

Abstract
Cytochrome P450 (P450) enzymes are mixed-function oxidases that catalyze the metabolism of xenobiotics and endogenous biochemicals. Selective inhibitors are needed to accurately distinguish the contributions of individual P450 enzymes in the metabolism of drugs and the activation of procarcinogens in human tissues, but very frequently these enzymes have substantial overlapping selectivity. We evaluated a chemically diverse set of nine previously identified CYP2A6 inhibitors to determine which are able to discriminate between human CYP2A enzymes CYP2A6 and the 94%-identical CYP2A13 enzyme. Inhibitor binding to recombinant purified enzyme was evaluated, and affinities were determined. Ki values were determined for inhibition of p-nitrophenol 2-hydroxylation, a reaction accomplished by CYP2A13 and CYP2A6 with more similar catalytic efficiencies (kcat/Km 0.19 and 0.12 μM−1 · min−1, respectively) than hydroxylation of the classic substrate coumarin (0.11 and 0.53 μM−1 · min−1, respectively). Of the nine compounds assayed, only tranylcypromine and (R)-(+)-menthofuran had a greater than 10-fold preference for CYP2A6 inhibition versus CYP2A13 inhibition. Most compounds evaluated [tryptamine, 4-dimethylaminobenzaldehyde, phenethyl isothiocyanate, β-nicotyrine, (S)-nicotine, and pilocarpine] demonstrated only moderate or no preference for inhibition of one CYP2A enzyme over the other. However, 8-methoxypsoralen has a 6-fold lower Ki for CYP2A13 than for CYP2A6. This information is useful to inform reinterpretation of previous data with these inhibitors and to guide future studies seeking to determine which human CYP2A enzyme is responsible for the in vivo metabolism of compounds in human tissues expressing both enzymes.
Introduction
The cytochrome P450 (P450) superfamily of mixed-function oxidases catalyzes metabolism of xenobiotics and endogenous biochemicals. Although xenobiotic metabolism is often a detoxifying event, toxins and procarcinogens can also be bioactivated. To evaluate the involvement of an individual P450 enzyme in drug metabolism or activation of harmful chemical agents in vivo, compounds are often incubated with microsomes in the presence and absence of potent and selective inhibitors for individual P450 enzymes. This phenotyping process is widely used but is critically dependent on inhibitor selectivity. Although xenobiotic-metabolizing P450 enzymes have substantial overlapping substrate selectivity, broad efforts have identified sufficiently selective inhibitors for the major human hepatic enzymes, including CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 (reviewed in Khojasteh et al., 2011).
However, in extrahepatic tissues, discrimination between the roles of individual enzymes is more difficult, both because information about P450 proteins present can be incomplete and because selective inhibitors are not as well developed for nonhepatic P450 enzymes. For example, in the human respiratory tract, at least 16 P450 enzymes may play a role in the balance between detoxification and activation of environmental pollutants, including industrial chemicals, therapeutic agents, and chemicals in cigarette smoke (Zhang et al., 2006; reviewed in Pavek and Dvorak, 2008). More than one protein from the same P450 subfamily is often present, further complicating dissection of individual contributions. This is the case for human CYP2A enzymes CYP2A6 and CYP2A13. Because commercially available antibodies for these proteins usually cross-react (Su et al., 2000), information about tissue expression is often derived from mRNA levels. CYP2A13 mRNA is highest in the respiratory tract but is only ∼5- and ∼9-fold higher than CYP2A6 mRNA in nasal mucosa and lung, respectively (Su et al., 2000). In contrast, in the liver where CYP2A6 levels are highest, CYP2A6 mRNA is ∼1900-fold higher than CYP2A13. Thus, in human liver microsomes, metabolism by and inhibition of CYP2A13 are likely to be negligible, but in respiratory tract microsomes, both CYP2A enzymes may have substantial contributions depending on the substrate or inhibitor. The CYP2A6 and CYP2A13 sequences are 94% identical, and although they have substantial overlapping substrate selectivity, they also have key functional differences (Su et al., 2000; Bao et al., 2005; Fukami et al., 2007, 2008). Perhaps the most physiologically relevant is the superior ability of CYP2A13 to activate the tobacco-specific procarcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (Su et al., 2000; Bao et al., 2005) to diazonium ions that generate DNA adducts and can initiate lung cancer (Hecht, 1998). However, dissecting the roles of CYP2A enzymes is also likely to be important to evaluate the metabolism of inhaled drugs such as volatile anesthetics or those used for asthma. Thus, CYP2A13- and CYP2A6-selective inhibitors are needed to evaluate their respective roles in tissues expressing both enzymes.
A number of compounds have been reported to be selective for CYP2A6 inhibition, but many were evaluated before CYP2A13 was known to be a functional CYP2A enzyme or have simply not been tested with CYP2A13. These include the following: phenethyl isothiocyanate (PEITC), 4-dimethylaminobenzaldehyde (DMABA), 8-methoxypsoralen (8-MOP; also known as methoxsalen or xanthotoxin), tranylcypromine, tryptamine, pilocarpine, (S)-nicotine, (R)-(+)-menthofuran, and β-nicotyrine (Koenigs et al., 1997; Khojasteh-Bakht et al., 1998; Nakajima et al., 2001; Zhang et al., 2001; Denton et al., 2004; Rahnasto et al., 2008). Work conducted to identify potent and selective CYP2A6 inhibitors (Yano et al., 2006) did not evaluate them for their ability to inhibit CYP2A13. Although a few of these inhibitors have now been tested for CYP2A13 inhibition (Bao et al., 2005; von Weymarn et al., 2005, 2006), these studies were conducted using a variety of methods and protein preparations that make comparison of CYP2A inhibition difficult.
Thus, we evaluated the ability of compounds traditionally used to identify CYP2A6-selective functions for their ability to differentially inhibit CYP2A6 and CYP2A13 using side-by-side assays. Compounds were evaluated for binding mode, binding constants, and Ki values with recombinant, purified CYP2A6 and CYP2A13. Comparisons suggested that only (R)-(+)-menthofuran and tranylcypromine demonstrate >10-fold preference for CYP2A6 inhibition over CYP2A13, and that only 8-MOP has a >5-fold selectivity for CYP2A13 over CYP2A6. These results may be used to guide selection of the most selective available inhibitors in future studies of drug metabolism or procarcinogen activation in the respiratory tract and suggest that new, more selective inhibitors are needed.
Materials and Methods
Chemicals and Reagents.
β-Nicotyrine was purchased from Toronto Research Chemicals Inc. (North York, ON, Canada). Other reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise specified.
Protein Expression and Purification.
CYP2A proteins were expressed in Escherichia coli TOPP-3 cells as a form encompassing truncation of the N-terminal transmembrane helix and addition of a C-terminal four-residue histidine tag as described previously (Smith et al., 2007; DeVore et al., 2008). Transformed colonies were grown in 5-ml cultures in lysogeny broth supplemented with ampicillin and tetracycline. Cultures were grown at 37°C for 7 to 8 h with shaking (250 rpm). Fifty microliters of the 5-ml culture were used to inoculate 200 ml of lysogeny broth supplemented with ampicillin and tetracycline. Cultures were again grown overnight at 37°C with shaking (250 rpm). Fifteen milliliters of the overnight culture were used to inoculate 250 ml of Terrific broth supplemented with ampicillin, which was then grown overnight with shaking (250 rpm) at 37°C to an optical density of 1 to 1.5 measured at a 600 nm. Expression was induced by adding isopropyl β-d-1-thiogalactopyranoside (Affymetrix, Santa Clara, CA) to a final concentration of 1 mM, and δ-aminolevulinic acid (5 mM) was added to promote heme production. Cultures were grown for an additional 72 h at 30°C while shaking at 190 rpm.
Cells were harvested by centrifugation at 6400g for 10 min and were resuspended in 200 ml of 20 mM potassium phosphate, pH 7.4, containing 20% glycerol. Spheroplasts were produced by treating the suspension with 0.3 mg/ml lysozyme for 30 min with stirring, followed by addition of an equal volume of ice-cold water. After 10 min, the spheroplasts were collected by centrifugation at 10,000g for 15 min, and the supernatant was discarded. The pellet was then frozen using either a liquid nitrogen bath or dry ice/ethanol slurry. Frozen pellets were thawed, suspended in 100 ml of 500 mM potassium phosphate, pH 7.4, containing 20% glycerol, and homogenized by hand. The resulting suspension was sonicated using three 30-s pulses with 60 s of cooling on ice between pulses and then was centrifuged at 10,000g for 15 min. The detergent Cymal-5 (Affymetrix) was added to the resulting supernatant to 4.8 mM with stirring. After centrifugation at 100,000g for 60 min., the crude membrane protein sample was purified using a two-step column chromatography scheme. Solubilized protein was first applied to a nickel-nitrilotriacetic acid (QIAGEN, Valencia, CA) column equilibrated with loading buffer (100 mM potassium phosphate, pH 7.4, 20% glycerol, 0.2 M NaCl, 4.8 mM Cymal-5) and then was washed with loading buffer followed by wash buffer (100 mM potassium phosphate, pH 7.4, 20% glycerol, 0.2 M NaCl, 4.8 mM Cymal-5, 8 mM histidine). The protein was eluted with 10 mM potassium phosphate, pH 7.4, with 20% glycerol, 0.1 M NaCl, 4.8 mM Cymal-5, 80 mM histidine, and 2 mM EDTA. Fractions with the most P450 (as evaluated by absorbance at 418 nm) were pooled, diluted 3-fold with 5 mM potassium phosphate, pH 7.4, with 20% glycerol, 4.8 mM Cymal-5, and 1 mM EDTA, and loaded onto a HiTrap CM-Sepharose Fast Flow column (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) equilibrated with the same buffer. The CM column was washed with the same buffer without the detergent, and protein was eluted with 50 mM potassium phosphate, pH 7.4, with 20% glycerol, 500 mM NaCl, and 1 mM EDTA. Protein was concentrated using centrifugal ultrafiltration. Purification was accomplished at 4°C and resulted in protein with an absorbance ratio at 417/280 nm of 1.3 to 1.9 and specific contents of 6.2 to 14.2 nmol P450/mg protein for various CYP2A6 and CYP2A13 preparations. Enzyme was quantitated using the reduced carbon monoxide difference spectrum as described previously (Schenkman and Jansson, 2006). Rat NADPH P450 oxidoreductase (Shen et al., 1989) and rat cytochrome b5 (Holmans et al., 1994) were expressed and purified as described previously.
Spectral Binding Assays.
Spectral binding assays were conducted at 20°C using a UV-visible scanning spectrophotometer (UV-2101; Shimadzu, Kyoto, Japan) as described previously (DeVore et al., 2009). Using Prism 5 (GraphPad Software Inc., San Diego, CA), equilibrium dissociation constants were determined from nonlinear least-squares fits, using the tight-binding equation as appropriate for high-affinity compounds. Kd values are the average of duplicate or triplicate independent titrations.
Inhibition Assays.
Purified CYP2A protein was reconstituted with NADPH P450 reductase and cytochrome b5 and was used to detect p-nitrophenol (pNP) and coumarin metabolism in the presence of CYP2A inhibitors as described previously (DeVore et al., 2012) with modifications to the mobile phase. For the pNP assay, the high-performance liquid chromatography mobile phase was slightly adjusted to 29% acetonitrile and 0.2% acetic acid and was run at 0.8 ml/min, resulting in slight adjustments in retention times (∼6 min for nitrocatechol and ∼9 min for pNP). However, DMABA required a mobile phase of 26% methanol and 0.2% acetic acid. For both assays, the amount of metabolite was determined by comparing the product peak area with a standard curve. Incubations were performed with at least four different inhibitor concentrations and at least eight different substrate concentrations. Ki values are the global fit to at least two separate inhibition experiments. The rate of metabolite formation plotted against substrate concentration was initially fit to the Michaelis-Menten equation using GraphPad Prism 5 (GraphPad Software Inc.). Km and Vmax values were determined and used to assess the inhibition mode. The data were then reanalyzed with the appropriate equation for the type of inhibition observed. A selectivity factor indicating preference for CYP2A6 versus CYP2A13 was calculated by taking the ratio of CYP2A13 Ki/CYP2A6 Ki.
Results
Binding Modes and Binding Affinity.
The nine CYP2A6 ligands shown in Table 1 were characterized for their binding mode, affinity, and selectivity for both CYP2A enzymes. Difference spectra collected upon ligand titration demonstrated that for both enzymes tranylcypromine, tryptamine, and pilocarpine clearly yielded increases at 431 to 432 nm and decreases at 406 to 412 nm, typical of type II interactions of nitrogen with the heme iron, as exemplified by tryptamine (Fig. 1A). CYP2A6 and CYP2A13 binding of the other six ligands caused increases in absorbance at 379 to 387 nm and decreases at 414 to 420 nm, typical of type I interactions, in which ligand binding disrupts water interaction with the heme iron, as exemplified by (R)-(+)-menthofuran (Fig. 1B).
TABLE 1.
Selectivity of inhibitors of CYP2A enzymes as evaluated by binding affinity and inhibitor dissociation constants
| Inhibitor | P450 | Kd (Binding Mode)a | 2A13 Kd/2A6 Kd | Kid | Mechanism (α Value) | 2A13 Ki/2A6 Ki |
|---|---|---|---|---|---|---|
| μM | μM | |||||
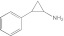 Tranylcypromine Tranylcypromine |
2A13 | 2.3 (II) | 1.2 | 6.5 ± 1.2 | Competitive | 49 |
| 2A6 | 2.0 (II) | 0.13 ± 0.02 | Mixed (α = 10) | |||
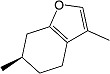 (R)-Menthofuran (R)-Menthofuran |
2A13 | 0.58 (I) | 0.13 | 54 ± 12 | Mixed (α = 4.3) | 27 |
| 2A6 | 4.5 (I) | 2.0 ± 0.44 | Mixed (α = 4.3) | |||
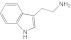 Tryptamine Tryptamine |
2A13 | 5.2 (II) | 2.3 | 16 ± 3.5 | Competitive | 9.4 |
| 2A6 | 2.3 (II) | 1.7 ± 0.12 | Competitive | |||
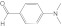 DMABA DMABA |
2A13 | 0.65 (I) | 0.96 | 17 ± 5.0 | Mixed (α = 13) | 4.6 |
| 2A6 | 0.68 (I) | 3.6 ± 0.83 | Mixed (α = 5.5) | |||
 PEITC PEITC |
2A13 | 0.43 (I)b | 0.064 | 3.8 ± 1.6 | Mixed (α = 0.20) | 2.2 |
| 2A6 | 6.2 (I)b | 1.7 ± 0.28 | Mixed (α = 270) | |||
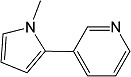 β-Nicotyrine β-Nicotyrine |
2A13 | 8.2 (I) | 0.12 | 5.6 ± 0.86 | Mixed (α = 6.6) | 0.74 |
| 2A6 | 71 (I) | 7.5 ± 2.9 | Mixed (α = 3.5) | |||
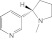 (S)-Nicotine (S)-Nicotine |
2A13 | 54 (I) | 0.11 | 72 ± 4.6 | Competitive | 0.55 |
| 2A6 | 470 (I) | 130 ± 8.8 | Competitive | |||
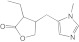 Pilocarpine Pilocarpine |
2A13 | 3.0 (II)c | 0.83 | 1.4 ± 0.12 | Competitive | 0.47 |
| 2A6 | 3.6 (II)c | 3.0 ± 0.45 | Mixed (α = 25) | |||
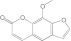 8-MOP 8-MOP |
2A13 | 1.6 (I) | 0.14 | 0.040 ± 0.009 | Mixed (α = 1.9) | 0.16 |
| 2A6 | 11 (I) | 0.25 ± 0.10 | Mixed (α = 3.3) |
The binding modes are shown in parentheses, where (I) represents type I binding and (II) represents type II binding.
Ki ± S.E.
Fig. 1.

Representative binding spectra for the type II ligand tryptamine (A) and the type I ligand (R)-(+)-menthofuran (B), both with CYP2A6. Increasing concentrations of ligands during the titrations are indicated by spectral scans colored from red (low concentration, 0.15–0.2 μM) to violet (high concentration, 400–500 μM). Insets represent the nonlinear regression analysis used to obtain Kd values.
Using titrations (e.g., Fig. 1, A and B), the equilibrium dissociation constants were determined for CYP2A6 and CYP2A13 for each compound (e.g., Fig. 1, insets). Affinities ranged from 0.43 to 54 μM for CYP2A13 and 0.68 to 470 μM for CYP2A6 (Table 1). The compounds with the highest affinity for CYP2A6 were tranylcypromine (Kd = 2.0 μM) and DMABA (Kd = 0.68 μM), whereas the compounds with the highest affinity for CYP2A13 were PEITC (Kd = 0.43 μM), (R)-(+)-menthofuran (Kd = 0.58 μM), and DMABA (Kd = 0.65 μM). β-Nicotyrine and (S)-nicotine had the lowest affinity for both enzymes.
Selectivity of each inhibitor for binding of the ferric, ligand-free, water-bound state of CYP2A enzymes was compared using the ratio of CYP2A13 Kd/CYP2A6 Kd (Table 1). This ratio varied from 0.064 to 2.3. The most selective compound for CYP2A6 binding was tryptamine, with a selectivity ratio of 2.3, i.e., only a ∼2-fold higher affinity for CYP2A6. The most selective ligand for CYP2A13 was PEITC, with 15.5-fold selectivity. β-Nicotyrine, (S)-nicotine, (R)-(+)-menthofuran, and 8-MOP showed >6.8- to 8.7-fold higher affinity for CYP2A13 over CYP2A6, but the overall affinities for β-nicotyrine and (S)-nicotine were much lower than those for (R)-(+)-menthofuran and 8-MOP.
Enzyme Inhibition.
Compounds were also tested for the ability to inhibit 2-hydroxylation of pNP. pNP was used instead of the more traditional CYP2A substrate coumarin because pNP has similar catalytic efficiencies for both enzymes, whereas coumarin is a much poorer substrate for CYP2A13 than for CYP2A6. CYP2A13 has a catalytic efficiency (kcat/Km) for pNP of 0.19 μM−1 · min−1 (kcat = 16.0 ± 1.1 min−1, Km = 83.5 ± 16 μM) compared with 0.11 μM−1 · min−1 for coumarin (kcat = 2.19 ± 0.07 min−1, Km = 19.6 ± 2.1 μM). CYP2A6 has a catalytic efficiency for pNP of 0.12 μM−1 · min−1 (kcat = 25.0 ± 3.4 min−1, Km = 206 ± 35 μM) compared with 0.53 μM−1 · min−1 for coumarin (kcat = 54.3 ± 5.1 min−1, Km = 102 ± 21 μM). This similar catalytic efficiency of pNP by both enzymes allows more straightforward analysis of the effects of inhibitors on selectivity.
Steady-state inhibition kinetics were evaluated for both CYP2A6 and CYP2A13 under conditions where inactivation would be minimized, and Ki values were determined (Table 1). For CYP2A6, Ki values varied from 0.13 to 130 μM, whereas for CYP2A13, Ki values ranged from 0.04 to 72 μM. The most potent inhibitors of CYP2A6 were tranylcypromine (Ki = 0.13 ± 0.02 μM) and 8-MOP (Ki = 0.25 ± 0.10 μM), both displaying submicromolar Ki values. 8-MOP was the only inhibitor with a submicromolar Ki value for CYP2A13 (Ki = 0.04 ± 0.009 μM), with the next closest as pilocarpine (Ki = 1.4 ± 0.12 μM). The least potent inhibitor for CYP2A6 was (S)-nicotine (Ki = 130 ± 8.8 μM). For CYP2A13, both (S)-nicotine (Ki = 72 ± 4.6 μM) and (R)-(+)-menthofuran (Ki = 54 ± 12 μM) were the least effective inhibitors.
The type of inhibition exhibited by each compound was competitive or predominantly competitive (mixed inhibition with α > 4 such that binding to the enzyme-substrate complex (ES) was >4-fold lower affinity than binding to the free enzyme). (S)-Nicotine and tryptamine exhibited purely competitive inhibition modes. DMABA, PEITC, (R)-(+)-menthofuran, β-nicotyrine, and 8-MOP demonstrated mixed inhibition. Although tranylcypromine and pilocarpine both exhibited competitive inhibition with CYP2A13, mixed inhibition was the best model for these compounds with CYP2A6; however, in both cases, the higher α values (10 and 25, respectively) indicated that binding to free enzyme was dominant. In addition, although PEITC demonstrated mixed inhibition of both enzymes (Fig. 2), this was of two extremes because with CYP2A6, PEITC inhibition suggested inhibitor binding much more tightly to the free enzyme (α = 270), and for CYP2A13, inhibition was dominated by tighter binding to the ES complex (α = 0.20).
Fig. 2.

PEITC inhibition of CYP2A13 and CYP2A6 exhibits the extremes of mixed inhibition. CYP2A6 inhibition by PEITC results in a much larger effect on the Km values, as shown in the Michaelis-Menten (A) and Lineweaver-Burk (B) plots, suggesting much tighter binding to the free enzyme (α = 270). However, for CYP2A13, Michaelis-Menten (C) and Lineweaver-Burk (D) plots illustrate that PEITC inhibition has a larger effect on the Vmax, suggesting much tighter binding to the ES state (α = 0.20). Bars represent the S.D.
Inhibition selectivity was compared by calculating the ratio of the CYP2A13 Ki with the CYP2A6 Ki (Table 1). Of the nine compounds, only (R)-(+)-menthofuran and tranylcypromine had >10-fold preference for CYP2A6 inhibition over CYP2A13. Tranylcypromine demonstrated the highest preference for CYP2A6, with a selectivity factor of 49, followed by (R)-(+)-menthofuran, with a selectivity factor of 27. DMABA and tryptamine had very moderate selectivity for CYP2A6, with selectivity factors ranging from ∼5 to 10. The other inhibitors had ≤2-fold preference for either of the CYP2A enzymes, with the exception of 8-MOP, which demonstrated a 6-fold preference for inhibition of CYP2A13 over CYP2A6. Finally, tranylcypromine, tryptamine, pilocarpine, and 8-MOP were selected for further evaluation because they spanned the range of selectivities observed for inhibition of pNP metabolism. These four inhibitors inhibited coumarin metabolism with complete conservation of the trend for inhibitor selectivity.
Discussion
Spectral ligand binding studies, coupled with kinetic analysis for the inhibition of pNP 2-hydroxylation, were used to evaluate a chemically diverse set of known CYP2A6 compounds to determine which were able to discriminate effectively between CYP2A6 and CYP2A13 (Table 1). For many of the compounds, the binding affinity (Kd values) and the Ki values for the same ligand with the same enzyme were within an order of magnitude. However, in other cases, the two values differed more widely, as would be expected if inhibitors interacted differently with the enzymes in various stages of the catalytic cycle compared with interactions with the ferric, substrate-free, water-bound enzyme evaluated in binding titrations. Thus, the selectivity evaluated from the ratio of the equilibrium binding constants differed from the selectivity based on enzyme inhibition. For example, (R)-(+)-menthofuran, β-nicotyrine, (S)-nicotine, and 8-MOP all had similar ∼8-fold selectivity for CYP2A13 over CYP2A6 binding to the enzyme resting state, but when inhibition was evaluated, (R)-(+)-menthofuran was one of the more selective inhibitors for CYP2A6, (S)-nicotine and β-nicotyrine showed little selectivity for one enzyme over the other, and 8-MOP retained its selectivity for CYP2A13. Because the contributions of various P450 enzymes to metabolism in tissues are evaluated via inhibition assays, further discussion focuses primarily on the inhibition characteristics to determine which might be useful for distinguishing the individual roles of CYP2A enzymes in tissue samples.
The most selective enzyme inhibitors tested were tranylcypromine and (R)-(+)-menthofuran, which demonstrate a 49-fold and 27-fold preference for CYP2A6 inhibition over CYP2A13, respectively. Tranylcypromine, a monoamine oxidase inhibitor, was a potent inhibitor of CYP2A6 and is commonly used for P450 phenotyping, but the selectivity is less than ideal (Khojasteh et al., 2011). The Ki value of tranylcypromine for CYP2A6 is 16 to 17 times lower than the next most potently inhibited enzymes found in liver microsomes, CYP2C19 and CYP2E1 (Taavitsainen et al., 2001). This is a narrower range than the 49-fold difference in CYP2A6 Ki versus CYP2A13 Ki reported herein. Thus, although tranylcypromine failed to discriminate very effectively among liver P450 enzymes from other subfamilies, this compound may have somewhat more utility when seeking to discriminate between CYP2A6 and CYP2A13 activities. Like tryptamine and pilocarpine, tranylcypromine was a type II inhibitor, which is often relatively nonselective among P450 enzymes. This was the case with tryptamine and pilocarpine, but tranylcypromine must have structural features that incorporate some selectivity for CYP2A6 versus CYP2A13. However, tranylcypromine also had a nanomolar affinity for CYP46A1 in the brain and can strongly inhibit cholesterol 24S-hydroxylase activity (Mast et al., 2010). This reinforces the idea that care must be taken when selecting an optimal CYP2A inhibitor depending on the tissue under evaluation.
(R)-(+)-Menthofuran is an inhibitor and inactivator of human CYP2A6 (Khojasteh-Bakht et al., 1998) often used in P450 phenotyping to selectively inhibit CYP2A6 (Diaz and Squires, 2000; Miyazawa and Haigou, 2011). Inhibition of pNP 2-hydroxylation herein showed that (R)-(+)-menthofuran was one of the more selective of the traditionally used CYP2A6 inhibitors when specifically discriminating enzyme activity within the CYP2A family. The observed Ki value of 2.0 μM agrees well with 2.5 and 0.84 μM reported for CYP2A6 assessed using coumarin hydroxylation and human liver microsomes or purified enzyme (Khojasteh-Bakht et al., 1998).
Tryptamine and DMABA were only moderately selective for CYP2A6 inhibition. Tryptamine, a precursor to serotonin and melatonin, demonstrated a competitive mode of inhibition for both enzymes. The Ki value of tryptamine for CYP2A6 of 1.7 μM was identical to the value reported previously for CYP2A6-expressing microsomes using coumarin 7-hydroxylase activity (Zhang et al., 2001). The 9.4-fold selectivity for inhibition of CYP2A6 over CYP2A13 is only slightly better than the 4.6-fold selectivity for the mixed mode inhibitor DMABA. Studies using human liver microsomes indicated >20-fold DMABA selectivity for CYP2A6 over all other hepatic P450 enzymes, with the exception of CYP2E1 where the DMABA is only 13-fold selective for CYP2A6 (Rahnasto et al., 2008). Thus, both DMABA and tryptamine demonstrated <10-fold preference for the inhibition of CYP2A6 over CYP2A13, making them undesirable candidates for effectively distinguishing between the activities of the two CYP2A enzymes.
PEITC, (S)-nicotine, β-nicotyrine, and pilocarpine demonstrated no real preference for inhibition of either enzyme. The only compound that demonstrated some selectivity for CYP2A13 inhibition, with a selectivity factor of 0.16, was 8-MOP. 8-MOP inhibition of both enzymes resulted in a decrease in the Vmax and an increase in the Km, suggesting a mixed mode of inhibition. However, the α values of 3.3 and 1.8 for CYP2A6 and CYP2A13, respectively, are similar. Previous reports have stated that 8-MOP was a noncompetitive inhibitor for both CYP2A enzymes (Zhang et al., 2001; von Weymarn et al., 2005). Our study also confirmed that 8-MOP more potently inhibits CYP2A13, with a Ki value of 0.04 versus 0.25 μM for CYP2A6. This 6-fold degree of selectivity observed was greater than the 1.8-fold difference reported previously (Zhang et al., 2001; von Weymarn et al., 2005). A structure of CYP2A6 with 8-MOP (Yano et al., 2005) showed that the exocyclic ketone oxygen is involved in a hydrogen bond with Asn297. This residue is conserved in the active site of CYP2A13 as well and has been shown to hydrogen bond with other ligands (Smith et al., 2007; DeVore et al., 2008). Thus, the affinity of 8-MOP is likely due, in part, to this key interaction. However, other ligands hydrogen bond with Asn297 yet have lower affinities (coumarin) or bind selectively (phenacetin binds CYP2A13 but not CYP2A6). In these cases, the steric contributions of other active site residues serve to substantially modulate binding affinity (DeVore et al., 2008, 2009). Thus, although 8-MOP does not have the highest affinity, it is clearly the most efficient inhibitor of the compounds investigated herein and appears to interact most favorably with CYP2A13 over CYP2A6.
In conclusion, the goal of this study was to compare a set of reportedly CYP2A6-selective compounds to determine their relative ability to discriminate between CYP2A6 and CYP2A13 and, thus, their utility in distinguishing between CYP2A activities in tissues where both enzymes are present. Tranylcypromine and (R)-(+)-menthofuran were the most selective inhibitors for CYP2A6, with 49- and 27-fold selectivity for CYP2A6 over CYP2A13, respectively. Even though the ability of tranylcypromine to distinguish between CYP2A enzymes is not ideal, this compound can be used as a benchmark for the development of more selective inhibitors. Six of the compounds tested demonstrated only moderate or no real selectivity between the human CYP2A enzymes. Thus, these compounds should not be used to evaluate individual CYP2A enzyme contributions to metabolism in tissues where both enzymes may be present. This study did not identify a suitably selective inhibitor for CYP2A13. The 6-fold selectivity of 8-MOP for CYP2A13 over CYP2A6 is marginal at best but provides a starting point for the development of a more selective inhibitor. Taken together, these results can be used as a guide for the selection of CYP2A-selective inhibitors in studies that seek to accurately distinguish between CYP2A6 and CYP2A13 activity.
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant GM076343].
This work was previously presented in poster form: Stephens ES and Scott EE (2011) Selectivity of cytochrome P450 2A6 inhibitors versus cytochrome P450 2A13 (Abstract). The University of Kansas Cancer Center Research Symposium; 2011 Nov 4; Kansas City, MO. University of Cancer Center, Kansas City, MO.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
- P450
- cytochrome P450
- PEITC
- phenethyl isothiocyanate
- DMABA
- 4-dimethylaminobenzaldehyde
- 8-MOP
- 8-methoxypsoralen
- pNP
- p-nitrophenol
- ES
- enzyme-substrate complex.
Authorship Contributions
Participated in research design: Stephens, Walsh, and Scott.
Conducted experiments: Stephens and Walsh.
Performed data analysis: Stephens, Walsh, and Scott.
Wrote or contributed to the writing of the manuscript: Stephens, Walsh, and Scott.
References
- Bao Z, He XY, Ding X, Prabhu S, Hong JY. (2005) Metabolism of nicotine and cotinine by human cytochrome P450 2A13. Drug Metab Dispos 33:258–261 [DOI] [PubMed] [Google Scholar]
- Denton TT, Zhang X, Cashman JR. (2004) Nicotine-related alkaloids and metabolites as inhibitors of human cytochrome P-450 2A6. Biochem Pharmacol 67:751–756 [DOI] [PubMed] [Google Scholar]
- DeVore NM, Smith BD, Urban MJ, Scott EE. (2008) Key residues controlling phenacetin metabolism by human cytochrome P450 2A enzymes. Drug Metab Dispos 36:2582–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVore NM, Smith BD, Wang JL, Lushington GH, Scott EE. (2009) Key residues controlling binding of diverse ligands to human cytochrome P450 2A enzymes. Drug Metab Dispos 37:1319–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVore NM, Meneely KM, Bart AG, Stephens ES, Battaile KP, Scott EE. (2012) Structural comparison of cytochromes P450 2A6, 2A13, and 2E1 with pilocarpine. FEBS J 279:1621–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz GJ, Squires EJ. (2000) Metabolism of 3-methylindole by porcine liver microsomes: responsible cytochrome P450 enzymes. Toxicol Sci 55:284–292 [DOI] [PubMed] [Google Scholar]
- Fukami T, Katoh M, Yamazaki H, Yokoi T, Nakajima M. (2008) Human cytochrome P450 2A13 efficiently metabolizes chemicals in air pollutants: naphthalene, styrene, and toluene. Chem Res Toxicol 21:720–725 [DOI] [PubMed] [Google Scholar]
- Fukami T, Nakajima M, Sakai H, Katoh M, Yokoi T. (2007) CYP2A13 metabolizes the substrates of human CYP1A2, phenacetin, and theophylline. Drug Metab Dispos 35:335–339 [DOI] [PubMed] [Google Scholar]
- Hecht SS. (1998) Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol 11:559–603 [DOI] [PubMed] [Google Scholar]
- Holmans PL, Shet MS, Martin-Wixtrom CA, Fisher CW, Estabrook RW. (1994) The high-level expression in Escherichia coli of the membrane-bound form of human and rat cytochrome b5 and studies on their mechanism of function. Arch Biochem Biophys 312:554–565 [DOI] [PubMed] [Google Scholar]
- Khojasteh SC, Prabhu S, Kenny JR, Halladay JS, Lu AY. (2011) Chemical inhibitors of cytochrome P450 isoforms in human liver microsomes: a re-evaluation of P450 isoform selectivity. Eur J Drug Metab Pharmacokinet 36:1–16 [DOI] [PubMed] [Google Scholar]
- Khojasteh-Bakht SC, Koenigs LL, Peter RM, Trager WF, Nelson SD. (1998) (R)-(+)-Menthofuran is a potent, mechanism-based inactivator of human liver cytochrome P450 2A6. Drug Metab Dispos 26:701–704 [PubMed] [Google Scholar]
- Koenigs LL, Peter RM, Thompson SJ, Rettie AE, Trager WF. (1997) Mechanism-based inactivation of human liver cytochrome P450 2A6 by 8-methoxypsoralen. Drug Metab Dispos 25:1407–1415 [PubMed] [Google Scholar]
- Mast N, Charvet C, Pikuleva IA, Stout CD. (2010) Structural basis of drug binding to CYP46A1, an enzyme that controls cholesterol turnover in the brain. J Biol Chem 285:31783–31795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa M, Haigou R. (2011) Determination of cytochrome P450 enzymes involved in the metabolism of (−)-terpinen-4-ol by human liver microsomes. Xenobiotica 41:1056–1062 [DOI] [PubMed] [Google Scholar]
- Nakajima M, Yoshida R, Shimada N, Yamazaki H, Yokoi T. (2001) Inhibition and inactivation of human cytochrome P450 isoforms by phenethyl isothiocyanate. Drug Metab Dispos 29:1110–1113 [PubMed] [Google Scholar]
- Pavek P, Dvorak Z. (2008) Xenobiotic-induced transcriptional regulation of xenobiotic metabolizing enzymes of the cytochrome P450 superfamily in human extrahepatic tissues. Curr Drug Metab 9:129–143 [DOI] [PubMed] [Google Scholar]
- Rahnasto M, Wittekindt C, Juvonen RO, Turpeinen M, Petsalo A, Pelkonen O, Poso A, Stahl G, Höltje HD, Raunio H. (2008) Identification of inhibitors of the nicotine metabolising CYP2A6 enzyme–an in silico approach. Pharmacogenomics J 8:328–338 [DOI] [PubMed] [Google Scholar]
- Schenkman JB, Jansson I. (2006) Spectral analyses of cytochromes P450. Methods Mol Biol 320:11–18 [DOI] [PubMed] [Google Scholar]
- Shen AL, Porter TD, Wilson TE, Kasper CB. (1989) Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J Biol Chem 264:7584–7589 [PubMed] [Google Scholar]
- Smith BD, Sanders JL, Porubsky PR, Lushington GH, Stout CD, Scott EE. (2007) Structure of the human lung cytochrome P450 2A13. J Biol Chem 282:17306–17313 [DOI] [PubMed] [Google Scholar]
- Su T, Bao Z, Zhang QY, Smith TJ, Hong JY, Ding X. (2000) Human cytochrome P450 CYP2A13: predominant expression in the respiratory tract and its high efficiency metabolic activation of a tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res 60:5074–5079 [PubMed] [Google Scholar]
- Taavitsainen P, Juvonen R, Pelkonen O. (2001) In vitro inhibition of cytochrome P450 enzymes in human liver microsomes by a potent CYP2A6 inhibitor, trans-2-phenylcyclopropylamine (tranylcypromine), and its nonamine analog, cyclopropylbenzene. Drug Metab Dispos 29:217–222 [PubMed] [Google Scholar]
- von Weymarn LB, Chun JA, Hollenberg PF. (2006) Effects of benzyl and phenethyl isothiocyanate on P450s 2A6 and 2A13: potential for chemoprevention in smokers. Carcinogenesis 27:782–790 [DOI] [PubMed] [Google Scholar]
- von Weymarn LB, Zhang QY, Ding X, Hollenberg PF. (2005) Effects of 8-methoxypsoralen on cytochrome P450 2A13. Carcinogenesis 26:621–629 [DOI] [PubMed] [Google Scholar]
- Yano JK, Denton TT, Cerny MA, Zhang X, Johnson EF, Cashman JR. (2006) Synthetic inhibitors of cytochrome P-450 2A6: inhibitory activity, difference spectra, mechanism of inhibition, and protein cocrystallization. J Med Chem 49:6987–7001 [DOI] [PubMed] [Google Scholar]
- Yano JK, Hsu MH, Griffin KJ, Stout CD, Johnson EF. (2005) Structures of human microsomal cytochrome P450 2A6 complexed with coumarin and methoxsalen. Nat Struct Mol Biol 12:822–823 [DOI] [PubMed] [Google Scholar]
- Zhang JY, Wang Y, Prakash C. (2006) Xenobiotic-metabolizing enzymes in human lung. Curr Drug Metab 7:939–948 [DOI] [PubMed] [Google Scholar]
- Zhang W, Kilicarslan T, Tyndale RF, Sellers EM. (2001) Evaluation of methoxsalen, tranylcypromine, and tryptamine as specific and selective CYP2A6 inhibitors in vitro. Drug Metab Dispos 29:897–902 [PubMed] [Google Scholar]