

Abstract
Oral submucous fibrosis (OSMF) is a chronic debilitating disease and a premalignant condition of the oral cavity characterized by generalized submucosal fibrosis. Despite its precancerous nature, the molecular biology regarding its malignant potential has not been extensively studied. PTEN, a known tumor suppressor gene is mutated in a majority of human cancers and has also been implicated in several fibrotic disorders. The present study aims to evaluate the expression of PTEN in OSMF and oral squamous cell carcinoma (OSCC) and correlate it with the pathogenesis and malignant transformation of OSMF. 60 cases total of OSMF (30) and OSCC (30) were subjected to immunohistochemistry using PTEN antibody. Ten normal oral mucosa (NOM) specimens were also stained as controls. There was progressive loss of PTEN expression from normal mucosa to OSMF and OSCC (p ≤ 0.001). Significant differences were observed for PTEN expression between NOM and OSMF, OSMF and OSCC as well as NOM and OSCC. Though a progressive loss of PTEN was noticed between early OSMF and advanced OSMF, the variation did not reach statistical significance (p ≥ 0.001). Data suggest that there is a significant loss of PTEN expression in OSMF as compared to normal oral mucosa and that this trend increased from OSMF to OSCC. Thus, alteration of PTEN is likely an important molecular event in OSMF pathogenesis and oral carcinogenesis.
Electronic supplementary material
The online version of this article (doi:10.1007/s12105-012-0341-z) contains supplementary material, which is available to authorized users.
Keywords: PTEN, Oral submucous fibrosis, Oral squamous cell carcinoma, Carcinogenesis, Pathogenesis, Malignant transformation
Introduction
Oral squamous cell carcinoma (OSCC) is the sixth most common cancer worldwide and the most frequent malignant tumor of the oral cavity. Despite availability of newer diagnostic and therapeutic strategies, the five-year survival rate is still low (~50%) making it a global health problem [1, 2]. Most oral squamous cell carcinomas are preceded by clinical premalignant lesions and conditions like oral leukoplakia, erythroplakia, and oral submucous fibrosis (OSMF) [3]. Among these, OSMF, a chronic fibrotic premalignant condition, demonstrates a regional distribution, being particularly prevalent in the Indian subcontinent [4, 5]. Scientific literature to date provides firm evidence that areca nut is the major etiological factor for this disease but the exact mechanism of action on the oral tissues remains to be elucidated [5, 6]. The disease is characterized by inflammation and progressive generalized submucosal fibrosis, leading to limitation of mouth opening. It exhibits characteristic histopathologic features that include juxtaepithelial hyalinization and excessive collagen deposition in the connective tissue, secondary to which the epithelium becomes atrophic [5, 6]. This atrophic epithelium is prone to injury by the areca nut extracts that predispose to the development of malignancy [5, 6]. The reported malignant transformation rate of OSMF to oral squamous cell carcinoma (OSCC) is 7–13% with a long-term follow-up study recording an annual malignant transformation rate of 0.5% [7]. The incidence of this disease is rising in India especially among the younger population due to increased access and improved marketing strategies for the availability of areca nut, which, in turn, predisposes this population to an increased risk for oral cancer [6].
Oral carcinogenesis is a multistep process involving the progressive acquisition of mutations and epigenetic abnormalities in the expression of multiple genes, especially the activation of oncogenes and inactivation of tumor suppressor genes. [8]. Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is a tumor suppressor gene and a negative regulator of PI3K/AKT (phosphotidyl inositol-3-kinase/AKT) pathway that controls various cellular processes including proliferation, apoptosis (cell death), cell cycle regulation, and cell adhesion and migration. Inactivation of PTEN results in unconditional proliferation and reduction in apoptosis, thereby predisposing to the development of cancer [9, 10]. Germline mutations of PTEN are found in patients with multiple hamartoma syndrome, a familial syndrome associated with a predisposition for multiple benign hamartomas, as well as malignant breast and thyroid neoplasms. Somatic mutation or deletion resulting in loss of PTEN has been reported in a variety of precancers and cancers, including glioblastomas, melanoma, breast, prostate, endometrial carcinomas, head and neck cancers, and oral squamous cell carcinoma [9–16]. Recently, PTEN has also been implicated in the pathogenesis of several fibrotic disorders, such as scleroderma [17], hepatic fibrosis [18], kidney [19], pulmonary [20, 21], and cardiac fibrosis [22]. However, the role of PTEN expression in the pathogenesis of oral submucous fibrosis and its relationship to malignant transformation has not been studied. The present study aims to evaluate the expression of PTEN in OSMF and OSCC to elucidate its possible association with the progression and malignant transformation of oral submucous fibrosis.
Materials and Methods
Tissue Material
Following approval of the our institutional review board, formalin-fixed paraffin-embedded tissue blocks (60 cases) of histopathologically proven cases of oral submucous fibrosis (30) and oral squamous cell carcinoma (30) were retrieved. Ten tissue blocks of normal oral mucosa obtained from gingival and vestibular mucosa after extraction of impacted teeth were used as controls. The hematoxylin and eosin (H&E) stained sections were reviewed and diagnosis confirmed by two oral pathologists. The oral submucous fibrosis tissue sections were further subdivided histopathologically into (1) very early, (2) early, (3) moderately advanced, and (4) advanced, using criteria of Pindborg and Sirsat [4] (Table 1). We combined the first two categories under (A) “early” (15 cases) and the last two categories under (B) “advanced” (15 cases). The OSCC cases were graded as well- (10 cases), moderate- (10 cases) and poorly-differentiated (10 cases). These cases of OSMF and OSCC along with normal oral mucosa were subjected to immunohistochemistry for PTEN. The patients with OSMF ranged from 17 to 50 years, with 29 males and only one female patient. Similarly, patients with OSCC were also predominantly male (M:F 28:2) in the age range of 35–65 years. All patients included in the study had a history of areca nut and tobacco chewing of varying duration, as revealed by their biopsy records. In patients with OSMF, the duration of areca nut with/without tobacco use varied from 1 to 10 years with an average frequency of at least 6 times/day; in OSCC patients, the history of areca nut/tobacco use ranged from 3 to 21 years with a frequency of 5 times a day on average.
Table 1.
Histopathologic grading criteria by Pindborg and Sirsat [4]
| Very early stage (grade I) | Early stage (grade II) | Moderately advanced stage (grade III) | Advanced stage (grade IV) |
|---|---|---|---|
| Presence of fine fibrillar collagen with marked edema | Early hyalinization in the juxtra epithelial region | Collagen is moderately hyalinised | Collagen is completely hyalinised |
| Extensive fibroblastic response | Plump young fibroblasts in moderate numbers | Fibroblastic response is less evident consisting of only adult fibrocytes | Hyalinised areas are devoid of fibroblasts |
| Blood vessels are normal or dilated and congested | Blood vessels are dilated and congested | Blood vessels are normal or constricted | Blood vessels are completely obliterated |
| Inflammatory cells usually neutrophils and occasional eosinophils | Inflammatory cells are lymphocytes, eosinophils and occasional plasma cells | Inflammatory cells are lymphocytes and plasma cells with occasional eosinophil | Inflammatory cells are lymphocytes and plasma cells |
Immunohistochemistry
The immunohistochemical analysis for PTEN was performed using super sensitive one step polymer-HRP technique (Biogenex Life Sciences, San Ramon, CA, USA). Paraffin-embedded tissue blocks were cut into 4–5 μm thick sections and taken onto 2%, 3-aminopropylethoxysilane solution (APES) (Sigma Aldrich, St. Louis, MO, USA) adhesive coated slides. The sections were then de-paraffinized and rehydrated through xylene and descending grades of alcohol. Antigen retrieval was done using commercial microwave antigen retrieval system where the sections were placed in a container containing 10 mM sodium citrate buffer (pH-6.0) at 96°C for 3 cycles of 6 min each (EZ-Retriever System, Biogenex life sciences, San Ramon, CA, USA). After rinsing in PBS, the sections were treated with peroxidase block consisting of 3% H2O2 in water for 15 min to block the endogenous peroxidase activity, followed by a 20 min power block to obstruct any nonspecific antigenic sites. The sections were then incubated for 1 h at room temperature with optimally pre-diluted antibody against PTEN (Mouse monoclonal: Clone 28H6, Biogenex, United States of America). After washing with PBS, the sections were then incubated with One-Step Polymer-HRP reagent for 30 min. Visualization was performed using freshly prepared DAB (diaminobenzidine tetrahydrochloride). The slides were counterstained with Harris hematoxylin, subsequent to which sections were dehydrated, cleared, and mounted with DPX. For each batch of staining, positive and negative controls were run simultaneously with the study specimens. Normal oral mucosa served as the positive control and the endothelial staining of blood vessels known to be reactive to PTEN were used as the internal positive control. The primary antibodies were replaced by non-immune mouse serum at the same dilutions for the negative controls.
Immunohistochemical Analysis
Evaluation of PTEN expression was based on the extent of positivity, modified from the method used by Sui Li et al. [23]. The quantity of stained epithelial cells was classified as follows: (0) 0–9 cells, (1) 10–49 cells, (2) ≥50 cells. All H&E- and immunohistochemical-stained sections were evaluated by two independent observers.
Statistical Analyses
The differences in PTEN immunoexpression between NOM, OSMF and OSCC was examined using Kruskal–Wallis and Chi-square tests. p Values of less than 0.05 were considered significant.
Results
Thirty histopathologically proven cases of OSMF and OSCC each, as well as NOM (10 cases), were stained with H&E and PTEN antibody. All NOM cases, which served as a positive control, exhibited strong nuclear PTEN expression in the epithelium (100%); the blood vessels within the connective tissue also served as a positive internal control (Fig. 1). The NOM, OSMF, and OSCC demonstrated immunostaining of the epithelial cells at varying levels for PTEN (Table 2).
Fig. 1.
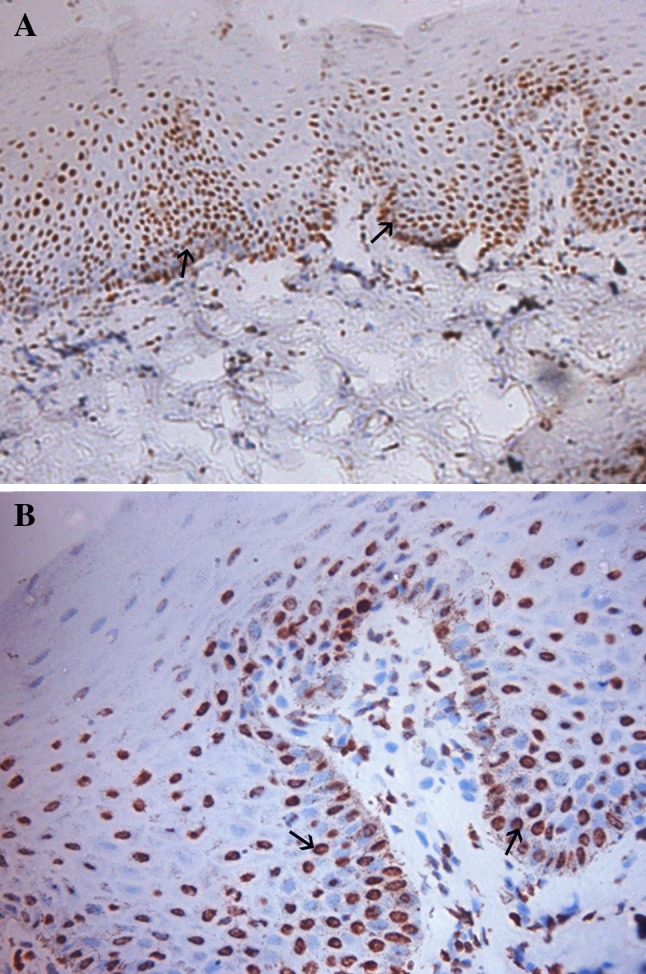
Intense PTEN positivity observed in normal epithelium (a ×100, b ×250) (black arrows)
Table 2.
Immunohistochemical staining of PTEN in NOM, OSMF, and OSCC
| Groups of cases | Total cases | PTEN scores | p Value | ||
|---|---|---|---|---|---|
| 0 | 1 | 2 | |||
| Normal Oral Mucosa | 10 | 0 | 1 | 9 | 0.000* |
| OSMF | 30 | 6 | 16 | 8 | |
| OSCC | 30 | 15 | 8 | 7 | |
PTEN phospatase tensin homologue deleted on chromosome 10, OSMF oral submucous fibrosis, OSCC oral squamous cell carcinoma
* Kruskal–Wallis test
There was progressive loss of PTEN expression from NOM to OSMF and OSCC (p ≤ 0.01) (Table 2). Loss of PTEN expression was observed in 6 out of 30 cases (20%) of OSMF. In 2 out of 15 cases (13.3%) of early OSMF and 4 out of 15 cases (26.6%) of advanced OSMF there was loss of PTEN expression. The degree of PTEN expression in the progression of OSMF was considerably different; however, no statistical significance was noted between PTEN expression and the histopathologic grade. Notably, loss of PTEN expression was significantly higher in OSMF as compared to NOM (p ≤ 0.01) (Table 3) (Fig. 2).
Table 3.
Chi square test for comparison of PTEN scores in the various study groups
| Groups of cases | Total cases | PTEN scores | p Value | ||
|---|---|---|---|---|---|
| 0 | 1 | 2 | |||
| NOM | 10 | 0 | 1 | 9 | 0.002(S) |
| OSMF | 30 | 6 | 16 | 8 | |
| NOM | 10 | 0 | 1 | 9 | 0.000(S) |
| OSCC | 30 | 15 | 8 | 7 | |
| OSMF | 30 | 6 | 16 | 8 | 0.037(S) |
| OSCC | 30 | 15 | 8 | 7 | |
| Early OSMF | 15 | 2 | 7 | 6 | 0.232(NS) |
| Advanced OSMF | 15 | 4 | 9 | 2 | |
PTEN phospatase tensin homologue deleted on chromosome 10, NOM normal oral mucosa, OSMF oral submucous fibrosis, OSCC oral squamous cell carcinoma, S significant, NS non-significant
Fig. 2.

PTEN expression seen in OSMF. Early (a) and advanced stages (b) of OSMF exhibiting intense PTEN expression (black arrows). Negative PTEN expression demonstrable in early (c) and advanced (d) stages of OSMF (black arrows)
Fifteen out of 30 cases (50%) of OSCC demonstrated loss of PTEN expression. This was significantly higher than NOM (p ≤ 0.01) and OSMF (p ≤ 0.01) (Table 2) (Fig. 3).
Fig. 3.

PTEN expression in OSCC. a and b demonstrate positive PTEN expression (black arrows); b and d depict loss of PTEN immunoexpression (black arrows)
Discussion
The pathogenesis of OSMF has been extensively studied, but only a few studies have examined the molecular aberrations in OSMF and their possible role in malignant transformation [5]. Certain reports of polymorphisms of heterochromatic chromosome regions, sister chromatid exchanges, aberrant p53 expression, alteration in Bcl-2 expression, and adenomatosis polyposis coli gene have been associated with OSMF carcinogenesis [24–28]. Recently, Teh et al. [29] have demonstrated genomic instability and loss of heterozygosity (LOH) in several chromosomal loci containing known oncogenes and tumor suppressor genes associated with head and neck carcinogenesis in OSMF tissues.
The role of PTEN in fibrosis and carcinogenesis has been investigated in recent literature [9–22]. As OSMF is a premalignant condition characterized by fibrosis and malignant transformation in the background of fibrosis, we hypothesized a possible role of PTEN in the progression and malignant transformation of OSMF. The PTEN gene encodes a protein able to dephosphorylate both proteins and lipid substrates and is notably referred to as “the most highly mutated gene in the post p53 era”. A pivotal role of PTEN is exerted through interference in signal transduction pathways of phosphoinositide second messengers. In fact, by dephosphorylating phosphoinositol 3, 4, 5-triphosphate (PIP3), PTEN decreases the cellular membrane translocation of AKT, a serine theonine kinase involved in proliferative, metabolic, and anti-apoptotic pathways, thereby reducing its phosphorylation and resulting in increased apoptosis. Additionally, by up regulating p27, PTEN induces down regulation of Cyclin D1, leading to cell cycle arrest. Further, PTEN may be important in cell-extracellular matrix (ECM) interactions by inhibiting cell adhesion, migration, and spreading by dephosphorylating focal adhesion kinase (FAK) and mitogen activated protein kinase (MAPK) [9–12].
Down regulation/loss of PTEN results in unconditional proliferation, increased cell survival, reduced adhesion, and increased cell migration and has been associated with the pathogenesis of numerous tumors, as previously mentioned [9–15, 30]. Furthermore, it has been linked to numerous fibrotic disorders where it negatively regulates cell survival and fibroblast proliferation [19–21].
In our study, PTEN expression was well maintained in NOM while there was significant loss of PTEN in OSMF (p ≤ 0.01), suggesting its altered expression in this condition. Additionally, the loss of PTEN expression progressively increased from early to advanced stages, though the results were not significant (p ≥ 0.05). A similar down-regulated expression of PTEN has been reported in hepatic tissues with increased fibrosis [18].
Fibrosis in OSMF is due to the imbalance between collagen deposition and degradation that is mediated by several diverse mechanisms [5]. One of the key molecules implicated is TGF-β, a proinflammatory and fibrogenic cytokine generally stimulated in response to arecoline [6]. In context with our study, TGF-β is known to be an important negative regulator of PTEN expression [11]. Increased TGF-β in OSMF may possibly cause decreased PTEN levels and resultant unrestrained AKT activity, which, in turn, leads to prolonged cell survival due to diminished apoptosis of the fibroblasts and increased extracellular matrix production, eventually resulting in fibrosis. This mechanism has been implicated in hepatic, pulmonary, kidney fibrosis, and scleroderma [31]. This is corroborated by reports of increased proliferative activity and aberrant Bcl-2 expression in OSMF [27–32]. Further, substantial research on OSMF has focused on the cellular and biochemical events in the connective tissue component. Nevertheless, the overlying epithelium may also play a significant role in inducing fibrosis in OSMF and is certainly involved in its malignant transformation. The reduced vasculature in the sub-epithelial connective tissue allows for long-term build-up of inflammatory cytokines, which aggravate inflammation and wounding of the overlying epithelium, in an intricate epithelial-mesenchymal interaction [33, 34]. This is substantiated by the demonstration of an altered keratinocyte phenotype [35], varied cytokeratin expression subsequent to chronic connective tissue pathology [36], and loss of E-cadherin in the epithelial cells of OSMF, all of which could signify the development of a mesenchymal /migratory phenotype, consistent with the model of epithelial-mesenchymal transition (EMT), a hypothesized model of cell development [37]. Moreover, recent studies have demonstrated that myofibroblasts are an integral component of OSMF and that their differentiation is facilitated by the betel quid alkaloid [38, 39]. This myofibroblast differentiation and increased collagen production in OSMF may be a result of EMT between the oral keratinocytes and fibroblasts via the integrin pathway [39, 40]. PTEN is considered to be a negative regulator for the induction of myofibroblast differentiation in fibro-proliferative disorders. The complex EMT signals involving TGF-β and integrin signaling could inactivate PTEN and induce myofibroblast differentiation via the FAK and MAPK pathways [41–44]. Reduced or loss of PTEN expression has been associated with increased myofibroblast levels in fibrosis, both in vitro and in vivo [21, 41]. What is more, the fibroblasts in fibrotic disorders have been reported to demonstrate features similar to malignant cells, i.e. resistance to apoptosis, increased migration and invasion of tissues, and increased proliferation [41]. It has, therefore, been proposed that PTEN expression in epithelial cells physiologically regulates the phenotype of these cells, but disruption of normal function of PTEN may contribute to the pathogenesis and exacerbation of fibrosis by preventing normal epithelial repair and allowing progression of abnormal fibroblast proliferation [21].
Additionally, in our study, a 50% loss of PTEN immunoexpression was observed in OSCC. The role of the PTEN gene in OSCC is still unclear. Chen et al. [45] and Mavros et al. [46] could not demonstrate any homozygous deletion of this gene in OSCC and concluded that PTEN gene alterations are rare in OSCC. The frequency of LOH in relation to this gene is also low i.e. 12 and 13% as reported by Mavros et al. and Chakraborthy et al., respectively [46, 47]. However, with regards to the loss of immunoexpression at the protein level, the occurrence ranges from 24.2 to 31.8% by various authors [15, 16, 48]. Consequently, the rate of PTEN inactivation at the protein level appears to be more common than that recognized at the genetic level, which could be attributed to reduced protein synthesis, increased protein degradation, or other posttranslational modifications. Another possibility could be epigenetic inactivation of the gene through hypermethylation of the promoter region [16]. This has been corroborated by Kurusawa et al. [16], who demonstrated significant reduction in PTEN mRNA in OSCC cell lines (77.8%). Thus, it can be understood that changes in epigenetic regulation of PTEN are more significant than genetic changes in OSCC. This may underscore the value of using PTEN immunohistochemistry as a tumor marker.
As previously mentioned, an important finding of this study was the loss of PTEN expression, which increased from NOM to OSMF to OSCC (p ≤ 0.001). Presumably, therefore, the loss of PTEN tumor suppressor function begins in the early stages of oral carcinogenesis, similar to what has been described in endometrial cancers [30]. The possibility that changes in PTEN expression actually represent a continuum from normal to precancer to cancer necessitates additional studies. Our results suggest that alteration of PTEN is likely an important molecular event in the pathogenesis of OSMF and oral carcinogenesis. The following mechanism is proposed for the role of PTEN in OSMF based on the downstream targets of PTEN/PI3K pathway studied in OSMF: Downregulation of PTEN results in: (1) increased AKT activity with effects on downstream targets of PTEN/PI3K pathway that include alteration of apoptotic factors, such as Bcl-2, caspase 9, p53, and Fas, all of which ultimately result in increased cell survival; (2) the activation of GSK-3β-β-catenin signaling coupled with decreased p27 leading to increased Cyclin D1 levels predisposing to increased cell proliferation; and (3) reduced cell adhesion, increased cell migration, invasion, and EMT by effects on FAK and integrin signaling via ERK and MAPK pathways [5, 6, 11, 19, 26–28, 31–34, 39, 43, 44] (Supplementary Fig 1). However, the precise molecular mechanism resulting in inactivation of PTEN, its potential significance, and the intricate epithelial mesenchymal transition in OSMF need further research. Importantly, this could aid in developing novel therapeutic strategies.
Conclusion
This is the first study demonstrating PTEN expression in OSMF. Our results further expand the molecular-biological spectrum of this entity. An insight into the probable association of PTEN in the evolution and malignant transformation of OSMF is presented, highlighting the need for additional studies.
Electronic supplementary material
{kind=link}
Diagram to demonstrate the possible mechanism of PTEN in the pathogenesis and malignant transformation of OSMF (JPEG 237 kb)
Acknowledgments
We would like to thank Mr. Mallapur for assisting us with statistical analyses, Mr. Justin from Biogenex Company for his technical support and gratefully acknowledge Patricia J. Kelly, Associate Dean for Research, University of Missouri-Kansas City for the assistance in manuscript preparation.
References
- 1.Scully C, Bagan J. Oral squamous cell carcinoma: an overview. Oral Oncol. 2009;45:301–308. doi: 10.1016/j.oraloncology.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 2.Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009;45:309–316. doi: 10.1016/j.oraloncology.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Waal I. Potentially malignant disorders of the oral and oropharyngeal mucosa: terminology, classification and present concepts of management. Oral Oncol. 2009;45:317–323. doi: 10.1016/j.oraloncology.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 4.Pindborg JJ, Sirasat SM. Oral Submucous fibrosis. Oral Surg Oral Med Oral Pathol. 1966;22:764–769. doi: 10.1016/0030-4220(66)90367-7. [DOI] [PubMed] [Google Scholar]
- 5.Tilakaratne WM, Klinikowski MF, Saku T, Peters TJ, Warnakulasuriya S. Oral submucous fibrosis: review on etiology and pathogenesis. Oral Oncol. 2006;42:561–568. doi: 10.1016/j.oraloncology.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Rajalalitha P, Vali S. Molecular pathogenesis of oral submucous fibrosis—a collagen metabolic disorder. J Oral Pathol Med. 2005;34:321–328. doi: 10.1111/j.1600-0714.2005.00325.x. [DOI] [PubMed] [Google Scholar]
- 7.Murti PR, Bhonsle RB, Pindborg JJ, Daftary DK, Gupta PC, Mehta FS. Malignant transformation rate in oral submucous fibrosis over a 17 year period. Commun Dent Oral Epidemiol. 1985;13:340–341. doi: 10.1111/j.1600-0528.1985.tb00468.x. [DOI] [PubMed] [Google Scholar]
- 8.Chen YJ, Chang JT, Liao CT, Wang HM, Yen TC, Chiu CC, Lu YC, Li HF, Cheng AJ. Head and neck cancer in the betel quid chewing area: recent advances in molecular carcinogenesis. Cancer Sci. 2008;99:1507–1514. doi: 10.1111/j.1349-7006.2008.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dahia PM. PTEN, a unique tumor suppressor gene. Endocr Relat Cancer. 2000;7:115–129. doi: 10.1677/erc.0.0070115. [DOI] [PubMed] [Google Scholar]
- 10.Yamada KM, Araki M. Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci. 2001;114:2375–2382. doi: 10.1242/jcs.114.13.2375. [DOI] [PubMed] [Google Scholar]
- 11.Stiles BL. Phosphatase and tensin homologue deleted on chromosome 10: extending its PTENtacles. Int J Biochem Cell Biol. 2009;41:757–761. doi: 10.1016/j.biocel.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leslie NR. Downes CP.PTEN function: how normal cells control it and tumor cells lose it. Biochem J. 2004;382:1–11. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954–2963. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 14.Shao X, Tandon R, Samara G, Kanki H, Yano H, Close LG, Parsons R, Sato T. Mutational analysis of the PTEN gene in head and neck squamous cell carcinoma. Int J Cancer. 1998;77:684–688. doi: 10.1002/(SICI)1097-0215(19980831)77:5<684::AID-IJC4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 15.Squarize CH, Castilho RM, Declo SPJ. Immunohistochemical evidence of PTEN in oral squamous cell carcinoma and its correlation with the histological malignancy grading system. J Oral Pathol Med. 2002;31:379–389. doi: 10.1034/j.1600-0714.2002.00142.x. [DOI] [PubMed] [Google Scholar]
- 16.Kurusawa Y, Shiba M, Nakamura M, Fushimi K, Ishigami T, Bukawa H, Yokoe H, Uzawa K, Tanzawa H. PTEN expression and methylation status in oral squamous cell carcinoma. Oncol Rep. 2008;19:1429–1434. [PubMed] [Google Scholar]
- 17.Bu S, Asano Y, Bujor A, Highland K, Hant F, Trojanowska M. Dihydrosphingosine 1-phosphate has a potent antifibrotic effect in scleroderma fibroblasts via normalization of phosphatase and tensin homolog levels. Arthr Rheum. 2010;62:2117–2126. doi: 10.1002/art.27463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hao LS, Zhang XL, An JY, Karlin J, Tian XP, Dun ZN, Xie SR, Chen S. PTEN expression is down-regulated in liver tissues of rats with hepatic fibrosis induced by biliary stenosis. APMIS. 2009;117:681–691. doi: 10.1111/j.1600-0463.2009.02515.x. [DOI] [PubMed] [Google Scholar]
- 19.Kato M, Putta S, Wang M, Yuan H, Lanting L, Nair I, Gunn A, Nakagawa Y, Shimano H, Todorov I, Rossi JJ, Natarajan R. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol. 2009;11:881–889. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xia H, Diebold D, Nho R, Perlman D, Kleidon J, Kahm J, Avdulov S, Peterson M, Nerva J, Bitterman P, Henke C. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med. 2008;205:1659–1672. doi: 10.1084/jem.20080001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuwano K. PTEN as a new agent in the fight against fibrogenesis. Am J Respir Crit Care Med. 2006;173:5–6. doi: 10.1164/rccm.2510001. [DOI] [PubMed] [Google Scholar]
- 22.Teunissen BE, Smeets PJ, Willemsen PH, Windt LJ, Vusse GJ, Bilsen M. Activation of PPAR delta inhibits cardiac fibroblast proliferation and the transdifferentiation into myofibroblasts. Cardiovasc Res. 2007;75:519–529. doi: 10.1016/j.cardiores.2007.04.026. [DOI] [PubMed] [Google Scholar]
- 23.Sui L, Dong Y, Watanabe Y, Yamaguchi F, Sugimoto K, Tokuda M. Alteration and clinical relevance of PTEN expression and its correlation with survivin expression in epithelial ovarian tumors. Oncol Rep. 2006;15:773–778. [PubMed] [Google Scholar]
- 24.Dave BJ, Trivedi AH, Adhvaryu SG. Role of areca nut consumption in cause of oral cancers—a cytogenetic assessment. Cancer. 1992;70:1017–1023. doi: 10.1002/1097-0142(19920901)70:5<1017::AID-CNCR2820700502>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 25.Trivedy C, Warnakulasuriya KAAS, Tavasoli M, Ateingrimsdottir H, Penhallow J, Maher R, Johnson NW. p53 aberrations in oral submucous fibrosis and oral squamous cell carcinoma detected by immunohistochemistry and PCR-SSCP. J Oral Pathol Med. 1998;27:72–77. doi: 10.1111/j.1600-0714.1998.tb02097.x. [DOI] [PubMed] [Google Scholar]
- 26.Cox SC, Walker DM. Epithelial growth fraction and expression of p53 tumor suppressor gene in oral submucous fibrosis. Aust Dent J. 1996;1996(41):91–96. doi: 10.1111/j.1834-7819.1996.tb05920.x. [DOI] [PubMed] [Google Scholar]
- 27.Teni T, Pawar S, Sanghvi V, Saranath D. Expression of bcl-2 and bax in chewing tobacco induced oral cancers and oral lesions in India. Pathol Oncol Res. 2002;8:109–114. doi: 10.1007/BF03033719. [DOI] [PubMed] [Google Scholar]
- 28.Liao PH, Lee TL, Yang LC, Yang SH, Chen SL, Chou MY. Adenomatous polyposis coli gene mutation and decreased wild—type p53 protein expression in oral submucous fibrosis: a preliminary investigation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2001;92:202–207. doi: 10.1067/moe.2001.116816. [DOI] [PubMed] [Google Scholar]
- 29.Teh TM, Tilakaratne WM, Chaplin T, Young BD, Ariyawardana A, Pitiyage G, et al. Fingerprinting genomic instability in oral submucous fibrosis. J Oral Pathol Med. 2008;37:430–436. doi: 10.1111/j.1600-0714.2008.00643.x. [DOI] [PubMed] [Google Scholar]
- 30.Mutter GL. Diagnosis of premalignant endometrial disease. J Clin Pathol. 2002;55:326–331. doi: 10.1136/jcp.55.5.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Willis BC, Borok Z. TGF-β induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–L534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 32.Chaing CP, Lang MJ, Liu BY, Wang JT, Leu JS, Hahn LJ, et al. Expression of proliferating cell nuclear antigen (PCNA) in oral submucous fibrosis, oral epithelial hyperkeratosis and oral epithelial dysplasia in Taiwan. Oral Oncol. 2000;36:353–359. doi: 10.1016/S1368-8375(00)00014-2. [DOI] [PubMed] [Google Scholar]
- 33.Yanjia H, Xinchun J. The role of epithelial-mesenchymal transition in oral squamous cell carcinoma and oral submucous fibrosis. Clinica Chemica Acta. 2007;383:51–56. doi: 10.1016/j.cca.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 34.Xia L, Ling TY, Gao YJ, Tang DS, Li WH. Arecoline and keratinocytes may affect the collagen metabolism of fibroblasts. J Oral Pathol Med. 2009;38:422–426. doi: 10.1111/j.1600-0714.2009.00758.x. [DOI] [PubMed] [Google Scholar]
- 35.Lalli A, Tilakaratne WM, Ariyawardana A, Fitchet C, Leigh IM, Hagi-Pavli E, Cruchley AT, Parkinson EK, The MT, Fortune F, Waseem A. An altered keratinocyte phenotype in oral submucous fibrosis: correlation of keratin K17 expression with disease severity. J Oral Pathol Med. 2008;37:211–220. doi: 10.1111/j.1600-0714.2007.00609.x. [DOI] [PubMed] [Google Scholar]
- 36.Ranganathan K, Kavitha R, Sawant SS, Vaidya MM. Cytokeratin expression in oral submucous fibrosis—an immunohistochemical study. J Oral Pathol Med. 2006;35:25–32. doi: 10.1111/j.1600-0714.2005.00366.x. [DOI] [PubMed] [Google Scholar]
- 37.Das RK, Pal M, Barui A, Paul RR, Chakraborthy C, Ray AK, Sengupta S, Chatterjee J. Assessment of malignant potential of oral submucous fibrosis through evaluation of p63, E-cadherin and CD 105 expression. J Clin Pathol. 2010;63(10):894–899. doi: 10.1136/jcp.2010.078964. [DOI] [PubMed] [Google Scholar]
- 38.Angadi PV, Kale AD, Hallikerimath S. Evaluation of myofibroblasts in oral submucous fibrosis: correlation with disease severity. J Oral Pathol Med. 2011;40(3):208–213. doi: 10.1111/j.1600-0714.2010.00995.x. [DOI] [PubMed] [Google Scholar]
- 39.Moutasim KA, Jenei V, Sapienza K, Marsh D, Weinreb PH, Violette SM, Lewis MP, Marshall JF, Fortune F, Tilakaratne WM, Hart IR, Thomas GJ. Betel-derived alkaloid up-regulates keratinocyte alphavbeta6 integrin expression and promotes oral submucous fibrosis. J Pathol. 2011;223(3):366–377. doi: 10.1002/path.2786. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Ling TY, Gao YJ. Effect of arecoline on the differentiation of myofibroblasts of oral mucosa (Abstract) Zhongua Kuo Qiang Yi Xue Za Zhi. 2007;42:423–427. [PubMed] [Google Scholar]
- 41.White ES, Atrasz RG, Hu B, Phan SH, Stambolic V, Mak TW, Hogaboam CM, Flaherty KR, Martinez FJ, Kontos CD, Toews GB. Negative regulation of myofibroblast differentiation by PTEN (phosphatase and tensin homolog deleted on chromosome 10) Am J Respir Crit Care Med. 2006;173:112–121. doi: 10.1164/rccm.200507-1058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by TGF-β1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- 43.Gu J, Tamura M, Yamada KM. Tumor suppressor PTEN inhibits integrin and growth factor mediated mitogen-activated protein (MAP) kinase. J Cell Biol. 1998;143:1375–1378. doi: 10.1083/jcb.143.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesion by tumor suppressor PTEN. Science. 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 45.Chen Q, Samaranayake LP, Zhou H, Xiao L. Homozygous deletion of PTEN tumor suppressor gene is not a feature of oral squamous cell carcinoma. Oral Oncol. 2000;36:95–99. doi: 10.1016/S1368-8375(99)00068-8. [DOI] [PubMed] [Google Scholar]
- 46.Mavros A, Hahn M, Weilang I, Koy S, Koufaki ON, Strelocke K, et al. Infrequent genetic alterations of the tumor suppressor gene PTEN/MMAC1 in squamous cell carcinoma of the oral cavity. J Oral Pathol Med. 2002;31:270–276. doi: 10.1034/j.1600-0714.2002.310504.x. [DOI] [PubMed] [Google Scholar]
- 47.Chakraborthy S, Azeem Mohiyuddin SM, Gopinath KS, Kumar A. Involvement of TSC genes and differential expression of other members of the mTOR signaling pathway in oral squamous cell carcinoma. BMC Cancer. 2008;8:163–169. doi: 10.1186/1471-2407-8-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JI, Soria JC, Hassan KA, El-Naggar AK, Tang X, Lui DD, Homg WK, Mao L. Loss of PTEN expression as a prognostic marker for tongue cancer. Arch Otolaryngol Head Neck Surg. 2001;127:1441–1445. doi: 10.1001/archotol.127.12.1441. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Diagram to demonstrate the possible mechanism of PTEN in the pathogenesis and malignant transformation of OSMF (JPEG 237 kb)