

Abstract
A widely used metric of substrate exposure in brain is the brain-to-serum partition coefficient (Kp,brain; Cbrain/Cserum), most appropriately determined at distribution equilibrium between brain tissue and serum. In some cases, Cbrain/Cserum can peak and then decrease, as opposed to monotonically increasing to a plateau, precluding accurate estimation of partitioning. This “overshoot” has been observed with compounds that undergo enterohepatic recycling (ER), such as valproic acid (VPA). Previous simulation experiments identified a relationship between overshoot in the Cbrain/Cserum versus time profile and distribution into a peripheral “compartment” (e.g., the ER loop). This study was conducted to evaluate model predictions of that relationship. Initial experiments tested the ability of activated charcoal, antibiotics, or Mrp2 deficiency to impair VPA ER in rats, thereby limiting the apparent volume of distribution associated with ER. Mrp2 deficiency (significantly) and antibiotics (moderately) interrupted VPA ER. Subsequently, brain partitioning was evaluated in the presence versus absence of ER modulation. Although overshoot was not eliminated completely, deconvolution revealed that overshoot was reduced in Mrp2-deficient and antibiotic-treated rats. Consistent with model predictions, overshoot was higher after antibiotic treatment (moderate ER interruption) than in Mrp2 deficiency (substantial ER interruption). Steady-state Kp,brain was unaffected by experimental manipulation, also consistent with model predictions. These data support the hypothesis that Cbrain/Cserum may overshoot Kp,brain based on the extent of peripheral sequestration. Consideration of this information, particularly for compounds that undergo significant extravascular distribution, may be necessary to avoid erroneous estimation of Kp,brain.
Introduction
A commonly used metric for substrate exposure in brain is the brain-to-serum partition coefficient, Kp,brain (Summerfield et al., 2007). Accurate determination of exposure is imperative for CNS-targeted therapeutics, for which extensive exposure is desirable, and non-CNS-targeted compounds or nontherapeutic xenobiotics, for which exposure could produce neurotoxicity. A network of physical and biochemical barriers between the brain and systemic circulation [the blood-brain barrier (BBB)] effectively attenuates CNS penetration of most compounds. Consequently, rate and extent of brain exposure are not easily predicted based upon physicochemical properties (Hammarlund-Udenaes et al., 1997; Liu et al., 2005). Therefore, experimental determination of penetration into brain is particularly important.
Kp,brain reflects the net influence of multiple factors on extent of brain exposure and often is used in lead development screening for CNS drug discovery (Raub, 2006). Compounds with Kp,brain > 1 are, somewhat arbitrarily, considered to exhibit “good” CNS exposure (Kalvass and Pollack, 2007). Tissue partitioning, which often is expressed as the tissue/serum concentration ratio at a fixed time point, is a time-dependent process; the Ctissue/Cserum ratio increases from zero (at the time of administration) until distributional equilibrium (DE) between the target tissue and the systemic circulation is attained (Gibaldi, 1969). Kp,brain, most appropriately, is determined at DE, either as the ratio of areas under the concentration-time curve (AUC) in brain versus serum or at steady state (Lin et al., 1982; Dallas et al., 1994). For practical reasons, the Cbrain/Cserum ratio under nonequilibrium conditions often is used as a surrogate for the “true” substrate partitioning.
Recent studies (Padowski and Pollack, 2011a,b) demonstrated via mathematical simulation that distribution into a “deep” peripheral pharmacokinetic compartment (i.e., prolonged sequestration in a nonbrain compartment) can impact distribution kinetics between brain and blood, resulting in a period of time during which the Cbrain/Cserum ratio overshoots Kp,brain. This phenomenon has been reported for valproic acid (VPA), an antiepileptic drug that exhibits a rapid peak in Cbrain/Cserum that decreases, rather than increases, with time (Hammond et al., 1982; Golden et al., 1993). VPA undergoes significant enterohepatic recycling (ER). The acyl glucuronide of VPA is excreted via bile into the intestines, hydrolyzed to VPA by intestinal β-glucuronidase, and reabsorbed into blood. This ER loop sequesters VPA outside of the systemic circulation, essentially functioning as a large peripheral compartment (Pollack and Brouwer, 1991).
The potential relationship between overshoot in VPA brain partitioning and ER is supported by two lines of evidence. VPA and two analogs, cyclohexanecarboxylic acid (CCA) and 1-methyl-1-cyclohexanecarboxylic acid, differ in the fraction of the dose that undergoes ER (φER); the rank order of φER corresponds to the rank order of overshoot in brain partitioning (Liu et al., 1992; Liu and Pollack, 1993, 1994). This observation suggests that ER is quantitatively important in the overshoot phenomenon. Second, VPA ER is not complete until at least 60 days of age in rats (Haberer and Pollack, 1994), and the appearance of ER during postnatal development corresponds to the appearance of overshoot in the VPA brain partitioning profile (Padowski and Pollack, 2011a).
Although simulation experiments and post hoc evaluation of data support the hypothesis that overshoot in Kp,brain can result from extensive peripheral distribution, this hypothesis has not been tested prospectively. VPA, by virtue of significant extravascular sequestration arising from ER, presents an interesting model compound for testing this hypothesis. Several methods for interrupting ER, and thus manipulating the apparent volume of the peripheral “compartment” because of ER, are available. Excretion of VPA-glucuronide in bile can be reduced by inhibiting the protein responsible for that excretion, Mrp2 (Wright and Dickinson, 2004); exteriorizing bile flow surgically diverts VPA glucuronide from the recycling loop (Nakajima et al., 2004). Liberation of VPA from the glucuronide conjugate in the intestines can be inhibited by killing gut flora that provide β-glucuronidase (Gott and Griffiths, 1987; Takasuna et al., 1996). Finally, reabsorption of VPA into portal blood could potentially be limited by adsorbing VPA-glucuronide and/or liberated VPA in the gut on activated charcoal (Neuvonen et al., 1983). Moreover, if the efficacy of such ER-interrupting strategies differs, it becomes possible to test whether the magnitude of overshoot is related to the degree to which peripheral sequestration is changed, as predicted by simulations (Padowski and Pollack, 2011a).
The objective of this study was to test the hypothesis that reduction of the apparent volume of a peripheral compartment will reduce overshoot in Kp,brain. Initial experiments were conducted to evaluate the ability of activated charcoal, pretreatment with antibiotics, or genetic Mrp2 deficiency to ablate VPA ER. Next, the effect of ER interruption on the Cbrain/Cserum ratio versus time profile was evaluated. Finally, negative control experiments were conducted to determine whether these treatments influenced aspects of VPA brain exposure in a manner unrelated to the interruption of ER.
Materials and Methods
Chemicals and Reagents.
[4,5-3H] VPA (51 Ci/mmol) was purchased from Moravek Biochemicals (Brea, CA), and [14C] inulin (2.1 mCi/g) was purchased from American Radiolabeled Chemicals (St. Louis, MO). VPA sodium salt, CCA, and streptomycin sulfate were obtained from Sigma-Aldrich (St. Louis, MO). All other chemicals and reagents used in this study were of the highest grade available from commercial sources.
Animals.
Male Sprague-Dawley rats (225–300 g), male Wistar rats (225–275 g), and male Mrp2-deficient rats [TR−; Abcc2(−/−), 225–250 g] were purchased from Harlan (Indianapolis, IN). With the exception of jugular vein-cannulated rats, which were housed individually after surgery, all rats were housed two to four per cage on a 12-h light/dark cycle, with ad libitum access to food and water. All procedures were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Interruption of VPA ER.
All rats were anesthetized with ketamine (160 mg/kg i.p.) and xylazine (8 mg/kg i.p.), and a silicone rubber cannula (0.047 o.d., 0.025 i.d.) was implanted in the right jugular vein at least 24 h before the experiment. On the day of experimentation, a single dose of VPA (75 mg/kg i.p.) was administered to all rats (n = 20). Three approaches were evaluated for ability to interrupt VPA ER. Rats in the first treatment group (n = 3, Sprague-Dawley) received a 200 mg/ml suspension of activated charcoal (100–400 mesh) in water via oral gavage. Two doses (1200 mg/kg each) were administered, the first at the time of VPA administration and the second 2 h later. Rats in the second treatment group (n = 5, Sprague-Dawley) received penicillin G (2 mg/ml) and streptomycin (4 mg/ml) in drinking water for 3 days (125 mg/kg penicillin G per day, 250 mg/kg streptomycin per day). At 24 h before experimentation, an additional dose of penicillin G and streptomycin (40 and 80 mg/kg, respectively) was administered by oral gavage. The third treatment group (n = 4) consisted of rats deficient in Mrp2 function due to an autosomal recessive mutation on locus Abcc2. Two control groups were evaluated concurrently: the first group (n = 4, Sprague-Dawley) received vehicle treatment only (water administered by oral gavage) as a strain-specific control for the charcoal- and antibiotic-treated animals, and the second group (n = 4, wild-type Wistar) served as strain-specific controls for the Mrp2-deficient rats. Both control groups received VPA doses as described above before experimentation. Upon VPA administration, 0.2- to 0.3-ml blood samples (replaced with an equal volume of saline) were collected from each rat via the implanted cannula at 0.25, 0.5, 1, 1.5, 2.5, 4, 5, 6, and 8 h after the dose. Serum was harvested from blood and stored at −20°C until analysis.
Effect of ER Interruption on VPA Brain Partitioning.
The VPA ER interruption experiment (see Results) indicated that partial interruption of VPA ER was achieved by pretreatment with antibiotics, and nearly complete interruption of VPA ER was observed in TR− rats. Thus, an experiment to evaluate the relationship between the shape of the VPA Cbrain/Cserum ratio versus time profile and the degree of VPA ER was conducted using antibiotic- versus vehicle-treated Sprague-Dawley rats and Mrp2-deficient versus wild-type Wistar rats. Penicillin G and streptomycin were administered in drinking water for 3 days before experimentation and as a bolus 24 h before experimentation, as described above. At the time of experimentation, all rats received a single dose of VPA (100 mg/kg, i.p.). This dose was selected to produce concentrations at the upper end of the linear range for VPA disposition in serum in rats (Liu and Pollack, 1993). Trunk blood and brain were collected by decapitation at 5, 10, 15, 20, 30, 45, 60, 75, 90, 120, 150, 180, 240, and 360 min after the dose from all Sprague-Dawley rats and at 5, 10, 15, 25, 40, 60, 90, and 120 min after dose from all Wistar rats (n = 3 per time point). Serum and brain samples were stored at −20°C until analysis.
VPA Brain Partitioning under Steady-State Conditions.
To evaluate whether manipulations to interrupt VPA ER altered the overall degree of VPA partitioning into brain tissue, a steady-state administration experiment was conducted using antibiotic- and vehicle-treated Sprague-Dawley rats and Mrp2-deficient and wild-type Wistar rats. VPA was administered in drinking water (6 mg/ml) to all rats for 3 days (∼700 mg/kg per day) before experimentation. This exposure condition was selected to produce steady-state VPA serum concentrations less than 100 mg/l, within the linear range of VPA disposition in rats. Antibiotic-treated rats received a combination of VPA, penicillin G, and streptomycin in drinking water for 3 days, followed by a bolus dose of penicillin G and streptomycin by oral gavage as described above. At the end of the 12-h dark cycle after 3 days of drug administration, trunk blood and brain were collected by decapitation. Serum and brain samples were stored at −20°C until analysis.
VPA Brain Uptake Index.
To identify any potential nonspecific alterations in BBB permeability to VPA resulting from manipulations to interrupt VPA ER, a brain uptake index experiment was conducted to compare uptake of VPA relative to the reference compound inulin in antibiotic- and vehicle-treated Sprague-Dawley rats and Mrp2-deficient and wild-type Wistar rats. Penicillin G and streptomycin were administered in drinking water for 3 days before experimentation and as a bolus dose 24 h before experimentation, as described above. At the time of experimentation, all rats were anesthetized with ketamine (160 mg/kg i.p.) and xylazine (8 mg/kg i.p.). The brain uptake index was determined as described previously (Hardebo and Nilsson, 1979). In brief, the right common carotid artery was exposed and injected (<0.5 s) with a 0.2-ml bolus of saline containing 1 μCi of 3H-VPA (50 μg/ml VPA) and 0.5 μCi of 14C-inulin. At 10 s after injection, brain tissue was collected by decapitation.
Quantitation of VPA.
VPA in serum and brain tissue samples was determined by gas chromatography with flame ionization detection (GC2014 series; Shimadzu, Kyoto, Japan) with a method modified from Liu et al. (2005). In brief, serum samples (50 μl), acidified 1:1 (v/v) with 2 N HCl, were extracted with 100 μl of ethyl acetate containing CCA as an internal standard. Brain samples were homogenized 1:1 (w/v) in saline, and 250 μl of the homogenate was acidified with 50 μl of 12 N HCl before extraction with ethyl acetate containing CCA. A 1-μl aliquot of the organic layer was injected onto a wide-bore, fused-silica capillary column (30 m × 0.53 mm id) with free fatty acid phase as the bonded stationary phase and helium as the carrier gas. For serum samples, isothermal chromatography was used, with the column oven maintained at 180°C and the injector and detector maintained at 200°C. For brain samples, these same column oven, injector, and detector temperatures were maintained for 5 min after injection. The column oven temperature then was increased to 230°C, and the injector and detector were increased to 250°C. Analyte versus internal standard peak area ratios were determined using GC Solution software (Shimadzu). The assay detection limit was 0.5 μg/g for brain and 0.1 μg/ml for serum, and standard curves were linear through the relevant range of concentrations for these studies.
Quantitation of 3H-VPA.
Upon collection, brain tissue was digested immediately with 3 ml of tissue solubilizer (Solvable; PerkinElmer Life and Analytical Sciences, Waltham, MA) overnight at 37°C. Samples were mixed with 15 ml of scintillation cocktail (Ultima Gold; PerkinElmer Life and Analytical Sciences) and stored at room temperature for 12 h before simultaneous determination of total radioactivity (3H and 14C) (1600TR liquid scintillation analyzer; PerkinElmer Life and Analytical Sciences). Brain tissue concentrations were corrected for residual blood contamination as described previously (Dagenais et al., 2000).
Data Analysis.
Data are presented as mean ± S.D. For evaluation of VPA ER interruption methods, AUC0–8 h was calculated by the trapezoidal method. The contribution of ER to AUC0–8 h was determined by subtracting from the total AUC0–8 h the AUC before ER, extrapolated through infinity using noncompartmental analysis (WinNonlin 5.0.1; Pharsight, Mountain View, CA) as described previously (Pollack and Brouwer, 1991). Because of the short half-life (<20 min) associated with the pre-ER phase of VPA disposition, in the absence of ER, VPA concentrations would become negligible well before 8 h. Therefore, the AUC pre-ER extrapolated through infinite time is identical to the AUC pre-ER that would be calculated through 8 h.
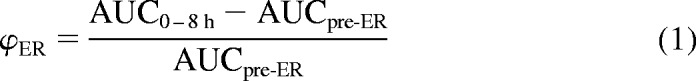 |
The degree of overshoot in the Cbrain/Cserum versus time profile in treated and untreated rats was compared by calculating the AUC above the last measurable Cbrain/Cserum ratio value for each treatment group, by the trapezoidal method. Integrated Cbrain/Cserum was calculated by normalizing the AUC for the concentration ratio by the total sampling time (t) for each strain (1.25 h for Sprague-Dawley, 1.5 h for Wistar).
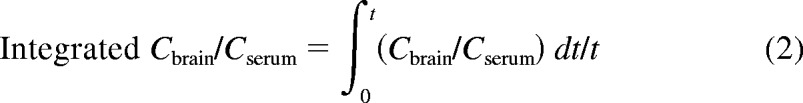 |
The brain uptake index was calculated as described by Hardebo and Nilsson (1979). In all cases, S.D. on AUC measurements was calculated by the method of Bailer (1988). Statistical comparisons between groups were accomplished by t test or analysis of variance with post hoc comparisons and correction for multiple comparisons, as appropriate, using SigmaStat software (Aspire Software International, Ashburn, VA). A P value less than 0.05 was considered to be statistically significant. Deconvolution analysis of brain partitioning data were conducted with PCDCON software (http://www.boomer.org/pkin/soft.html).
Results
Interruption of ER.
No interruption of VPA ER was observed in charcoal- versus vehicle-treated Sprague-Dawley rats, based upon comparison of serum concentrations after a 75 mg/kg dose of VPA (Fig. 1a). Concentration-time profiles for the two groups were nearly superimposable, with appearance of a secondary peak in VPA concentration, a hallmark of the ER process, beginning at 2 h and becoming maximal at approximately 4 h after the dose. By subtracting the pre-ER AUC0-inf from the observed AUC0–8 h, for each treatment group it was determined that the contribution of ER to the AUC0–8 h did not differ significantly between the charcoal-treated (39.9 μg · ml−1 · h−1) and vehicle-treated (43.5 μg · ml−1 · h−1) groups; 91.8% of the AUC contributed by ER in vehicle-treated animals remained after charcoal treatment (Table 1). The fraction of the dose undergoing recycling through 8 h (ΦER; eq. 1) also did not differ with charcoal treatment.
Fig. 1.

Serum VPA concentrations in Sprague-Dawley control (a; filled symbols) and charcoal-treated (a; open symbols) rats, Sprague-Dawley control (b; filled symbols) and antibiotic-treated (b; open symbols) rats, and Wistar wild-type (c; filled symbols) and Mrp2-deficient (c; open symbols) rats after a 75 mg/kg bolus dose of VPA. Symbols indicate sample mean, and bars represent S.D. Lines are included to emphasize temporal relationships.
TABLE 1.
Influence of experimental manipulations on the enterohepatic recycling of VPA in rats
Data are presented as mean (S.D.).
| Sprague-Dawley |
Wistar |
||||
|---|---|---|---|---|---|
| Vehicle (n = 4) | Charcoal (n = 3) | Antibiotics (n = 5) | Wild Type (n = 4) | Mrp2(−/−) (n = 4) | |
| AUC0–8 h | 124 (6.54) | 133 (14.3) | 145 (30.2) | 102 (10.5) | 94.4 (10.5) |
| AUCpre-ER* | 80.1 (4.73) | 92.8 (8.11) | 126** (26.8) | 54.9 (3.42) | 89.4*** (10.8) |
| AUCER* | 43.5 (4.44) | 39.9 (11.5) | 19.2*** (3.54) | 47.1 (13.6) | 4.91*** (0.862) |
| ΦER* | 0.352 (0.0274) | 0.298 (0.0605) | 0.133*** (0.00894) | 0.456 (0.0868) | 0.0530*** (0.0127) |
| Percentage of control | 91.8 | 44.1 | 10.4 | ||
AUCpre-ER, area under the concentration-time profile before the secondary increase in concentrations and extrapolated through infinite time; AUCER, difference between AUC0–8 h and AUCpre-ER.
P < 0.05 (ANOVA), significantly different among Sprague-Dawley groups.
P < 0.01, significantly different from respective control.
P < 0.001, significantly different from respective control.
A partial interruption of ER was observed after antibiotic treatment. In antibiotic-treated rats, the beginning of a secondary VPA concentration peak occurred slightly later (4 h) but at lower concentrations than in control Sprague-Dawley rats (Fig. 1b). This secondary rise in serum concentrations did not peak within the time frame of the experiment but rose slowly through 8 h, with concentrations remaining consistently lower than those in the control group. Antibiotic treatment substantially reduced the ER-contributed AUC relative to Sprague-Dawley controls (P < 0.001), with antibiotic-treated rats exhibiting approximately 44% of the ER-contributed AUC observed in vehicle-treated animals (Table 1). The fraction of the dose undergoing recycling through 8 h (ΦER) was also significantly reduced (P < 0.001) with charcoal treatment. Antibiotic treatment also increased the pre-ER AUC significantly (Table 1), suggesting that systemic clearance of VPA in the absence of ER was reduced.
As illustrated in Fig. 1c, a nearly complete attenuation of ER was observed in Mrp2-deficient rats relative to wild-type Wistar controls. Whereas no marked secondary VPA serum concentration peak was apparent in Mrp2-deficient rats, a minor contribution of ER (or some other undefined distributional process) to the AUC0–8 h was calculated based on the functional definition underlying the area analysis, with ER accounting for 4.91 μg · ml−1 · h−1 of the 94.4-μg · ml−1 · h−1 AUC0–8 h. Therefore, Mrp2-deficient rats exhibited only 10.4% of the ER-contributed AUC0–8 h observed in wild-type controls (Table 1). Loss of Mrp2 function increased the pre-ER AUC significantly (Table 1), suggesting that in the absence of ER, systemic clearance of VPA was reduced in Mrp2-deficient rats.
Brain Partitioning of VPA.
Because pilot studies indicated that brain concentrations fell below the lower limit of detection (0.5 μg/g) earlier than serum concentrations, the sampling schedule for this experiment was limited to 4 h. Serum VPA concentrations (total and unbound) in antibiotic-treated and vehicle-treated control Sprague-Dawley rats and in Mrp2-deficient and wild-type Wistar rats are illustrated in Fig. 2. The beginning of a secondary VPA concentration peak was evident in Sprague-Dawley control animals by 120 min after the dose. A small apparent secondary rise in the concentration-time profile in antibiotic-treated rats was observed at 150 min, and no apparent secondary increase in concentration was observed for the Mrp2-deficient animals, consistent with the preceding experiment. The unbound fraction in serum varied from 0.3 to 0.5 and tended to increase with increasing concentration. Brain VPA concentrations determined in antibiotic- and vehicle-treated Sprague-Dawley rats, and in Mrp2-deficient and wild-type Wistar rats, after administration of 100 mg/kg VPA, are depicted in Fig. 3.
Fig. 2.

Total (left) and unbound (right) serum VPA concentrations in control (a; filled symbols) and antibiotic-treated (a; open symbols) Sprague-Dawley rats and wild-type (b; filled symbols) and Mrp2-deficient (b; open symbols) Wistar rats after a 100 mg/kg bolus dose of VPA. Symbols indicate sample mean, and bars represent S.D. Lines are included to emphasize temporal relationships.
Fig. 3.

Brain VPA concentrations in control (a; filled symbols) and antibiotic-treated (a; open symbols) Sprague-Dawley rats and wild-type (b; filled symbols) and Mrp2-deficient (b; open symbols) Wistar rats after a 100 mg/kg bolus dose of VPA. Symbols indicate sample mean, and bars represent S.D. Lines are included to emphasize temporal relationships.
Brain-to-blood partitioning of VPA, represented as Cbrain/Cserum versus time, is illustrated in Fig. 4. Consistent with previous studies (Hammond et al., 1982; Golden et al., 1993), an early overshoot in the Cbrain/Cserum profile was apparent in vehicle-treated Sprague-Dawley rats (Fig. 4a). Cbrain/Cserum peaked at 10 min and then decreased throughout the remainder of the experiment, although the rate of decline decreased after approximately 45 min after the dose. In antibiotic-treated rats, an early overshoot also was observed, peaking within 10 min at a Cbrain/Cserum of 0.0810, which was slightly lower than that in control rats (0.0953). Notably, this early peak in the concentration ratio decreased more rapidly in antibiotic-treated rats and remained lower through 45 min, when the rate of change became low in both groups of animals.
Fig. 4.

Brain partitioning of VPA in Sprague-Dawley control (a; filled symbols) and antibiotic-treated (a; open symbols) rats and Wistar wild-type (b; filled symbols) and Mrp2-deficient (b; open symbols) rats after a 100 mg/kg bolus dose of VPA. Symbols indicate sample mean, and bars represent S.D. Lines are included to emphasize temporal relationships.
Wild-type Wistar rats also exhibited an early overshoot in the Cbrain/Cserum versus time profile (Fig. 4b). This overshoot peaked slightly later than that in Sprague-Dawley rats, at 25 min, with a Cbrain/Cserum value of 0.0733. An overshoot was observed in the Cbrain/Cserum profile for Mrp2-deficient rats, with a magnitude of overshoot similar to that in the control animals (0.0726). However, as with the antibiotic-treated Sprague-Dawley rats, this overshoot was truncated, with the partition ratio decreasing more quickly and remaining lower than that in the corresponding ratio in ER-unimpaired animals.
In both of the ER-interrupted groups, a brief (spanning 10–25 min in the Wistar group and 5–15 min in the Sprague-Dawley group) early peak in the Cbrain/Cserum versus time profile was observed. To evaluate the relationship between the appearance of this peak and the VPA ER process, deconvolution analysis was conducted. Deconvolution was selected as an analytical approach because of the possibility that multiple processes unrelated to ER might influence the kinetics of brain-to-serum partitioning of VPA. By deconvolving data obtained in ER-interrupted rats from data obtained in ER-intact animals, it is possible to isolate the specific effect of ER on brain partitioning kinetics. Ideally, the influence of ER on these Cbrain/Cserum versus time profiles would be evaluated by comparing brain partitioning profiles in the presence compared with the complete absence of ER and all other peripheral distribution mechanisms that might contribute to the overshoot phenomenon. However, the fact that VPA ER interruption was nearly complete in the Mrp2-deficient Wistar rats, whereas antibiotic treatment in Sprague-Dawley rats provided only partial interruption of ER, provided an opportunity to test the hypothesis that the magnitude of overshoot in the VPA Cbrain/Cserum ratio is related to the extent of VPA ER. Deconvolving the Cbrain/Cserum versus time profile in ER-ablated (or ER-diminished) rats from the profile in ER-intact controls allowed direct assessment of this hypothesis. As illustrated in Fig. 5, the deconvolved Cbrain/Cserum versus time profile in Wistar rats (Mrp2-deficient versus control) evidenced a clear peak, consistent with overshoot, approximately 30 min after dose. The peak in the deconvolution profile represents the peak of the overshoot phenomenon resulting specifically from ER and is unrelated to the time at which the brain/serum concentration ratio is maximal, which may be confounded by additional processes unrelated to ER. Sprague-Dawley rats (antibiotic treated versus controls) also evidenced a peak at the same time. The area bounded by the partitioning rate versus time profile in Wistar rats was approximately two times larger than that in Sprague-Dawley rats, consistent with the fact that Mrp2 deficiency essentially ablated ER (reducing the apparent volume of the recycling compartment to zero), whereas antibiotic treatment reduced ER by only about 50% (reducing the apparent volume of the recycling compartment by 50%).
Fig. 5.

Results of deconvolving Cbrain/Cserum versus time data in ER-interrupted rats from control animals. The solid line indicates Mrp2-deficient/control Wistar rats; the dashed line indicates antibiotic-treated/control Sprague-Dawley rats. The deconvolved partitioning rate is the rate of change in the Cbrain/Cserum ratio. The area under the partitioning rate versus time profile is a unitless number that serves as a metric of the degree of overshoot and was 7.95 × 10−4 for Mrp2-deficient/control Wistar rats and 3.38 × 10−4 for antibiotic-treated/control Sprague-Dawley rats.
As a result of the rapid decrease of brain VPA concentrations below the limit of detection, it was not possible to evaluate the brain partitioning profile through the attainment of distribution equilibrium. Thus, the magnitude of overshoot in the Cbrain/Cserum versus time profile was quantitated through the last time point at which brain concentrations remained detectable by calculating the AUC (normalized for sampling time) for Cbrain/Cserum versus time above the last measureable value for each treatment group. This final observed concentration ratio was used as a surrogate for the eventual (at distribution equilibrium) partition coefficient. The resulting AUC values (±S.D.), as calculated by the method for destructive sampling (Bailer, 1988) through the final sampling time, are presented in Fig. 6. Antibiotic-treated Sprague-Dawley (P < 0.05) and Mrp2-deficient Wistar (P < 0.01) rats exhibited significantly reduced overshoot areas, 32.7 and 21.3% of the size of the overshoot area calculated in each control group, respectively.
Fig. 6.

Area under the Cbrain/Cserum versus time profile normalized for sampling duration (eq. 2) after a 100 mg/kg bolus dose of VPA. Filled bars indicate ER-competent control (vehicle-treated Sprague-Dawley and vehicle-treated wild-type Wistar) rats, and open bars indicate ER-interrupted (antibiotic-treated Sprague-Dawley and Mrp2-deficient Wistar) rats. Error bars indicate S.D.; asterisks indicate statistically significant difference within each strain (P < 0.001).
VPA Brain Partitioning under Steady-State Conditions.
To rule out a generalized effect of ER-interrupting treatments on VPA brain partitioning, Cbrain/Cserum values were calculated for each treatment group under steady-state conditions. As depicted in Fig. 7, antibiotic- versus vehicle-treated Sprague-Dawley rats evidenced similar serum and brain concentrations and did not differ significantly in Cbrain/Cserum. Although serum VPA concentrations were significantly higher in wild-type versus Mrp2-deficient Wistar rats, brain concentrations were also moderately higher (data not shown). Thus, Cbrain/Cserum did not differ significantly between the two groups.
Fig. 7.

Brain/blood partition ratios calculated after administration of VPA to steady state. Filled bars indicate ER-competent control (vehicle-treated Sprague-Dawley and wild-type Wistar) rats, and open bars indicate ER-interrupted (antibiotic-treated Sprague-Dawley and Mrp2-deficient Wistar) rats. Error bars indicate S.D.
VPA Brain Uptake Index.
To determine whether ER-interrupting conditions affected BBB permeability to VPA, the initial rate of VPA uptake relative to uptake of the brain vascular space marker inulin was compared between each strain of ER-interrupted and control rats (Fig. 8). In antibiotic-treated rats, 14C-inulin uptake was slightly lower than in vehicle-treated Sprague-Dawley rats, whereas 3H-VPA uptake was similar. The ratio of VPA to inulin uptake did not differ statistically significantly between the two groups. Although 14C-inulin uptake was somewhat lower in Mrp2-deficient versus wild-type Wistar rats, 3H-VPA uptake was also slightly lower. Thus, the ratio of VPA to inulin uptake did not differ significantly between these groups.
Fig. 8.

Percentage uptake of the injected dose of 14C inulin (a) and 3H VPA(b) and ratios of VPA to inulin uptake (c), as determined by the brain uptake index method. Error bars indicate S.D.
Discussion
Interruption of VPA ER.
The present study demonstrated the various efficiencies of three different approaches to experimental ablation of VPA ER in rats. Oral administration of charcoal has been reported to eliminate more than half of the ER-related absorption of VPA in humans (Neuvonen et al., 1983), and administration of activated charcoal can be used to treat acute VPA overdose (Manoguerra et al., 2008). However, activated charcoal did not impact VPA ER in this study. Failure of this treatment to reduce VPA ER may have resulted from saturation of binding sites on the activated charcoal due to relatively high VPA concentrations under the conditions of the current experiment. A brief in vitro experiment demonstrated that the capacity of activated charcoal to bind VPA was saturable within a relevant range of VPA concentrations (data not shown). Although an increased dose of charcoal may have been effective, it was not viewed as a viable solution for the purposes of this experiment.
The 3-day regimen of oral penicillin G and streptomycin was selected based upon several reports documenting loss of enzyme-mediated hydrolysis of a β-glucuronide metabolite after exposing rats to 1 or 2 mg/ml penicillin and streptomycin in drinking water (Takasuna et al., 1996). In the current study, this treatment significantly reduced ER-associated systemic VPA exposure (by 56% relative to controls). Although other antibiotic regimens, including neomycin, bacitracin, and tetracycline, are efficient inhibitors of ER for various compounds, including VPA (Gott and Griffiths, 1987; Kojima et al., 1998; Takasuna et al., 2006), penicillin G and streptomycin were chosen because these compounds can be administered in drinking water (i.e., they do not influence water consumption in rats). This regimen furnished a simple and noninvasive means of reducing ER relative to other antibiotic administration routes. It also provided an intermediate degree of VPA recycling, less than in untreated controls but more than in Mrp2-deficient rats (Table 1).
The impact of lack of Mrp2 function on VPA ER was examined in Mrp2-deficient Wistar rats. VPA glucuronide is a substrate of Mrp2 (Khemawoot et al., 2007), and Mrp2 deficiency almost completely eliminates active secretion of the acyl glucuronide metabolite of VPA across the canalicular membrane, with a nearly complete reduction of choleresis associated with movement of VPA glucuronide into bile (Wright and Dickinson, 2004). Absence of Mrp2 function resulted in a nearly complete abolition of the VPA AUC attributable to ER relative to Mrp2-competent controls (Table 1), consistent with a central role of Mrp2 in the biliary excretion of VPA-glucuronide.
The fact that interruption of VPA ER by antibiotic treatment was incomplete, whereas VPA ER was essentially ablated in TR− rats, was viewed as an experimental advantage in that it allowed us to address that hypothesis that the magnitude of overshoot in VPA brain partitioning is related to the magnitude of ER. This hypothesis is different from, but compatible with, the broader hypothesis that overshoot in brain partitioning of VPA is a consequence of peripheral “distribution” into a recycling unit.
Brain Partitioning of VPA.
As predicted based upon previous simulations (Padowski and Pollack, 2011a,b) and the abundance of examples in the literature (Hammond et al., 1982; Liu et al., 1992; Golden et al., 1993; Liu and Pollack, 1993, 1994; Haberer and Pollack, 1994; Padowski and Pollack, 2011a), overshoot in the brain/serum concentration ratio was observed in Sprague-Dawley and wild-type Wistar rats as an early peak, followed by a decline in the Cbrain/Cserum versus time profile (Fig. 4). Although antibiotic treatment and Mrp2 deficiency elicited moderate and nearly complete ablation of VPA ER, notable, although brief, peaks were observed at early time points in both ER-reduced groups. This observation suggests that mechanisms other than ER also contribute to the VPA overshoot phenomenon.
Previous simulation experiments (Padowski and Pollack, 2011a) predicted that increasing the apparent volume of a peripheral compartment would result in a proportionate increase in the duration of overshoot. Visual comparisons of the brain partitioning profiles in the current study are consistent with these predictions. In contrast, simulations predicted that a “ceiling” would exist for the magnitude of overshoot. In the current study, the overshoot magnitude was similar in partially ER-interrupted (Sprague-Dawley) and completely ER-interrupted (Wistar) rats (Fig. 6), suggesting that overshoot magnitude is maximum at a peripheral volume represented by 50% of normal valproate recycling.
Because of deficiencies associated with simple visual comparisons, deconvolution analysis was performed to isolate the influence of ER on the brain/serum concentration ratio versus time profile. Deconvolution abolished the initial early peak (Fig. 5), indicating that this peak was a component of both profiles and was independent of ER. This analysis suggests that a mechanism unrelated to ER may be responsible for the brief overshoot observed in the ER-interrupted rats. The mechanism leading to this non-ER-related overshoot is unclear and will require additional experimentation to explore. However, it could be related to a process as simple as distribution into another (non-ER-related) peripheral compartment, resulting in peripheral sequestration that is much less substantial than that associated with ER. Moreover, deconvolution analysis confirmed that incomplete interruption of ER (antibiotic treatment) had a smaller effect on the Cbrain/Cserum versus time profile than nearly complete ablation of ER (Mrp2-deficient rats).
Although mathematical descriptors of the overshoot phenomenon have been developed previously (Padowski and Pollack, 2011a,b), these metrics of the magnitude (peak Cbrain/Cserum value as a percentage of the Kp,brain value) and duration (time at which the system reaches a stable plateau value, Kp,brain) of the overshoot could not be used in the current study. Both metrics depend upon confirming the plateau Cbrain/Cserum value (i.e., Kp,brain, which is calculated after attainment of distribution equilibrium). Despite good sensitivity of the VPA assay (0.1–0.5 μg/ml limit of detection), brain concentrations could not be quantified beyond 2 h after dose. A survey of reported VPA brain partitioning profiles failed to yield an example of attainment of distribution equilibrium after a bolus dose of VPA.
An additional consequence of the inability to determine the plateau Kp,brain for VPA in the current study is the inability to measure total overshoot in each treatment group. Comparison of the measurable portions of the overshoot area between ER-inhibited and control groups necessarily overpredicts the ER-interruption-associated difference between the two groups, although, depending on the true Kp,brain value, the magnitude of this overprediction could be large or small. Although Kp,brain for VPA was measured in all treatment groups under steady-state conditions, this value, while relatively consistent across groups, was not used as the baseline Kp,brain value for comparison of overshoot area in the brain partitioning versus time profiles because they were generated under fundamentally different conditions and may have been affected by the well established nonstationary kinetics of VPA (Arens and Pollack, 2001).
Nonspecific Effects of ER-Interrupting Conditions on VPA CNS Kinetics.
The current study was intended to compare the effect of manipulation of the apparent volume of a peripheral pharmacokinetic compartment (as degree of ER) on the kinetics of brain partitioning. To rule out nonspecific effects of ER-interrupting conditions, two endpoints were evaluated. One endpoint was the steady-state Kp,brain value. Although no experimental evidence in the literature would suggest that steady-state Kp,brain values should differ in the presence versus absence of ER, a previous simulation study (Padowski and Pollack, 2011a) illustrated several situations under which a small effect (typically <10% difference) might be observed. No statistical differences between Kp,brain in ER-interrupted versus control rats were observed in the present study, consistent with predictions. The other endpoint evaluated for potential nonspecific influence of the ER interruption was initial uptake clearance into brain, which was assessed with the brain uptake index technique. No significant differences were observed between antibiotic- and vehicle-treated Sprague-Dawley rats. Therefore, we can conclude that changes in the Cbrain/Cserum ratio versus time profile were likely due to changes in peripheral distribution kinetics of VPA and not to nonspecific effects of VPA translocation across the BBB.
Although VPA-glucuronide is a substrate for Mrp2 in liver, mediating biliary excretion of the conjugate (Wright and Dickinson, 2004), VPA does not seem to be a substrate for this transport system (Baltes et al., 2007; Luna-Tortós et al., 2010). Thus, we would not anticipate a direct influence of Mrp2 deficiency on the kinetics of brain-to-serum partitioning of VPA. This expectation is consistent with the results of both the steady-state (Fig. 7) and brain uptake index (Fig. 8) experiments. VPA does seem to undergo carrier-mediated translocation at the blood-brain and blood-cerebrospinal fluid barriers by monocarboxylic acid (Terasaki et al., 1991; Takasuna et al., 2006) and/or organic anion transporting systems (Fischer et al., 2008). However, there is no evidence that either Mrp2 deficiency or antibiotic treatment would affect VPA transport into or out of the CNS by these systems or by either passive diffusion or electrostatic effects known to impede flux of anions across the endothelial cells that compose the BBB (Yuan et al., 2010).
Conclusions
The current study demonstrated that overshoot in the VPA Cbrain/Cserum versus time profile occurs in the presence of active ER and that attenuation of ER (and thus reduction of peripheral distribution volume) reduced the degree of overshoot. The nearly complete ablation of VPA ER in Mrp2-deficient versus wild-type Wistar rats did not completely abolish overshoot. The persistence of a diminished magnitude of overshoot in the nearly complete absence of ER may be due to non-ER-related peripheral sequestration of VPA (e.g., binding to proteins on tissue) or to another as-yet identified mechanism. Future simulation or experimental studies may be able to probe the origin of the brief, early overshoot peak in ER-interrupted animals. Finally, although the present study is consistent with simulations (Padowski and Pollack, 2011), the identification of additional CNS-distributing compounds with significant ER-related overshoot would be desirable.
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant GM61191]; the National Institutes of Health National Institute of Environmental Health Sciences [Grant T32-ES007126]; and Eli Lilly and Company.
Part of this work was presented in partial fulfillment of a doctoral degree: Padowski JM (2008) A Multi-Factorial Approach to Understanding and Predicting Brain Exposure to Pharmacologic Agents. Doctoral dissertation, University of North Carolina at Chapel Hill, Chapel Hill, NC.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
- CNS
- central nervous system
- Kp,brain and Cbrain/Cserum
- brain/serum partition coefficient
- ER
- enterohepatic recycling
- VPA
- valproic acid
- CCA
- cyclohexanecarboxylic acid
- DE
- distribution equilibrium
- φER
- fraction of the dose undergoing enterohepatic recirculation
- BBB
- blood-brain barrier
- AUC
- area under the concentration-time curve
- AUC0–8 h
- area under the concentration-time profile calculated by the trapezoidal method.
Authorship Contributions
Participated in research design: Padowski and Pollack.
Conducted experiments: Padowski and Pollack.
Performed data analysis: Padowski and Pollack.
Wrote or contributed to the writing of the manuscript: Padowski and Pollack.
References
- Arens TL, Pollack GM. (2001) Nonstationary disposition of valproic acid during prolonged intravenous infusion: contributions of unbound clearance and protein binding. Biopharm Drug Dispos 22:243–249 [DOI] [PubMed] [Google Scholar]
- Bailer AJ. (1988) Testing for the equality of area under the curves when using destructive measurement techniques. J Pharmacokinet Biopharm 16:303–309 [DOI] [PubMed] [Google Scholar]
- Baltes S, Fedrowitz M, Tortós CL, Potschka H, Löscher W. (2007) Valproic acid is not a substrate for P-glycoprotein or multidrug resistance proteins 1 and 2 in a number of in vitro and in vivo transport assays. J Pharmacol Exp Ther 320:331–343 [DOI] [PubMed] [Google Scholar]
- Dagenais C, Rousselle C, Pollack GM, Scherrmann JM. (2000) Development of an in situ mouse brain perfusion model and its application to mdr1a P-glycoprotein-deficient mice. J Cereb Blood Flow Metab 20:381–386 [DOI] [PubMed] [Google Scholar]
- Dallas CE, Chen XM, O'Barr K, Muralidhara S, Varkonyi P, Bruckner JV. (1994) Development of a physiologically based pharmacokinetic model for perchloroethylene using tissue concentration-time data. Toxicol Appl Pharmacol 128:50–59 [DOI] [PubMed] [Google Scholar]
- Fischer W, Praetor K, Metzner L, Neubert RH, Brandsch M. (2008) Transport of valproate at intestinal epithelial (Caco-2) and brain endothelial (RBE4) cells: mechanism and substrate specificity. Eur J Pharm Biopharm 70:486–492 [DOI] [PubMed] [Google Scholar]
- Gibaldi M. (1969) Effect of mode of administration on drug distribution in a two-compartment open system. J Pharm Sci 58:327–331 [DOI] [PubMed] [Google Scholar]
- Golden PL, Brouwer KR, Pollack GM. (1993) Assessment of valproic acid serum-cerebrospinal fluid transport by microdialysis. Pharm Res 10:1765–1771 [DOI] [PubMed] [Google Scholar]
- Gott DM, Griffiths LA. (1987) Effects of antibiotic pretreatments on the metabolism and excretion of [U14C](+)-catechin [(U14C](+)-cyanidanol-3) and its metabolite, 3′-0-methyl-(+)-catechin. Xenobiotica 17:423–434 [DOI] [PubMed] [Google Scholar]
- Haberer LJ, Pollack GM. (1994) Disposition and protein binding of valproic acid in the developing rat. Drug Metab Dispos 22:113–119 [PubMed] [Google Scholar]
- Hammarlund-Udenaes M, Paalzow LK, de Lange EC. (1997) Drug equilibration across the blood-brain barrier—pharmacokinetic considerations based on the microdialysis method. Pharm Res 14:128–134 [DOI] [PubMed] [Google Scholar]
- Hammond EJ, Perchalski RJ, Villarreal HJ, Wilder BJ. (1982) In vivo uptake of valproic acid into brain. Brain Res 240:195–198 [DOI] [PubMed] [Google Scholar]
- Hardebo JE, Nilsson B. (1979) Estimation of cerebral extraction of circulating compounds by the brain uptake index method: influence of circulation time, volume injection, and cerebral blood flow. Acta Physiol Scand 107:153–159 [DOI] [PubMed] [Google Scholar]
- Kalvass JC, Pollack GM. (2007) Kinetic considerations for the quantitative assessment of efflux activity and inhibition: implications for understanding and predicting the effects of efflux inhibition. Pharm Res 24:265–276 [DOI] [PubMed] [Google Scholar]
- Khemawoot P, Maruyama C, Tsukada H, Noda H, Ishizaki J, Yokogawa K, Miyamoto K. (2007) Influence of chronic hepatic failure on disposition kinetics of valproate excretion through a phase II reaction in rats treated with carbon tetrachloride. Biopharm Drug Dispos 28:331–338 [DOI] [PubMed] [Google Scholar]
- Kojima S, Nadai M, Kitaichi K, Wang L, Nabeshima T, Hasegawa T. (1998) Possible mechanism by which the carbapenem antibiotic panipenem decreases the concentration of valproic acid in plasma in rats. Antimicrob Agents Chemother 42:3136–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Sugiyama Y, Awazu S, Hanano M. (1982) In vitro and in vivo evaluation of the tissue-to-blood partition coefficient for physiological pharmacokinetic models. J Pharmacokinet Biopharm 10:637–647 [DOI] [PubMed] [Google Scholar]
- Liu MJ, Brouwer KL, Pollack GM. (1992) Pharmacokinetics and pharmacodynamics of valproate analogs in rats. III. Pharmacokinetics of valproic acid, cyclohexanecarboxylic acid, and 1-methyl-1-cyclohexanecarboxylic acid in the bile-exteriorized rat. Drug Metab Dispos 20:810–815 [PubMed] [Google Scholar]
- Liu MJ, Pollack GM. (1993) Pharmacokinetics and pharmacodynamics of valproate analogs in rats. II. Pharmacokinetics of octanoic acid, cyclohexanecarboxylic acid, and 1-methyl-1-cyclohexanecarboxylic acid. Biopharm Drug Dispos 14:325–339 [DOI] [PubMed] [Google Scholar]
- Liu MJ, Pollack GM. (1994) Pharmacokinetics and pharmacodynamics of valproate analogues in rats. IV. Anticonvulsant action and neurotoxicity of octanoic acid, cyclohexanecarboxylic acid, and 1-methyl-1-cyclohexanecarboxylic acid. Epilepsia 35:234–243 [DOI] [PubMed] [Google Scholar]
- Liu X, Smith BJ, Chen C, Callegari E, Becker SL, Chen X, Cianfrogna J, Doran AC, Doran SD, Gibbs JP, et al. (2005) Use of a physiologically based pharmacokinetic model to study the time to reach brain equilibrium: an experimental analysis of the role of blood-brain barrier permeability, plasma protein binding, and brain tissue binding. J Pharmacol Exp Ther 313:1254–1262 [DOI] [PubMed] [Google Scholar]
- Luna-Tortós C, Fedrowitz M, Löscher W. (2010) Evaluation of transport of common antiepileptic drugs by human multidrug resistance-associated proteins (MRP1, 2 and 5) that are overexpressed in pharmacoresistant epilepsy. Neuropharmacology 58:1019–1032 [DOI] [PubMed] [Google Scholar]
- Manoguerra AS, Erdman AR, Woolf AD, Chyka PA, Caravati EM, Scharman EJ, Booze LL, Christianson G, Nelson LS, Cobaugh DJ, et al. (2008) Valproic acid poisoning: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol (Phila) 46:661–676 [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Mizobuchi M, Nakamura M, Takagi H, Inagaki H, Kominami G, Koike M, Yamaguchi T. (2004) Mechanism of the drug interaction between valproic acid and carbapenem antibiotics in monkeys and rats. Drug Metab Dispos 32:1383–1391 [DOI] [PubMed] [Google Scholar]
- Neuvonen PJ, Kannisto H, Hirvisalo EL. (1983) Effect of activated charcoal on absorption of tolbutamide and valproate in man. Eur J Clin Pharmacol 24:243–246 [DOI] [PubMed] [Google Scholar]
- Padowski JM, Pollack GM. (2011a) The influence of distributional kinetics into a peripheral compartment on the pharmacokinetics of substrate partitioning between blood and brain tissue. J Pharmacokinet Pharmacodyn 38:743–767 [DOI] [PubMed] [Google Scholar]
- Padowski JM, Pollack GM. (2011b) Influence of time to achieve substrate distribution equilibrium between brain tissue and blood on quantitation of the blood-brain barrier P-glycoprotein effect. Brain Res 1426:1–17 [DOI] [PubMed] [Google Scholar]
- Pollack GM, Brouwer KL. (1991) Physiologic and metabolic influences on enterohepatic recirculation: simulations based upon the disposition of valproic acid in the rat. J Pharmacokinet Biopharm 19:189–225 [DOI] [PubMed] [Google Scholar]
- Raub TJ. (2006) P-glycoprotein recognition of substrates and circumvention through rational drug design. Mol Pharm 3:3–25 [DOI] [PubMed] [Google Scholar]
- Summerfield SG, Read K, Begley DJ, Obradovic T, Hidalgo IJ, Coggon S, Lewis AV, Porter RA, Jeffrey P. (2007) Central nervous system drug disposition: the relationship between in situ brain permeability and brain free fraction. J Pharmacol Exp Ther 322:205–213 [DOI] [PubMed] [Google Scholar]
- Takasuna K, Hagiwara T, Hirohashi M, Kato M, Nomura M, Nagai E, Yokoi T, Kamataki T. (1996) Involvement of beta-glucuronidase in intestinal microflora in the intestinal toxicity of the antitumor camptothecin derivative irinotecan hydrochloride (CPT-11) in rats. Cancer Res 56:3752–3757 [PubMed] [Google Scholar]
- Takasuna K, Hagiwara T, Watanabe K, Onose S, Yoshida S, Kumazawa E, Nagai E, Kamataki T. (2006) Optimal antidiarrhea treatment for antitumor agent irinotecan hydrochloride (CPT-11)-induced delayed diarrhea. Cancer Chemother Pharmacol 58:494–503 [DOI] [PubMed] [Google Scholar]
- Terasaki T, Takakuwa S, Moritani S, Tsuji A. (1991) Transport of monocarboxylic acids at the blood-brain barrier: studies with monolayers of primary cultured bovine brain capillary endothelial cells. J Pharmacol Exp Ther 258:932–937 [PubMed] [Google Scholar]
- Wright AW, Dickinson RG. (2004) Abolition of valproate-derived choleresis in the Mrp2 transporter-deficient rat. J Pharmacol Exp Ther 310:584–588 [DOI] [PubMed] [Google Scholar]
- Yuan W, Li G, Gil ES, Lowe TL, Fu BM. (2010) Effect of surface charge of immortalized mouse cerebral endothelial cell monolayer on transport of charged solutes. Ann Biomed Eng 38:1463–1472 [DOI] [PubMed] [Google Scholar]