

Abstract
Indole-3-carbinol (I3C), a naturally occurring component of Brassica vegetables, such as cabbage, broccoli, and Brussels sprouts, induces a G1 cell cycle arrest of human breast cancer cells. Structure-activity relationships of I3C that mediate this anti-proliferative response were investigated using synthetic and natural I3C derivatives that contain substitutions at the indole nitrogen. Nitrogen substitutions included N-alkoxy substituents of one to four carbons in length, which inhibit dehydration and the formation of the reactive indolenine. Analysis of growth and cell cycle arrest of indole-treated human breast cancer cells revealed a striking increase in efficacy of the N-alkoxy I3C derivatives that is significantly enhanced by the presence of increasing carbon lengths of the N-alkoxy substituents. Compared to I3C, the half maximal growth arrest responses occurred at 23-fold lower indole concentration for N-methoxy-I3C, 50-fold lower concentration for N-ethoxy-I3C, 217-fold lower concentration for N-propoxy-I3C, and 470-fold lower concentration for N-butoxy-I3C. At these lower concentrations, each of the N-alkoxy substituted compounds induced the characteristic I3C response in that CDK6 gene expression, CDK6 promoter activity, and CDK2 specific enzymatic activity for its retinoblastoma protein substrate were strongly down-regulated. 3-Methoxymethylindole and 3-ethoxymethylindole were approximately as bioactive as I3C, whereas, both tryptophol and melatonine failed to induce the cell cycle arrest, showing the importance of the C-3 hydroxy methyl substituent on the indole ring. Taken together, our study establishes the first I3C structure activity relationship for cytostatic activities, and implicates I3C-based N-alkoxy derivatives as a novel class of potentially more potent experimental therapeutics for breast cancer.
Keywords: I3C, synthetic derivatives, N-alkoxy constituents, breast cancer cells, cell cycle arrest
1. Introduction
One of the complexities of breast cancer is the formation of distinct classes of tumors that differ in their proliferative responses to hormonal signals and other environmental cues. Approximately one-third of all breast cancers are estrogen responsive, and endocrine therapy targeting the estrogen receptor directly, such as with the nonsteroidal anti-estrogen tamoxifen, or indirectly, such as with aromatase inhibitors, are the main adjuvant therapies used to control the growth of estrogen responsive breast cancers [1–4]. Current options for treatment of most other breast cancers include surgical removal of tumors, general chemotherapy and/or radiation therapy. Thus, a critical problem in the clinical management of breast cancer is the need to develop new classes of chemotherapeutics that can effectively target estrogen-independent as well as estrogen-dependent mammary tumors. Epidemiological findings show that an increased consumption of phytochemicals from whole grains, vegetables, and fruits is directly associated with a decreased risk for certain human cancers, including breast cancer [4–7]. These studies suggest that dietary plants produce unique compounds that represent a largely untapped source of potentially potent chemotherapeutic molecules. One such phytochemical is indole-3-carbinol (I3C), a naturally occurring component of Brassica vegetables, such as cabbage, broccoli, and Brussels sprouts [8–12].
Early studies focusing on the chemopreventative properties of I3C established that high doses of I3C in the diet of rodents greatly reduced the incidence of spontaneous and carcinogen-induced tumors of the mammary gland, endometrium and other cancer types [13–15]. For example, I3C treatment prevented the formation of 7,12-dimethyl-benz(a)anthracene (DMBA)-induced mammary tumors in rats [15], and of benzo(a)pyrene-induced tumors of the forestomach and pulmonary adenomas in mice [13, 15, 16]. Consistent with these studies, I3C tested positive as a chemopreventative agent in several short-term bioassays relevant to carcinogen-induced DNA damage, tumor initiation and promotion, and oxidative stress [17].
When ingested, the low pH of the stomach converts I3C into several products, including the diindole products indole[3,2-b]carbazole (ICZ) and 3,3′-diindolylmethane (DIM), and the trimerization product 5,6,11,12,17,18-hexahydrocyclonona[1,2-b:4,5-b′:7,8b″]triindole (CTr) [11, 18–22]. A general picture has emerged that a subset of these acid-catalyzed products have distinct anti-proliferative and anti-tumorigenic physiological bioactivities. For example, DIM can induce apoptosis in breast cancer cells and endometrial tumor cells independent of the effects of estrogen [23]. Other acid catalyzed products of I3C likely account for the ability of dietary I3C to markedly reduce the incidence of estrogen-responsive mammary tumors in rodents. Several studies have shown that ICZ binds to the aromatic hydrocarbon (Ah) receptor and induces P450 CYP1A1 gene expression, which can alter estrogen metabolism [18, 24, 25]. However, at least one of the acid catalyzed products, CTr, is a strong estrogen receptor agonist capable of increasing the proliferation rate of cultured human breast cancer cells [22]. In contrast, I3C has little affinity for either the Ah or estrogen receptors [10, 25, 26]. Thus, the overall effect of oral I3C may result from a complex balance of the proliferative and anti-proliferative effects of the acid catalyzed products.
We have previously documented that direct exposure of cultured human breast cancer cells to I3C inhibits cell growth by inducing a G1 cell cycle arrest of both estrogen responsive and estrogen nonresponsive cells [27]. In cultured cells a significant portion of I3C is converted into its natural dimerization product DIM [28], and we have shown that the anti-proliferative effects of I3C are distinct from and complement the bioactivities of DIM [10, 29–32]. The I3C mediated G1 cell cycle arrest of human breast cancer cells is mediated in part by an I3C activated transcriptional cascade that leads to the rapid inhibition in expression of the G1-acting cyclin dependent kinase 6 (CDK6) transcripts and protein [27]. I3C down regulates CDK6 transcription by inhibiting Sp1 binding and function at a Sp1-Ets composite DNA element in the CDK6 promoter [29]. In contrast, the cell cycle arrest and apoptosis induced by DIM is not accompanied by CDK6 down regulation [23, 33]. I3C treatment of breast cancer cells also results in an inhibition of CDK2 specific enzymatic activity due to a disruption in composition and subcellular localization of the CDK2 protein complex [30], whereas, CDK4 enzymatic activity and protein levels remained relatively unaffected [34]. We have also shown that I3C, but not DIM, down regulates estrogen receptor-alpha expression [32] and acts synergistically with tamoxifen to inhibit the growth of estrogen responsive MCF7 breast cancer cells and suppress CDK2 specific activity [34].
Because there is only limited information about the potential role of natural indoles in the treatment breast cancer, a necessary first experimental step for the development of novel I3C-based therapeutic compounds, is to uncover I3C derivatives that have a more potent growth inhibitory effect than I3C itself. The natural indole product, N-methoxy-I3C, was shown to be a more potent inducer of cytochrome P450 activity in cultured cells than I3C [35]. This observation suggests that synthetic I3C derivatives with more potent anti-proliferative and transcriptional effects in human breast cancer cells could be produced. Therefore, as a starting point to understand I3C structure-activity relationships, we investigated a series of I3C derivatives that either contain substitutions at the indole nitrogen or at the 3-hydroxymethyl oxygen. Our results demonstrate that the efficacy of the I3C-mediated G1 cell cycle arrest and transcriptional control of CDK6 expression is significantly enhanced by more hydrophobic 1-alkoxy substitution at nitrogen, and requires the presence of the C-3 hydroxy methyl substituent on the indole ring. This study implicates the potential use of I3C-based 1-alkoxy derivatives as a novel class of experimental therapeutics for breast cancer.
2. Materials and Methods
2.1 Chemicals and biological materials
Indole-3-carbinol (I3C), N-methylindole-3-carboxaldehyde, tryptophol, and melatonin were purchased from Aldrich (Milwaukee, WI). I3C was recrystallized from toluene prior to use. Dulbecco’s Modified Eagle Medium (DMEM), fetal bovine serum (FBS), calcium and magnesium-free phosphate buffered saline (PBS), L-glutamine, and trypsin-EDTA were supplied by BioWhittaker (Walersville, MD). [γ-32P]ATP (3,000 Ci/mmol) and [3H]thymidine (84 Ci/mmol), were obtained from NEN Life Science Products (Boston, MA). Salts and other chemicals used were of the highest purity available, and generally purchased from Sigma Chemical Corp. (St. Louis, MO).
2.2 Synthesis of I3C derivatives
N-Alkoxy-3-formylindoles, used for the preparation of N-alkoxy-indole-3-carbinols, were obtained by the following reaction sequence as previously published for N-methoxy-indole-3-carbinol [36, 37]. N-Hydroxyindole was obtained from indoline by oxidation with H2O2 in the presence of Na2WO4 [38]. The oxidation mixture was diluted with water, extracted with ethyl acetate, dried over Na2SO4, evaporated to dryness in vacuo, and the residue was dissolved in tetrahydrofuran for for treatment with an alkyl halide and NaH. Standard formylation reaction conditions (dimethylformamide/POCl3) were used for the preparation of N-substituted-3-formylindoles. The formyl derivatives were converted to the hydroxymethyl products by reduction with substituted by HPLC to confirm the complete conversion of the aldehydye to the alcohol. The final product was diluted with water, extracted with diethyl ether, dried over sodium sulfate, and evaporated to dryness under nitrogen gas. NMR spectra for N-methyl I3C and N-methoxy I3C were similar to previously published data [18].
3-Methoxy-I3C and 3-ethoxy-I3C were prepared from N,O-diacetyl-I3C and recrystallized. Briefly, N,O-diacetyl-I3C was prepared by treatment of gramine with dry sodium acetate in refluxing acetic anhydride. The product was recrystallized from ethanol to give pale yellow needles, m.p. 88–90 °C (lit. 89–90 °C). The O-alkyl ether derivatives were prepared by dissolution of the diacetate in methanol or ethanol and treatment with 10% sodium hydroxide at room temperature. Recrystallization of the precipitated products provided the 3-methoxy-I3C (m.p. 93–94 °C, lit. 94–95 °C) and 3-ethoxy-I3C (m.p. 63–65 °C, lit. 63–64 °C) as colorless needles. HPLC analysis of these products indicated that they were greater than 95% pure.
2.3 Characterization of N-alkoxy Derivatives
HPLC
Reverse phase liquid chromatography was performed using a HPLC system (model SCL-10A, Shimadzu Scientific Instruments, Inc., Japan) equipped with a C-18 bonded-phase column (Ultrasphere-ODS, 4.6 × 250 mm, 5 mm; Beckman, San Ramon, CA) and Shimadzu UV/VIS detector (model SPD-10AV) monitoring wavelength 280 nm. The products were eluted using a gradient of acetonitrile in water at constant 0.05% TEA with an initial 0% acetonitrile for 2 min followed by linear gradients of 0–40% acetonitrile over 8 min, 40–60% over 10 min, 60–95% over 10 min, and then constant 95% for 5 min before returning to initial conditions, all at 1.0 mL/min. Retention times for purified, non-crystalline products was as follows: N-methoxy I3C 17.0 min, N-ethoxy I3C 18.5 min, N-propoxy I3C 20.8 min, N-butoxy I3C 23.3 min. The known MMI and EMI were purified by recrystallization.
FAB/MS
FAB/MS spectra were acquired with the ZAB2-EQ double focusing mass spectrometer (Micromass, Manchester, U.K.). FAB analyses were performed by the mass spectrometry facility at the University of California in Berkeley.
Spectroscopic Measurements
1H-NMR measurements were performed on a Bruker AMX 300 (300MHz) spectrometer in deuterated chloroform (CDCL3).
N-methoxy I3C
FAB m/z 177 [M+], 160 [ArN(1-OCH3)CHC=CH2+]; δ (ppm) [acetonitrile-D3] 2.67 (3 H, s, OCH3), 3.36 (2 H, s, CH2), 5.68 (1 H, t, J 7 Hz, ArH), 5.82 (1 H, t, J 7 Hz, ArH), 5.99 (1 H, s, CH), 6.01 (1 H, d, J 7 Hz, ArH), 6.28 (1 H, d, J 7 Hz, ArH). Yellowish oil.
N-ethoxy I3C
FAB m/z 191 [M+] (23%), 174 [ArN(1-OCH2CH3)CHC=CH2+] (100%), 146 (8%), 130 (10%); δ (ppm) [CDCL3] 1.41 (3 H, t, J = 7 Hz, CH3), 1.75 (1 H, broad s, OH), 4.29 (2 H, q, J = 7 Hz, OCH2), 4.83 (2 H, s, CH2), 7.15 (1 H, t, J = 8 Hz, ArH), 7.27 (1 H, t, J = 7 Hz, ArH), 7.43 (1 H, d, J = 8 Hz, ArH), 7.70 (1 H, d, J = 8 Hz, ArH). Yellowish oil.
N-propoxy I3C
FAB m/z 205 [M+] (20%), 188 [ArN(1-OCH2CH2CH3)CHC=CH2+] (100%), 146 (13%), 130 (7%); δ (ppm) [CDCl3] 1.09 (3 H, t, J = 7 Hz, CH3), 1.52 (1 H, broad s, OH), 1.82 (2 H, m, J = 7 Hz, CH2), 4.19 (2 H, t, J = 7 Hz, OCH2), 4.84 (2 H, s, CH2), 7.15 (1 H, t, J = 8 Hz, ArH), 7.27 (1 H, t, J = 7 Hz, ArH), 7.43 (1 H, d, J = 8 Hz, ArH), 7.71 (1 H, d, J = 8 Hz, ArH). Yellowish oil.
N-butoxy I3C
FAB m/z 219 [M+] (32%), 202 [ArN(1-OCH2CH2CH2CH3)CHC=CH2+] (100%), 163 (9%), 146 (22%), 130 (10%); δ (ppm) [CDCl3] 0.99 (3 H, t, J = 7 Hz, CH3), 1.51 (3 H, m, J = 7 Hz, CH2 and OH), 1.76 (2 H, m, J = 6 Hz, CH2), 4.22 (2 H, t, J = 7 Hz, OCH2), 4.83 (2 H, s, CH2), 7.13 (1 H, t, J = 7 Hz, ArH), 7.25 (1 H, t, J = 7 Hz, ArH), 7.40 (1 H, d, J = 8 Hz, ArH), 7.69 (1 H, d, J = 8 Hz, ArH). Yellowish oil.
2.4 Cell Culture
The MCF-7 (estrogen responsive) and the MDA-MB-231 (estrogen nonresponsive) human breast adenocarcinoma cell lines were obtained from the American Type Culture Collection (Rockville, MD). MCF-7 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 μg/ml insulin (Bovine), 50 units/ml penicillin, 50 units/ml streptomycin, and 2 mM L-glutamine. The MDA-MB-231 cells were cultured as previously described [27]. Both cell lines were maintained at 37 °C in humidified air containing 5% CO2 at subconfluency. I3C, tryptophol, I3C N-substituted, and I3C O-substituted derivatives were dissolved in dimethyl sulfoxide (DMSO, 99.9% HPLC grade, Aldrich) at a concentration 1000-fold higher than the final medium concentration. In all experiments, 1 μl of the concentrated agent was added per ml of DMEM. For the vehicle control, 1 μl DMSO was added per ml medium.
2.5 [3H]Thymidine Incorporation
Breast cancer cells were plated onto 24-well Corning tissue culture dishes. Quadruplicate samples of asynchronously growing mammary cells were treated for 72 hours, changing the media every 24 hours, with addition of either a vehicle control (1 μl DMSO/ml medium) or varying concentrations of I3C, tryptophol, I3C derivative. The cells were pulsed for three hours with 3 μCi [3H]thymidine (84 Ci/mmol), washed three times with ice cold 10% trichloroacetic acid, and lysed with 500 μl 0.3N NaOH. Aliquotes of lysates (250 μl) were transferred into plastic scintillation vials. To each vial 4 ml of Scintiverse scintillation fluid (Fisher Scientific) was added and each sample vortexed for 3–5 seconds. Radioactive thymidine incorporation was quantified using a Beckman LS 1801 scintillation counter. Results of quadruplicate experiments were averaged and normalized per derivative, and expressed as percent maximum thymidine incorporation per well.
2.6 Flow Cytometric Analyses of DNA Content
Breast cancer cells were plated onto Corning 100 mm tissue culture dishes at approximately 20% starting confluency. Cells were treated with varying concentrations of I3C, tryptophol, N-methoxy I3C, N-ethoxy I3C, N-propoxy I3C, N-butoxy I3C, or the vehicle control (DMSO) at the indicated concentrations. In one set of experiments, cells were treated with I3C, 3-methoxymethylindole-I3C, 3-ethoxymethylindole-I3C or N-methyl-I3C. Cells were incubated for 72 hours, changing the media every 24 hours, and hypotonically lysed in 1 ml of DNA staining solution (0.5 mg/ml propidium iodide, 0.1% sodium citrate, 0.05% Triton X-100). Lysates were filtered using 60μ Nitrex flow mesh (Sefar America Inc., Kansas City, MO) to remove a majority of impurities. Propidium iodide stained nuclei were picked up using a PL-2 detector with a 575 nm band pass filter on a Beckman-Coulter FACS analyzer with laser output adjusted to deliver 15 mW at 488 nm. Nuclei (10,000) were analyzed from each sample at a rate of 300–500 cells/second. The percentages of cells within the G1, S, and G2/M phases of the cell cycle were determined by analysis with the Multicycle computer program provided by Phoenix Flow Systems in the Cancer Research Laboratory Microchemical Facility of the University of California, Berkeley.
2.7 Western Blot Analysis
After the indicated treatments, cells were harvested in RIPA buffer (150 mM NaCl, 0.5% deoxycholate, 0.1% NP-40, 0.1% SDS, 50 mM Tris) containing protease and phosphatase inhibitors (50 μg/ml PMSF, 10 μg/ml aprotinin, 5 μg/ml leupeptin, 0.1 μg/ml NaF, 1mM dithiothreitol, 0.1 mM sodium orthovanadate). Equal amounts of total cellular protein were mixed with loading buffer (25% glycerol, 0.075% SDS, 1.25 ml 14.4 M 2-mercaptoethanol, 10% bromophenol blue, 3.13% stacking gel buffer) and fractionated by electrophoresis on 10% polyacrylamide/0.1% SDS gels. Rainbow marker (Amersham Life Sciences, Arlington Heights, IL) was used as the molecular weight standard. Proteins were electrically transferred to nitrocellulose membranes (Micron Separations, Inc., Westborough, MA) and blocked overnight at 4 °C with western wash buffer/5% non-fat dry milk (10 mM Tris HCl pH 8.0, 150 mM NaCl, 0.05% Tween-20). Blots were subsequently incubated for 1 hour at room temperature for rabbit anti-CDK2 and CDK6 antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA Cat. #sc-748 and #sc-177). Working concentration for all antibodies was 1 μg/ml western wash buffer. Immunoreactive proteins were detected after 1 hour incubation at room temperature with horseradish peroxidase-conjugated secondary goat anti-rabbit (BioRad, Hercules, CA Cat. #170–6515) diluted to 3 × 10−4 in western wash buffer/1% non-fat dry milk. Blots were treated with ECL reagents (NEN Life Science Products) and all proteins were detected by autoradiography. Equal protein loading was ascertained by Ponceau-S staining of blotted membranes.
2.8 Assay of immunoprecipitated CDK-2 enzymatic activity
Breast cancer cells were cultured for 72 hours in growth medium with either individual indoles (200 μM I3C, 200 μM tryptophol, 10 μM N-methoxy I3C, 5 μM N-ethoxy I3C, 1 μM N-propoxy I3C, 0.5 μM N-butoxy I3C), or with the DMSO vehicle control. Cells were rinsed with 5 ml PBS and harvested in PBS then stored as dry pellets at −70°C. For the immunoprecipitation, cells were lysed for 15 minutes in immunoprecipitation (IP) buffer (50 mM Tris HCl pH 7.4, 200 mM NaCl, 0.1% Triton X-100) containing protease and phosphatase inhibitors (50 μg/ml PMSF, 10 μg/ml aprotinin, 5 μg/ml leupeptin, 0.1 μg/ml NaF, 10 μg/ml β-glycerophosphate, and 0.1 mM sodium orthovanadate). Samples were diluted to 500 μg protein in 1 ml IP buffer. Samples were pre-cleared for 2 hours at 4°C with 40 μl of a 1:1 slurry of protein-A sepharose beads (Pharmacia Biotech, Sweden) in IP buffer and 1 μg rabbit IgG. After a brief centrifugation to remove precleared beads, 0.5 μg anti-CDK2, -CDK4, or -CDK6 antibody was added to each sample and incubated on a rocking platform at 4 °C for 2 hours. Next, 10 μl protein-A sepharose beads were added to each sample and the slurries incubated on the rocking platform at 4 °C for 30 minutes. The beads were then washed five times with IP buffer and twice with kinase buffer (50 mM HEPES, 10 mM MgCl2, 5 mM MnCl2, 0.1 μg/ml NaF, 10 μg/ml β-glycerophosphate, and 0.1 mM sodium orthovanadate). Half of the immunoprecipitated sample was checked by western blot analysis to confirm the immunoprecipitation and compare protein loading.
For the kinase assay, the remaining half of the immunoprecipitated sample was resuspended in 25 μl kinase buffer containing 20 mM ATP, 5 mM DTT, 0.21 μg Rb carboxy-terminal domain protein substrate (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and 10 μCi [γ33P] dATP (3000 Ci/mmol). Reactions were incubated for 15 minutes at 30 °C and stopped by adding an equal volume of 2X loading buffer (10% glycerol, 5% β-mercaptoethanol, 3% SDS, 6.25 mM Tris HCl pH 6.8, and bromophenol blue). Reaction products were boiled for 10 minutes and then electrophoretically fractionated in SDS-10% polyacrylamide gels. Gels were stained with Coomassie blue to monitor loading and de-stained overnight with 3% glycerol. Subsequently, gels were dried and quantitated by Phosphor-Imager analysis (Molecular Dynamics, Sunnyvale, CA) and then visualized by autoradiography.
2.9 Luciferase reporter assay for CDK6 promoter activity
For luciferase assays, MCF-7 cells stably transfected with a −920 bp CDK6 promoter-luciferase reporter plasmid [29] were harvested by washing twice in PBS and lysed in 100–200 μl of 1X Promega reporter lysis buffer. Cell lysate (20 μl) was added to 12 × 75 mm cuvettes (Analytical Luminescence Laboratory, San Diego, CA) and subsequently loaded into a luminometer (Monolight 2010, Analytical Luminescence Laboratory). Luciferase substrate buffer (100 μl) (20 mM Tricine, 1.07 mM (MgCO3)4 Mg (OH)2 5H20, 2.67 mM MgSO4, 0.1 mM EDTA, 33.3 mM DTT, 270 μM coenzyme A, 470 μM D-luciferin sodium salt, 530 μM ATP disodium salt, pH 7.8) was injected automatically into each sample and luminescence was measured in relative light units. The luciferase specific activity was expressed as an average of relative light units produced per Pg of protein present in the corresponding cell lysates as measured by Bradford assay (Biorad, Hercules, CA).
3. Results
3.1 Synthesis of N-alkoxy derivatives of Indole-3-carbinol
As a starting point to understand the structure features of indole-3-carbinol (I3C) that are responsible for its potent anti-proliferative effects on human breast cancer cells, we tested a series on derivatives with substitutions on the N-1 and C-3 positions. As shown in Figure 1, synthetic N-1 substitutions included a series of electron withdrawing alkoxy moieties (N-methoxy, N-ethoxy, N-propoxy and N-butoxy derivatives), and the electron donating N-methyl substituent of the natural indole product N-methyl I3C). Substitutions at the C-3 position included components that maintained the reactive benzylic oxygen moiety (methyl and ethyl I3C ethers), and the tryptophol (See Fig. 1) and melatonine hydroxy-indole derivatives in which this reactive center was absent. Substitution of the indole nitrogen deactivates the system to dehydration with indolenine formation, and lipophilicy is increased with increasing length of the alkoxy side chain. Ether formation increases lipophilicity but should not affect reactivity of the indole nucleus.
Fig. 1.
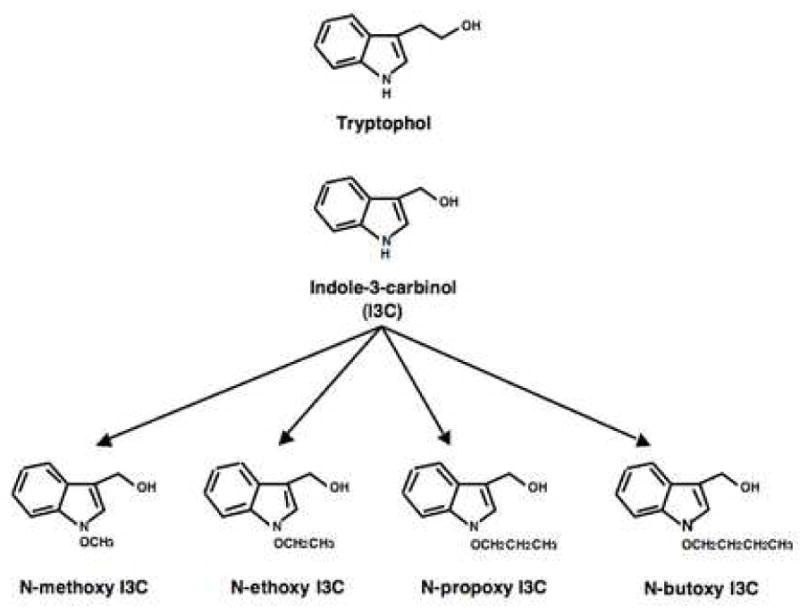
Structures of N-alkoxy I3C derivatives and tryptophol. The I3C parent compound contains a hydrogen at the N 1 position. Four N-alkoxy I3C derivatives were synthesized containing the N-methoxy, N-ethoxy, N-propoxy, and N-butoxy substituents at the N 1 position (denoted by an X in the indole ring). Tryptophol contains a ethanol substituent off the C-3 position in the indole ring instead of a methanol group.
3.2 Efficacy of the anti-proliferative responses of the N-alkoxy substituted I3C derivatives
To determine whether the N-alkoxy substituents alter the efficacy of the I3C anti-proliferative response, human MCF-7 breast cancer cells were treated for 72 hours with various concentrations of I3C, N-methoxy I3C, N-ethoxy I3C, N-propoxy I3C, or N-butoxy I3C, with DMSO vehicle control, as well as with the control tryptophol compound. In each set of cultures, the effects on cell growth were monitored in comparison to the vehicle control treated cells as a function of DNA synthesis (pulse-labeled with [3H]thymidine for 3 hours), and by the increase in cells arrested in the G1 phase of the cell cycle (monitored by flow cytometry of propidium iodide stained nuclei). As shown in Fig 2 (upper panel), the dose response effects of these N-substituted I3C derivatives on DNA synthesis displayed a significant left shift in the curve compared to I3C. For example, half maximal inhibition of [3H]thymidine incorporaton was 94 μM for I3C, 3.5 μM for N-methoxy-I3C, 1.19 μM for N-ethoxy I3C, 0.36 μM for N-propoxy I3C and 0.21 μM for N-butoxy I3C (Figure 4 insert). Tryptophol had no effect on the growth of MCF7 cells compared to the vehicle (DMSO) treated cells. Substitution of a methyl group at the nitrogen position caused a small, but reproducible, left shift in the growth inhibition dose response compared to the I3C parent compound (data not shown). The concentrations of the N-alkoxy derivatives that maximally inhibited DNA synthesis without any cytotoxic effects were 200 μM I3C, 10 μM N-methoxy I3C, 5 μM N-ethoxy I3C, 1 μM N-propoxy I3C, and 0.5 μM N-butoxy I3C (Fig. 2, upper panel). These maximal concentrations were used in some of the subsequent experiments described in this report.
Fig. 2.
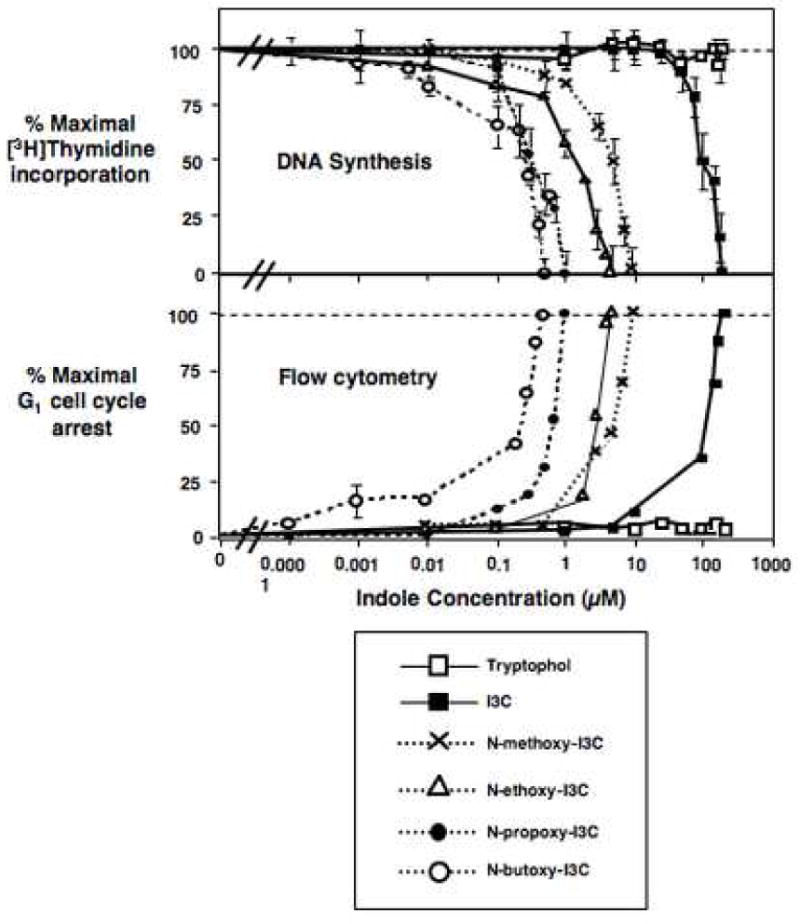
Indole dose response effects on DNA synthesis and G1 cell cycle arrest of MCF-7 breast cancer cells. Cells were treated with the indicated concentrations of I3C, N-alkoxy I3C derivatives, tryptophol, or with the DMSO vehicle control for 48 hrs. Upper panel: The level of DNA synthesis was determined as a function of acid precipitable radioactivity from cells pulse labeled for 3 hrs with [3H]thymidine. The % maximal [3H]thymidine incorporation determine by comparing the level of DNA synthesis observed in indole treated cells to that of DMSO vehicle control treated cells (which corresponds to the 100% value). Lower panel: Nuclei in cell extracts were stained with propidium iodide, and the number of cells arrested with a G1 phase DNA content determined by flow cytometry using a Beckman-Coulter Fluorescence Activated Cell Sorter as described in the Methods and Materials section. The Percent maximal G1 cell cycle arrest was calculated as the number of nuclei with a G1 DNA content compared to cells maximally arrested in G1. Cells treated with the DMSO vehicle control represents the 0% level of G1 arrested cells.
Fig. 4.
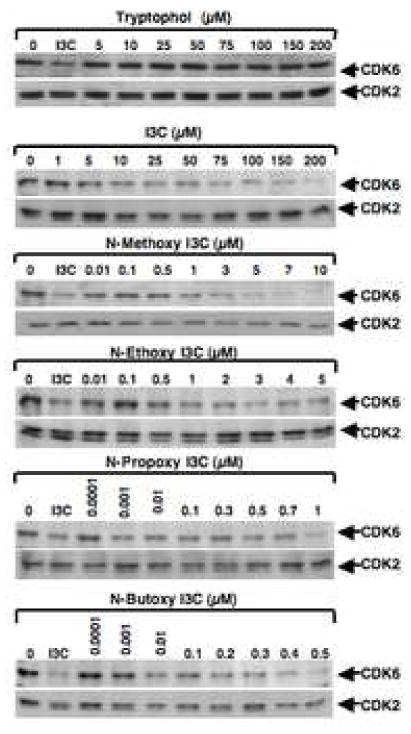
Western blot analysis of the effects of the N-alkoxy I3C derivatives on CDK6 and CDK2 protein expression. MCF-7 cells were treated with 200 μM I3C, 10 μM N-methoxy I3C, 5 μM N-ethoxy I3C, 1 μM N-propoxy I3C, and 0.5 μM N-butoxy I3C, or with 100 μM tryptophol, and harvested at the indicated time points. Total cell extracts were electrophoretically fractionate in SDS polyacrylamide gels, and western blots were probed with specific antibodies to either CDK6 or CDK2. Equal gel loading was confirmed using Ponceau S staining of the western blot membrane.
We previously established that the I3C parent compound induced a G1 cell cycle arrest of human MCF-7 breast cancer cells [27, 30]. To quantify the efficacy of the cell cycle arrest of the N-alkoxy I3C derivatives in comparison to I3C, tryptophol, and the DMSO vehicle control, MCF-7 breast cancer cells were treated with varying concentrations of each derivative for 72 hours, and the nuclear DNA content analyzed by flow cytometry analysis of propidium iodide stained nuclei. As also shown in figure 2, each of N-alkoxy derivatives as well as the I3C parent compound induced a significant dose dependent enhancement in the number of cells displaying a G1 phase level of DNA content. The efficacy of the G1 cell cycle arrest mediated by I3C and each of the N-alkoxy I3C derivatives was consistent with the [3H]thymidine incorporation results. The maximal increase in number G1 phase cells for each of N-alkoxy derivatives was similar to that of I3C but at their respective lower concentrations, suggesting that the N-alkoxy substitutients did not qualitatively alter the I3C specific cell cycle arrest. Cells treated with the DMSO vehicle control or with tryptophol, grew as an asynchronous population in all phases of the cell cycle.
The growth inhibitory responses of each N-alkoxy I3C derivative and I3C were completely reversible (data not shown), suggesting that at the concentrations utilized, these indoles compounds do not affect cell viability. Interestingly, we observed a significant increase in the efficacy of the N-alkoxy I3C derivatives to inhibit DNA synthesis and induce a G1 cell cycle arrest that directly correlated with the carbon length of the substituent. Compared to I3C, the half maximal growth arrest responses occurred at 23-fold lower indole concentration for N-methoxy-I3C, 50-fold lower concentration for N-ethoxy-I3C, 217-fold lower concentration for N-propoxy-I3C, and 470-fold lower concentration for N-butoxy-I3C (Fig. 3). The half-maximal effects on the G1 cell cycle arrest for compounds were 138 μM for I3C, 6.5 μM for N-methoxy-I3C, 3.5 μM for N-ethoxy I3C, 0.71 μM for N-propoxy I3C, and 0.29 μM for N-butoxy I3C (Figure 3 insert).
Fig. 3.
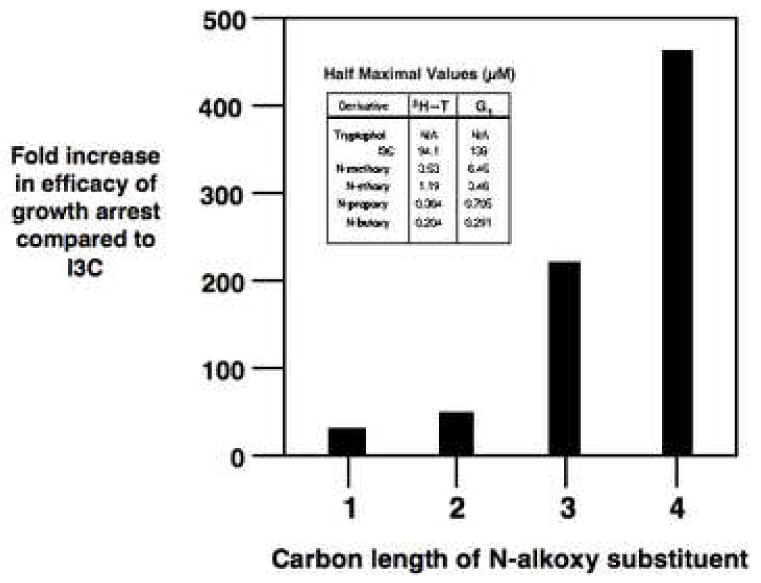
Correlation of the efficacy of the anti-proliferative effects of the N-alkoxy I3C derivatives with the carbon length of the N-alkoxy substituents. The insert shows the indole concentrations for I3C and its N-alkoxy derivatives that induce the half maximal inhibition of DNA synthesis and half maximal G1 cell cycle arrest. The fold efficacy was calculated as the ratio of the half maximal response for each N-alkoxy derivative to the half maximal response observed with the I3C parent compound. The graph correlates the fold efficacy of the growth arrest with the carbon length of the N-alkoxy derivatives. These data were based on three independent DNA synthesis experiments and on three independent determinations of half maximal G1 cell cycle arrest. The approximate standard deviation is −/+ 5% of the final calculated fold efficacies of growth arrest for each N-alkoxy I3C derivative.
To demonstrate that the increased efficacy of the N-alkoxy I3C derivatives occurs in breast cancer cells other than the estrogen responsive MCF-7 cells, the anti-proliferative effects of I3C and the N-alkoxy I3C derivatives were assessed in MDA-MB-231 cells, which do not express estrogen receptor-alpha and are not estrogen responsive [27]. We have previously shown that I3C, but not the anti-estrogen tamoxifen, induces a G1 cell cycle arrest of MDA-MB-231 cells [27]. Therefore, cultures of MDA-MB-231 cells were treated with I3C or each of the four N-alkoxy I3C derivatives at concentrations slightly higher than their half-maximal response (200 μM I3C, 10 μM N-methoxy I3C, 5 μM N-ethoxy I3C, 1 μM N-propoxy I3C, and 0.5 μM N-butoxy I3C). Parallel cell cultures were treated with either the DMSO vehicle control or 100 PM tryptophol for 72 hours. The nuclear DNA content was analyzed by flow cytometric analysis of propidium iodide stained nuclei, and the percentage of G1 phase cells used to monitor the anti-proliferative effects of the indole compounds. As shown in Table 1, MDA-MB-231 cells treated with the DMSO vehicle control or with tryptophol displayed approximately 54%–57% cells in the G1 phase, which is indicative of their proliferating states. The total cell numbers in culture continue to increase under these conditions (data not shown). In contrast, treatment with I3C or the N-alkoxy I3C derivatives caused a G1 cell cycle arrest with the total percentage of cells in the G1 phase induced to approximately 68%–71% depending on the tested indole. Under these conditions, I3C and the N-alkoxy I3C derivatives caused a significant drop in S phase cells compared to the vehicle control or tryptophol-treated cells, and the total number cells in the culture did not significantly increase after the first 24 hours of indole treatment (data not shown). Taken together, our results show that N-alkoxy I3C derivatives maintain the I3C-specific property of inhibiting the growth of breast cancer cells that are not estrogen responsive and also maintain the their increased efficacy of response.
Table 1.
Cell cycle effects of I3C and N-alkoxy derivatives in MDA-MB-231 human breast cancer cells.
| Indole treatment
|
Percentage G1 phase cells
|
|---|---|
| None (vehicle control) | 57.4% |
| Indole-3-carbinol (200 μM) | 70.8% |
| N-methoxy-I3C (10 μM) | 68.0% |
| N-ethoxy-I3C (5 μM) | 71.5% |
| N-propoxy-I3C (1 μM) | 71.1% |
| N-butoxy-I3C (0.5 μM) | 67.1% |
| Tryptophol (100 μM) | 54.6% |
MDA.MB-231 human breast cancer cells, which are not responsive to estrogen because of a lack of estrogen receptor-alpha expression, were treated with each of the indole compounds listed above, or with only the DMSO vehicle control (none) for 72 hours. Harvested cells were prepared for propidium iodide staining of the nuclei and flow cytometry as described in the methods section. The number of cells with a G1 phase DNA content after the treatment was determined as an average of at least three independent experiments with the standard deviations ranging between ± 2–5%. Under these conditions, the vehicle control treated and tryptophol-treated cells are proliferating, whereas, cells treated with I3C or N-alkoxy I3C derivatives are growth arrested.
3.3 Anti-proliferative responses of I3C methyl and ethyl ethers
As a complementary approach, the anti-proliferative properties of several synthetic or natural derivatives of I3C with altered substituents at the C-3 position were examined. MCF-7 cells were treated with 100 μM of I3C or with the indicated I3C derivatives, as well as the DMSO control for 72 hours. The harvested cells were hyptonically lysed in a propidium iodide solution to stain the nuclear DNA, and effects on the cell cycle monitored by flow cytometry analysis of the nuclear DNA content. As shown in Table 2, treatment with 3-methoxymethylindole (MMI) or 3-ethoxymethylindole (EMI) increased the number of breast cancer cells with a G1 phase DNA content to approximately the same extent as I3C (Table 2). Thus, I3C methyl or ethyl ethers did not alter the cell cycle arrest response compared to I3C. Both tryptophol and melatonin, failed to induce the cell cycle arrest (Table 2).
Table 2.
Cell cycle effects of I3C derivatives with altered substituents at the C-3 ring position of I3C.
| Indole treatment
|
Percentage maximal G1 arrested cells
|
|---|---|
| None (vehicle control) | 0% |
| Indole-3-carbinol | 100% |
| 3-methoxymethylindole | 93% |
| 3-ethoxymethylindole | 98% |
| N,O-diacetyl indole-3-carbinol | 41% |
| Melatonin | 3% |
| Tryptophol | 1% |
MCF-7 breast cancer cells were treated with 100 μM of each listed indole compound, or with only the DMSO vehicle control (none) for 72 hours. Harvested cells were prepared for propidium iodide staining of the nuclei and flow cytometry as described in the methods section. The increase in number of growth arrested cells with a G1 phase DNA content in indole-3-carbinol treated cells relative to vehicle control treated cells is the maximal cell cycle arrest. The percent maximal G1 arrested cells was calculated as the number of nuclei with a G1 DNA content compared to cells maximally arrested in G1. Cells treated with I3C represents the 100% level of G1 arrested cells, and the DMSO vehicle control represents the 0% level of G1 arrested cells. The results are an average at least three independent experiments the standard deviation ranging between ± 2–5% with each indole.
3.4 N-alkoxy I3C derivatives selectively down-regulate CDK6 gene expression
A characteristic feature of the I3C anti-proliferative response is the inhibition of cyclin dependent kinase-6 (CDK6) gene expression [10, 27, 29], which is a key G1 acting cell cycle component. MCF-7 cells were treated with various concentrations of the N-alkoxy I3C derivatives, I3C, tryptophol, or the DMSO vehicle for 72 hours, total cell extracts were fractionated in SDS polyacrylamide gels, and the corresponding western blots were probed for either CDK6 or, as a control, for CDK2 protein production. As shown in Figure 4, I3C and each of the N-alkoxy derivatives reduced CDK6 protein levels to approximately the same extent and with approximately the same kinetics of down-regulation. RT-PCR analysis showed a similar effect on CDK6 transcripts (data not shown). Consistent with their anti-proliferative responses, the efficacy of the CDK6 down-regulation responses increased in relationship to the size of the alkoxy side chain. The effectiveness of the N-alkoxy derivatives can therefore be ordered based on the reduction in concentration needed for their maximal down-regulation of CDK6 production as N-butoxy I3C ≥ N-propoxy I3C, >N-ethoxy I3C, ≥ N-methoxy I3C > I3C. In comparison, CDK6 protein production was unresponsive to tryptophol treatment (Fig. 4). Importantly, as previously reported for I3C [10, 27], no effect was observed for any of the N-substituted derivatives on the protein levels of the two other G1-acting cyclin dependent kinases, CDK2 (Fig. 4) or CDK4 (data not shown), or on either cyclin D1 or cyclin E (data not shown). This lack of effect on the G1-acting CDK2 or CDK4 expression demonstrates the selectivity of the N-alkoxy I3C response for the G1-acting CDK6.
3.5 N-Alkoxy-I3C derivatives down-regulate CDK6 promoter activity
We have developed a sensitive cellular assay to examine the regulation of CDK6 promoter activity that responds to the inhibitory effects of I3C [29], and which can be used to assess the effects of I3C derivatives. A 1 kb fragment of the CDK6 gene promoter was cloned, linked to the luciferase reporter gene, and stably transfected into MCF-7 breast cancer cells. The luciferase protein has a short 4 hour half life, and therefore is highly sensitive to inhibitory effects and/or rapid changes in CDK6 promoter activity [29]. The transfected cells were treated with 200 μM I3C, 10 μM N-methoxyI3C, 5 μM N-ethoxy I3C, 1 μM N-propoxy I3C, 0.5 μM N-butoxy I3C, 100 μM tryptophol, or with the DMSO vehicle control for 24 hours. As shown in figure 5, each of the N-alkoxy I3C derivatives down-regulated CDK6 promoter activity to approximately the extent as I3C but at the corresponding lower concentrations. Tryptophol-treated cells were indistinguishable from the DMSO treated vehicle controls. In the presence of actinomycin D, the level of CDK6-luciferase reporter assay was approximately 75 RLU/μg protein (data not shown), which represents the background activity of this plasmid in the absence of de novo RNA synthesis, and suggests that I3C and its N-alkoxy derivatives nearly quantitatively inhibit CDK6 promoter activity to same extent at the corresponding concentrations. These results indicate that similar to the I3C parent compound, the N-alkoxy derivatives of I3C suppress CDK6 gene expression by reducing CDK6 promoter activity, but at concentrations lower than I3C.
Fig. 5.
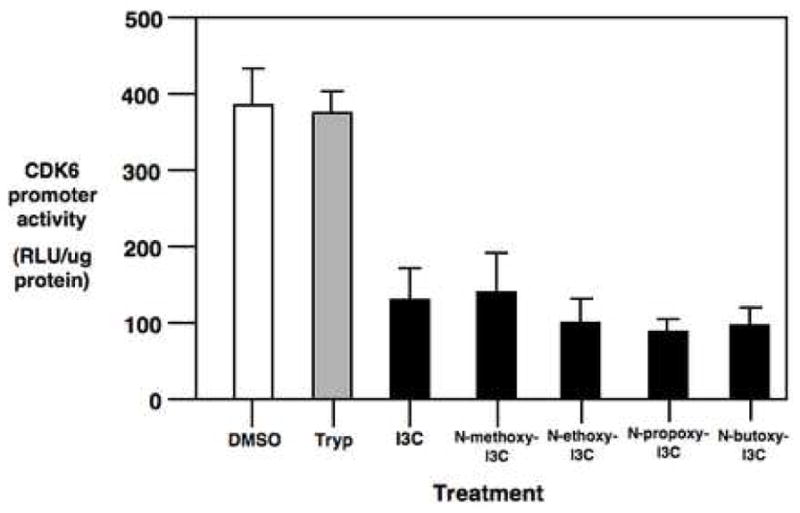
Down regulation of CDK6 promoter activity by I3C and N-alkoxy I3C derivatives. MCF-7 cells stably transfected with the −920 bp CDK6-luciferase reporter plasmid were treated with 200 μM I3C, 100 μM tryptophol, 10 μM N-methoxy I3C, 5 μM N-ethoxy I3C, 1 μM N-propoxy I3C, or 0.5 μM N-butoxy I3C, or with the vehicle DMSO control for 48 hours and assayed for luciferase activity as described in the Methods and Materials. The relative light units per μg protein are representative of three independent experiments of triplicate samples, and the error bars indicate the standard deviation.
3.6 N-alkoxy I3C derivatives inhibit CDK2 protein kinase activity
We have established that the inhibition of CDK2 specific enzymatic activity without changes in the level of CDK2 protein is another cellular marker for the cell cycle effects of I3C [10, 30, 34]. To determine the efficacy of this response for the N-alkoxyl substituents, MCF-7 breast cancer cells were treated with I3C, individual N-alkoxy I3C derivatives, tryptophol, or DMSO vehicle control for 48 hours, and CDK2 immunoprecipitated from isolated nuclear extracts using conditions that maintain the CDK-cyclin interaction [30, 34]. One set of control immunoprecipitations utilized non-immune antibodies (No IP). Each immunoprecipitated sample was then incubated with [gamma-32P]ATP in the presence of the same amount of glutathione S-transferase-retinoblastoma (GST-Rb) fusion protein substrate, and the level of [32P]GST-Rb monitored electrophoretically in SDS polyacrylamide gels. A western blot of the same set of reactions was monitored for the level of immunoprecipitated CDK2 protein. As shown in figure 6, each of the N-alkoxy derivatives of I3C inhibited CDK2 specific enzymatic activity to approximately the same extent as I3C, but at reduced concentrations. Cells treated either with the DMSO vehicle control or tryptophol maintained a high level of enzymatically active CDK2, as well as express the faster migrating form of CDK2 protein, which is the enzymatically active form of CDK2. As expected, immunoprecipitations carried out with non-immune antibodies did not contain any CDK2 protein nor display Rb phosphorylating activity.
Fig. 6.
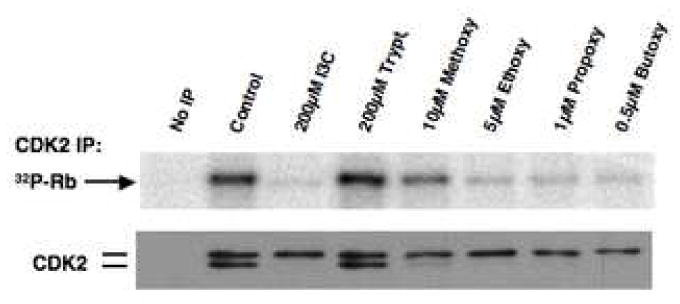
Effects of the N-alkoxy I3C derivatives on CDK2 specific kinase activity. MCF-7 cells were treated with 200 μM I3C, 10 μM N-methoxy I3C, 5 μM N-ethoxy I3C, 1 μM N-propoxy I3C, 0.5 μM N-butoxy I3C, 100 μM tryptophol, or with the DMSO vehicle control for 48 hr, and CDK2 was immunoprecipitated from total cell extracts and immunoprecipated with anti-CDK2 antibodies, one control sample did not receive any CDK2 antibodies (No IP). The immunoprecipitated CDK2 protein was assayed for in vitro kinase activity using 0.21 μg Rb carboxy-terminal domain protein as a substrate in each reaction mixture. The kinase reactions were electrophoretically fractionated and the level of [32P]Rb was visualized by autoradiography. The efficiency of immunoprecipitation for each sample was confirmed by western blot analysis using antibodies specific to CDK2.
4. Discussion
The consumption of Brassica vegetables is directly associated with decreased risk of various cancers in human subjects and reduced tumor incidence in experimental animals [6, 11]. We previously established that I3C, which is one the major active compounds in Brassica vegetables, induces a G1 cell cycle arrest of human breast cancer cell lines through a cellular pathway that leads to the down-regulation of CDK6 transcription and inhibition of CDK2 kinase activity [10, 27, 29, 30]. When added to cultured breast cancer cells, I3C is surprisingly stable, and a significant fraction of I3C is converted into its self-condensation product DIM [28], and a combination of I3C and the DIM that is produced in cells has a strong anti-proliferative effect. DIM inhibits the growth of reproductive cancer cells through a pathway that is distinct from I3C [10], and an analysis of the cellular response to indole derivatives must take into account the bifurcation of the anti-proliferative pathways. Most notably, I3C, but not DIM, inhibits CDK6 gene expression by down-regulating its promoter activity, and inhibits CDK2 enzymatic specific activity by altering the cyclin E products associated with the CDK2 protein complex without any detectable changes in the level of CDK2 protein [10, 27, 29, 30, 34]. In contrast, DIM induces expression of p21 CDK inhibitor that leads to an inhibition of CDK2 activity [33]. Thus, the overall profile of the I3C-specific effects on the G1-acting CDKs provides a bioassay for testing the effects of more potent synthetic I3C derivatives.
In order to initially elucidate the structure-activity relationships associated with anti-proliferative specific response of the I3C parent compound, we examined the effects of altered reactivities of the indole nucleus on biological activity. The I3C-specific cell cycle effects of a series of N-alkoxy derivatives of I3C that contain chains of one to four carbons in length were characterized. Dose response experiments examining the inhibition of DNA synthesis and G1 block in cell cycle progression demonstrated that the efficacy of the I3C-mediated anti-proliferative effects on human breast cancer cells is significantly enhanced by N-alkoxy substitution and by the presence of increasing carbon lengths of the N-alkoxy substituents. Half maximal growth arrest observed in DNA synthesis and cell cycle analyses with the N-methoxy-I3C occurred at approximately 23-fold lower concentrations than I3C. The corresponding half maximal responses of N-ethoxy-I3C, N-propoxy-I3C and N-butoxy I3C were approximately 50-fold, 217-fold and 470-fold lower than that of I3C. This increase in efficacy of the N-alkoxy I3C derivatives was observed in estrogen responsive MCF-7 human breast cancer cells as well as in estrogen nonresponsive MDA-MB-231 human breast cancer cells demonstrating that the effect is not a cell line specific phenomena. Importantly, each of the N-alkoxy substituted compounds functioned similarly to I3C, but at lower concentrations, in that each indole reduced CDK6 gene expression, down-regulated CDK6 promoter activity and inhibited CDK2 specific enzymatic activity. Importantly, the transcriptional responses mediated by the I3C parent compound [10, 27, 29] are maintained in this series of N-alkoxy derivatives. Our work has also established a direct link between I3C signaling and the control of CDK6 gene expression by targeting Sp1 transcription factor binding to a composite Ets-Sp1 DNA site within the I3C regulated region of the CDK6 promoter [29]. It is likely that the down-regulation of CDK6 promoter activity in stably transfected MCF-7 cells by the N-alkoxy derivatives of I3C is due to a similar mechanism.
A trend was observed among the four tested N-substituted I3C derivatives in which the concentration of each derivative needed to inhibit breast cancer cell growth decreased in direct correlation to the number of carbons in the N-alkoxy substituent. Chemical reactivity to indolenine formation of the N-alkoxy compounds should be similar, whereas, the lipid solubilities of these compounds increase with chain length. Thus, the increasing activities in this series may be due to increasing cellular concentrations. I3C can be converted into one of several dimeric or trimeric forms under conditions similar to the intracellular environment. Thus, another possibility is that the longer chain lengths may slow the intracellular oligomerization or metabolism of the N-alkoxy derivatives and thereby potentially maintain the higher concentrations of active compounds. The N-alkoxy substitution and the longer carbon chain lengths may also stabilize the interactions of the derivatives with putative intracellular I3C target protein(s) needed for the cell cycle arrest. The direct I3C target proteins are unknown and when isolated would allow a direct test of whether the increased efficacies of the N-alkoxy derivatives are due to higher binding affinity or cellular stability.
The C-3 hydroxymethyl group of I3C is highly reactive, and therefore the cell cycle effects of I3C derivatives with altered substituents at this position were examined. Both tryptophol and melatonin, in which the C-3 hydroxyl is lost, failed to induce the cell cycle arrest. These results suggest that the indole-3-carbinol mediated cell cycle arrest requires the presence of the hydroxymethyl substituent at the C-3 position. The addition of methyl or ethyl groups to the C-3 oxygen would be expected to increase lipophilicity but not retard indolenine formation under aqueous conditions. Consistent with this expectation, treatment of breast cancer cells with I3C derivatives containing 3-methoxymethylindole or 3-ethoxymethylindole substituents did not alter the indole cell cycle arrest response compared to I3C. Taken together, our results suggest that the addition of electron withdrawing substituents on the indole nitrogen, as exemplified by the N-alkoxy derivatives, increase the efficacy of the I3C cell cycle arrest. Interestingly, introduction of an electron donating N-methyl group on I3C had little effect on the cytostatic activity compared to I3C. This suggests that the electron withdrawing effects of the N-alkoxy substituents or possibly altered metabolism compared to the N-methyl derivative, are important to their biological activities.
Our results implicate N-alkoxy derivatives of I3C as a particularly promising class of experimental therapeutics for breast cancer because of their increased potency compared to I3C and ability to maintain the I3C-specific cell cycle response. One previous study analyzed a tetrameric I3C derivative that can inhibit CDK6 expression [39], although this derivative is only 5-fold more effective than I3C in inhibiting cell growth. In this regard, our results show that the N-ethoxy-I3C derivative is 10-fold more effective and the N-butyl derivative is approximately 90-fold more effective than this tetrameric derivative. This difference may be due to the bioactivity of adding electron donating N-alkoxy groups with increasing hydrophobicity onto the I3C structure. One other study developed a synthetic derivative of I3C, SR13668, that resembles the structure of DIM, the self condensation product of I3C [40]. The investigators did not characterize I3C-specific responses and showed that SR13668 inhibits growth factor stimulated Akt enzymatic activity, which is a DIM response in cultured reproductive cancer cells [41]. Thus, our work as established the first systematic structure-activity relationships for I3C-specific cellular effects in human breast cancer cells.
The development of I3C derivatives as targeted chemotherapeutic agents to control human breast cancer depends upon precise information on the structure activity relationships for the anti-proliferative functions of these indoles in vitro in a cellular context, as well as eventually in vivo in animal systems. Given the limited studies about the role of indoles derived from vegetables in the treatment of breast cancer, the information from our study provides the necessary first experimental steps that are crucial for the development of novel chemotherapeutic, or perhaps chemopreventative, strategies that effectively utilize potent derivatives of I3C alone or in combination with known anti-breast cancer treatments. We are expanding our studies to understand the in vivo effects of the N-alkoxy I3C derivatives, and to generate additional synthetic derivatives of I3C with an increased potency of their anti-proliferative response in cultured breast cancer cells and in vivo in breast cancer cell-derived tumors.
Acknowledgments
This study was supported by an award from the Department of Defense Army Breast Cancer Research Program, and from the California Breast Cancer Research Program during the early stages of the work. This study was also supported by NIH Public Service Grant CA102360 award from the National Cancer Institute. R.S. was supported by NIEHS training grant ES07075–23.
Abbreviations
- I3C
indole-3-carbinol
- DIM 3
3′-diindolylmethane
- CDK
cyclin-dependent kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Muss HB. Endocrine therapy for advanced breast cancer: a review. Breast Cancer Res Treat. 1992;21:15–26. doi: 10.1007/BF01811960. [DOI] [PubMed] [Google Scholar]
- 2.Pennisi E. Drug’s link to genes reveals estrogen’s many sides. Science. 1996;273:1171. doi: 10.1126/science.273.5279.1171. [DOI] [PubMed] [Google Scholar]
- 3.Forbes JF. The control of breast cancer: the role of tamoxifen. Semin Oncol. 1997;24(Suppl 1):S1–5–S1–19. [PubMed] [Google Scholar]
- 4.Jordan VC. SERMs: meeting the promise of multifunctional medicines. J Natl Cancer Inst. 2007;99:350–56. doi: 10.1093/jnci/djk062. [DOI] [PubMed] [Google Scholar]
- 5.Birt DF, Pelling JC, Nair S, Lepley D. Diet intervention for modifying cancer risk. Prog Clin Biol Res. 1996;395:223–34. [PubMed] [Google Scholar]
- 6.Lopez-Otin C, Diamandis EP. Breast and prostate cancer: an analysis of common epidemiological, genetic, and biochemical features. Endocr Rev. 1998;19:365–96. doi: 10.1210/edrv.19.4.0337. [DOI] [PubMed] [Google Scholar]
- 7.Freudenheim JL, Marshall JR, Vena JE, Laughlin R, Brasure JR, Swanson MK, Nemoto T, Graham S. Premenopausal breast cancer risk and intake of vegetables, fruits, and related nutrients. J Natl Cancer Inst. 1996;88:340–8. doi: 10.1093/jnci/88.6.340. [DOI] [PubMed] [Google Scholar]
- 8.Wattenberg LW, Loub WD. Inhibition of polycyclic aromatic hydrocarbon-induced neoplasia by naturally occurring indoles. Cancer Res. 1978;38:1410–3. [PubMed] [Google Scholar]
- 9.Bradfield CA, Bjeldanes LF. Effect of dietary indole-3-carbinol on intestinal and hepatic monooxygenase, glutathione S-transferase and epoxide hydrolase activities in the rat. Food Chem Toxicol. 1984;22:977–82. doi: 10.1016/0278-6915(84)90147-9. [DOI] [PubMed] [Google Scholar]
- 10.Firestone GL, Bjeldanes LF. Indole-3-Carbinol (I3C) and 3-3′diindolylmethane (DIM) anti-proliferative signaling pathways control cell cycle gene transcription in human breast cancer cells by regulating promoter-Sp1 tanscription factor interactions. J Nutrition. 2003;133:2448S–55S. doi: 10.1093/jn/133.7.2448S. [DOI] [PubMed] [Google Scholar]
- 11.Aggarwal BB, Ichikawa H. Molecular targets and anti-cancer potential of indole-3-carbinol and its derivatives. Cell Cycle. 2005;4:1201–15. doi: 10.4161/cc.4.9.1993. [DOI] [PubMed] [Google Scholar]
- 12.Kim YS, Milner JA. Targets for indole-3-carbinol in cancer prevention. Journal of Nutritional Biochemistry. 2005;16:65–73. doi: 10.1016/j.jnutbio.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Morse MA, LaGreca SD, Amin SG, Chung FL. Effects of indole-3-carbinol on lung tumorigenesis and DNA methylation induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and on the metabolism and disposition of NNK in A/J mice. Cancer Res. 1990;50:2613–7. [PubMed] [Google Scholar]
- 14.Bradlow HL, Sepkovic DW, Telang NT, Osborne MP. Indole-3-carbinol. A novel approach to breast cancer prevention. Ann N Y Acad Sci. 1995;768:180–200. doi: 10.1111/j.1749-6632.1995.tb12121.x. [DOI] [PubMed] [Google Scholar]
- 15.Grubbs CJ, Steele VE, Casebolt T, Juliana MM, Eto I, Whitaker LM, Dragnev KH, Kelloff GJ, Lubet RL. Chemoprevention of chemically-induced mammary carcinogenesis by indole-3-carbinol. Anticancer Res. 1995;15:709–16. [PubMed] [Google Scholar]
- 16.Bradlow HL, Michnovicz J, Telang NT, Osborne MP. Effects of dietary indole-3-carbinol on estradiol metabolism and spontaneous mammary tumors in mice. Carcinogenesis. 1991;12:1571–4. doi: 10.1093/carcin/12.9.1571. [DOI] [PubMed] [Google Scholar]
- 17.Sharma S, Stutzman JD, Kelloff GJ, Steele VE. Screening of potential chemopreventive agents using biochemical markers of carcinogenesis. Cancer Res. 1994;54:5848–55. [PubMed] [Google Scholar]
- 18.Bradfield CA, Bjeldanes LF. Structure-activity relationships of dietary indoles: a proposed mechanism of action as modifiers of xenobiotic metabolism. J Toxicol Environ Health. 1987;21:311–23. doi: 10.1080/15287398709531021. [DOI] [PubMed] [Google Scholar]
- 19.De Kruif CA, Marsman JW, Venekamp JC, Falke HE, Noordhoek J, Blaauboer BJ, Wortelboer HM. Structure elucidation of acid reaction products of indole-3-carbinol: detection in vivo and enzyme induction in vitro. Chem Biol Interact. 1991;80:303–15. doi: 10.1016/0009-2797(91)90090-t. [DOI] [PubMed] [Google Scholar]
- 20.Grose KR, Bjeldanes LF. Oligomerization of indole-3-carbinol in aqueous acid. Chem Res Toxicol. 1992;5:188–93. doi: 10.1021/tx00026a007. [DOI] [PubMed] [Google Scholar]
- 21.Chang YC, Riby J, Chang GH, Peng BC, Firestone G, Bjeldanes LF. Cytostatic and antiestrogenic effects of 2-(indol-3-ylmethyl)-3,3′-diindolylmethane, a major in vivo product of dietary indole-3-carbinol. Biochem Pharmacol. 1999;58:825–34. doi: 10.1016/s0006-2952(99)00165-3. [DOI] [PubMed] [Google Scholar]
- 22.Riby JE, Feng C, Chang YC, Schaldach CM, Firestone GL, Bjeldanes LF. The major cyclic trimeric product of indole-3-carbinol is a strong agonist of the estrogen receptor signaling pathway. Biochemistry. 2000;39:910–8. doi: 10.1021/bi9919706. [DOI] [PubMed] [Google Scholar]
- 23.Hong C, Firestone GL, Bjeldanes LF. Bcl-2 family-mediated apoptotic effects of 3,3′-diindolylmethane (DIM) in human breast cancer cells. Biochem Pharmacol. 2002;63:1085–97. doi: 10.1016/s0006-2952(02)00856-0. [DOI] [PubMed] [Google Scholar]
- 24.Chen YH, Riby J, Srivastava P, Bartholomew J, Denison M, Bjeldanes L. Regulation of CYP1A1 by indolo[3,2-b]carbazole in murine hepatoma cells. J Biol Chem. 1995;270:22548–55. doi: 10.1074/jbc.270.38.22548. [DOI] [PubMed] [Google Scholar]
- 25.Bjeldanes LF, Kim JY, Grose KR, Bartholomew JC, Bradfield CA. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc Natl Acad Sci U S A. 1991;88:9543–7. doi: 10.1073/pnas.88.21.9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu H, Wormke M, Safe SH, Bjeldanes LF. Indolo[3,2-b]carbazole: a dietary-derived factor that exhibits both antiestrogenic and estrogenic activity. J Natl Cancer Inst. 1994;86:1758–65. doi: 10.1093/jnci/86.23.1758. [DOI] [PubMed] [Google Scholar]
- 27.Cover CM, Hsieh SJ, Tran SH, Hallden G, Kim GS, Bjeldanes LF, Firestone GL. Indole-3-carbinol inhibits the expression of cyclin-dependent kinase-6 and induces a G1 cell cycle arrest of human breast cancer cells independent of estrogen receptor signaling. J Biol Chem. 1998;273:3838–47. doi: 10.1074/jbc.273.7.3838. [DOI] [PubMed] [Google Scholar]
- 28.Staub RE, Feng C, Onisko B, Bailey GS, Firestone GL, Bjeldanes LF. Fate of indole-3-carbinol in cultured human breast tumor cells. Chem Res Toxicol. 2002;15:101–9. doi: 10.1021/tx010056m. [DOI] [PubMed] [Google Scholar]
- 29.Cram EJ, Liu BD, Bjeldanes LF, Firestone GL. Indole-3-carbinol inhibits CDK6 expression in human MCF-7 breast cancer cells by disrupting Sp1 transcription factor interactions with a composite element in the CDK6 gene promoter. J Biol Chem. 2001;276:22332–40. doi: 10.1074/jbc.M010539200. [DOI] [PubMed] [Google Scholar]
- 30.Garcia HH, Brar GA, Nguyen DH, Bjeldanes LF, Firestone GL. Indole-3-carbinol (I3C) inhibits cyclin-dependent kinase-2 function in human breast cancer cells by regulating the size distribution, associated cyclin E forms, and subcellular localization of the CDK2 protein complex. J Biol Chem. 2005;280:8756–64. doi: 10.1074/jbc.M407957200. [DOI] [PubMed] [Google Scholar]
- 31.Gong Y, Sohn H, Xue L, Firestone GL, Bjeldanes LF. 3,3′-Diindolylmethane is a novel mitochondrial H(+)-ATP synthase inhibitor that can induce p21(Cip1/Waf1) expression by induction of oxidative stress in human breast cancer cells. Cancer Res. 2006;66:4880–7. doi: 10.1158/0008-5472.CAN-05-4162. [DOI] [PubMed] [Google Scholar]
- 32.Sundar SN, Kerekatte V, Equinozio CN, Doan VB, Bjeldanes LF, Firestone GL. Indole-3-carbinol selectively uncouples expression and activity of estrogen receptor subtypes in human breast cancer cells. Mol Endocrinol. 2006;20:3070–82. doi: 10.1210/me.2005-0263. [DOI] [PubMed] [Google Scholar]
- 33.Hong C, Kim HA, Firestone GL, Bjeldanes LF. 3,3′-Diindolylmethane (DIM) induces a G1 cell cycle arrest in human breast cancer cells that is accompanied by Sp1-mediated activation of p21(WAF1/CIP1) expression. Carcinogenesis. 2002;23:1297–305. doi: 10.1093/carcin/23.8.1297. [DOI] [PubMed] [Google Scholar]
- 34.Cover CM, Hsieh SJ, Cram EJ, Hong C, Riby JE, Bjeldanes LF, Firestone GL. Indole-3-carbinol and tamoxifen cooperate to arrest the cell cycle of MCF-7 human breast cancer cells. Cancer Res. 1999;59:1244–51. [PubMed] [Google Scholar]
- 35.Stephensen PU, Bonnesen C, Schaldach C, Andersen O, Bjeldanes LF, Vang O. N-methoxyindole-3-carbinol is a more efficient inducer of cytochrome P-450 1A1 in cultured cells than indol-3-carbinol. Nutr Cancer. 2000;36:112–21. doi: 10.1207/S15327914NC3601_15. [DOI] [PubMed] [Google Scholar]
- 36.Yudina LN, Korolev AM, Reznikova MI, Preobrazhenskaya MN. Study of Neoascorbigen. Chemistry of Heterocyclic Compounds (Russ) 2000;2:178–86. [Google Scholar]
- 37.Korolev AM, Yudina LN, Rozhkov II, Lysenkova LN, Lazhko EI, Luzikov YN, Preobrazhenskaya MN. The formation of 2-hydroxy-4-hydroxymethyl-3-(indol-3-yl)-cyclopent-2-enone derivatives from ascorbigens. Carbohydr Res. 2001;330:469–77. doi: 10.1016/s0008-6215(00)00310-4. [DOI] [PubMed] [Google Scholar]
- 38.Somei M, Kawasaki S. A new and simple synthesis of N-hydroxyindole derivatives. Heterocycles. 1989;29:1251–54. [Google Scholar]
- 39.Brandi G, Paiardini M, Cervasi B, Fiorucci C, Filippone P, De Marco C, Zaffaroni N, Magnani M. A new indole-3-carbinol tetrameric derivative inhibits cyclin-dependent kinase 6 expression, and induces G1 cell cycle arrest in both estrogen-dependent and estrogen-independent breast cancer cell lines. Cancer Res. 2003;63:4028–36. [PubMed] [Google Scholar]
- 40.Chao WR, Yean D, Amin K, Green C, Jong L. Computer-Aided Rational Drug Design: A Novel Agent (SR13668) Designed to Mimic the Unique Anticancer Mechanisms of Dietary Indole-3-Carbinol to Block Akt Signaling. J Med Chem. 2007;50:3412–5. doi: 10.1021/jm070040e. [DOI] [PubMed] [Google Scholar]
- 41.Garikapaty VP, Ashok BT, Tadi K, Mittelman A, Tiwari RK. 3,3′-Diindolylmethane downregulates pro-survival pathway in hormone independent prostate cancer. Biochem Biophys Res Commun. 2006;340:718–25. doi: 10.1016/j.bbrc.2005.12.059. [DOI] [PubMed] [Google Scholar]