

Abstract
The ability of pathogenesis-related proteins of family 10 to bind a broad spectrum of ligands is considered to play a key role for their physiological and pathological functions. In particular, Bet v 1, an archetypical allergen from birch pollen, is described as a highly promiscuous ligand acceptor. However, the detailed recognition mechanisms, including specificity factors discriminating binding properties of naturally occurring Bet v 1 variants, are poorly understood.
Here, we report crystal structures of Bet v 1 variants in complex with an array of ligands at a resolution of up to 1.2 Å. Residue 30 within the hydrophobic pocket not only discriminates in high and low IgE binding Bet v 1 isoforms but also induces a drastic change in the binding mode of the model ligand deoxycholate. Ternary crystal structure complexes of Bet v 1 with several ligands together with the fluorogenic reporter 1-anilino-8-naphthalene sulfonate explain anomalous fluorescence binding curves obtained from 1-anilino-8-naphthalene sulfonate displacement assays. The structures reveal key interaction residues such as Tyr83 and rationalize both the binding specificity and promiscuity of the so-called hydrophobic pocket in Bet v 1.
The intermolecular interactions of Bet v 1 reveal an unexpected complexity that will be indispensable to fully understand its roles within the physiological and allergenic context.
Abbreviations: ANS, 1-anilino-8-naphthalene sulfonate; BRA, brassinolide; DXC, deoxycholate; iDXC, inner deoxycholate; oDXC, outer deoxycholate; LPS, lipopolysaccharide; MPD, 2-methyl-2,4-pentanediol; NDSB-256, non-detergent sulfobetaine 256; PR-10, pathogenesis-related protein 10; PDB, Protein Data Bank
Keywords: molecular allergenicity, ANS displacement assay, structure–allergenicity relationship, binding specificity and promiscuity, dressed allergens
Graphical Abstract

Highlights
► Ligand binding to Bet v 1 may contribute to explain its allergenicity. ► High-resolution structures reveal the binding mode of diverse ligands to Bet v 1. ► Residue 30 starkly influences the binding properties of different Bet v 1 isoforms. ► Ternary complexes with diverse ligands explain anomalous fluorescence binding curves. ► Betv1 isoforms differ in ligand binding, which may translate into their allergenicity.
Introduction: Background and Significance
Proteins belonging to the pathogenesis-related protein 10 (PR-10) family are found in virtually all studied plants.1 They are assumed to play key roles in diverse plant defense mechanisms, because they are commonly upregulated in response to diverse danger signals (e.g., pathogen infection).2–5
The 17.5 kDa protein Bet v 1 is the best known representative of the PR-10 family. It consists of different naturally occurring isoforms, encoded by seven genes, sharing more than 95% sequence identity.6 Despite being, by far, the most abundant protein in birch pollen, the physiological function of Bet v 1 still remains elusive. As the major pollen allergen from the White birch tree (Betula pendula), Bet v 1 represents one of the best characterized model allergens in immunology. It is the causative agent of diverse allergic reactions such as pollinosis and allergic rhinitis. Worldwide, an estimated 100 million people are sensitized to Bet v 1, especially in Northern America and Western Europe. In some countries, more than 90% of all birch-pollen-sensitized patients exhibit serum IgE reactivity towards Bet v 1.7
Based on their IgE-binding capacity, Bet v 1 isoforms were divided into hyper- and hypoallergenic variants.8 Despite their high sequence similarity, such differences in IgE-binding activity are a common characteristic within the PR-10 family: some of these structurally, closely related proteins are potent allergens, whereas others are in fact nonallergenic. Crystal structures of both high and low IgE binding Bet v 1 isoforms have been determined.9,10 However, the molecular mechanisms rendering these proteins allergenic remained so far elusive.
The determination of the crystal structure of a Bet v 1a variant in 1996 and the identification of the hydrophobic cavity marked a milestone in understanding of its function.9 In 2002, the affinity of Bet v 1 towards a rather broad spectrum of ligands was described using a fluorescent assay.11
Hereby, the strong fluorescence increase of 1-anilino-8-naphthalene sulfonate (ANS) upon binding to hydrophobic protein patches is exploited. With the use of an ANS displacement assay, the binding of several—potentially—physiologically relevant ligands to the hydrophobic cavity of Bet v 1 could be identified. In 2003, the crystal structure of Bet v 1l in complex with the steroid deoxycholate (DXC) was solved.10 These results suggested that the physiological role of Bet v 1 is most likely the binding, transport, or storage of small lipidic plant mediators such as steroids, cytokinins, or flavonoids. In accordance with that, several crystal structures of structurally closely related proteins with ligands bound in the hydrophobic cavity were determined, including PR-10 protein LIPR-10.2B from Lupinus luteus in complex with trans-zeatin and N,N′-diphenylurea,12,13 as well as the structural homologue VrCSBP from Vigna radiata in complex with trans-zeatin.14
Further support for the importance of its ligand binding properties was deduced from structural relation of Bet v 1 with the START domain of human MLN64 protein, which is involved in steroid binding.15
The accumulated, massive evidence for physiologically relevant ligand binding of Bet v 1 is contrasted by a lack of structural and mechanistic understanding of the binding properties: There is nothing known about differences in ligand acceptance to distinct Bet v 1 isoforms. Such studies would be particularly important as the natural isoforms also differ in amino acids forming the surface of the hydrophobic binding pocket. Similarly, it is unknown whether ligand binding induces (possibly isoform-specific) conformational changes within Bet v 1, which may again influence its allergenic activity. Moreover, ANS displacement assay data were controversial for some ligands, calling for detailed structural information.
To address such questions, we determined a set of high-resolution complex structures comprising the high IgE binding Bet v 1 isoforms a and j. Using DXC as a model ligand, we identified residue 30 as crucial for ligand coordination differences in high and low IgE binding Bet v 1 isoforms. We report ternary binding of different ligands simultaneously to Bet v 1a, resulting in changes in both ligand conformation and binding sites compared to the binary complexes. These findings result in a detailed chemical mapping of the binding pocket, illustrating the complexity of ligand binding to Bet v 1.
Results
Quality of the structure
The presented Bet v 1 structures are solved in a resolution range from 1.16 to 2.10 Å with similar overall crystal characteristics (Tables 1 and 2). All structures exhibit the classical Bet v 1 fold, consisting of a seven-stranded antiparallel β-sheet (β1–β7), two short α-helices (α1 and α2) connecting β1 and β2, a long C-terminal α-helix (α3), and the glycine-rich loop motif between β2 and β3 (Fig. 1). The arrangement of these structural features builds up a large hydrophobic cavity (Fig. 1a and b). Ligand binding does not affect the overall fold of the protein. However, minor local rearrangements were observed upon ligand binding, as described below.
Table 1.
Overview of crystal structures and ligands
| Bet v 1 isoform | PDB ID | Resolution (Å) | Ligand | Ligand structure |
|---|---|---|---|---|
| Bet v 1a | 4A88 | 1.51 | — | — |
| Bet v 1j | 4A8U | 1.16 | — | |
| Bet v 1a | 4A80 | 1.96 | ANS | 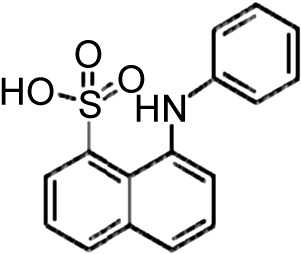 |
| Bet v 1j | 4A8V | 1.23 | ANS | |
| Bet v 1a | 4A81 | 2.05 | ANS and DXC | |
| Bet v 1a | 4A83 | 1.54 | DXC | 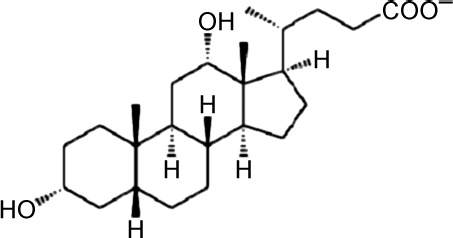 |
| Bet v 1a F30V | 4A84 | 1.50 | DXC | |
| Bet v 1a | 4A87 | 1.24 | Naringenin | 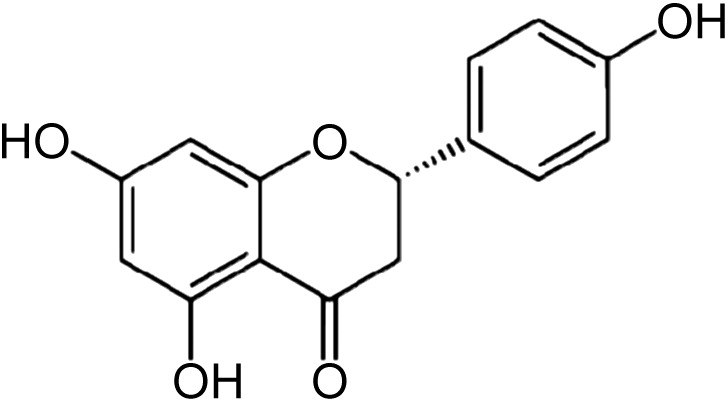 |
| Bet v 1a | 4A85 | 1.40 | Kinetin | 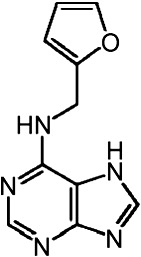 |
| Bet v 1a | 4A86 | 1.58 | ANS and kinetin | |
| Bet v 1a | 4A8G | 2.10 | NDSB-256 | 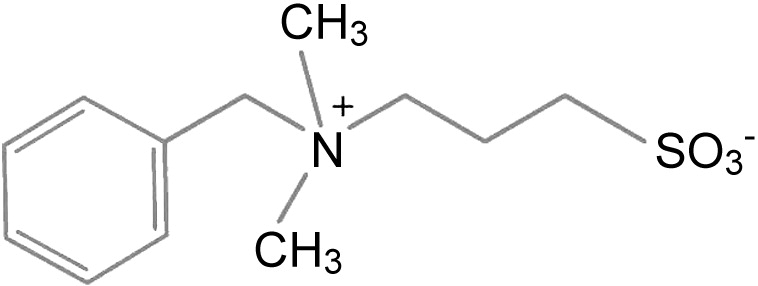 |
Table 2.
X-ray data collection and model refinement statistics
| Bet v 1 isoform and ligand | A | J | A DXC | A F30V DXC | A ANS | J ANS | A Naringenin | A Kinetin | A ANS kinetin | A ANS DXC | A NDSB-256 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accession number | 4A88 | 4A8U | 4A83 | 4A84 | 4A80 | 4A8V | 4A87 | 4A85 | 4A86 | 4A81 | 4A8G |
| Data collection | |||||||||||
| Space groupCell | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 |
| X-ray source | Synchro | Synchro | Synchro | Synchro | Synchro | Synchro | Synchro | Synchro | Synchro | Rot. anode | Rot. anode |
| Cell dimensions | |||||||||||
| 1000 | |||||||||||
| a (Å) | 32.6 | 32.7 | 32.6 | 32.6 | 32.7 | 32.8 | 32.6 | 32.7 | 32.6 | 32.6 | 32.7 |
| b (Å) | 55.4 | 55.7 | 56.1 | 56.0 | 55.6 | 56.0 | 55.4 | 55.9 | 55.6 | 55.8 | 55.7 |
| c (Å) | 37.9 | 38.0 | 38.1 | 38.0 | 38.0 | 38.1 | 37.9 | 38.0 | 38.0 | 37.8 | 38.0 |
| α/β/γ (°) | 90/93/90 | 90/93/90 | 90/93/90 | 90/93/90 | 90/93/90 | 90/93/90 | 90/93/90 | 90/93/90 | 90/93/90 | 90/93/90 | 90/93/90 |
| Wavelength (Å) | 0.91841 | 0.91841 | 0.91841 | 0.91841 | 0.91841 | 0.91841 | 0.91841 | 0.91841 | 0.91841 | 1.5418 | 1.5418 |
| Number of unique reflections | 20,772 | 46,196 | 20,001 | 21,271 | 8488 | 39,178 | 37,419 | 22,328 | 17,743 | 7579 | 7052 |
| Resolution (Å) | 37.8–1.51 | 38.0–1.16 | 38.0–1.54 | 32.5–1.50 | 37.9–1.96 | 28.2–1.23 | 25.2–1.24 | 32.6–1.40 | 37.9–1.59 | 37.8–2.05 | 38.0–2.10 |
| Rmerge (%) | 0.050 (0.130) | 0.040 (0.140) | 0.040 (0.090) | 0.110 (0.740) | 0.040 (0.060) | 0.080 (0.150) | 0.060 (0.170) | 0.050 (0.220) | 0.050 (0.460) | 0.050 (0.160) | 0.060 (0.140) |
| Completeness (%) | 97.9 (85.6) | 98.1 (88.1) | 98.3 (88.8) | 97.3 (96.0) | 90.5 (98.8) | 98.3 (88.8) | 97.9 (88.4) | 82.8 (85.8) | 96.2 (93.6) | 88.3 (90.5) | 87.9 (93.7) |
| Redundancy | 3.6 (2.8) | 3.6 (3.2) | 3.7 (3.2) | 3.1 (2.9) | 3.3 (3.3) | 3.6 (2.5) | 3.6 (2.8) | 3.0 (2.8) | 3.7 (3.8) | 2.6 (2.4) | 3.5 (3.5) |
| I/σ(I) | 16.7 (6.6) | 17.0 (7.3) | 19.3 (9.7) | 4.90 (2.10) | 11.1 (21.8) | 11.5 (5.3) | 12.7 (5.2) | 11.8 (3.9) | 15.9 (2.8) | 13.0 (5.9) | 16.6 (8.0) |
| Wilson B-factor | 13.5 | 6.7 | 14.1 | 14.2 | 10.3 | 8.2 | 10.1 | 11.9 | 18.5 | 18.7 | 15.0 |
| Refinement statistics | |||||||||||
| Resolution range (Å) | 37.8–1.51 | 38.0–1.16 | 38.0–1.54 | 37.9–1.50 | 37.9–1.96 | 38.0–1.23 | 37.8–1.24 | 38.0–1.40 | 37.9–1.59 | 37.8–2.05 | 38.0–2.10 |
| Number of unique reflections | 19,696 | 43,861 | 18,973 | 20,173 | 8487 | 37,186 | 35,510 | 21,237 | 16,818 | 7189 | 6705 |
| Rwork/Rfree (%) | 11.12/16.79 | 11.90/14.86 | 12.28/16.82 | 16.49/24.31 | 14.43/20.52 | 14.43/20.52 | 12.64/16.23 | 11.86/17.97 | 13.547/20.67 | 16.19/23.01 | 16.25/24.90 |
| Number of atoms | |||||||||||
| Protein | 1319 | 1338 | 1369 | 1267 | 1267 | 1364 | 1368 | 1298 | 1304 | 1245 | 1277 |
| Water | 215 | 207 | 140 | 188 | 115 | 185 | 191 | 150 | 145 | 108 | 12 |
| Ligands | 51 | 80 | 89 | 69 | 41 | 65 | 67 | 89 | 57 | 71 | 32 |
| rmsd from ideal values | |||||||||||
| Bond lengths (Å) | 0.012 | 0.010 | 0.009 | 0.014 | 0.007 | 0.015 | 0.014 | 0.020 | 0.012 | 0.016 | 0.015 |
| Bond angles (°) | 1.453 | 1.576 | 1.485 | 1.846 | 1.102 | 1.751 | 1.698 | 1.027 | 1.461 | 1.851 | 1.681 |
| Average B-factors (Å2) | 13.7 | 11.0 | 13.4 | 14.1 | 13.4 | 12.1 | 14.1 | 14.3 | 18.4 | 20.6 | 16.5 |
| Protein | 14.2 | 11.4 | 14.6 | 15.2 | 14.6 | 12.5 | 14.9 | 14.9 | 19.8 | 22.8 | 14.9 |
| Solvent | 34.4 | 27.4 | 32.5 | 19.1 | 34.8 | 28.4 | 31.5 | 36.0 | 35.6 | 44.9 | 51.4 |
| Ligands | — | — | 17.8 | 32.8 | 27.8 | 30.9 | 34.9 | 33.2 | 37.9 | 32.4 | 27.3 |
Fig. 1.

Structure of Bet v 1j. (a) Topology model of Bet v 1j. α-Helices are indicated as cylinders, flanking the seven-stranded β-sheet. Throughout the article, the identical standard orientation is maintained, unless indicated otherwise. (b) Stereo representation of the overall structure, oriented and colored according to the topology model. The hydrophobic cavity is shown as blue mesh. Cavity entries (ɛ1–ɛ3) are indicated by arrows. (c) Solvent-accessible surface color coded by its electrostatic potential. Red and blue colors indicate negative and positive (partial) charges, respectively. (d) Cut-open surface presentation of the hydrophobic cavity. By removing the C-terminal helix, solvent molecules bound to the hydrophobic pocket become visible. The cut-open interface is indicated as semitransparent surface in gray.
High-resolution structures of high IgE binding variants Bet v 1a and j
Apo Bet v 1a could now be solved at unmatched accuracy, to a resolution of 1.51 Å. Additionally, we could determine for the first time the structure of variant Bet v 1j, at near atomic resolution (1.16 Å). The electron density maps are of outstanding quality, allowing reliable modeling of alternative conformations and solvent molecules. Remarkably, within the loop connecting β7 with the C-terminal helix α3, the backbone adopts a continuous spectrum of conformation, resulting in a blurred density distribution (in particular residues G124, D125, and H126). We observed this flexibility of the loop consistently in nearly all solved structures. This is in line with the crystal packing, which offers space for almost unrestricted movements in that region of the protein.
Bet v 1 isoform relation
There are six amino acid differences, mostly conservative, between the crystallized isoforms Bet v 1a and j (j bracketed): T(A)9, N(S)82, I(V)91, D(N)109, S(N)117, and V(I)133. Since both isoforms reportedly strongly bind IgE,8 the exhibited mutations seem to be rather neutral in this context.
Solvent molecules map the overall chemical properties of the protein
The following description of the solvent represents apo Bet v 1j, the highest-resolution structure at 1.16 Å. Besides the protein, we modeled 207 water molecules into the asymmetric unit as well as several molecules derived from the crystallization buffer, including four 2-methyl-2,4-pentanediols (MPDs), one Tris buffer molecule, and eight sulfate ions. This allowed us to map the solvent structure surrounding and permeating the protein to unprecedented accuracy. The negatively charged sulfate ions were located at the protein surface, whereas the MPDs and several water molecules were found within the hydrophobic cavity. Tris was located at the glycine-rich loop, masking this opening to the cavity. The solvent molecules built up a complex hydrogen-bonding network, highlighting the existence of solvent-accessible polar anchor sites within the hydrophobic cavity (Fig. 1d).
Three access routes connect to distinct binding sites within the cavity
The hydrophobic cavity of Bet v 1 was accessible to solvent and small molecules at three distinct entry sites (Fig. 1b). One opening (entry ɛ1) was located at the flexible loop connecting β7 with α3. This entry was bordered by eight residues (F62, P63, F64, P90, Q132, A135, S136, and M139), creating a relatively nonpolar access route to the cavity. A second small opening (entry ɛ2) with a diameter of ∼ 6 Å was surrounded by residues Y5, T7, V133, and K137. The third entry (entry ɛ3) was located at the opposite side of the protein. It is partly formed by the glycine-rich loop and framed by the residues I23, L24, D25, D27, T52, K54, Y81, and I102. The residues bordering the three entries are equally conserved in high and low IgE binding Bet v 1 isoforms.
Relating binding properties to high and low IgE binding Bet v 1 variants: Phe30 versus Val30 discriminates high and low IgE binding isoforms
Despite crystal structure information of high and low IgE binding Bet v 1 isoforms,9,10 a structural–mechanistic basis rationalizing the respective allergenic properties remained elusive so far. The classification of naturally occurring Bet v 1 variants into “hyperallergenic” and “hypoallergenic” isoforms is based on their IgE-binding activity.8 We hypothesized that there is a correlation between these allergenic subtypes and differentiated ligand binding properties to the hydrophobic pocket of Bet v 1. Thus, we searched for residues that influence the topology of the binding pocket, and subsequently the ligand binding, to gain a detailed understanding of isoform-dependent binding properties of Bet v 1. Sequence alignment of high IgE binding (Bet v 1a, e, and j) with the low IgE binding variants (d, g, and l) revealed five hallmark residues strictly discriminating these two classes (Supplementary Fig. 1). These are F30V/I, S57N, S112C, I113V, and D125N for the high and low IgE binding variants, respectively. From these, only residue 30 is directly influencing the topology of the hydrophobic cavity.
Identification of a suitable model compound to identify distinguishing binding determinants in the hydrophobic pocket
To investigate the possible implications of F30V on ligand binding to the hydrophobic pocket in Bet v 1, we analyzed all Bet v 1 and homologue structures, as deposited in the Protein Data Bank (PDB IDs 3E85,12 2QIM,13 2FLH,14 3IE5,16 1IFV,17 and 1FM410)†. DXC appeared as a candidate ligand to discriminate amino acid exchanges at position 30 in Bet v 1, as this ligand was expected to clash with Phe at this position. Thus, we chose DXC as a model substance to investigate the role of residue 30 in ligand binding to different Bet v 1 isoforms.
Crystal structure of Bet v 1a in complex with DXC
Analogously as in the Bet v 1l–DXC complex structure, we identified two DXC molecules bound in tandem to Bet v 1a, occupying equivalent positions as reported in isoform l,10 with one critical difference as detailed below (Fig. 2a). One molecule is bound deeply in the cavity (inner deoxycholate, further referred to as iDXC), with almost all solvent molecules in the cavity being displaced by it (Fig. 2b). The other DXC was found close to the loop between β7 and α3, capping entry ɛ1 (outer deoxycholate, oDXC, Fig. 2c). Here, the binding of oDXC caused conformational changes of residues K129–Q132 compared to the apo structure, representing the end of the C-terminal loop between β7 and α3. This loop thus adapts to the bound ligand, whereas the residual Bet v 1 structure proved rather rigid, as also confirmed in other ligand complex structures described below.
Fig. 2.

DXC binding to Bet v 1a. (a) Bet v 1a in complex with two DXC molecules, iDXC and oDXC. iDXC is bound deep in the hydrophobic cavity; oDXC caps the cavity opening ɛ1. (b) Close-up view of the iDXC binding site with the experimental 2Fo − Fc electron density, contoured at 1 σ. Hydrogen bonds are shown as broken lines. Here, as well as in other views onto the cavity, the C-terminal helix is removed for clarity. (c) Close-up view of the oDXC binding site. Coordinating residues and solvent molecules (H2O as blue spheres) are shown. Ligands are shown with their corresponding 2Fo − Fc electron density, contoured at 1 σ over the mean.
oDXC adopted a virtually identical orientation in both Bet v 1 isoforms a and l, with comparable polar and hydrophobic contacts facilitating the binding. Slight differences result from isoform-specific loop conformations that are involved in binding.
Contrast oDXC, we found iDXC to be approximately 180° rotated in both Bet v 1 isoforms along an axis perpendicular to the sterane C ring (Fig. 3a). Consequently, the detailed interaction pattern changed drastically; iDXC interacts with the protein via several hydrogen bonds (e.g., with hydroxyl group of Tyr83) and hydrophobic contacts (e.g., with Phe22). iDXC further exhibits a different orientation of its alkyl side chain, which is now pointing towards the glycine-rich loop.
Fig. 3.

Residue 30 decides the coordination of the iDXC. (a) Comparison of iDXC in Bet v 1a (yellow) and Bet v 1l (PDB ID 1fm4; pink), with the corresponding residues 30 being highlighted. The F30 side chain would clash with iDXC binding in the orientation as it was observed in Bet v 1l; these contacts apparently induced a different orientation of iDXC in Bet v 1a. (b) Comparison of iDXC in Bet v 1a (yellow) and Bet v 1a F30V (blue). In F30V mutant, iDXC showed the same binding as in Bet v 1l, except for minor changes in its alkyl side-chain orientation. This clearly identifies residue 30 as single determinant for iDXC binding mode. (c) iDXC in Bet v 1a F30V with its corresponding electron density.
Bet v 1a mutant F30V in complex with DXC
To proof that phenylalanine 30 is exclusively responsible for the differences in iDXC binding to Bet v 1a compared to Bet v 1l, we engineered a Bet v 1a F30V mutant18 and solved its crystal structure in complex with DXC (Fig. 3b and c). Similar to the wild-type Bet v 1a structure, we identified two DXC molecules in the cavity, with the loop preceding the C-terminal helix being displaced by the carboxylic acid moiety of oDXC.
Detailed analysis of both DXC binding sites revealed the same polar and hydrophobic interactions as described for low IgE binding Bet v 1l.10 Consequently, iDXC featured exactly the same arrangement as in Bet v 1l, that is, rotated around the axis perpendicular to the sterane C as compared to Bet v 1a. This unambiguously proves that residue 30 is the crucial determinant for the orientation of iDXC within the hydrophobic core of Bet v 1.
Mechanistic interpretation of ANS displacement assay by ligand crystal structures
Mogensen et al. established an assay for the detection of ligand binding to Bet v 1, based on the increasing fluorescence signal of ANS at 474 nm when binding to a hydrophobic patch or cavity of a protein.11 Displacement of ANS out of the cavity by another ligand comes along with a decrease in the fluorescent signal. Herewith, interaction between Bet v 1 and a number of ligands was shown.
Bet v 1 in complex with ANS
We have selected a set of ligands that were characterized by the ANS assay11 to structurally investigate the detection mechanism; in particular, we aimed (a) to reliably map the putative binding sites within the cavity, (b) to explain ligand-specific changes in the ANS fluorescence signal, and thus (c) to reconcile conflicting assay results. As a reference, we solved the binary complex structures of Bet v 1a and Bet v 1j in complex with ANS (Fig. 4a).
Fig. 4.

ANS binding site. (a) Bet v 1a with ANS bound to the hydrophobic core. The binding site is located beneath the entry ɛ2, cf., Fig. 1b. (b–d) Series of close-up views on different ligands to Bet v 1 near the ANS binding site. Hydrogen-bond coordinating residues and solvent molecules (H2O as blue spheres) are displayed. Ligands are superimposed by their corresponding electron density (2Fo − Fc, contoured at different σ levels). (b) ANS bound to Bet v 1j, coordinated by H-bonds between its sulfonate group and Y83 and N118. (c) Naringenin bound to Bet v 1a. In addition to hydrogen bonds and hydrophobic interactions, naringenin is sodium bridged with Asn118. The sodium ion is shown in purple, with the bond lengths given in angstroms. (d) Kinetin bound to Bet v 1a. Kinetin is predominantly coordinated by hydrophobic contacts. An alternate kinetin conformation (with approximately 20% occupancy) is shown in pink.
Details on the binding site
For both isoforms, we identified one ANS molecule bound into the cavity, located beneath entry ɛ2. ANS formed two H-bonds with the side chains of Y83 and N118 (Fig. 4b). It further interacted with the protein via hydrophobic contacts (e.g., F22, I102).
In the apo-form (uncomplexed) of Bet v 1a, eight water molecules were located near the ANS binding site, which were displaced upon ANS binding. ANS is mostly harbored by hydrophobic residues, with only limited access to bulk solvent via entry ɛ1 (Figs. 1b and 4a). ANS binding induced only very limited changes in Bet v 1 structure, for example, a shift in the side-chain conformation of K137, enlarging entry ɛ2. ANS binding to Bet v 1j was found basically the same as for Bet v 1a.
The identified residues coordinating the ligand are in accordance with a model proposed by Mogensen et al.;11 using two-dimensional NMR, they correctly identified two of the interacting residues (Y83 and I102). Our findings further suggest that ANS enters the cavity via ɛ2.
Bet v 1a in complex with naringenin
Naringenin is a plant pigment hormone belonging to the family of the flavonoids, representing a ligand class that can be of physiological relevance. Mogensen et al. demonstrated interaction with Bet v 1 by ANS displacement assay.11 According to these results, naringenin has to bind at the same site or in close proximity to the binding site occupied by ANS.
Confirmation of the expected binding site
We determined the structure of the binary complex of Bet v 1a with one naringenin molecule. The identified binding site for naringenin overlapped the ANS site, consistent with the results of the ANS displacement assay.
Overall comparison of the binding of naringenin with that of ANS revealed that its benzopyran double ring system was occupying the binding site of the ANS naphthalene. Interestingly, its hydroxyphenyl pointed towards the inner part of the hydrophobic cavity, whereas the allegedly more hydrophobic phenyl ring of ANS pointed towards the solvent (Fig. 4b and c).
Naringenin was coordinated by several hydrogen bonds and hydrophobic contacts. Additionally, the benzopyran keto and hydroxyl oxygens were sodium bridged to the Oδ of N118. The changes in protein structure caused by naringenin binding corresponded to those observed for ANS binding, in particular a change in the side-chain conformation of K137.
Bet v 1a in complex with kinetin
Kinetin is a plant hormone of the group of cytokinins. In ANS displacement assays, it caused an anomalous effect, increasing the fluorescent signal of ANS upon kinetin binding (Fig. 5).11 This indicates that kinetin is not displacing ANS completely from the protein but induces an even more apolar ANS binding environment. Alternative models were proposed in order to account for the observed anomalous fluorescence effect, based on in silico modeling studies or known binding patterns in the related P-loop motif.11,19 Contrasting these suggestions, crystal structures of two Bet v 1 homologous proteins in complex with zeatin, a plant hormone highly similar to kinetin, revealed cytokinin binding sites within the cavity (PDB IDs 2QIM13 and 2FLH14). We wanted to identify the binding site of kinetin to Bet v 1 to explain the increasing ANS fluorescence upon kinetin binding.
Fig. 5.

ANS displacement assay. Changes in the fluorescence (ΔF/F0) of ANS signal induced by three ligands (kinetin, NDSB-256, and DXC) in different molar ratios. Bet v 1a (10 μM final concentration) was incubated at 4 °C overnight with the corresponding ligand. Fluorescence emission intensities of ANS (50 μM final concentration) at 486 nm are shown. Kinetin titration (pink) initially increased the fluorescent signal. Very high kinetin concentrations (10:1 ratio) led to a decrease of fluorescence, but the signal remained still higher compared to Bet v 1a with ANS. NDSB-256 titration (green) decreased the fluorescence signal and remained stable for all tested ligand:protein ratios. DXC (cyan) titration led to a loss in fluorescence signal. After an intermediate plateau, a further decrease in fluorescence could be observed at 10-fold excess of DXC.
Identification of the binding site
We found electron density corresponding to one kinetin molecule beneath entry ɛ2, overlapping the binding site of ANS (Fig. 4d). Kinetin occupied two alternative conformations related by a 180° rotation, with the furan ring pointing into the cavity and towards the protein surface, respectively. Kinetin binding was predominantly mediated by hydrophobic interactions (e.g., F22, I102). The lack of directed polar interactions helps to explain why the kinetin conformation is not locked into a unique conformation but may wobble around an average orientation, consistent with blurred electron density. Notably, this binary Bet v 1 kinetin complex structure cannot explain the ANS assay results but would rather suggest a decrease of the fluorescence signal due to ANS displacement.
Bet v 1a in ternary complex with kinetin and ANS
These apparently conflicting findings prompted us to determine the ternary complex structure of Bet v 1a with ANS and kinetin (Fig. 6a). We found ANS to occupy a binding site identical with the binary Bet v 1–ANS complex. Accordingly, kinetin bound to a Bet v 1 site that differed from the binary Bet v 1–kinetin complex (Fig. 6b and c). We found kinetin flipped about 180° around the furan ring, pointing with its adenine system towards ɛ3 and the glycine-rich loop. The furan ring binding site turned out to be invariant in both the binary and ternary complex and thus seems to determine the kinetin position (Fig. 6d).
Fig. 6.

Ternary complex of Bet v 1a with ANS and kinetin. Ligands bound in the ternary complex are shown in yellow. For comparison, ligands from binary complexes are shown in pink (ANS) and blue (kinetin). (a) Overall view of Bet v 1a with ANS and kinetin bound in the hydrophobic core. (b) Superposition of ANS and kinetin as bound in the respective binary complexes (cf., Fig. 4b and c) results in overlapping binding modes. For clarity, only kinetin conformation A is shown. (c) Close-up view of ANS and kinetin bound simultaneously to Bet v 1a. ANS is coordinated similar to the binary complex. Thus, the presence of ANS in the cavity displaces kinetin to a secondary binding site. (d) Comparison of kinetin coordination in the binary and ternary complex. The furan ring was located at the same site, indicating a role as anchor for kinetin in the cavity.
This structure underscores that kinetin does not displace ANS but rather adapts to a lipophilic binding site conferred by Bet v 1 and ANS. Conversely, kinetin binding changes the immediate environment of ANS; the hydrophobic kinetin is displacing waters in the neighborhood of ANS, as found in the binary Bet v 1–ANS structure. This stripping of water molecules decreases the polarity of the ANS environment and explains the observed increase of the ANS fluorescence signal in the presence of kinetin.
The conclusion that the presence of kinetin in close proximity to ANS, rather than changes of the protein itself, is responsible for the increased ANS signal is further supported by the fact that binding of different ligands had no influence on the conformation of Bet v 1.
Our crystallographic findings demonstrate a higher affinity of ANS than kinetin towards its primary binding site within Bet v 1, in agreement with ligand affinities as deduced from fluorescence measurments.11 When kinetin concentrations were further increased, a reduction of the fluorescence was detected; this biphasic behavior is consistent with the law of mass, where a partial ANS displacement by massive excess of kinetin is to be expected.
Bet v 1a in ternary complex with DXC and ANS
ANS assay with DXC resulted in a decrease of fluorescence (Fig. 5), which could indicate a (partial) displacement of ANS. However, a comparison of the ligand binding sites from the respective binary complex structures solved in this study revealed that it could be sterically possible for Bet v 1 to bind ANS and DXC simultaneously.
We solved the structure of Bet v 1a in ternary complex with ANS and iDXC (Fig. 7a). While we confirmed both ligands to occupy the binding sites that were to be expected from the respective binary complexes (Fig. 7b), after careful refinement, we identified an important change in the ANS orientation: The ANS was rotated for almost 180° at C11 of the aniline ring as compared to the orientation found in the binary complex (Figs. 7c and 4b). As a consequence, the sulfonate group occupied the position of the naphthalene ring linked with aniline in the secondary ANS complex and vice versa.
Fig. 7.

Ternary complex of Bet v 1a with ANS and DXC. (a) Bet v 1a with ANS and DXC bound in the hydrophobic cavity. (b) Close-up view of ANS and kinetin simultaneously bound to Bet v 1a with the corresponding electron density. Both ligands occupy the same binding sites as identified in the corresponding binary complexes. (c) Comparison of ANS coordination in the binary (pink) and ternary (yellow) complex. The presence of DXC caused a change in the relative orientation of ANS, pointing now towards the protein surface with its sulfonate group.
Careful inspection and structural analysis revealed why in the ternary complex (Bet v 1a–ANS–DXC) was a reorientation of the ANS relative to the binary Bet v 1a–ANS structure induced: The ligand conformations, as found in the binary complexes, would bring three oxygens in close proximity (Y83 hydroxyl, iDXC C12 hydroxyl, and ANS sulfonate oxygen). Such an arrangement, while sterically acceptable, would cause electrostatic repulsions that caused the ANS to flip over, as observed in the ternary complex.
As revealed by the binary and ternary complex crystal structures, ANS bound in two alternative modes depending on the presence of DXC. This observation provides a possible explanation for the loss in ANS fluorescence signal upon DXC binding, albeit ANS is still bound to Bet v 1. The alternative ANS binding modes exhibit different environments that are likely to give rise to different fluorescence signals. Additionally, DXC may reduce the binding affinity, and consequently the occupancy, of ANS, which may also reduce its fluorescence signal.
As with kinetin, we found a biphasic change in fluorescence signal with high DXC concentrations (Fig. 5). This further reduction may relate to DXC binding to the oDXC binding site, which was not occupied in the ternary complex crystal structure. This finding suggests that binding of oDXC may negatively cooperate with the binding of ANS via long-range interactions. This long-range effect may possibly be mediated by conformational changes in the C-terminal loop that are caused upon oDXC binding. Hence, as described for kinetin, a massive excess of ligand could lead to partial ANS displacement and consequently a loss of fluorescence signal.
Bet v 1a in complex with a sulfobetaine molecule
In an unbiased approach to further map the Bet v 1 ligand binding, we screened a commercially available compound collection with affinity to hydrophobic proteins (“Detergent screen”, Hampton Research) by native PAGE. From the initial hits, a sulfobetaine derivative [non-detergent sulfobetaine 256 (NDSB-256)] could be confirmed in the ANS displacement assay (Fig. 5). We identified one NDSB-256 molecule in the cavity, partly overlapping the binding sites of ANS (with its sulfonate group) and iDXC (with its phenyl group) (Fig. 8). As for kinetin in its binary structure, the binding of this ligand is not promoted by polar contacts. This structure of Bet v 1 complexed with another synthetic compound underscores the promiscuity of the lipophilic binding site of Bet v 1 towards a broad spectrum of amphiphilic ligands.
Fig. 8.

NDSB-256 binding to Bet v 1a. (a) Bet v 1a in complex with the sulfobetaine NDSB-256. (b) Close-up view of NDSB-256 molecule with its corresponding electron density. The ligand is coordinated predominantly by hydrophobic contacts. (c) Comparison of ligand binding sites in the corresponding binary complexes. NDSB-256 (yellow) overlaps the binding sites of both ANS and iDXC (purple). Interestingly, the sulfate moieties of NDSB-256 and ANS almost overlay.
Discussion
Physicochemical mechanisms and driving forces underlying Bet v 1 ligand binding
The presented structures allow a detailed description of the characteristics that make Bet v 1 a very potent and flexible ligand acceptor. The excellent resolution (better than 1.2 Å) provides details of both the protein and the included solvent and ligands with unmatched accuracy. Thus, we can describe ligand-induced changes in the hydrophobic cavity in detail.
In apo Bet v 1, the cavity is occupied by solvent molecules, comprising water as well as molecules from crystallization buffers. In all complex structures presented, several solvent molecules are displaced by ligands. In contrast to the massive influence on the hydration shell of the protein, the structure of the protein itself, and particularly of the residues creating the inner surface of the cavity, is not affected by binding of any of the presented ligands. From a physicochemical perspective, these findings enable us to draw to important conclusions: (i) ligand binding to Bet v 1 is primarily entropically driven, whereby clusters of solvent molecules are stripped off the protein surface, releasing according degrees of freedom; (ii) the so-called hydrophobic binding pocket of Bet v 1 is actually amphiphilic.
Biological implications
DXC has been employed previously as a model system to study ligand binding to Bet v 1.10 We similarly capitalized on DXC as a model to specifically investigate the impact of residue 30 on ligand binding towards variable Bet v 1 isoforms. Hence, it is particularly tempting to translate our results to physiologically and immunologically relevant ligands that possibly discriminate high and low IgE binding Bet v 1.
Binding of steroids
Bet v 1 is assumed to act as a steroid carrier in plants. Markovic-Housley et al. reported brassinolide (BRA) binding to Bet v 1l by mass spectrometry. Furthermore, they docked BRA into the binding pocket of Bet v 1l using DXC as template.10 BRA is present in plant pollen at significant levels20 and regulates growth processes and plant development.21 Although BRA differs from DXC in parts of the polycyclic ring structure and the alkyl side chain, it could be fitted to both the iDXC and oDXC binding site in Bet v 1l. Steroid binding to PR-10 proteins was further corroborated by the binding of castasterone, a phytosteroid highly similar to BRA, to the hydrophobic pocket of the major cherry allergen Pru av 1.15 Pru av 1 exhibits Val30, similar to low IgE binding Bet v 1 variants.
The iDXC binding site in high IgE binding isoforms such as Bet v 1a is considerably constricted by the characteristic Phe30 as compared to low IgE binding Bet v 1 isoforms or Pru av 1 with V30. To test whether phytosteroids could bind to high IgE binding isoforms in a conformation as suggested for Bet v 1l and Pru av 1, we modeled BRA (coordinates used from PDB ID 3RJ022) into Bet v 1a by superimposing its ring systems with iDXC (Supplementary Fig. 2). In this orientation, the alkyl side chain of BRA points towards the glycine-rich loop (ɛ3). Compared to DXC, BRA exhibits an extension of the alkyl side chain by one carbon and two additional hydroxyl groups, together making it much more bulky than DXC. The cavity builds up a narrow tunnel towards ɛ3, which is further restrained in high IgE binding Bet v 1 by Phe30. Thus, the cavity in Bet v 1a does not provide the required space for BRA binding similarly to DXC, leading to steric clashes between its side chain and the protein. Consequently, we can exclude BRA or related phytosteroids to bind to the inner binding niche of high IgE binding Bet v 1 variants (F30) as suggested for Bet v 1l and Pru av 1.10,15 However, Bet v 1a is, by far, the most abundant isoform in birch pollen.23 Thus, a physiological function alternative to plant steroid carriage or storage for this Bet v 1 variant should be considered.
Interaction of allergens with lipopolysaccharide
Lipopolysaccharide (LPS) strongly stimulates an innate immune response, which can induce an acquired immune response, in particular via TH1 cells.24,25 Therefore, it is interesting that a majority of allergens are thought to be lipid binding proteins.26 This suggests that an intrinsic adjuvant activity by allergens relates to the bound lipidic compounds and may thus provide a mechanism that contributes to allergenicity.27 For Bet v 1, binding of various fatty acids into the cavity was shown by ANS assay.11 Further, it can interact with phospholipidic vesicles at low pH.28 A model proposed by Mattila and Renkonen for Bet v 1–phospholipid interaction19 can easily be translated to LPS. Accordingly, fatty acids could enter the cavity via ɛ3 at the glycine-rich loop.
The loop should serve as anchor point for the polar head group, consistent with the experimentally observed coordination of Tris observed in our crystal structures. Interestingly, Phe30 is located beneath the glycine-rich loop and thus directly influences the topology of this particular cavity opening.
Currently, there is no experimental data for Bet v 1–LPS interaction available. Albeit speculative, Phe/Val30 could act as specific trigger for binding of LPS or other large hydrophobic molecules, thus influencing the polarization of subsequent immune response.
Ligand binding promiscuity and ternary complexes of Bet v 1
We can ascertain that (1) several ligands can share the same binding site within Bet v 1; (2) Bet v 1 can coordinate the same ligand in different orientations at one distinct binding site; (3) Bet v 1 can bind one ligand at different binding sites, depending on the presence of another ligand; (4) differences in the binding pocket topology of Bet v 1 isoforms are mainly caused by residue 30 and result in remarkable alterations in ligand coordination; (5) Bet v 1 is capable of binding two different ligands simultaneously.
For the latter, we identified two mechanisms: (a) non-overlapping primary binding sites sterically allow the presence of two ligands; (b) overlapping primary sites result in the displacement of one ligand to a secondary anchor point within the cavity. However, other than the structurally observed ternary complexes are realistic. For instance, DXC and kinetin show no sterical conflicts in their primary binding sites, Figs. 2 and 4. This ternary complex is in accordance with Koistinen et al., describing distinct, non-overlapping anchor points for these ligands in homologues birch protein PR-10c.29 Furthermore, concomitant binding of phytosteroids and other plant mediators such as cytokinins and flavonoids is thinkable in low IgE binding Bet v 1 variants.
Conclusion
The architecture of the Bet v 1 binding pocket is optimized for its promiscuous ligand binding that is probably related with storage and/or transport of a spectrum of lipidic mediators. Importantly, the binding promiscuity is complemented by specific interactions that discern high and low IgE binding Bet v 1 isoforms. The combination of isoform-dependent and context-dependent binding modes for a single defined ligand suggests that the physiological function of this remarkable allergen is even more complex as thought hitherto. In the light of its complex interactome, we propose to study Bet v 1 as a “dressed allergen”, where isoform-dependent ligand complexes may prime the immune response to Bet v 1.
Materials and Methods
Protein preparation
Recombinant Bet v 1a and Bet v 1j were expressed in Escherichia coli strain BL21(DE3), using a modified pET-28b vector, lacking the N-terminal 6His-tag. Cells grew in 600 ml LB medium supplemented with 20 μg/ml kanamycin at 37 °C to an OD600 (optical density at 600 nm) of 1.0. After adding 1 mM IPTG, expression was performed for 4 h at 37 °C. Protein was purified as described elsewhere,30 with some minor changes of the protocol. As a final polishing step, size-exclusion chromatography was performed using a Superdex75 column with a running buffer of 20 mM imidazole, pH 7.4, and 50 mM NaCl.
Crystallization of Bet v 1
Bet v 1a crystals were obtained by sitting drop vapor diffusion, using 5 mg/ml protein concentration. The crystallization buffer was composed of 2.0 M ammonium sulfate as main precipitant and 1.5% MPD. High-quality Bet v 1j crystals were obtained using micro-seeding.31 Micro-seeds were obtained by low-quality Bet v 1j crystals, grown in 2.5 M ammonium sulfate and 1% MPD. Crystals were seeded into equilibrated drops of the same crystallization conditions as used for Bet v 1a.
Crystallization of Bet v 1–ligand complexes
Bet v 1–ligand complexes were obtained using three techniques: soaking, co-crystallization of protein with ligand, and micro-seeding of apo-Bet crystals into equilibrated co-crystallization drops.
Data collection and structure determination
Crystals were flash frozen in a stream of nitrogen gas at 100 K. X-ray diffraction data sets were collected at beamline 14.1 at Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung (BESSY II, Berlin/Adlershof), at beamline 14-4 at the European Synchrotron Radiation Facility (ESRF, Grenoble), and in-house using a Bruker Microstar rotating anode generator mounted with a Mar345dtb detector. Diffraction data were indexed, scaled, and further processed using CCP4 software suite.32 The 1.16 Å structure of apo-Bet v 1j was solved by molecular replacement, using Phaser.33 As a search model, Bet v 1l (PDB ID 1FM410) was used. For all subsequently solved structures, Bet v 1j was used as a search model. Refinement was performed using Refmac534 and monitored throughout using an Rfree calculated with 5% of the unique reflections. Model building was performed in Coot.35 All figures were generated using PyMOL.36 Data collection and processing statistics are summarized in Table 2.
ANS displacement assay
Fifty microliters of protein solution (10 μM final concentration) was mixed with ligands in a 96-well UV-Star plate in different molar ratios, typically 1:1, 1:2, 1:3, and 1:10. Mixtures were incubated overnight at 4 °C. Prior to the measurements, 50 μl of ANS (50 μM final concentration) was added, and the mixtures were incubated for another 5 min at room temperature. ANS was excited at 350 nm, and measurements were performed from 400 to 600 nm (in 2-nm steps). Blanks of ligand alone as well as ligand with ANS were subtracted.
Structure validation
Prior to deposition, the quality of all models was checked using MolProbity,37 NQ-Flipper,38 and PROCHECK.39
Bet v 1 nomenclature
The designation of Bet v 1 variants as originally published was used for consistency.40 In parentheses, the official http://www.iuisonline.org International Union of Immunological Societies nomenclature is given: Bet v 1a (Bet v 1.0101), Bet v 1d (Bet v 1.0401), Bet v 1e (Bet v 1.0501), Bet v 1g (Bet v 1.0701), Bet v 1j (Bet v 1.0801), and Bet v 1l (Bet v 1.1001).
Accession numbers
Coordinates have been deposited in the PDB with accession codes 4A80, 4A81, 4A83, 4A84, 4A85, 4A86, 4A87, 4A88, 4A8G, 4A8V, and 4A8U.
Acknowledgements
Excellent technical support by the staff at ESRF and BESSY II is very much appreciated. We thank Elfriede Dall, Esther Schönauer, Thomas Zögg, and Peter Briza for help with X-ray data collection, processing, and mass spectrometry. We appreciate the support by the Austrian Science Funding Agency under the projects W_01213 and P_23417 and the Priority Program of the University of Salzburg.
Edited by R. Huber
Footnotes
Supplementary data to this article can be found online at doi:10.1016/j.jmb.2012.05.016
Appendix A.
Supplementary materials
References
- 1.Liu J.J., Ekramoddoullah A.K.M. The family 10 of plant pathogenesis-related proteins: their structure, regulation, and function in response to biotic and abiotic stresses. Physiol. Mol. Plant Pathol. 2006;68:3–13. [Google Scholar]
- 2.Somssich I.E., Schmelzer E., Bollmann J., Hahlbrock K. Rapid activation by fungal elicitor of genes encoding “pathogenesis-related” proteins in cultured parsley cells. Proc. Natl Acad. Sci. USA. 1986;83:2427–2430. doi: 10.1073/pnas.83.8.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walter M.H., Liu J.W., Grand C., Lamb C.J., Hess D. Bean pathogenesis-related (PR) proteins deduced from elicitor-induced transcripts are members of a ubiquitous new class of conserved PR proteins including pollen allergens. Mol. Gen. Genet. 1990;222:353–360. doi: 10.1007/BF00633840. [DOI] [PubMed] [Google Scholar]
- 4.Walter M.H., Liu J.W., Wunn J., Hess D. Bean ribonuclease-like pathogenesis-related protein genes (Ypr10) display complex patterns of developmental, dark-induced and exogenous-stimulus-dependent expression. Eur. J. Biochem. 1996;239:281–293. doi: 10.1111/j.1432-1033.1996.0281u.x. [DOI] [PubMed] [Google Scholar]
- 5.Warner S.A., Scott R., Draper J. Characterisation of a wound-induced transcript from the monocot asparagus that shares similarity with a class of intracellular pathogenesis-related (PR) proteins. Plant Mol. Biol. 1992;19:555–561. doi: 10.1007/BF00026782. [DOI] [PubMed] [Google Scholar]
- 6.Schenk M.F., Gilissen L.J., Esselink G.D., Smulders M.J. Seven different genes encode a diverse mixture of isoforms of Bet v 1, the major birch pollen allergen. BMC Genomics. 2006;7:168. doi: 10.1186/1471-2164-7-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moverare R., Westritschnig K., Svensson M., Hayek B., Bende M., Pauli G. Different IgE reactivity profiles in birch pollen-sensitive patients from six European populations revealed by recombinant allergens: an imprint of local sensitization. Int. Arch. Allergy Immunol. 2002;128:325–335. doi: 10.1159/000063855. [DOI] [PubMed] [Google Scholar]
- 8.Ferreira F., Hirtenlehner K., Jilek A., Godnik-Cvar J., Breiteneder H., Grimm R. Dissection of immunoglobulin E and T lymphocyte reactivity of isoforms of the major birch pollen allergen Bet v 1: potential use of hypoallergenic isoforms for immunotherapy. J. Exp. Med. 1996;183:599–609. doi: 10.1084/jem.183.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gajhede M., Osmark P., Poulsen F.M., Ipsen H., Larsen J.N., Joost van Neerven R.J. X-ray and NMR structure of Bet v 1, the origin of birch pollen allergy. Nat. Struct. Biol. 1996;3:1040–1045. doi: 10.1038/nsb1296-1040. [DOI] [PubMed] [Google Scholar]
- 10.Markovic-Housley Z., Degano M., Lamba D., von Roepenack-Lahaye E., Clemens S., Susani M. Crystal structure of a hypoallergenic isoform of the major birch pollen allergen Bet v 1 and its likely biological function as a plant steroid carrier. J. Mol. Biol. 2003;325:123–133. doi: 10.1016/s0022-2836(02)01197-x. [DOI] [PubMed] [Google Scholar]
- 11.Mogensen J.E., Wimmer R., Larsen J.N., Spangfort M.D., Otzen D.E. The major birch allergen, Bet v 1, shows affinity for a broad spectrum of physiological ligands. J. Biol. Chem. 2002;277:23684–23692. doi: 10.1074/jbc.M202065200. [DOI] [PubMed] [Google Scholar]
- 12.Fernandes H., Bujacz A., Bujacz G., Jelen F., Jasinski M., Kachlicki P. Cytokinin-induced structural adaptability of a Lupinus luteus PR-10 protein. FEBS J. 2009;276:1596–1609. doi: 10.1111/j.1742-4658.2009.06892.x. [DOI] [PubMed] [Google Scholar]
- 13.Fernandes H., Pasternak O., Bujacz G., Bujacz A., Sikorski M.M., Jaskolski M. Lupinus luteus pathogenesis-related protein as a reservoir for cytokinin. J. Mol. Biol. 2008;378:1040–1051. doi: 10.1016/j.jmb.2008.03.027. [DOI] [PubMed] [Google Scholar]
- 14.Pasternak O., Bujacz G.D., Fujimoto Y., Hashimoto Y., Jelen F., Otlewski J. Crystal structure of Vigna radiata cytokinin-specific binding protein in complex with zeatin. Plant Cell. 2006;18:2622–2634. [Google Scholar]
- 15.Neudecker P., Schweimer K., Nerkamp J., Scheurer S., Vieths S., Sticht H., Rosch P. Allergic cross-reactivity made visible: solution structure of the major cherry allergen Pru av 1. J. Biol. Chem. 2001;276:22756–22763. doi: 10.1074/jbc.M101657200. [DOI] [PubMed] [Google Scholar]
- 16.Michalska K., Fernandes H., Sikorski M., Jaskolski M. Crystal structure of Hyp-1, a St. John's wort protein implicated in the biosynthesis of hypericin. J. Struct. Biol. 2010;169:161–171. doi: 10.1016/j.jsb.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 17.Biesiadka J., Bujacz G., Sikorski M.M., Jaskolski M. Crystal structures of two homologous pathogenesis-related proteins from yellow lupine. J. Mol. Biol. 2002;319:1223–1234. doi: 10.1016/S0022-2836(02)00385-6. [DOI] [PubMed] [Google Scholar]
- 18.Ferreira F., Ebner C., Kramer B., Casari G., Briza P., Kungl A.J. Modulation of IgE reactivity of allergens by site-directed mutagenesis: potential use of hypoallergenic variants for immunotherapy. FASEB J. 1998;12:231–242. doi: 10.1096/fasebj.12.2.231. [DOI] [PubMed] [Google Scholar]
- 19.Mattila K., Renkonen R. Modelling of Bet v 1 binding to lipids. Scand. J. Immunol. 2009;70:116–124. doi: 10.1111/j.1365-3083.2009.02277.x. [DOI] [PubMed] [Google Scholar]
- 20.Takatsuto S. Brassinosteroids: distribution in plants, bioassays and microanalysts by gas chromatography–mass spectrometry. J. Chromatogr., A. 1994;658:3–15. [Google Scholar]
- 21.Clouse S.D., Sasse J.M. BRASSINOSTEROIDS: essential regulators of plant growth and development. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998;49:427–451. doi: 10.1146/annurev.arplant.49.1.427. [DOI] [PubMed] [Google Scholar]
- 22.Hothorn M., Belkhadir Y., Dreux M., Dabi T., Noel J.P., Wilson I.A., Chory J. Structural basis of steroid hormone perception by the receptor kinase BRI1. Nature. 2011;474:467–471. doi: 10.1038/nature10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erler A., Hawranek T., Kruckemeier L., Asam C., Egger M., Ferreira F., Briza P. Proteomic profiling of birch (Betula verrucosa) pollen extracts from different origins. Proteomics. 2011;11:1486–1498. doi: 10.1002/pmic.201000624. [DOI] [PubMed] [Google Scholar]
- 24.Akira S., Takeda K., Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 25.Beutler B., Rietschel E.T. Innate immune sensing and its roots: the story of endotoxin. Nat. Rev., Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 26.Thomas W.R., Hales B.J., Smith W.A. Structural biology of allergens. Curr. Allergy Asthma Rep. 2005;5:388–393. doi: 10.1007/s11882-005-0012-1. [DOI] [PubMed] [Google Scholar]
- 27.Trompette A., Divanovic S., Visintin A., Blanchard C., Hegde R.S., Madan R. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature. 2009;457:585–588. doi: 10.1038/nature07548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mogensen J.E., Ferreras M., Wimmer R., Petersen S.V., Enghild J.J., Otzen D.E. The major allergen from birch tree pollen, Bet v 1, binds and permeabilizes membranes. Biochemistry. 2007;46:3356–3365. doi: 10.1021/bi062058h. [DOI] [PubMed] [Google Scholar]
- 29.Koistinen K.M., Soininen P., Venalainen T.A., Hayrinen J., Laatikainen R., Perakyla M. Birch PR-10c interacts with several biologically important ligands. Phytochemistry. 2005;66:2524–2533. doi: 10.1016/j.phytochem.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 30.Hoffmann-Sommergruber K., Susani M., Ferreira F., Jertschin P., Ahorn H., Steiner R. High-level expression and purification of the major birch pollen allergen, Bet v 1. Protein Expr. Purif. 1997;9:33–39. doi: 10.1006/prep.1996.0671. [DOI] [PubMed] [Google Scholar]
- 31.Bergfors T. Seeds to crystals. J. Struct. Biol. 2003;142:66–76. doi: 10.1016/s1047-8477(03)00039-x. [DOI] [PubMed] [Google Scholar]
- 32.Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 33.McCoy A.J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murshudov G.N., Skubak P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeLano W.L. The case for open-source software in drug discovery. Drug Discov. Today. 2005;10:213–217. doi: 10.1016/S1359-6446(04)03363-X. [DOI] [PubMed] [Google Scholar]
- 37.Chen V.B., Arendall W.B., III, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weichenberger C.X., Sippl M.J. NQ-Flipper: recognition and correction of erroneous asparagine and glutamine side-chain rotamers in protein structures. Nucleic Acids Res. 2007;35:W403–406. doi: 10.1093/nar/gkm263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laskowski R.A., Moss D.S., Thornton J.M. Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 40.Swoboda I., Jilek A., Ferreira F., Engel E., Hoffmann-Sommergruber K., Scheiner O. Isoforms of Bet v 1, the major birch pollen allergen, analyzed by liquid chromatography, mass spectrometry, and cDNA cloning. J. Biol. Chem. 1995;270:2607–2613. doi: 10.1074/jbc.270.6.2607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials