

Abstract
Glutathione (GSH) is the most abundant antioxidant and a major detoxification agent in cells. It is synthesized through two-enzyme reaction catalyzed by glutamate cysteine ligase and glutathione synthetase, and its level is well regulated in response to redox change. Accumulating evidence suggests that GSH may play important roles in cell signaling. This review will focus on the biosynthesis of GSH, the reaction of S-glutathionylation (the conjugation of GSH with thiol residue on proteins), GSNO, and their roles in redox signaling.
Keywords: glutathione, redox signaling, glutathionylation, GSNO, nitrosylation
Introduction
Several reactive species derived from oxygen and nitrogen are produced in cells from a variety sources such as NADPH oxidases (NOX) [1], leaks from the mitochondrial electron transport chain [2], redox cycling of quinones [3], and nitric oxide synthases (NOS) [4]. The principal reactive species produced enzymatically are superoxide (O2• −), hydrogen peroxide (H2O2), and nitric oxide (•NO). These can be involved in cell injury but are also the principal actors in cell signaling, particularly the latter two [5–7]. Other species derived from these, hydroxyl radical (•OH) and peroxynitrous acid (ONOOH), are extremely potent oxidants and are more likely involved in cell injury than signaling. Environmental exposure adds additional reactive species such as ozone and nitrogen dioxide [8]. Together these species are often referred to as reactive oxygen (ROS) and nitrogen species [9], terms we will use sparingly as they are not helpful in understanding mechanism. Indeed, an argument has been made for considering only H2O2 as the actual species involved among the ROS while •NO and nitrosoglutathione (GSNO), a conjugate of •NO and glutathione (GSH) that is produced by an as yet not definitively demonstrated oxidative mechanism, are responsible for ROS and RNS signaling. Finally, other reactive species, lipid hydroperoxides (ROOH) and α, β-unsaturated aldehydes, that are derived from lipid peroxidation can also participate in both injury and cell signaling [7].
As mentioned, O2•− , H2O2, and •NO can contribute to pathology as well as signaling. Therefore, it is necessary for cells to maintain a narrow rate of production of these reactive species that is not directly harmful yet effective in mediating diverse physiologic functions through signaling. Our focus here will be on the involvement of H2O2 and GSNO in signaling. The propensity of H2O2 to modify protein function is attributed to its ability to react with particular cysteine residues within proteins [10]. The possible oxidative modification of cysteine residues within proteins includes sulfenic acid (RSOH), intra- or intermolecular disulfide (RSSR), and S-glutathionylation (RSSG), sulfinic acid (RS(=O)OH) and sulfonic acid (RS(=O)2OH) acids, thiyl radicals (RS•), sulfenyl-amides, thiosulfinates, disulfide-S-monoxides; however, physiological signaling probably only involves the first three. Formation of the other species would influence signaling by producing species that are far more difficult to reduce back to a thiol than RSOH, RSSR and RSSG but are also less likely to form at low H2O2 concentration. Formation of RSOH, RSSG and RSSG can be transient and reversible, allowing these species to participate in biochemical functions, such as redox sensing and responding, catalysis, and signal transduction [11]. This redox-modification-based mode of signal transduction is called redox signaling. Only some select cysteine residues are involved in reactions with H2O2 or ROOH as even the more nucleophilic thiolate (−S−) form is not strong enough to act in the absence of a nearby proton donor or metal to remove the OH− that would be the leaving group if a thiolate alone reacted with ROOH or H2O2. This complex chemistry has been recently reviewed [5].
The concentration of GSH, γ-L-glutamyl-L-cysteinyl-glycine, which is the most abundant non-protein thiol in cells, is in the range of 1–10 mM in most mammalian cells [12]. GSH is also the most abundant antioxidant and a major detoxification agent in cells. Enzymes such as glutathione peroxidases (GPx) and one of the peroxiredoxins (Prdx VI) catalyze the reduction of H2O2 (or ROOH) by GSH into H2O (or the corresponding alcohol (ROH) and GSSG. GSSG is reduced back to GSH by glutathione reductase using NADPH to maintain a steady state GSH/GSSG ratio that is almost all GSH but varies with cell type and disease state [13]. GSH also conjugates with electrophiles and thus participates in the metabolism and detoxification of endogenous compounds and xenobiotic toxicants [14–16]. In addition, GSH is involved in many other metabolic reactions [9, 17, 18]. Therefore, it is not surprising that GSH plays roles in various cellular processes such as cell growth, proliferation, and apoptosis. Thus, it is clear that GSH is also involved in cell signaling through which it affects these important cell functions.
GSH participates in cell signaling through at least two mechanisms, protein S-glutathionylation and cysteine S-nitrosylation through thiol exchange with GSNO. The former is formed when GSH conjugates with reactive cysteine residues within proteins to form protein mixed disulfides (PSSG), and the latter is formed by reaction of a thiolate (−S−) with GSNO. Emerging evidence suggest that both are controlled redox signaling mechanisms. How GSNO is formed has been the subject of intense investigation and several mechanisms have been proposed [6, 19]. Of course, GSH may also indirectly participate in the redox signaling by changing cellular redox homeostasis. The rest of this review will focus on the participation of protein S-glutathionylation and GSNO in signaling and on how cells regulate GSH homeostasis.
Regulation of glutathione content
In most cells, GSH is synthesized de novo through a two-step reaction. First γ-glutamylcysteine is formed from glutamate and cysteine catalyzed by glutamate cysteine ligase (GCL). Then glycine is added by glutathione synthetase to form GSH. GCL activity and cysteine availability are two rate-limiting factors in GSH synthesis. It is apparently so important for cells to maintain redox homeostasis and normal cellular function that both its concentration and GSH/GSSG ratio are tightly regulated. Some authors insist on expressing this as 2GSH/GSSG or as the redox potential (−RT ln [GSH]2/[GSSG] where R is the universal gas constant and T is the temperature on the Kelvin scale [13]. First, GSSG is quickly reduced back by GSH reductase or exported to the outside of cells. Second, the import and export of cysteine and export of GSSG are also regulated to maintain the GSH/GSSG ratio. Third, cells can up-regulate the expression of γ-glutamyl transpeptidase (GGT) [20], an enzyme on the outer surface of cells that transfers the glutamate from GSH to an acceptor amino acid. GGT prefers to transfer the glutamate to cystine, the product (γ-glutamylcystine) is transported back into cells where it is rapidly reduced providing both γ-glutamylcysteine and cysteine in what is called the scavenger pathway [21, 22]. More significantly however, the activity of GCL, the rate-limiting enzyme for GSH synthesis, is regulated by GSH through a negative feedback loop [23], by phosphorylation [24], and by protein expression. In response to oxidative stress or other situations when more GSH is required, the expression of GCL is regulated through two redox sensitive signaling pathways comprised of Nrf2-EpRE system and AP-1 [25, 26]. There is more debate regarding a role of a third redox sensitive system, NF-κB [27].
Regulation of GCL expression
The regulation of both subunits of GCL, the catalytic subunit (GCLC) and the modifier subunit (GCLM), has been extensively studied in the past 20 years. A variety of signaling pathways, such as ERK1/2 [28], JNK1/2 [29], p38MAPK [30], PKC [31], and PI3K [32, 33], and transcription factors, such as c-Jun [29], NF-κB [27], JunD [34], and Nrf2 [25], etc., have been found to be involved in the regulation of GCLC and GCLM genes. The most significant and intriguing finding though, is that both genes are regulated via a redox mechanism involving Nrf2-EpRE pathway.
NF-E2 related factor 2 (Nrf2) is a protein that is usually sequestered in the cytosol via associating with Keap1, which is tethered to β-actin. Under unstressed conditions, Nrf2 is rapidly turned over in the cytosol via ubiquitin-dependent degradation mediated by Keap1 [35–37]. Upon exposure to electrophiles or other mediators, Nrf2 dissociates from Keap1 and escapes ubiquitination/degradation [38, 39]. Nrf2 is then translocated to the nucleus, forms heterodimers with other transcription factors such as c-Jun and small Maf proteins (G/F/K), binds to the electrophile response element (EpRE) (also known as the antioxidant response element) in the promoters of GCLC and GCLM, and regulates gene transcription. Studies have demonstrated that Nrf2-EpRE pathway is involved in both the basal and inducible expression of GCL genes [25]. The Nrf2-EpRE pathway is regulated at several levels. 1) The availability of free Nrf2 protein. This is mainly regulated by the interaction of Nrf2 with Keap1 [35–37]. Recently it was found that c-Myc could also decrease Nrf2 half-life via an unidentified mechanism [40]. In addition, it was reported that phosphorylation of Nrf2 by GSK-3β pathway could lead to Nrf2 nuclear transportation and degradation [41–43]. Nrf2 phosphorylation by GSK-3β is also found to lead to Nrf2 degradation through a Keap1-independent manner [42, 44]. 2) Nrf2 partners. Several nuclear proteins are found to be Nrf2 heterodimerization partners, such as c-Jun and small Maf proteins (Maf G/F/K). Our work [45] and that of Jaiswal group [46] suggest that c-Jun is the predominant activating partner and that the small Mafs as well as c-Myc are inactivators of transcription when bound to Nrf2 [40, 47]. How the amount and/or activity of these proteins are regulated under various conditions though, needs to be further determined. 3) Other binding proteins in the Nrf2-EpRE complex. Studies have demonstrated that in addition to Nrf2 and its dimerization partners c-Jun, small Mafs and c-Myc, many other proteins exist in the EpRE complex, including JunD, JunB, Nrf1 [48], Bach1[49], Nrf3 [50], ATF4 [51], CBP/P300 [52], etc. Among them, Bach1 is consistently recognized as a competitor of Nrf2 while the role of other proteins in Nrf2-EpRE activity though remains obscure and needs further determined. 4) Phosphorylation of Nrf2 and its associated proteins. Although the exact mechanism of how signaling kinases, such as MAPK, PKC, and PI3K, regulate the gene expression of GCLC and GCLM remains unclear, it is generally thought that these kinases act through direct phosphorylation of Nrf2, or other proteins involved in Nrf2-EpRE complex formation.
Another important oxidant responsive cis element regulating GCL genes is the AP-1 binding site (also called the TRE element). There are several TREs in the human GCLC and GCLM promoters. In fact, a TRE is embedded in the EpRE consensus sequence. Studies have shown that TREs are involved in the basal and inducible expression of GCL genes [53]. The main transcription factors binding to TRE are members of the Jun and Fos family [54, 55]. Studies suggest that c-Jun phosphorylation is required for TRE-mediated GCL induction, since inhibiting JNK, which phosphorylate c-Jun, abolished GCL induction and c-Jun binding to TRE [56]. A recent study shows that inactivation of PTP1B may also be involved in the activation of JNK/c-Jun pathway [57].
Protein S-glutathionylation
In a recent review [58], several reaction mechanisms of protein S-glutathionylation (PSSG) formation were proposed. We have added to and modified these in accord with the chemistry of cysteine oxidation in signaling proteins that was discussed in our recent review [5] (Figure 1). Proton catalyzed nucleophilic substitution: To break the O-O bond in ROOH bond (where R is either a H or a lipid) and form a protein-cysteine sulfenic acid and ROH, there must be two conditions met; (a) the cysteine must be in the nucleophilic thiolate form [59] to donate electrons to the O-O bond in ROOH and (b) a hydrogen donor must be close enough to protonate the very poor leaving group RO− to form ROH, a good leaving group. The sulfenic acid, which is formed would presumably dissociate to the sulfenate (RSO−) as the pKa of sulfenic acid is lower than the corresponding thiol and the proton would go back to the hydrogen donor. The RSO− would then react with GSH to form PSSG and return the proton to the proton donor. Metal catalyzed nucleophilic substitution: Here, instead of a proton donor, a metal, which could be Zn2+, binds to the cysteine sulfur giving it a negative charge so it is equivalent to a thiolate. Alternatively, the metal may be in proximity but not bound to a thiolate. When the ROOH is given electron by the thiolate, the RO− then binds to the metal. ROH is then released by reaction of the metal-OR with water. As in the proton-catalyzed mechanism, RSO− then reacts with GSH in the second step. Recently it was demonstrated that the sulfenic acid moiety in peroxiredoxin 6 was S-glutathionylated via glutathione S-transferase Pi (GSTP), indicating that a non-enzymatic reaction between sulfenic acid and GSH did not occur [60, 61]; however, in other proteins evidence suggests that non-enzymatic reaction occurs and that RSO− is a much stronger nucleophile than is RS− [62]. Other proposals from the Pimental et al. review, which we consider less likely under physiological conditions include: PSSG formation from reaction of a PS− and GS-sulfenic acid (GSO−); PSSG formation from reaction of PS− and GSSG via a thiol transferase activity if GSSG concentration is high enough; PSSG formation from condensation of protein sulfenic acid and GSH disulfide S-oxide (GS(O)SG), a dimer of GS(OH) the formation of which is catalyzed by metals [63, 64]; PSSG formation from reaction of GSH and a cyclic sulfenylamide bond, which is generated from reaction of a protein sulfenate and a protein amide under oxidative stress. An example model of this reaction can be demonstrated with PTP1B in vitro, although its physiological significance has been questioned due to the reaction being too slow to occur under physiological conditions. Thus, protein S-glutathionylation can be formed through multiple possible reactions. To determine the exact mechanism is experimentally challenging and probably actually varies among target proteins. In keeping with the general rules of physiological signaling in which regulation is an essential requirement in contrast with oxidative stress, one would expect that glutathionylation associated with signaling is often catalyzed by enzymes that use the target protein cysteine as a substrate replacing one of the GSH in the glutathione peroxidase reaction. Indeed, while there are already some examples in the literature [65], the identification of some of these enzymes is expected in the near future.
Figure 1.
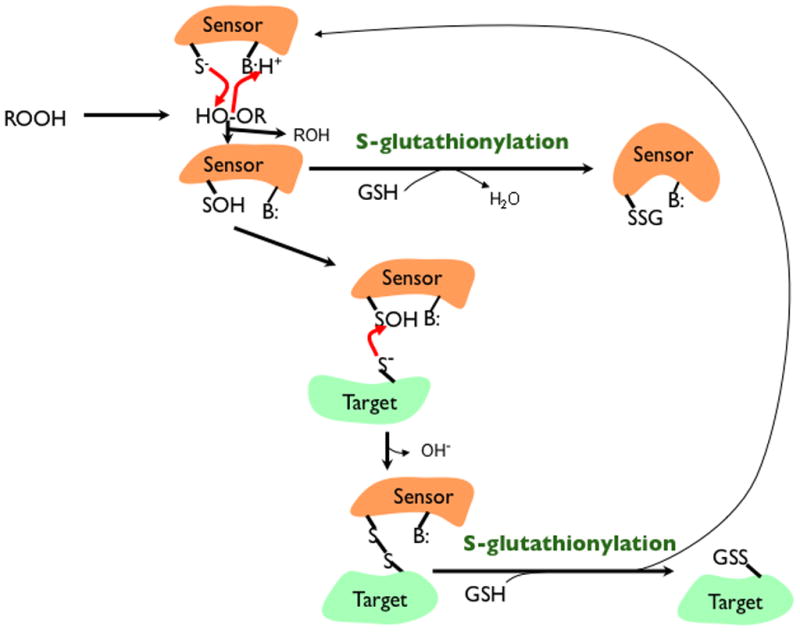
Glutathionylation in signaling. Some proteins, including peroxiredoxins, may be either sensors or direct targets for glutathionylation. When acting as a direct target, glutathionylation would occur on a protein thiolate (S−) after it has been oxidized by a hydroperoxide (ROOH) to a sulfenic acid (SOH) with the assistance of a proton donating amino acid (BH+) that allows ROH to leave. GSH can then react with the SOH; however, either the SOH or GSH would need to be in the anionic form to be a reasonably good nucleophile in that reaction. When acting as a sensor that then assists in glutathionylation, the formation of a disulfide with a target thiolate is a reasonable possibility. This would be followed by disulfide exchange where again the GSH would need assistance from a base (B:) to remove its proton forming GS−.
S-glutathionylation of cysteine residues on protein needs to be reversed to maintain homeostasis. Accumulating evidence suggests this is mainly catalyzed by a thiol-disulfide oxidoreductase called glutaredoxin (Grx) [66, 67]. There are two isoforms of Grx in mammalian cells: Grx1 in the cytoplasm, and Grx 2 in mitochondria [68]. Evidence for the importance of deglutathionylation by Grx1 has been accumulating [69–71]. In overt oxidative stress however, Grx1 may also promote PSSG formation via a mechanism involving a thiyl radical [72]; however, this is unlikely to be part of signaling. In addition to Grx1, other proteins such as sulfiredoxin (Srx) are also reported to deglutathionylate some specific PSSGs [73, 74]. Thus, as with their formation, many aspects of the exact molecular mechanisms of deglutathionylation remain elusive.
Many proteins have been found to be targets of S-glutathionylation in response to various stimuli [75]. As PSSG may evoke specific changes in the structure and function of target proteins, it can affect many cellular signaling pathways. Table 1 lists some identified signaling proteins that are affected by S-glutathionylation. These targeted signaling molecules are either part of a signaling cascade, or transcription factor, or an enzyme that further generates signaling mediators (e.g. eNOS). Of course, S-glutathionylation of other proteins, such as proteins involved in electron transfer chain, may also indirectly affect signaling pathways by increasing or decreasing H2O2 and O2•− generation from mitochondria [76].
Table.
| Target proteins | Effect on activity | Signaling pathways | References |
|---|---|---|---|
| Ryanodine receptors | increase | Ca2+ | [105] [106] |
| eNOS | inhibition | NO signaling | [89] |
| Na+-K+ ATPase | inhibition | voltage dependend signaling | [107] |
| p21Ras | increase | p21Ras-MEK-ERK | Clavereul et al [108] |
| p50 | inhibition | NF-κB signaling | [77] |
| RelA/p65 | inhibition | NF-κB signaling | [72] |
| IKKβ | inhibition | NF-κB signaling | [78] |
| TRAF 6 | inhibition | NF-κB signaling | [79] |
| c-Jun | inhibition | AP-1 signaling | [109] |
| GAPDH | inhibition | Ca2+signaling | [110] |
| p53 | inhibition | p53 signaling | [111] |
| Adenosine transporter (ANT) | varies | mitochondrial function | [90] |
| Ubiquitin ligases E 1,E2 | inhibition | protein degradation | [112] |
| Rpn2 (regulator of 26 proteosome) | inhibition | protein degradation | [113] |
| Fas | increase | apoptosis | [114] |
| caspase 3 | inhibition | apoptosis | [115] |
| PTP1B | inhibition | various signaling pathways | [116] |
Through regulating the function of signaling molecules (Table 1), protein S-glutathionylation may play important roles in various cell processes and diseases, such as apoptosis, inflammation, mitochondrial function, cancers, and neurodegenerative diseases. As examples of how S-glutathionylation is involved in regulating cell signaling and function, we will now discuss its involvement in inflammation and its regulation of peroxiredoxin I (Prdx I).
The NF-κB pathway controls expression of a variety of genes involved in inflammation. NF-κB is a transcription factor that is normally sequestered in cytosol by IκB. Inflammatory signals activate IκB kinase (IKK), which then phosphorylates IκB leading to IκB degradation and NF-κB activation. Studies have demonstrated that the most common NF-κB form, the heterodimer of p50 and p65, along with IKK-β are targets of S-glutathionylation upon stimulation [69, 77, 78]. S-glutathionylation of p50 at Cys-62 inhibits DNA binding of NF-κB and production of cytokines [77]. In addition, S-glutathionylation of IKK-β at Cys-179 causes inactivation and subsequent attenuation of LPS-induced NF-κB activation and cytokine induction [78]. Other proteins involved in inflammation are also targets for S-glutathionylation. S-glutathionylation of tumor necrosis factors receptor-associated factor 6 (TRAF6) inhibits its function and attenuates NF-κB activation [79]. Although it is generally accepted that oxidative stress can activate NF-κB, it is found that oxidative stress may also decrease the DNA binding activity of NF-κB [80]. Whether S-glutathionylation of p50 and p65 and its inhibitory effect on NF-κB activity is involved in the differences in NF-κB regulation under oxidative stress remains to be determined.
Peroxiredoxin I (Prdx I) is the most abundant and ubiquitously expressed 2-Cys Prdx, a family of peroxidases that are capable of reducing H2O2, ROOH and peroxynitrite [81]. Prdx I normally exists in the equilibrium between homodimers and decamers, depending on the oxidation status of the conserved N-terminal low pKa Cys (peroxidatic Cys) in Prdx I [82]. Under reducing condition, the peroxidatic Cys exists as a thiolate anion and Prdx I forms homodimers predominantly. During reduction of H2O2 or ROOH, the peroxidatic Cys is oxidized to sulfenic acid, which will cause formation of intermolecular disulfide bond and Prdx I oligomers. If the sulfenic acid derivative is further oxidized to form sulfinic acid, the Prdx I will form decamers and lose its peroxidases activity. Cells have evolved mechanisms to maintain the structural equilibrium of Prdx I under oxidative stress, these include the reduction of intermolecular disulfide bond by Trx system [83] and reduction of the peroxidatic Cys from sulfinic status back to sulfenic acid by sulfiredoxin (Srx) [84]. Recent studies suggest that glutathionylation is another important mechanism to regulate Prdx structure and function under oxidative stress. Proteomic analysis shows that Prdx I undergoes glutathionylation when cells are treated with low concentration of H2O2 [74, 85]. In vitro assays found that Prdx1 is glutathionylated at Cys 52, 83, and 173 [74]. Using sedimentation velocity methods, Park et al found that glutathionylation of Prdx I could efficiently disrupt the Prdx I oligomerization and shift the equilibrium toward Prdx I dimers [86]. Further study revealed that glutathionylation of single cysteine (Cys 83) was sufficient to convert Prdx I from a decamer to dimer structure. Nevertheless, the significance of glutathionylation of Prdx I remain elusive as it is a less-studied pathophysiologic function of Prdx I. According to a study by Morinaka et al [87], oligomeric structure is essential for Prdx I to bind and activate mammalian ste20-like kinase-1 (MST1) kinase, which should subsequently activate the P53 pathway and cause apoptosis. Therefore, it seems that Cys 83 glutathionylation induced by a low concentration of H2O2 would facilitate Prdx I dimer structure and inhibit apoptosis while higher range of H2O2 would cause Prdx I oligomerization and induce apoptosis. Cao et al also reported that Prdx I could bind and protect phosphatase (PTEN) from oxidation-induced inactivation and thus inhibit cancer cell growth [88], indicating that Prdx I glutathionylation may protect PTEN from inactivation and thus inhibit cell growth.
It should be noted that S-glutathionylation might affect protein function (activity) in diverse manners. For example, S-glutathionylation alters eNOS function and causes it to produce superoxide instead of •NO [89]. In addition, the degree of PSSG formation can also alter protein activities just as do differences in the degree of phosphorylation. For example, low-level S-glutathionylation of ANT increased translocation of ADP/ATP, while higher-level S-glutathionylation of ANT increased ADP/ATP translocation [90]. One must be cautious to separate what happens under physiological redox signaling versus oxidative stress and further what degree of oxidative stress realistically can occur in vivo [91].
GSNO: reaction and regulation
•NO is involved in a wide range of biochemical reactions and cell signaling pathways through protein modification. Although •NO can diffuse freely, it is readily oxidized and this limits its function as a second messenger. Formation of S-nitrosothiols (−SNO) from •NO and cysteine can protect •NO from oxidative consumption and thereby extend •NO bioavailability, both temporally and spatially. Protein S-nitrosylation can alter protein function and mediate signaling events and it has been considered as a non-classical mechanism of •NO signaling [92], compared to the classical mechanism. In the classical mechanism •NO activates soluble guanylate cyclase (sGC) by binding to its heme and leading to activation of the cGMP/PKG pathway [93]. Small mass SNOs (products of •NO and cysteine or GSH), are relatively stable and resistant to oxidation and can act as •NO carriers to allow •NO signaling to remote places from •NO synthetases.
The formation of GSNO from •NO or peroxynitrite (−ONOOH) and GSH occurs in cells but requires an oxidation step; however, it remains unclear which of several potentially physiologically relevant reactions is responsible [7, 94–96]. GSNO is an intermediate in •NO metabolism and plays a critical role in fulfilling •NO biofunction and cell signaling either as an •NO donor or through mediating protein S-glutathionylation [97]. The latter however also requires either a reaction of glutathione radical (GS•) with •NO or some other oxidative mechanism. After intracellular formation, GSNO could form GSH and another S-nitrosothiol through transnitrosation, be reduced by GSNO reductase to release •NO and mediate •NO signaling locally [98, 99], or be exported out of the cells via a GSH transporter. Extracellular GSNO can be decreased by a transnitrosation reaction with cystine [100] or by GGT to generate S-nitrosocysteinylglycine that can be cleaved by a membrane dipeptidase to generate S-nitrosocysteine. S-nitrosocysteine can be up taken directly by cells through L-type amino-acid transporter (L-AT) [101]. It appears that S-nitrosocysteinylglycine cannot enter cells directly, but may transfer its •NO to cysteine or cystine through transnitrosation [102, 103]. Upon entering cells, S-nitrosocysteine can be reduced to release •NO or transfer •NO to proteins and thus mediate •NO signaling.
There is no supportive evidence available that extracellular GSNO can enter cells directly. The system composed of GSNO reductase, GGT, dipeptidase, GSNO exporters and importer L-AT, along with cysteine and cystine, play an important role in GSNO metabolism and in regulating its local concentration. It is reported that the extracellular concentration of GSNO in normal brain tissue is 6–8 μM while undetectable in cytosolic extracts of brain tissue [104]. This heterogeneously distribution of GSNO suggests that its metabolism is tightly regulated.
Future direction
After decades of studies, it has been well established that redox-dependent reversible oxidative modification of proteins can occur under various pathophysiologic conditions, and is an important cell-signaling mode analogous to phosphorylation-based cell signaling mechanism. As the most abundant non-protein antioxidant in the cells, GSH is critical to maintain redox homeostasis and thus indirectly involved in redox signaling. Accumulating evidence suggest that GSH may directly participate in signaling process through protein S-glutathionylation and/or GSNO-mediated protein S-nitrosylation. Both S-glutathionylation and S-nitrosylation are reverse post-translational modifications and underlie the functional change of many proteins under various redox conditions. Further research will be needed to elucidate the biochemistry of protein S-glutathionylation, including the contexts, the compartmentalization, catalyzing enzymes, and the selectivity of target proteins and cysteine residues. How Grx1 is involved in deglutathionylation is also required for further study. Additional investigation into the interplay between S-glutathionylation and S-nitrosylation of proteins such as the selectivity of both modifications under conditions with concurrent oxidative and nitrosative stress is also critically needed. Regarding to GSNO, the regulation of GSNO metabolism needs to be elucidated, which include its synthesis, transportation, degradation, and regulating enzymes under various conditions. The elucidation of how protein S-glutathionylation and GSNO are involved in various physiological and pathological processes, remains as a challenging focus of future studies.
Highlights.
Glutathione (GSH) is the most abundant antioxidant and a major detoxification agent in cells.
Glutamate cysteine ligase, which catalyzes the first step in GSH synthetase is regulated by redox signaling.
Accumulating evidence suggests that GSH plays important roles in cell signaling.
Protein S-glutathionylation and S-nitrosylation play major roles in redox signaling.
Acknowledgments
Supported by ES005511 from the National Institutes of Health
Abbreviations list
- ANT
adenine nucleotide translocator
- AP-1
activator protein 1
- ATF4
activating transcription factor 4
- CBP
CREB binding protein
- EpRE
electrophile response element
- ERK
extracellular signal regulated protein kinase
- GSK-3β
glycogen synthase kinase 3β
- Grx
glutaredoxin
- GSH
glutathione
- GSSG
glutathione dioxide
- GSNO
nitrosoglutathione
- GPx
glutathione peroxidases
- GCL
glutamate cysteine ligase
- GGT
γ-glutamyl transpeptidase
- GCLC
catalytic subunit of GCL
- GCLM
modifier subunit of GCL
- H2O2
hydrogen peroxide
- JNK
c-Jun N-terminal kinase
- MST1
kinase mammalian ste20-like kinase-1
- •NO
nitric oxide
- NOS
nitric oxide synthases
- NOX
NADPH oxidase
- Nrf2
nuclear factor erythroid 2-related factor 2
- NF-κB
nuclear factor κB
- O2•−
superoxide
- •OH
hydroxyl radical
- ONOOH
peroxynitrous acid
- OH
sulfinic acid
- Prdx
peroxiredoxin
- PKC
protein kinase C
- p38MAPK
p38 mitogen-activated protein kinase
- PSSG
protein mixed disulfides
- PI3K
phosphoinositide-3 kinase
- PTEN
Phosphatase and tensin homolog
- RSOH
sulfenic acid
- RSSR
intra- or intermolecular disulfide
- RSSG
S-glutathionylation
- RS(
O)2OH, sulfonic acid
- RS•
thiyl radicals
- Srx
sulfiredoxin
- -SNO
S-nitrosothiols
- sGC
guanylate cyclase
- TRE
TBP response element
- TRAF6
tumor necrosis factors receptor-associated factor 6
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lambeth JD. Nox/Duox family of nicotinamide adenine dinucleotide (phosphate) oxidases. Curr Opin Hematol. 2002;9(1):11–7. doi: 10.1097/00062752-200201000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552(Pt 2):335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCord JM, Fridovich I. The utility of superoxide dismutase in studying free radical reactions. II. The mechanism of the mediation of cytochrome c reduction by a variety of electron carriers. J Biol Chem. 1970;245(6):1374–7. [PubMed] [Google Scholar]
- 4.Wink DA, Mitchell JB. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med. 1998;25(4–5):434–56. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 5.Forman HJ, Maiorino M, Ursini F. Signaling functions of reactive oxygen species. Biochemistry. 2010;49(5):835–42. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol. 2004;287(2):C246–56. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 7.Forman HJ, et al. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch Biochem Biophys. 2008;477(2):183–95. doi: 10.1016/j.abb.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kampa M, Castanas E. Human health effects of air pollution. Environ Pollut. 2008;151(2):362–7. doi: 10.1016/j.envpol.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 9.Stjernschantz J. The leukotrienes. Med Biol. 1984;62(4):215–30. [PubMed] [Google Scholar]
- 10.Jones DP. Redox sensing: orthogonal control in cell cycle and apoptosis signalling. J Intern Med. 2010;268(5):432–48. doi: 10.1111/j.1365-2796.2010.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacob C, Knight I, Winyard PG. Aspects of the biological redox chemistry of cysteine: from simple redox responses to sophisticated signalling pathways. Biol Chem. 2006;387(10–11):1385–97. doi: 10.1515/BC.2006.174. [DOI] [PubMed] [Google Scholar]
- 12.Meister A. Glutathione metabolism and its selective modification. Journal of Biological Chemistry. 1988;263:17205–17208. [PubMed] [Google Scholar]
- 13.Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8(9–10):1865–79. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 14.Bock KW, et al. The role of conjugation reactions in detoxication. Arch Toxicol. 1987;60(1–3):22–9. doi: 10.1007/BF00296941. [DOI] [PubMed] [Google Scholar]
- 15.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 16.Ketterer B, Coles B, Meyer DJ. The role of glutathione in detoxication. Environ Health Perspect. 1983;49:59–69. doi: 10.1289/ehp.834959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy RC, Zarini S. Glutathione adducts of oxyeicosanoids. Prostaglandins Other Lipid Mediat. 2002;68–69:471–82. doi: 10.1016/s0090-6980(02)00049-7. [DOI] [PubMed] [Google Scholar]
- 18.Sies H. [Biochemistry of thiol groups: the role of glutathione] Naturwissenschaften. 1989;76(2):57–64. doi: 10.1007/BF00396705. [DOI] [PubMed] [Google Scholar]
- 19.Chen L, et al. Mechanisms of cystic fibrosis transmembrane conductance regulator activation by S-nitrosoglutathione. J Biol Chem. 2006;281(14):9190–9. doi: 10.1074/jbc.M513231200. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Forman HJ. Redox regulation of gamma-glutamyl transpeptidase. Am J Respir Cell Mol Biol. 2009;41(5):509–15. doi: 10.1165/rcmb.2009-0169TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meister A. New developments in glutathione metabolism and their potential application in therapy. Hepatology. 1984;4(4):739–42. doi: 10.1002/hep.1840040431. [DOI] [PubMed] [Google Scholar]
- 22.Griffith OW, Bridges RJ, Meister A. Formation of gamma-glutamycyst(e)ine in vivo is catalyzed by gamma-glutamyl transpeptidase. Proc Natl Acad Sci U S A. 1981;78(5):2777–81. doi: 10.1073/pnas.78.5.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang C-S, et al. Catalytic and regulatory properties of the heavy subunit of rat kidney κ-glutamylcysteine synthetase. Journal of Biological Chemistry. 1993;268:19675–19680. [PubMed] [Google Scholar]
- 24.Sun W-M, Huang Z-Z, Lu SC. Regulation of κ-glutamylcysteine synthetase by protein phosphorylation. Biochemical Journal. 1996;320:321–328. doi: 10.1042/bj3200321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H, Court N, Forman HJ. Submicromolar concentrations of 4-hydroxynonenal induce glutamate cysteine ligase expression in HBE1 cells. Redox Rep. 2007;12(1):101–6. doi: 10.1179/135100007X162266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iles KE, Liu RM. Mechanisms of glutamate cysteine ligase (GCL) induction by 4-hydroxynonenal. Free Radic Biol Med. 2005;38(5):547–56. doi: 10.1016/j.freeradbiomed.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 27.Yang H, et al. Cloning and characterization of the 5'-flanking region of the rat glutamate-cysteine ligase catalytic subunit. Biochem J. 2001;357(Pt 2):447–55. doi: 10.1042/0264-6021:3570447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan KP, et al. NRF2 as a determinant of cellular resistance in retinoic acid cytotoxicity. Free Radic Biol Med. 2008;45(12):1663–73. doi: 10.1016/j.freeradbiomed.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 29.Dickinson DA, et al. 4-hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free Radic Biol Med. 2002;33(7):974. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 30.Zipper LM, Mulcahy RT. Inhibition of ERK and p38 MAP kinases inhibits binding of Nrf2 and induction of GCS genes. Biochem Biophys Res Commun. 2000;278(2):484–92. doi: 10.1006/bbrc.2000.3830. [DOI] [PubMed] [Google Scholar]
- 31.Rushworth SA, et al. Role of protein kinase C delta in curcumin-induced antioxidant response element–mediated gene expression in human monocytes. Biochem Biophys Res Commun. 2006;341(4):1007–16. doi: 10.1016/j.bbrc.2006.01.065. [DOI] [PubMed] [Google Scholar]
- 32.Kim SK, et al. Insulin signaling regulates gamma-glutamylcysteine ligase catalytic subunit expression in primary cultured rat hepatocytes. J Pharmacol Exp Ther. 2004;311(1):99–108. doi: 10.1124/jpet.104.070375. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, et al. Essential roles of the PI3 kinase/Akt pathway in regulating Nrf2-dependent antioxidant functions in the RPE. Invest Ophthalmol Vis Sci. 2008;49(4):1671–8. doi: 10.1167/iovs.07-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dickinson DA, et al. Curcumin alters EpRE and AP-1 binding complexes and elevates glutamate-cysteine ligase gene expression. FASEB J. 2003;17(3):473–5. doi: 10.1096/fj.02-0566fje. [DOI] [PubMed] [Google Scholar]
- 35.Itoh K, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13(1):76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cullinan SB, et al. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23(20):7198–209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J Biol Chem. 2003;278(45):44675–82. doi: 10.1074/jbc.M307633200. [DOI] [PubMed] [Google Scholar]
- 38.Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med. 2004;36(10):1208–13. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- 39.Giudice A, Arra C, Turco MC. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. Methods Mol Biol. 2010;647:37–74. doi: 10.1007/978-1-60761-738-9_3. [DOI] [PubMed] [Google Scholar]
- 40.Levy S, Forman HJ. C-Myc is a Nrf2-interacting protein that negatively regulates phase II genes through their electrophile responsive elements. IUBMB Life. 2010;62(3):237–46. doi: 10.1002/iub.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jain AK, Jaiswal AK. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem. 2007;282(22):16502–10. doi: 10.1074/jbc.M611336200. [DOI] [PubMed] [Google Scholar]
- 42.Rojo AI, Sagarra MR, Cuadrado A. GSK-3beta down-regulates the transcription factor Nrf2 after oxidant damage: relevance to exposure of neuronal cells to oxidative stress. J Neurochem. 2008;105(1):192–202. doi: 10.1111/j.1471-4159.2007.05124.x. [DOI] [PubMed] [Google Scholar]
- 43.Niture SK, et al. Src subfamily kinases regulate nuclear export and degradation of transcription factor Nrf2 to switch off Nrf2-mediated antioxidant activation of cytoprotective gene expression. J Biol Chem. 2011;286(33):28821–32. doi: 10.1074/jbc.M111.255042. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Rada P, et al. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31(6):1121–33. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levy S, Jaiswal AK, Forman HJ. The role of c-Jun phosphorylation in EpRE activation of phase II genes. Free Radic Biol Med. 2009;47(8):1172–9. doi: 10.1016/j.freeradbiomed.2009.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeyapaul J, Jaiswal AK. Nrf2 and c-Jun regulation of antioxidant response element (ARE)-mediated expression and induction of gamma-glutamylcysteine synthetase heavy subunit gene. Biochem Pharmacol. 2000;59(11):1433–9. doi: 10.1016/s0006-2952(00)00256-2. [DOI] [PubMed] [Google Scholar]
- 47.Dhakshinamoorthy S, Jaiswal AK. Small maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:Quinone oxidoreductase1 gene. J Biol Chem. 2000;275(51):40134–41. doi: 10.1074/jbc.M003531200. [DOI] [PubMed] [Google Scholar]
- 48.Myhrstad MC, et al. TCF11/Nrf1 overexpression increases the intracellular glutathione level and can transactivate the gamma-glutamylcysteine synthetase (GCS) heavy subunit promoter. Biochim Biophys Acta. 2001;1517(2):212–9. doi: 10.1016/s0167-4781(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 49.Dhakshinamoorthy S, et al. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. 2005;280(17):16891–900. doi: 10.1074/jbc.M500166200. [DOI] [PubMed] [Google Scholar]
- 50.Sankaranarayanan K, Jaiswal AK. Nrf3 negatively regulates antioxidant-response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. J Biol Chem. 2004;279(49):50810–7. doi: 10.1074/jbc.M404984200. [DOI] [PubMed] [Google Scholar]
- 51.He CH, et al. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem. 2001;276(24):20858–65. doi: 10.1074/jbc.M101198200. [DOI] [PubMed] [Google Scholar]
- 52.Zhu M, Fahl WE. Functional characterization of transcription regulators that interact with the electrophile response element. Biochem Biophys Res Commun. 2001;289(1):212–9. doi: 10.1006/bbrc.2001.5944. [DOI] [PubMed] [Google Scholar]
- 53.Rahman I, Antonicelli F, MacNee W. Molecular mechanism of the regulation of glutathione synthesis by tumor necrosis factor-alpha and dexamethasone in human alveolar epithelial cells. J Biol Chem. 1999;274(8):5088–5096. doi: 10.1074/jbc.274.8.5088. [DOI] [PubMed] [Google Scholar]
- 54.Ofir R, et al. Phosphorylation of the C terminus of Fos protein is required for transcriptional transrepression of the c-fos promoter. Nature. 1990;348(6296):80–2. doi: 10.1038/348080a0. [DOI] [PubMed] [Google Scholar]
- 55.Binetruy B, Smeal T, Karin M. Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature. 1991;351(6322):122–7. doi: 10.1038/351122a0. [DOI] [PubMed] [Google Scholar]
- 56.Dickinson DA, et al. 4-hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free Radic Biol Med. 2002;33(7):974–987. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 57.Rinna A, Forman HJ. SHP-1 Inhibition by 4-hydroxynonenal Activates Jun N-terminal Kinase and Glutamate Cysteine Ligase. Am J Respir Cell Mol Biol. 2008 doi: 10.1165/rcmb.2007-0371OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pimentel D, et al. Regulation of cell physiology and pathology by protein S-glutathionylation. Lessons learned from the cardiovascular system. Antioxid Redox Signal. 2011 doi: 10.1089/ars.2011.4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leisinger T, Braus-Stromeyer SA. Bacterial growth with chlorinated methanes. Environ Health Perspect. 1995;103(Suppl 5):33–6. doi: 10.1289/ehp.95103s433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manevich Y, Feinstein SI, Fisher AB. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc Natl Acad Sci U S A. 2004;101(11):3780–5. doi: 10.1073/pnas.0400181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ralat LA, et al. Direct evidence for the formation of a complex between 1-cysteine peroxiredoxin and glutathione S-transferase pi with activity changes in both enzymes. Biochemistry. 2006;45(2):360–72. doi: 10.1021/bi0520737. [DOI] [PubMed] [Google Scholar]
- 62.Klomsiri C, Karplus PA, Poole LB. Cysteine-based redox switches in enzymes. Antioxid Redox Signal. 2011;14(6):1065–77. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J, Huang FL, Huang KP. Glutathiolation of proteins by glutathione disulfide S-oxide derived from S-nitrosoglutathione. Modifications of rat brain neurogranin/RC3 and neuromodulin/GAP-43. J Biol Chem. 2001;276(5):3098–105. doi: 10.1074/jbc.M008260200. [DOI] [PubMed] [Google Scholar]
- 64.Huang KP, Huang FL. Glutathionylation of proteins by glutathione disulfide S-oxide. Biochem Pharmacol. 2002;64(5–6):1049–56. doi: 10.1016/s0006-2952(02)01175-9. [DOI] [PubMed] [Google Scholar]
- 65.Townsend DM, et al. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J Biol Chem. 2009;284(1):436–45. doi: 10.1074/jbc.M805586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gallogly MM, Starke DW, Mieyal JJ. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid Redox Signal. 2009;11(5):1059–81. doi: 10.1089/ars.2008.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7(3–4):348–66. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 68.Kalinina EV, Chernov NN, Saprin AN. Involvement of thio-, peroxi-, and glutaredoxins in cellular redox-dependent processes. Biochemistry (Mosc) 2008;73(13):1493–510. doi: 10.1134/s0006297908130099. [DOI] [PubMed] [Google Scholar]
- 69.Mieyal JJ, et al. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Signal. 2008;10(11):1941–88. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ho YS, et al. Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia. Free Radic Biol Med. 2007;43(9):1299–312. doi: 10.1016/j.freeradbiomed.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu H, et al. Glutaredoxin 2 knockout increases sensitivity to oxidative stress in mouse lens epithelial cells. Free Radic Biol Med. 2011;51(11):2108–17. doi: 10.1016/j.freeradbiomed.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qanungo S, et al. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J Biol Chem. 2007;282(25):18427–36. doi: 10.1074/jbc.M610934200. [DOI] [PubMed] [Google Scholar]
- 73.Findlay VJ, Tapiero H, Townsend DM. Sulfiredoxin: a potential therapeutic agent? Biomed Pharmacother. 2005;59(7):374–9. doi: 10.1016/j.biopha.2005.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Park JW, et al. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J Biol Chem. 2009;284(35):23364–74. doi: 10.1074/jbc.M109.021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fratelli M, et al. Identification of proteins undergoing glutathionylation in oxidatively stressed hepatocytes and hepatoma cells. Proteomics. 2003;3(7):1154–61. doi: 10.1002/pmic.200300436. [DOI] [PubMed] [Google Scholar]
- 76.Hurd TR, et al. Glutathionylation of mitochondrial proteins. Antioxid Redox Signal. 2005;7(7–8):999–1010. doi: 10.1089/ars.2005.7.999. [DOI] [PubMed] [Google Scholar]
- 77.Pineda-Molina E, et al. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40(47):14134–42. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 78.Reynaert NL, et al. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci U S A. 2006;103(35):13086–91. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chantzoura E, et al. Glutaredoxin-1 regulates TRAF6 activation and the IL-1 receptor/TLR4 signalling. Biochem Biophys Res Commun. 2010;403(3–4):335–9. doi: 10.1016/j.bbrc.2010.11.029. [DOI] [PubMed] [Google Scholar]
- 80.Flescher E, et al. Oxidative stress suppresses transcription factor activities in stimulated lymphocytes. Clin Exp Immunol. 1998;112(2):242–7. doi: 10.1046/j.1365-2249.1998.00548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Immenschuh S, Baumgart-Vogt E. Peroxiredoxins, oxidative stress, and cell proliferation. Antioxid Redox Signal. 2005;7(5–6):768–77. doi: 10.1089/ars.2005.7.768. [DOI] [PubMed] [Google Scholar]
- 82.Wood ZA, et al. Dimers to doughnuts: redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry. 2002;41(17):5493–504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- 83.Chae HZ, et al. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Res Clin Pract. 1999;45(2–3):101–12. doi: 10.1016/s0168-8227(99)00037-6. [DOI] [PubMed] [Google Scholar]
- 84.Woo HA, et al. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300(5619):653–6. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 85.Fratelli M, et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci U S A. 2002;99(6):3505–10. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Park JW, et al. Glutathionylation of peroxiredoxin I induces decamer to dimers dissociation with concomitant loss of chaperone activity. Biochemistry. 2011;50(15):3204–10. doi: 10.1021/bi101373h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Morinaka A, et al. Oligomeric peroxiredoxin-I is an essential intermediate for p53 to activate MST1 kinase and apoptosis. Oncogene. 2011;30(40):4208–18. doi: 10.1038/onc.2011.139. [DOI] [PubMed] [Google Scholar]
- 88.Cao J, et al. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 2009;28(10):1505–17. doi: 10.1038/emboj.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen CA, et al. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468(7327):1115–8. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Queiroga CS, et al. Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. J Biol Chem. 2010;285(22):17077–88. doi: 10.1074/jbc.M109.065052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Forman HJ. Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radic Biol Med. 2007;42(7):926–32. doi: 10.1016/j.freeradbiomed.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Martinez-Ruiz A, Cadenas S, Lamas S. Nitric oxide signaling: classical, less classical, and nonclassical mechanisms. Free Radic Biol Med. 2011;51(1):17–29. doi: 10.1016/j.freeradbiomed.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 93.Ignarro LJ, et al. Activation of purified guanylate cyclase by nitric oxide requires heme. Comparison of heme-deficient, heme-reconstituted and heme-containing forms of soluble enzyme from bovine lung. Biochim Biophys Acta. 1982;718(1):49–59. doi: 10.1016/0304-4165(82)90008-3. [DOI] [PubMed] [Google Scholar]
- 94.Wink DA, et al. Reaction kinetics for nitrosation of cysteine and glutathione in aerobic nitric oxide solutions at neutral pH. Insights into the fate and physiological effects of intermediates generated in the NO/O2 reaction. Chem Res Toxicol. 1994;7(4):519–25. doi: 10.1021/tx00040a007. [DOI] [PubMed] [Google Scholar]
- 95.Clancy RM, et al. Nitric oxide reacts with intracellular glutathione and activates the hexose monophosphate shunt in human neutrophils: evidence for S-nitrosoglutathione as a bioactive intermediary. Proc Natl Acad Sci U S A. 1994;91(9):3680–4. doi: 10.1073/pnas.91.9.3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Moro MA, et al. Paradoxical fate and biological action of peroxynitrite on human platelets. Proc Natl Acad Sci U S A. 1994;91(14):6702–6. doi: 10.1073/pnas.91.14.6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chiueh CC. Neuroprotective properties of nitric oxide. Ann N Y Acad Sci. 1999;890:301–11. doi: 10.1111/j.1749-6632.1999.tb08007.x. [DOI] [PubMed] [Google Scholar]
- 98.Liu L, et al. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410(6827):490–4. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 99.Fernandez MR, Biosca JA, Pares X. S-nitrosoglutathione reductase activity of human and yeast glutathione-dependent formaldehyde dehydrogenase and its nuclear and cytoplasmic localisation. Cell Mol Life Sci. 2003;60(5):1013–8. doi: 10.1007/s00018-003-3025-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zeng H, Spencer NY, Hogg N. Metabolism of S-nitrosoglutathione by endothelial cells. Am J Physiol Heart Circ Physiol. 2001;281(1):H432–9. doi: 10.1152/ajpheart.2001.281.1.H432. [DOI] [PubMed] [Google Scholar]
- 101.Hogg N, et al. S-Nitrosoglutathione as a substrate for gamma-glutamyl transpeptidase. Biochem J. 1997;323( Pt 2):477–81. doi: 10.1042/bj3230477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang Y, Hogg N. S–Nitrosothiols: cellular formation and transport. Free Radic Biol Med. 2005;38(7):831–8. doi: 10.1016/j.freeradbiomed.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 103.Matsumoto A, Gow AJ. Membrane transfer of S-nitrosothiols. Nitric Oxide. 2011;25(2):102–7. doi: 10.1016/j.niox.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Carver J, et al. S-nitrosothiol formation. Methods Enzymol. 2005;396:95–105. doi: 10.1016/S0076-6879(05)96010-2. [DOI] [PubMed] [Google Scholar]
- 105.Aracena P, et al. S-glutathionylation decreases Mg2+ inhibition and S-nitrosylation enhances Ca2+ activation of RyR1 channels. J Biol Chem. 2003;278(44):42927–35. doi: 10.1074/jbc.M306969200. [DOI] [PubMed] [Google Scholar]
- 106.Sanchez G, et al. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J Mol Cell Cardiol. 2005;39(6):982–91. doi: 10.1016/j.yjmcc.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 107.Bundgaard H, et al. beta(3) adrenergic stimulation of the cardiac Na+-K+ pump by reversal of an inhibitory oxidative modification. Circulation. 2010;122(25):2699–708. doi: 10.1161/CIRCULATIONAHA.110.964619. [DOI] [PubMed] [Google Scholar]
- 108.Clavreul N, et al. S-glutathiolation by peroxynitrite of p21ras at cysteine-118 mediates its direct activation and downstream signaling in endothelial cells. FASEB J. 2006;20(3):518–20. doi: 10.1096/fj.05-4875fje. [DOI] [PubMed] [Google Scholar]
- 109.Klatt P, Molina EP, Lamas S. Nitric oxide inhibits c-Jun DNA binding by specifically targeted S-glutathionylation. J Biol Chem. 1999;274(22):15857–64. doi: 10.1074/jbc.274.22.15857. [DOI] [PubMed] [Google Scholar]
- 110.Mohr S, et al. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1999;274(14):9427–30. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- 111.Velu CS, et al. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry. 2007;46(26):7765–80. doi: 10.1021/bi700425y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jahngen-Hodge J, et al. Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J Biol Chem. 1997;272(45):28218–26. doi: 10.1074/jbc.272.45.28218. [DOI] [PubMed] [Google Scholar]
- 113.Zmijewski JW, Banerjee S, Abraham E. S-glutathionylation of the Rpn2 regulatory subunit inhibits 26 S proteasomal function. J Biol Chem. 2009;284(33):22213–21. doi: 10.1074/jbc.M109.028902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Anathy V, et al. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J Cell Biol. 2009;184(2):241–52. doi: 10.1083/jcb.200807019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Huang Z, et al. Inhibition of caspase-3 activity and activation by protein glutathionylation. Biochem Pharmacol. 2008;75(11):2234–44. doi: 10.1016/j.bcp.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rinna A, Torres M, Forman HJ. Stimulation of the alveolar macrophage respiratory burst by ADP causes selective glutathionylation of protein tyrosine phosphatase 1B. Free Radic Biol Med. 2006;41(1):86–91. doi: 10.1016/j.freeradbiomed.2006.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]