,28 and increases T-cell infiltration in responding tumors.29 Therefore, we hypothesized that IFN-α and tremelimumab may have an additive or synergistic effect in the promotion of tumor elimination, which led to our additional hypothesis that the combination as tested in our phase II study would improve the clinical outcome of patients with metastatic melanoma.
PATIENTS AND METHODS
Patients
Patients 18 years of age or older were enrolled in the study if they had inoperable American Joint Committee on Cancer stage IV melanoma (cutaneous, uveal, or mucosal) and measurable disease. All patients were required to have an Eastern Cooperative Oncology Group performance status of 0 or 1 and adequate tests of hematologic, renal, and liver function, including lactate dehydrogenase (LDH) of no more than 2× the upper limit of normal (ULN). Previous adjuvant therapy or systemic therapy for advanced melanoma was allowed except for anti–CTLA-4. Patients with treated brain metastasis were eligible. Patients were ineligible if they had serious illnesses specified in the study protocol such as inflammatory bowel disease and diverticulitis. An institutional review board–approved written informed consent form was obtained from all patients.
Study Design and Treatment
This was a study of the safety and efficacy of the combination of HDI and tremelimumab. One course of therapy consisted of three cycles (one cycle = 28 days). Tremelimumab 15 mg/kg was administered intravenously at the start of the first cycle. For cycle 1, IFN-α-2b was administered intravenously at 20 MU/m2/d for 5 d/wk for 4 weeks. For cycle 2 onward, IFN-α-2b was administered subcutaneously at 10 MU/m2/d for 3 d/wk for 4 weeks. Patients without evidence of PD or limiting toxicities were offered additional courses of therapy up to a maximum of four courses.
Toxicity and Response Assessments
Descriptions and grading scales found in the National Cancer Institute Common Terminology Criteria for Adverse Events (version 3.0) were used for grading and reporting of adverse events (AEs). For the purpose of response assessment (Response Evaluation Criteria in Solid Tumors [RECIST] version 1), imaging staging studies were carried out at the end of each course (three cycles) or earlier if clinically indicated. Patients were classified as having a complete response (CR), partial response (PR), stable disease (SD), or PD.
Dose Modifications
For tremelimumab, there were no dose modifications but only delays or permanent discontinuation. Dose modifications were adopted for IFN-α-2b on the basis of published criteria.30,31
Statistical Methods
The study size was based on the therapeutic target of achieving, with acceptable toxicity, a 20% or better objective RR, CR, or PR by RECIST. We assumed a RR of 7% because the standard of care would have been too low to generate interest to continue investigation of this regimen. The study sample size was based on an optimal two-stage design in which 16 patients were to be enrolled onto stage 1. If toxicity was acceptable and two or more responses occurred in the first stage, an additional 21 patients were to be enrolled onto stage 2 (N = 37 patients). If five or more responses occurred by the end of stage 2, then we would have considered the regimen to be potentially worthy of further investigation. The type I error was 10%, and the power was 80%.
Secondary objectives included estimation of progression-free survival (PFS) and OS. Survival times of patients were measured from the initial date of treatment to the recorded date of death. Survival and PFS were estimated by using the the Kaplan-Meier method. The number of patients who experienced AEs (National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0) in each cycle of treatment and for 30 days after the last protocol treatment administered were characterized by the type of AE and grade and the time of onset in relation to the first day of therapy for each cycle. Associations between biomarkers and the primary clinical end points (response, survival, and PFS) were explored by using statistical graphics, Fisher's exact test, Wilcoxon two-sample test, or log-rank test. The statistical analysis of these biomarkers was conducted as exploratory data analyses.
Biomarkers
Selected serum biomarkers (ie, interleukin [IL]-1α, -1β, -2, -2R, -6, -8, -10, and -17, tumor necrosis factor α, IFN-α, macrophage inflammatory protein (MIP) –1α, MIP-1β, IFN-γ inducible protein 10 (IP-10), and vascular endothelial growth factor) were evaluated at baseline in 33 patients with available samples by using a multiplex system (Luminex 100 Bio-Plex System and Human 16-Plex Custom Kit; Bio-Rad, Hercules, CA). Human CRP (Invitrogen, La Jolla, CA) was assayed separately as a result of required dilution differences. In addition, serum samples of patients were tested (at baseline, 3, 6, 9, 12 months, and progression) for the presence of the following autoantibodies (enzyme-linked immunosorbent assay immunoassay kits; DiaSorin, Saluggia, Itally): antinuclear antibody, antithyroglobulin, antithyroperoxidase, and anticardiolipin antibody. Induced autoimmunity (present/absent) was defined by either the existence of antibody (during treatment) above the threshold to any one of four different antigens or the existence of immune-related AEs during treatment (ie, rash, diarrhea/colitis, hepatitis, or endocrinopathies; Table 1).
Table 1.
Adverse Events Considered Possibly, Probably, or Definitely Related to the Study Regimen (CTCAE version 3)
| Type | All Grades |
Grade 3 |
Grade 4 |
|||
|---|---|---|---|---|---|---|
| No. of Patients | % | No. of Patients | % | No. of Patients | % | |
| Immune mediated | ||||||
| Diarrhea/colitis | 21 | 57.0 | 3 | 8.0 | 1 | 2.7 |
| Hyper/pothyroidism | 2 | 5.4 | 0 | 0 | 0 | 0 |
| Hypogonadism | 1 | 2.7 | 0 | 0 | 0 | 0 |
| Hepatitis-increased AST/ALT/AP/GGT | 8 | 21.6 | 3 | 8.0 | 1* | 2.7 |
| Skin rash | 23 | 62.0 | 4 | 11.0 | 0 | 0 |
| Constitutional | ||||||
| Fatigue | 37 | 100 | 15 | 40.5 | 0 | 0 |
| Gastrointestinal | ||||||
| Nausea | 27 | 73.0 | 1 | 2.7 | 0 | 0 |
| Vomiting | 17 | 46.0 | 1 | 2.7 | 0 | 0 |
| Hematologic | ||||||
| Neutropenia | 19 | 51.4 | 5 | 13.5 | 1 | 2.7 |
| Neuropsychiatric | ||||||
| Depression/anxiety | 9 | 24.3 | 4 | 11.0 | 0 | 0 |
| Renal | ||||||
| Increased Cr/dehydration | 2 | 5.4 | 1 | 2.7 | 0 | 0 |
| Respiratory | ||||||
| Bronchospasm | 1 | 2.7 | 1 | 2.7 | 0 | 0 |
| Other | ||||||
| Cardiac arrhythmia (atrial fibrillation) | 1 | 2.7 | 1 | 2.7 | 0 | 0 |
| Increased CPK | 9 | 24.3 | 2 | 5.4 | 1 | 2.7 |
Abbreviations: AP, alkaline phosphatase; Cr, creatinine; CPK, creatine phosphokinase; CTCAE, Common Terminology Criteria for Adverse Events; GGT, gamma-glutamyltransferase.
GGT.
RESULTS
Patient Characteristics
Thirty-seven patients (23 men and 14 women), age 28 to 76 years were enrolled between November 2006 and March 2010. All patients had stage IV melanoma (nine, M1a; six, M1b; and 22, M1c), and most patients had previously received therapy (zero to five regimens). Two patients had previously treated brain metastases (Table 2).
Table 2.
Patient Demographics and Baseline Disease Characteristics (N = 37 patients)
| Variable | No. of Patients | % |
|---|---|---|
| Age, years | ||
| Median | 56 | |
| Range | 28-76 | |
| Cutaneous/unknown primary | 29 | 78 |
| Ocular | 8 | 22 |
| Sex | ||
| F | 14 | 38 |
| M | 23 | 62 |
| Performance status | ||
| 0 | 18 | 49 |
| 1 | 19 | 51 |
| Previous therapy | 22 | 60 |
| No. of previous regimens (range) | 1-5 | |
| Adjuvant IFN | 14 | 38 |
| High-dose IL-2 | 7 | 19 |
| Previous brain metastases | 2 | 5.4 |
| AJCC stage | ||
| M1a | 9 | 24 |
| M1b | 6 | 16 |
| M1c | 22 | 60 |
Abbreviations: AJCC, American Joint Committee on Cancer; IFN, interferon; IL-2, interleukin-2.
Treatment Details
Seventy-two courses of tremelimumab were administered (average of two courses per patient) as summarized in (Appendix Table A1, online only).
Efficacy
At the end of stage I enrollment (n = 16), the study met the interim analysis criterion of at least two objective responses and, therefore, moved into stage II enrollment.
Response (stages I and II)
Response data were available for 35 patients. One patient with poor performance status had worsening fatigue and depression during the HDI induction phase and was discontinued from the study without imaging studies or objective assessment of PD. Another patient with baseline symptoms of nausea and vomiting and biopsy-proven gastric metastases received 4 days of intraveneous HDI and was discontinued as a result of poor tolerance (grade 2 vomiting was the worst AE). The patient/physician decision was to resect the gastric mass that contributed to the symptoms. The best objective RR (35 evaluable patients) was 26% (90% CI, 0.14% to 0.38%; four CRs and five PRs that lasted 6, 6, > 12, > 14, > 18, 20, > 28, 30, and 37 months, respectively), including M1a (five patients), M1b (two patients), and M1c (three patients, including one uveal primary). Among these nine responders, four patients had received previous adjuvant IFN-α. It is noteworthy that seven patients had received previous high-dose IL-2, but none of these patients had a response in this study. For one additional patient, the PR status was not confirmed (unconfirmed PR) then had PD (RECIST) after which the patient was rendered disease-free (no evidence of disease [NED]) surgically. This patient continued to be NED postoperatively for greater than 16 months. Fourteen patients (38%; including four patients with uveal melanoma) had SD (which lasted 1.5 to 21 months). The disease control rate was 66% (90% CI, 0.53% to 0.79%). Another patient who had PD as the best response went on to receive 2 weeks of temozolomide and decitabine in a study and was discontinued as a result of toxicities and transferred to hospice care. This patient presented NED by positron emission tomography– computed tomography PET-CT 15 months later with no other treatment for melanoma in the interim. With the use of intention-to-treat analysis (N = 37), the RR was 24% (90% CI, 13% to 36%). The efficacy by tumor response is summarized in Table 3. The durability of responses and SD in the individual patients are summarized in Table 4. We conducted a one-tailed binomial test that the observed RR (nine of 35 patients [26%]) was better than the comparison rate of 7%. This test yielded a P < .001, and thus, we rejected the null hypothesis and claim the therapy was significantly better than the assumed uninteresting rate of 7%.
Table 3.
Efficacy Summary (stages I and II): Best Response on the Basis of 37 Patients Enrolled
| Response Status | No. of Patients | Duration (months) | Primary |
Classification |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cutaneous |
Ocular |
Unknown |
M1a |
M1b |
M1c |
|||||||||
| No. of Patients | % | No. of Patients | % | No. of Patients | % | No. of Patients | % | No. of Patients | % | No. of Patients | % | |||
| RR | ||||||||||||||
| Overall | 9* | 8 of 9 | 89 | 1 of 9 | 11 | 0 | 5 of 9 | 56 | 2 of 9 | 22 | 3 of 9 | 33 | ||
| CR | 4 of 9 | > 14 to 30 | 3 of 4 | 75 | 1 of 4 | 25 | 0 | 1 of 4 | 25 | 1 of 4 | 25 | 2 of 4 | 50 | |
| PR* | 5 of 9 | 3 to > 37 | 5 of 5 | 100 | 0 | 0 | 3 of 5 | 60 | 1 of 5 | 20 | 1 of 5 | 20 | ||
| SD | 14 | 1.5-21 | 7 of 14 | 50 | 4 of 14 | 29 | 2 of 14 | 3 of 14 | 21 | 3 of 14 | 21 | 7 of 14 | 50 | |
| PD | 12 | 8 of 12 | 67 | 3 of 12 | 25 | 0 | 1 of 12 | 8 | 1 of 12 | 8 | 9 of 12 | 75 | ||
| No response data | 2† | |||||||||||||
Abbreviations: CR, complete response; NED, no evidence of disease; PD, progression of disease; PR, partial response; RR, response rate; SD, stable disease.
One additional responder was not confirmed per Response Evaluation Criteria in Solid Tumors, had PD, and rendered surgically NED with no progression at greater than 16 months. This patient was designated SD.
Two (one cutaneous, M1c; one unknown, M1c) unknown responses.
Table 4.
Durability of Responses and Stable Disease (best radiologic response)
| Primary | Classification | Duration (months) | Comment |
|---|---|---|---|
| Responders | |||
| 1. Cutaneous | M1a | > 37 | PR → surgical CR* |
| 2. Cutaneous | M1c | 30 | CR |
| 3. Ocular | M1c | > 28 | CR |
| 4. Cutaneous | M1c | 20 | PR → PD → surgical NED > 4 months |
| 5. Cutaneous | M1a | > 18 | CR |
| 6. Cutaneous | M1b | > 12 | PR† |
| 7. Cutaneous | M1a | 6 | PR |
| 8. Cutaneous | M1a | 6 | PR |
| 9. Cutaneous | M1b | > 14 | CR |
| Durable stable disease (≥ 3 months) | |||
| 1. Unknown | M1a | 4 | |
| 2. Cutaneous | M1c | 9 | |
| 3. Ocular | M1c | 4.5 | |
| 4. Ocular | M1c | 13 | |
| 5. Cutaneous | M1b | 21 | |
| 6. Cutaneous | M1b | 4 | |
| 7. Ocular | M1c | 7 | |
| 8. Cutaneous | M1b | 4.5 | |
| 9. Cutaneous | M1a | 10.5 | |
| 10. Unknown | M1c | 4 | → Surgical NED for 5 months |
| 11. Cutaneous | M1a | 3 | Unconfirmed PR → PD → surgical NED > 16 months |
Abbreviations: CR, complete response; NED, no evidence of disease; PD, progression of disease; PR, partial response.
NED.
Likely CR; residual 4-mm lung nodule.
Survival
The median follow-up was 21 months (range, 9 to 33 months) for patients at risk of progression and 22 months (range, 15 to 44 months) for patients who were still alive. The median PFS was 6.4 months (95% CI, 3.3 to 13.1 months). The Kaplan-Meier plot of the probability of PFS is shown in Figure 1. The median OS was 21 months (95% CI, 9.5 months to not reached). The Kaplan-Meier plot of the probability of OS is shown in Figure 2.
Fig 1.

Kaplan-Meier plot of the probability of progression-free survival (PFS; N = 37). The estimated median was 6.4 months (95% CI, 3.3 to 12.1).
Fig 2.

Kaplan-Meier plot of the probability of overall survival (N = 37). The estimated median was 21 months (95% CI, 9.5 months to not reached).
We estimated the 1-year OS rate by the model proposed by Korn et al.32 The distribution of prognostic factors for 37 patients is shown in Appendix Table A2 (online only) along with the observed and predicted 1-year survival rates for each prognostic category. The predicted 1-year OS rate was 21%, although the observed rate was 62% (95% CI, 46% to 78%; P < .001).
Safety
AEs that were considered related to the study regimen are summarizes by severity in Table 1. Autoimmune toxicities as a result of tremelimumab were successfully managed with corticosteroids. Overall, toxicities were not worse than those expected with HDI or tremelimumab monotherapy.30,33
Biomarkers
The baseline lymphocyte count (absolute lymphocyte count [ALC]) and CRP were found to be weakly associated with therapeutic benefit (CR/PR/SD v PD). Similarly, baseline vascular endothelial growth factor and IL-6 were also found to be weakly associated with clinical benefit but with even lesser significance. CRP was examined at the cutoff value of 1.5× the ULN (on the basis of the report by Marshall et al34), but no significant association was detected with clinical response, clinical benefit, survival, or PFS. Because the distribution of our data suggested that CRP at 2.7× the ULN might be a cutoff value, we explored baseline CRP at ≤2.7× the ULN and found a weak association with clinical benefit (P = .05; Fisher's exact test) but not with clinical response, and we found an association with improved probability of survival (P = .003; log-rank test) but not with PFS as illustrated in Appendix Figure A1 (online only). The baseline ALC of at least 1,000/μL (n = 34 patients) was associated with response (CR/PR v SD/PD; P = .02) and clinical benefit (CR/PR/SD v PD; P = .03; Wilcoxon two-sample test) but not survival end points (Appendix Fig A2, online only). In addition, post-therapy evidence of autoimmunity was associated with clinical benefit (CR/PR/SD v PD; P = .006; Fisher's exact test; Appendix Fig A3, online only). All of these associations lost significance (all P > .05) when multiple comparisons were corrected for. We found no correlations between baseline LDH or S100B levels and any of the efficacy outcomes. Similarly, no associations were found with outcomes for baseline measurements of IL-1α, -1β, -2, -2R, -6, -8, -10, and -17, tumor necrosis factor α, IFN-α, MIP-1α, MIP-1β or IP-10.
DISCUSSION
The tested combination of HDI and tremelimumab was relatively well tolerated with AEs that were expected and manageable. The frequency of AEs was not worse than those reported with HDI, tremelimumab, or ipilimumab monotherapy.3,30
The clinical activity appeared promising by all measures that we assessed, including durable RR, PFS, OS, and 1-year OS rates as analyzed by the model proposed by Korn et al.32 Interestingly, one patient with unconfirmed PR (designated SD), who went on to progress at 3 months after grade 3 diarrhea that was managed with steroids, was rendered NED surgically. This patient remained NED at greater than 16 months. This observation suggested that surgery to render patients NED is appropriate for those who respond and then progress at solitary sites that are operable. Another patient, who had PD, received 2 weeks of temozolomide/decitabine, and was transferred to hospice care, presented NED by positron emission tomography– computed tomography 15 months later. This finding was similar to other observations reported with this class of drugs in patients with melanoma.35 These results compare favorably to monotherapy with HDI, tremelimumab,33 or ipilimumab.3 IFN-α was the first recombinant cytokine to be investigated for the therapy of metastatic melanoma and yielded RRs of approximately 16%. However, the median duration of response was only approximately 4 months. The ipilimumab-gp100 phase III study that led to recent approval of the US Food and Drug Administration of ipilimumab for metastatic melanoma randomly assigned 676 pretreated patients. The RR was 5.7% (ipilimumab plus gp100), 10.9% (ipilimumab plus placebo), and 1.5% (gp100 plus placebo). The median OS was 10.0 months (ipilimumab plus gp100), 10.1 months (ipilimumab plus placebo), and 6.4 months (gp100 plus placebo). One-year survival rates were 44% (ipilimumab plus gp100), 46% (ipilimumab plus placebo), and 25% (gp100 plus placebo).3 Our data also compare favorably to the recently reported CA184-024 phase III trial in which ipilimumab plus dacarbazine had a significant survival benefit over dacarbazine alone as a first-line treatment in metastatic melanoma (median OS, 11.2 v 9.1 months; median PFS, 2.8 v 2.6 months; RR, 15% v 10%).2 Tremelimumab 15 mg/kg every 90 days (up to four cycles) was tested in a second-line phase II study (A3671008) in inoperable, American Joint Committee on Cancer stage III/IV melanoma (N = 246).36 The objective RR was 7%, median OS was 10.1 month, and 1-year survival rate was 41%.36 In a subsequent phase III trial (A3671009) in treatment-naive advanced melanoma that compared tremelimumab to dacarbazine/temozolomide, the median OS was 11.8 months.37 Therefore, we concluded that the level of activity noted in our single-arm phase II study warrants additional testing in a randomized trial. Because of the interval approval of the US Food and Drug Administration of ipilimumab for therapy of advanced melanoma, this study, by extension, argues for the evaluation of ipilimumab in combination with IFN-α in a randomized phase II study.
The identification of biomarkers that predict therapeutic benefits of these agents would enable the improved selection of patients so that only those patients who are most likely to benefit from therapy would be treated, which would spare patients who are less likely to benefit from the significant toxicities associated with treatment. This is especially important for anti–CTLA-4 monoclonal antibody therapy and for IFN-α treatment, both of which induced durable clinical benefits in a group of patients. We have explored a panel of candidate biomarkers at baseline for their prognostic and predictive value. These biomarkers were selected on the basis of previous supporting reports. We found a weak association between autoimmunity and clinical benefit. This association lost significance when multiple comparisons were corrected for. Our hypothesis on the basis of similar observations38,39 was that the prevention of melanoma relapse and mortality with IFN is associated with immune modulation that may increase resistance to melanoma.38,40–52 Furthermore, the immunotherapeutic induction of autoimmunity may provide a useful surrogate biomarker of therapeutic benefit in studies of autoimmunity and its genetic determinants that may help identify patients more likely to benefit from immunotherapies associated with autoimmunity. We believe that this association needs to be explored further in larger studies that are adequately powered for this purpose.
For the first detection of melanoma stage IV disease, CRP has been shown to be potentially superior to conventional LDH measurement.53 The potential role of CRP as a mediator of immune tolerance is also interesting. CRP binds to phosphocholine in damaged membranes where it increases clearance of apoptotic cells and binds to nuclear antigens, and by masking autoantigens from the immune system or enhancing their clearance, CRP may prevent autoimmunity.54 Interestingly, a study that used a human hepatoma cell line showed that IFN-α inhibited CRP promoter activity and CRP secretion.55 In addition, CRP was reported to predict response in a phase III study that tested tremelimumab.34 In this study, the association we found with clinical benefit and improved survival lost significance when multiple comparisons were corrected for. Therefore, additional exploration in larger studies that are adequately powered for this purpose is important to better define the role of CRP.
In a pooled analysis of three studies that tested ipilimumab in metastatic melanoma, a higher ALC was significantly associated with clinical activity.56,57 Similarly, in another analysis of 51 patients who received ipilimumab, the ALC also correlated with clinical benefit. Patients with an ALC of at least 1,000/μL after two ipilimumab doses had a significantly improved clinical benefit rate and median OS.58 In this study, no patient with an ALC less than 1,550/μL had an objective response, and no patient with an ALC less than 1,200/μL had either an objective response or SD by RECIST. A baseline ALC ≥ 1,000/μL showed an association with response and clinical benefit that lost significance when multiple comparisons were corrected for. Again, additional exploration of the predictive role of the ALC in larger studies is indicated. To date, the reports on the ALC are interesting but weak, and it is important to explore the impact of this and similar regimens on specific T-cell components, including helper, cytotoxic, and regulatory, tumor antigen-specific T-cell reactivity, and myeloid-derived suppressor cell activity.59,60 Through a deepening of our understanding mechanistically, our work may lead to improved designs of therapeutic combinations in our quest to safely and definitively overcome melanoma immune tolerance.
In conclusion, this study met our phase II criteria for efficacy. The combination of HDI and tremelimumab had tolerable and manageable toxicity that was acceptable in relation to the significant therapeutic benefit observed. Therefore, testing in a randomized setting is warranted.
Acknowledgment
We thank Pfizer Oncology for their support and Pawel Kalinski, MD, and Lisa Butterfield, PhD, for their valuable critical editorial input.
Appendix
Fig A1.
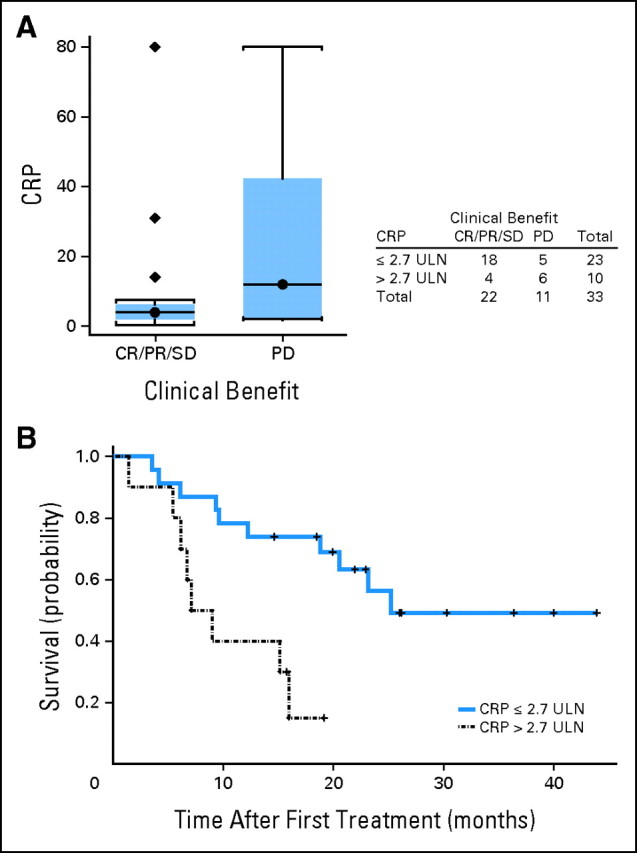
(A) Distribution of baseline C-reactive protein (CRP) by clinical benefit (complete response [CR]/partial response [PR]/stable disease [SD] v progression of disease [PD]) and (B) the probability of survival (CRP, ≤ 2.7× v > 2.7× the upper limit of normal [ULN]).
Fig A2.

Distribution of the baseline absolute lymphocyte count (ALC) by (A) response (complete response [CR]/partial response [PR] v stable disease [SD] v progression of disease [PD]) and by (B) clinical benefit (CR/PR/SD v PD).
Fig A3.

Distribution of patients with therapy-induced autoimmunity by clinical benefit (complete response [CR]/partial response [PR]/stable disease [SD] v progression of disease [PD]). Among 33 patients tested, 23 patients had evidence of induced autoimmunity, of whom, 19 patients had CR, PR, or SD.
Table A1.
Treatment Details
| Course | No. of Patients Treated | % | No. of Patients Off Study After Treatment | % | PD as Reason for DC |
Toxicity as Reason for DC |
Other Reason for DC* |
|||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of Patients | % | No. of Patients | % | No. of Patients | % | |||||
| 1 | 37 of 37 | 100 | 20 of 37 | 54 | 12 of 20 | 60 | 4 of 20 | 20 | 4 of 20 | 20 |
| 2 | 17 of 37 | 46 | 7 of 17 | 41 | 6 of 7 | 86 | 0 | 1 of 7 | 14 | |
| 3 | 10 of 37 | 27 | 2 | 2 of 2 | 100 | 0 | 0 | |||
| 4 | 8 of 37 | 22 | ||||||||
Abbreviations: DC, discontinued treatment; PD, progression of disease.
Included poor performance status (one patient), patient/physician decision to pursue surgery (two patients), second malignancy (one patient), and early (ie, still receiving course two of therapy at time of data cutoff; one patient).
Table A2.
One-Year Survival Rate: Observed v Predicted as of February 16, 2011
| Sex | PS | Visceral Disease | Total | No. of Patients Alive at 1 Year | Observed Rate (%) | Predicted Rate (%) |
|---|---|---|---|---|---|---|
| M | 0 | N | 3 | 2 | 67 | 35 |
| M | 0 | Y | 11 | 9 | 82 | 22 |
| M | 1 | N | 3 | 3 | 100 | 17 |
| M | 1 | Y | 6 | 2 | 33 | 10 |
| F | 0 | N | 0 | 0 | 49 | |
| F | 0 | Y | 4 | 2 | 50 | 33 |
| F | 1 | N | 3 | 2 | 76 | 27 |
| F | 1 | Y | 7 | 3 | 43 | 16 |
NOTE: The 1-year survival rate was estimated according to the model proposed by Korn et al (Korn EL, Liu PY, Lee SJ, et al: J Clin Oncol 26:527-534, 2008). The 1-year survival rate predicted by the model of Korn et al was 21%. Thirty-seven patients were analyzed (23 patients were alive at 1 year, and 14 patients were dead at 1 year). The observed 1-year survival rate was 62% (95% CI, 46% to 78%). By using the one-tailed hypothesis test, the observed rate was better than predicted (21%; P < .001).
Abbreviations: N, no; PS, performance status; Y, yes.
Footnotes
Supported by a grant from Pfizer Oncology.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical trial information can be found for the following: NCT00610857.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Ahmad A. Tarhini, Pfizer (C); Stergios J. Moschos, Genentech/Roche (C); John M. Kirkwood, GlaxoSmithKline (C), Merck (C) Stock Ownership: None Honoraria: John M. Kirkwood, Genentech/Roche, Merck Research Funding: Ahmad A. Tarhini, Merck, Pfizer; Hussein A. Tawbi, Merck Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Ahmad A. Tarhini, John M. Kirkwood
Administrative support: Ahmad A. Tarhini
Provision of study materials or patients: Ahmad A. Tarhini, Stergios J. Moschos, Hussein A. Tawbi, John M. Kirkwood
Collection and assembly of data: Ahmad A. Tarhini, John Cherian, Stergios J. Moschos, Hussein A. Tawbi, Yongli Shuai, Cindy Sander, John M. Kirkwood
Data analysis and interpretation: Ahmad A. Tarhini, William E. Gooding, John M. Kirkwood
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 3.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tatsumi T, Kierstead LS, Ranieri E, et al. Disease-associated bias in T helper type 1 (Th1)/Th2 CD4(+) T cell responses against MAGE-6 in HLA-DRB10401(+) patients with renal cell carcinoma or melanoma. J Exp Med. 2002;196:619–628. doi: 10.1084/jem.20012142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishimura T, Nakui M, Sato M, et al. The critical role of Th1-dominant immunity in tumor immunology. Cancer Chemother Pharmacol. 2000;46(suppl):S52–S61. doi: 10.1007/pl00014051. [DOI] [PubMed] [Google Scholar]
- 6.Kirkwood JM, Strawderman MH, Ernstoff MS, et al. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: The Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996;14:7–17. doi: 10.1200/JCO.1996.14.1.7. [DOI] [PubMed] [Google Scholar]
- 7.Kirkwood JM, Richards T, Zarour HM, et al. Immunomodulatory effects of high-dose and low-dose interferon alpha2b in patients with high-risk resected melanoma: The E2690 laboratory corollary of intergroup adjuvant trial E1690. Cancer. 2002;95:1101–1112. doi: 10.1002/cncr.10775. [DOI] [PubMed] [Google Scholar]
- 8.Wang W, Edington HD, Rao UN, et al. Modulation of signal transducers and activators of transcription 1 and 3 signaling in melanoma by high-dose IFNalpha2b. Clin Cancer Res. 2007;13:1523–1531. doi: 10.1158/1078-0432.CCR-06-1387. [DOI] [PubMed] [Google Scholar]
- 9.Paquette RL, Hsu NC, Kiertscher SM, et al. Interferon-alpha and granulocyte-macrophage colony- stimulating factor differentiate peripheral blood monocytes into potent antigen-presenting cells. J Leukoc Biol. 1998;64:358–367. doi: 10.1002/jlb.64.3.358. [DOI] [PubMed] [Google Scholar]
- 10.Parlato S, Santini SM, Lapenta C, et al. Expression of CCR-7, MIP-3beta, and Th-1 chemokines in type I IFN-induced monocyte-derived dendritic cells: Importance for the rapid acquisition of potent migratory and functional activities. Blood. 2001;98:3022–3029. doi: 10.1182/blood.v98.10.3022. [DOI] [PubMed] [Google Scholar]
- 11.Brinkmann V, Geiger T, Alkan S, et al. Interferon alpha increases the frequency of interferon gamma-producing human CD4+ T cells. J Exp Med. 1993;178:1655–1663. doi: 10.1084/jem.178.5.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wenner CA, Güler ML, Macatonia SE, et al. Roles of IFN-gamma and IFN-alpha in IL-12-induced T helper cell-1 development. J Immunol. 1996;156:1442–1447. [PubMed] [Google Scholar]
- 13.Rogge L, Barberis-Maino L, Biffi M, et al. Selective expression of an interleukin-12 receptor component by human T helper 1 cells. J Exp Med. 1997;185:825–831. doi: 10.1084/jem.185.5.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palmer KJ, Harries M, Gore ME, et al. Interferon-alpha (IFN-alpha) stimulates anti-melanoma cytotoxic T lymphocyte (CTL) generation in mixed lymphocyte tumour cultures (MLTC) Clin Exp Immunol. 2000;119:412–418. doi: 10.1046/j.1365-2249.2000.01159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carballido JA, Moltó LM, Manzano L, et al. Interferon-alpha-2b enhances the natural killer activity of patients with transitional cell carcinoma of the bladder. Cancer. 1993;72:1743–1748. doi: 10.1002/1097-0142(19930901)72:5<1743::aid-cncr2820720538>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 16.Yurkovetsky ZR, Kirkwood JM, Edington HD, et al. Multiplex analysis of serum cytokines in melanoma patients treated with interferon-alpha2b. Clin Cancer Res. 2007;13:2422–2428. doi: 10.1158/1078-0432.CCR-06-1805. [DOI] [PubMed] [Google Scholar]
- 17.Tough DF. Type I interferon as a link between innate and adaptive immunity through dendritic cell stimulation. Leuk Lymphoma. 2004;45:257–264. doi: 10.1080/1042819031000149368. [DOI] [PubMed] [Google Scholar]
- 18.Moschos SJ, Edington HD, Land SR, et al. Neoadjuvant treatment of regional stage IIIB melanoma with high-dose interferon alfa-2b induces objective tumor regression in association with modulation of tumor infiltrating host cellular immune responses. J Clin Oncol. 2006;24:3164–3171. doi: 10.1200/JCO.2005.05.2498. [DOI] [PubMed] [Google Scholar]
- 19.Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily–CTLA-4. Nature. 1987;328:267–270. doi: 10.1038/328267a0. [DOI] [PubMed] [Google Scholar]
- 20.Khattri R, Auger JA, Griffin MD, et al. Lymphoproliferative disorder in CTLA-4 knockout mice is characterized by CD28-regulated activation of Th2 responses. J Immunol. 1999;162:5784–5791. [PubMed] [Google Scholar]
- 21.Tivol EA, Borriello F, Schweitzer AN, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 22.Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 23.Lin H, Rathmell JC, Gray GS, et al. Cytotoxic T lymphocyte antigen 4 (CTLA4) blockade accelerates the acute rejection of cardiac allografts in CD28-deficient mice: CTLA4 can function independently of CD28. J Exp Med. 1998;188:199–204. doi: 10.1084/jem.188.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hurwitz AA, Foster BA, **Kwon ED, et al. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res. 2000;60(9):2444–2448. [PubMed] [Google Scholar]
- 26.Kwon ED, Foster BA, Hurwitz AA, et al. Elimination of residual metastatic prostate cancer after surgery and adjunctive cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) blockade immunotherapy. Proc Natl Acad Sci U S A. 1999;96:15074–15079. doi: 10.1073/pnas.96.26.15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190:355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanson DC, Canniff PC, Primiano MJ, et al. Preclinical in vitro characterization of anti-CTLA4 therapeutic antibody CP-675,206. Proc Am Assoc Cancer Res. 2004;45 abstr 3802. [Google Scholar]
- 29.Ribas A, DT, Comin-Anduix B, de la Rocha P, et al. Changes in intratumoral immune cell infiltrates, Foxp3 and indoleamine 2, 3-dioxygenase (IDO) expression with the CTLA4 blocking MAB CP-675,206. J Immunother. 2006;29:636. [Google Scholar]
- 30.Kirkwood JM, Bender C, Agarwala S, et al. Mechanisms and management of toxicities associated with high-dose interferon alfa-2b therapy. J Clin Oncol. 2002;20:3703–3718. doi: 10.1200/JCO.2002.03.052. [DOI] [PubMed] [Google Scholar]
- 31.Hauschild A, Gogas H, Tarhini A, et al. Practical guidelines for the management of interferon-alpha-2b side effects in patients receiving adjuvant treatment for melanoma: Expert opinion. Cancer. 2008;112:982–994. doi: 10.1002/cncr.23251. [DOI] [PubMed] [Google Scholar]
- 32.Korn EL, Liu PY, Lee SJ, et al. Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. J Clin Oncol. 2008;26:527–534. doi: 10.1200/JCO.2007.12.7837. [DOI] [PubMed] [Google Scholar]
- 33.Tarhini AA, Kirkwood JM. Tremelimumab (CP-675,206): A fully human anticytotoxic T lymphocyte-associated antigen 4 monoclonal antibody for treatment of patients with advanced cancers. Expert Opin Biol Ther. 2008;8:1583–1593. doi: 10.1517/14712598.8.10.1583. [DOI] [PubMed] [Google Scholar]
- 34.Marshall M, Ribas A, Huang B. Evaluation of baseline serum C-reactive protein (CRP) and benefit from tremelimumab compared to chemotherapy in first-line melanoma. J Clin Oncol. 2010;28:231s. abstr 2609. [Google Scholar]
- 35.Wolchok JD, Ibrahim R, DePril V, et al. Antitumor response and new lesions in advanced melanoma patients on ipilimumab treatment. J Clin Oncol. 2008;26(suppl; abstr 3020):137s. [Google Scholar]
- 36.Kirkwood JM, Lorigan P, Hersey P, et al. A phase II trial of tremelimumab (CP-675,206) in patients with advanced refractory or relapsed melanoma. J Clin Oncol. 2008;26(suppl; abstr 9023):488s. doi: 10.1158/1078-0432.CCR-09-2033. [DOI] [PubMed] [Google Scholar]
- 37.Ribas A, Hauschild A, Kefford R, et al. Phase III, open-label, randomized, comparative study of tremelimumab (CP-675,206) and chemotherapy (temozolomide [TMZ] or dacarbazine [DTIC]) in patients with advanced melanoma. J Clin Oncol. 2008;26(suppl; abstr LBA9011):485s. [Google Scholar]
- 38.Gogas H, Ioannovich J, Dafni U, et al. Prognostic significance of autoimmunity during treatment of melanoma with interferon. N Engl J Med. 2006;354:709–718. doi: 10.1056/NEJMoa053007. [DOI] [PubMed] [Google Scholar]
- 39.Krauze MT, Tarhini A, Gogas H, et al. Prognostic significance of autoimmunity during treatment of melanoma with interferon. Semin Immunopathol. 2011;33:385–391. doi: 10.1007/s00281-011-0247-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Atkins MB, Mier JW, Parkinson DR, et al. Hypothyroidism after treatment with interleukin-2 and lymphokine-activated killer cells. N Engl J Med. 1988;318:1557–1563. doi: 10.1056/NEJM198806163182401. [DOI] [PubMed] [Google Scholar]
- 41.Weijl NI, van der Harst D, Brand A, et al. Hypothyroidism during immunotherapy with interleukin-2 is associated with antithyroid antibodies and response to treatment. J Clin Oncol. 1993;11:1376–1383. doi: 10.1200/JCO.1993.11.7.1376. [DOI] [PubMed] [Google Scholar]
- 42.Scalzo S, Gengaro A, Boccoli G, et al. Primary hypothyroidism associated with interleukin-2 and interferon alpha-2 therapy of melanoma and renal carcinoma. Eur J Cancer. 1990;26:1152–1156. doi: 10.1016/0277-5379(90)90275-x. [DOI] [PubMed] [Google Scholar]
- 43.Krouse RS, Royal RE, Heywood G, et al. Thyroid dysfunction in 281 patients with metastatic melanoma or renal carcinoma treated with interleukin-2 alone. J Immunother Emphasis Tumor Immunol. 1995;18:272–278. doi: 10.1097/00002371-199511000-00008. [DOI] [PubMed] [Google Scholar]
- 44.Phan GQ, Attia P, Steinberg SM, et al. Factors associated with response to high-dose interleukin-2 in patients with metastatic melanoma. J Clin Oncol. 2001;19:3477–3482. doi: 10.1200/JCO.2001.19.15.3477. [DOI] [PubMed] [Google Scholar]
- 45.Becker JC, Winkler B, Klingert S, et al. Antiphospholipid syndrome associated with immunotherapy for patients with melanoma. Cancer. 1994;73(6):1621–1624. doi: 10.1002/1097-0142(19940315)73:6<1621::aid-cncr2820730613>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 46.Rosenberg SA, White DE. Vitiligo in patients with melanoma: Normal tissue antigens can be targets for cancer immunotherapy. J Immunother Emphasis Tumor Immunol. 1996;19:81–84. [PubMed] [Google Scholar]
- 47.Franzke A, Peest D, Probst-Kepper M, et al. Autoimmunity resulting from cytokine treatment predicts long-term survival in patients with metastatic renal cell cancer. J Clin Oncol. 1999;17:529–533. doi: 10.1200/JCO.1999.17.2.529. [DOI] [PubMed] [Google Scholar]
- 48.Phan GQ, Yang JC, Sherry RM, Hwu P, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2003;100:8372–8377. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanderson K, Scotland R, Lee P, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. 2005;23:741–750. doi: 10.1200/JCO.2005.01.128. [DOI] [PubMed] [Google Scholar]
- 50.Dranoff G, Jaffee E, Lazenby A, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A, 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ribas A, Camacho LH, Lopez-Berestein, et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J Clin Oncol. 2005;23:8968–8977. doi: 10.1200/JCO.2005.01.109. [DOI] [PubMed] [Google Scholar]
- 52.Stuckert JJ, Tarhini AA, Lee S, et al. Interferon alfa-induced autoimmunity and serum S100 levels as predictive and prognostic biomarkers in high-risk melanoma in the ECOG-intergroup phase II trial E2696. J Clin Oncol. 2007;25(suppl; abstr 8506):473s. [Google Scholar]
- 53.Deichmann M, Kahle B, Moser K, et al. Diagnosing melanoma patients entering American Joint Committee on Cancer stage IV, C-reactive protein in serum is superior to lactate dehydrogenase. Br J Cancer. 2004;91:699–702. doi: 10.1038/sj.bjc.6602043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marnell L, Mold C, Du Clos TW. C-reactive protein: Ligands, receptors and role in inflammation. Clin Immunol. 2005;117:104–111. doi: 10.1016/j.clim.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 55.Enocsson H, Sjöwall C, Skogh T, et al. Interferon-alpha mediates suppression of C-reactive protein: Explanation for muted C-reactive protein response in lupus flares? Arthritis Rheum. 2009;60:3755–3760. doi: 10.1002/art.25042. [DOI] [PubMed] [Google Scholar]
- 56.Hamid O, Chasalow SD, Tsuchihashi Z, et al. Association of baseline and on-study tumor biopsy markers with clinical activity in patients (pts) with advanced melanoma treated with ipilimumab. J Clin Oncol. 2009;27(suppl; abstr 9008):463s. [Google Scholar]
- 57.Hamid O. Dose effect of ipilimumab in patients with advanced melanoma: Results from a phase II, randomized, dose-ranging study. J Clin Oncol. 2008;26(suppl; abstr 9025):489s. [Google Scholar]
- 58.Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: A randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155–164. doi: 10.1016/S1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]
- 59.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Strauss L, Bergmann C, Szczepanski M, et al. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin Cancer Res. 2007;13:4345–4354. doi: 10.1158/1078-0432.CCR-07-0472. [DOI] [PubMed] [Google Scholar]