

Abstract
Indole-3-carbinol (I3C), a natural autolysis product of a gluccosinolate present in Brassica vegetables such as broccoli and cabbage, has anti-proliferative and anti-estrogenic activities in human breast cancer cells. A new and significantly more potent I3C analogue, 1-benzyl-I3C was synthesized, and in comparison to I3C, this novel derivative displayed an approximate 1000-fold enhanced potency in suppressing the growth of both estrogen responsive (MCF-7) and estrogen independent (MDA-MB-231) human breast cancer cells (I3C IC50 of 52 μM, and 1-benzyl-I3C IC50 of 0.05 μM). At significantly lower concentrations, 1-benzyl-I3C induced a robust G1 cell cycle arrest and elicited the key I3C-specific effects on expression and activity of G1 acting cell cycle genes including the disruption of endogenous interactions of the Sp1 transcription factor with the CDK6 promoter. Furthermore, in estrogen responsive MCF-7 cells, with enhanced potency 1-benzyl-I3C down regulated production of estrogen receptor-alpha protein, acts with tamoxifen to arrest breast cancer cell growth more effectively than either compound alone, and inhibited the in vivo growth of human breast cancer cell-derived tumor xenografts in athymic mice. Our results implicate 1-benzyl-I3C as a novel, potent inhibitor of human breast cancer proliferation and estrogen responsiveness that could potentially be developed into a promising therapeutic agent for the treatment of indole-sensitive cancers.
Keywords: Indole-3-carbinol, 1-benzyl-indole-3-carbinol, breast cancer cells, anti-proliferative signaling, disrupted estrogen receptor responsiveness, high potency indole derivatives
1. Introduction
A critical problem in the clinical management of cancer is the need to identify new classes of highly potent therapeutic agents that act specificity on their cellular targets with reduced side effects, especially after prolonged treatments. Epidemiological studies have shown that diets high in fruits and vegetables is directly associated with decreased risk for a variety of human cancers, including breast and other reproductive cancers [1-5]. These studies suggest that dietary plants produce unique compounds that represent a largely untapped source of starting molecules to synthetically develop new chemotherapeutic molecules with potent anti-cancer properties. One such phytochemical is indole-3-carbinol (I3C), a naturally occurring autolysis product of glucobrassicin, a gluccosinolate present in Brassica vegetables, such as cabbage, broccoli, and Brussels sprouts that exhibits anti-proliferative activity in many types of human cancer cells [6-10] including estrogen responsive and estrogen independent breast cancer cells [11-15] and human prostate cancer cells [16-21]. Furthermore, I3C inhibits cellular adhesion [22] and cell motility [23], and has been shown to reduce the incidence of spontaneous and chemically induced mammary tumor formation in vivo in experimental animal model systems [24]. I3C tested positive as a chemopreventative agent in several short-term bioassays relevant to carcinogen-induced DNA damage, tumor initiation and promotion, and oxidative stress [25], and is a chemopreventative agent for human breast and prostate cancer cells [26].
I3C treatment of human reproductive cancer cells activates or dampens specific transcriptional, signal transduction, and metabolic cascades that lead to a cell cycle arrest, apoptosis, down-regulation of cancer cell migration, and modulation of hormone receptor signaling [6-10, 27]. Down stream targets of I3C signaling include transcription factors such as Sp1 and NFκB and their target genes [28, 29], the p53 tumor suppressor protein [12], and specific cell cycle components such as the G1 acting CDKs, CDK inhibitors, cyclin E and cyclin D [11, 13, 14, 28, 30]. In human breast cancer cells, a response specific to the I3C cell cycle arrest is the disruption of Sp1 interactions with the CDK6 promoter, which accounts for the loss of CDK6 promoter activity and gene expression [28]. We have also observed that I3C inhibits expression of estrogen receptor-alpha [27] and cooperates with tamoxifen [31] to inhibit growth of estrogen responsive breast cancer cells, and has both transcriptional and non-transcriptional effects in human breast cancer cells that together control cell proliferation. One such critical non-transcriptional effect of I3C is the direct inhibition of elastase enzymatic activity that accounts for the indole-dependent disruption of cyclin E protein processing in human breast cancer cells [30].
Although the cellular and physiological evidence demonstrating the anti-cancer effects of I3C is compelling, several factors may limit the use of I3C as a therapeutic agent for the treatment of human breast cancer. I3C is unstable and is readily converted into a number of products in vivo [32], which have some common targets but have also been demonstrated to have distinct biological effects on breast cancer cells [7, 10]. When ingested, the low pH of the stomach converts I3C into six major dimeric and trimeric condensation products, including the diindole compound 3,3’-diindolylmethane (DIM), and the trimerization product 5,6,11,12,17,18-hexahydrocyclonona[1,2-b:4,5-b’:7,8b”]triindole (CTr), as well as roughly a dozen minor condensation products including the well characterized diindole product indole[3,2-b]carbazole (ICZ) [4, 6, 33, 34]. Approximately 20% of I3C is converted into DIM, which is the most prevalent condensation product. A general picture has emerged that a subset of these acid-catalyzed reaction products have distinct anti-proliferative and anti-tumorigenic physiological bioactivities. Little is known about the exact ratio of conversion of I3C into these condensation products and under what conditions I3C is stable, thus estimating the effective dose for I3C in vivo may become problematic.
Further complications in the use of I3C as an anti-cancer agent are the relatively high concentrations (50-200 μM range) of I3C are needed to suppress the growth of human breast cancer cells [11, 13, 22, 28, 31] and that in indole treated cells, a significant fraction of I3C is converted into its natural dimerization product, DIM [35]. However, the effectiveness of I3C in controlling expression and activity of specific cell cycle components in many types of breast cancer cells, and other cancer cell types, suggests that this phytochemical would make a useful lead compound to develop highly potent anti-cancer agents. Several previous studies have used different synthetic strategies to develop new classes of I3C derivatives, however, these studies have been largely disappointing because of the low enhancement of potency of the biological activity. For example, analysis of a tetrameric I3C derivative that can inhibit CDK6 expression showed that this compound is only 5-fold more effective than I3C in inhibiting cell growth [36], whereas, detailed characterization of a large number of I3C derivatives resulting in the finding that the most potent derivative, OSU-A9, is only 100-fold more effective than I3C in apoptotic response in human prostate cancer cells [21]. One other study developed a synthetic derivative of I3C, SR13668 that resembles the structure of DIM, which is a self-condensation dimerization product of I3C, and displays only a 10-fold enhancement of potency [37]. In a systematic structure-activity analysis of I3C, we previously reported that addition of substituents to the nitrogen at position 1 in the indole ring with the more hydrophobic alkyl groups showing the highest potency with an increased potency of the growth arrest response up to several hundred fold [38], suggesting that substituents that increase lipophilicity and modify the indole ring nitrogen may improve activity. Based on these structure-activity relationships, we now report that 1-benzyl-I3C is a novel and the most efficient synthetic derivative of I3C to date with an approximately 1000-fold increased potency for the growth arrest of both human estrogen responsive and estrogen independent human breast cancer cells. Moreover, 1-benzyl-I3C displays significantly more potent effects on estrogen responsiveness and on in vivo growth of breast cancer cell xenografts in athymic mice.
2. Materials and Methods
2.1. Materials
Iscove’s modified Dulbecco’s medium (IMDM) and Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum, calcium and magnesium-free phosphate-buffered saline, L-glutamine, penicillin/streptomysin and typsin-EDTA were supplied by Cambrex/Biowhittaker (Walersville, MD). I3C and DIM were purchased from LKT laboratory Inc (St. Paul, Minnesota). [3H]Thymidine (84 Ci/mmol), [γ-32P]ATP (3,000 Ci/mmol) was purchased from NEN Life Science Products (Boston, MA). Insulin (bovine) and tryptophol were purchased from Sigma Chemical Corp. (St. Louis, MO). The anti-Rb antibodies were purchased from Pharmigen (San Diego, CA) and all other antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
2.2. Synthesis and characterization of 1-benzyl-I3C
The synthesis of 1-Benzyl-indole-3-carbinol (Figure 1) first involves the benzylation of indole, then formylation to the aldehyde, and finally reduction to the carbinol. Benzylation of indole followed the procedure of Heaney and Ley [39]. Potassium hydroxide (6.5 g) was added to DMSO (50 mL) in a 250 mL flask and stirred for 5 minutes. Indole (2.93 g) was added and stirred an additional 45 minutes. The reaction was placed in an ice bath, then benzyl bromide (8.55 g, 5.95 mL) was added and the reaction was again stirred for 45 minutes. Water (50 mL) was added and the reaction was partitioned with ether (100 mL, 3x). The ether phases were combined, back extracted with water (100 mL, 3x), and taken to dryness by rotary evaporation.
Fig. 1.
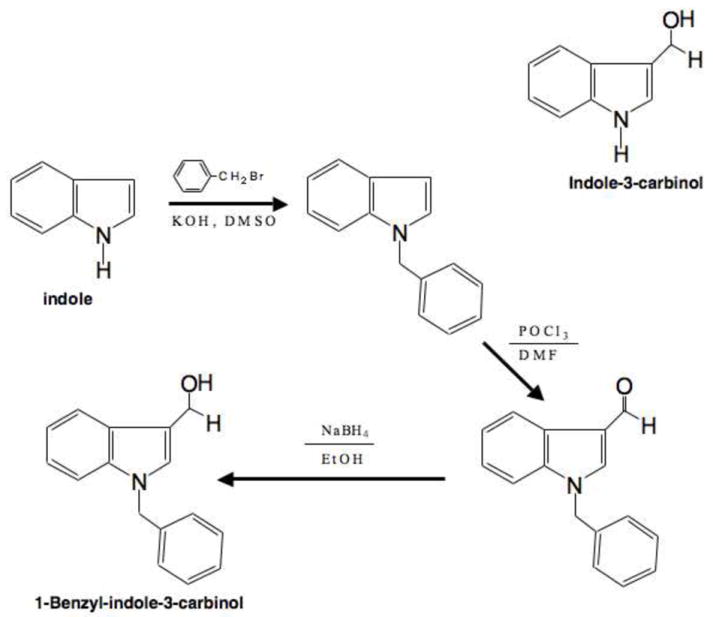
Synthesis and structure of 1-Benzyl-indole-3-carbinol. The upper diagram shows the structure of indole-3-carbinol. The synthesis of 1-Benzyl-indole-3-carbinol was accomplished by the benzylation of indole, then formylation to the aldehyde, and finally reduction to the carbinol. The structure of 1-Benzyl-indole-3-carbinol is shown in the lower diagram.
Conversion of 1-benzyl indole to 1-benzyl indole-3-aldehyde followed previously outlined procedure for formylation of indole [40]. Under very dry conditions, phosphorous oxychloride (2.6 mL, 0.028 mole) was added to 8.7 mL freshly distilled dimethylformamide in a 3-neck flask while stirring on an ice bath under a drying column and purged with nitrogen gas. 1-Benzyl indole (0.025 mole) in 3 mL dry dimethyl formamide was slowly added to the reaction mixture and stirred for 1 hour at room temperature, then an additional hour at 35 °C. Crushed ice was added (9 g) to the solution before transfer to another flask with 6 g ice in 5 mL water. Sodium hydroxide (10 g in 30 mL water) was added slowly at first with the final 5-7 mL added rapidly. The reaction was heated rapidly to a boil for 2-3 minutes, then cooled to room temperature and refrigerated overnight. Water (50 mL) was added and the solution was partitioned with ethyl acetate (3 × 50 mL). The organic phase was back extracted with (2 × 100 mL), dried with sodium sulfate, and concentrated to dryness.
The formyl derivative was converted to the hydroxymethyl product by reduction with NaBH4 in ethanol [41] and monitoring by HPLC to confirm the complete reduction of the formylindole. Sodium borohydride (2.7 mmoles) was added to 1-benzyl indole-3-aldehyde (2.25 mmoles) in ethanol (25 mL) and stirred for 4-6 h at room temperature. Water (50 mL) was added and the solution was partitioned with ether (3 × 50 mL). The combined organic phases were back extracted with water (2 × 100 mL), dried with sodium sulfate, and concentrated to dryness. Purity of 1-Benzyl-indole-3-carbinol was greater than 95% as determined by HPLC analysis.
1-benzyl-I3C: FAB m/z 237 [M+], (35 %), 220 [ArN(1-CH2Ph)CHC=CH2+] (100), 133 (11), 185 (10), 130 (7); δ (ppm) [acetonitrile-D3] 2.81 (1 H, t, , J 5.5 Hz, OH), 4.73 (2 H, d, J 2.8 Hz, 3-CH2), 5.32 (2 H, s, N-CH2), 7.04-7.33 (9 H, m, ArH), 7.63 (1 H, d, J 3.9 Hz, ArH), Yellowish solid. FAB/MS: FAB/MS spectra were acquired with the ZAB2-EQ double focusing mass spectrometer (Micromass, Manchester, U.K.). FAB analyses were performed by the mass spectrometry facility at the University of California in Berkeley.
2.3. Spectroscopic Measurements
1H-NMR measurements were performed on a Bruker AMX 300 (300MHz) spectrometer in deuterated acetonitrile.
2.4. Tissue Culture and Cell Lines
Estrogen responsive MCF-7 and estrogen independent MDA-MB-231 human breast cancer cell lines were cultured in DMEM or IMDM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 10 μg/mL insulin, 1.25 ml of 20,000 units/ml penicillin and streptomycin respectively. Cells were maintained at subconfluency at 37°C in humidified air containing 5% CO2. 1-Benzyl-I3C was dissolved in DMSO (99.9% high performance liquid chromatography grade; Aldrich) at concentrations 1000-fold higher than the final concentration used. In all experiments, 1 μL of the concentrated 1-benzyl-I3C or I3C in DMSO were added per ml of media and for the vehicle control, 1 μL of DMSO was added per ml of media. Similarly, 1 μL of concentrated tamoxifen in DMSO was added per ml of media.
2.5. [3H]Thymidine Incorporation
Cells were plated onto 24-well Corning tissue culture dishes with 30,000 cells/well. Triplicate samples of asynchronously growing breast cancer cells were treated with the indicated times with either vehicle control (1 μl DMSO/ml media) or varying concentrations of either 1-benzyl-I3C or I3C. The cells were pulsed for three hours with 3 μCi [3H]thymidine (84 Ci/mmol), washed three times with ice cold 10% trichloroacetic acid, and lysed with 300 μl of 0.3N NaOH. Lysates (150 μl) were transferred into vials containing liquid scintillation cocktail and radioactivity was quantified by scintillation counting. Triplicates were averaged and expressed as counts per minute per well.
2.6. Flow Cytometry Analysis of DNA Content
MCF-7 and MDA-MB-231 human breast cancer cells were plated at 30% confluency on 100 mm tissue culture dishes and treated for the indicated time points with varying concentrations of 1-benzyl-I3C or I3C in complete media. The media was changed every 24 hours. An aliquot of harvested cells were than hypotonically lysed in 0.5-1 ml of DNA staining solution (0.5 mg/ml propidium iodide, 0.1% sodium citrate, 0.05% Triton X-100). Cell debris was filtered out using a 60 um nylon mesh (Sefar America Inc., Kansas city, MO). Nuclear-emitted fluorescence with wavelengths of >585 nm was measured using a Coulter Elite Instrument. Ten thousand nuclei were analyzed from each sample at a rate of 300-500 nuclei/s. The percentage of the cells within the G1, S, and G2/M phases of the cell cycle were determined by analysis with the Multicycle software MPLUS (Phoenix Flow Systems) in the Cancer Research Laboratory Microchemical Facility of the University of California, Berkeley.
2.7. Western Blot Analysis
After the indicated treatments, cells were washed once with cold PBS, harvested and lysed in either CDK2 lysis buffer (250 mM NaCl, 0.1% triton X-100, 50 mM Tris/HCl, pH 7.3) or RIPA buffer (150 mM NaCl, 0.5% deoxycholate, 0.1% NP-40, 0.1% SDS, 50 mM Tris) containing protease and phosphatase inhibitors (50 μg/mL phenylmethylsulfonyl fluoride (PMSF), 10 μg/mL aprotinin, 5 μg/ml leupeptin, 0.1% sodium fluoride (NaF), 10 μg/mL β-glycerophosphate, and 0.1 mM sodium orthovanadate). Equal amounts of proteins were mixed with protein loading buffer (25% glycerol, .075% SDS, 10% bromophenol blue, 14.4 mM β-mercaptoethanol, 3.13% 0.5 M Tris/HCl) and fractionated on 10% polyacrylamide/0.1% SDS gels by electrophoresis. A rainbow marker was used as the molecular standard (Amersham Life Sciences, Arlington Heights, IL). Proteins were electronically transferred to nitrocellulose membranes (Micron Separations, Inc., Westboro, MA) and blocked either overnight at 4 °C or at room temperature for 1 hour with 5% NFDM in TBST (10 mM Tris/HCl (pH 8.0), 150 mM NaCl, 0.05% TweeN-20). Blots were subsequently incubated at room temperature for an hour with primary antibodies to CDK2, CDK4, cyclin D1, cyclin E1, Rb or ER-alpha. Western blots probed for p21 or p27 were incubated at room temperature for 2-3 hours or overnight, respectively. The antibodies were used at a concentration of 1 μg/ml in TBST. Immunoreactive proteins were detected after incubation with horseradish-peroxidase-conjugated goat anti-rabbit, goat anti-mouse, and rabbit anti-goat (Bio-Rad, Hercules, CA) secondary antibodies diluted to 3 × 10−4 in TBST-1%NFDM. Blots were incubated with ECL reagents (NEN Life Science Products) and all proteins were detected by autoradiography. Equal protein loading was confirmed by Ponceau S staining of blotted membranes or probed for actin antibodies.
2.8. Immunoprecipitation and protein kinase assays
After the indicated treatments, cells were lysed for 15 minutes at 4 °C in CDK2 lysis buffer with protease and phosphatase inhibitors (50 μg/ml phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 5 μg/mL leupeptin, 0.1% sodium fluoride, 10 μg/mL β-glycerophosphate, and 0.1 mM sodium orthovanadate). Samples were diluted to 500-800 μg of protein in 1 ml of CDK2 lysis buffer and pre-cleared for 30 minutes at 4 °C with 30 μL of 1:1 slurry of protein G-beads (Pharmacia Biotech, Sweden) with 1 μg of rabbit IgG (Sigma). After a brief centrifugation to remove the precleared beads, 0.5 μg of anti-CDK2, anti-CDK6, or anti-CDK4 antibodies were added to each sample and incubated on a rocking platform at 4 °C for 2 hours. Thirty microliters of the 1:1 bead slurry was added to each sample and was left on a rocking platform for 30 minutes. The beads were than washed four times in CDK2 lysis buffer (with protease and phosphatase inhibitors as described previously) and twice with kinase buffer (50 mM Hepes pH 7.3, 5 mM MnCl2, 10 mM MgCl2) with protease and phosphatase inhibitors. Half of the immunoprecipitation was checked by western blot analysis to confirm the efficiency of the immunoprecipitation.
The other half of the immunoprecipitation was resuspended in 25 μL of kinase buffer containing 20 mM ATP, 5 mM DTT, 0.21 μg of Rb COOH-terminal domain protein substrate (Santa Cruz Biotechnology) and 10 μCi of ([γ-32P]-ATP (3000 Ci/mmol)). Each reaction was incubated for 15 minutes at 30°C and stopped by the addition of equal volume of 2× protein loading buffer. Reaction products were boiled for 5 minutes and than fractionated in SDS-10% polyacrylamide gels. Gels were stained with Coommassie blue (Sigma) and destained overnight with 3% glycerol and 10% glacial acetic acid to monitor loading. Subsequently, gels were dried and quantified on a phosphoimager (Molecular Dynamics, Sunnyvale, CA) and/or visualized by autoradiograph.
2.9. Quantification of autoradiography
Autoradiographic exposures were scanned with a UMAX UC630 scanner, and band intensities were quantified using the NIH Image program. Autoradiographs from a minimum of three independent experiments were scanned per time point.
2.10. Reverse Transcription Polymerase Chain Reaction
MCF-7 cells were treated with the DMSO vehicle control, 1 μM 1-benzyl-I3C or 200 μM I3C for 48 hours, and then harvested in 1 ml of Trizol reagent (Invitrogen, San Diego, CA), and the recommended protocol was followed to extract total RNA. In brief, chloroform was added to extract the RNA-containing fraction, and isopropyl alcohol was used to precipitate the total RNA. After washing with 75% ethanol, the RNA pellets were air-dried, dissolved in DEPC water, and quantified using spectroscopy. The quality of RNA was confirmed using A260/A280, and by electrophoresis on a 1% agarose gel. 8 μg of total RNA was subjected to reverse transcription using random hexamers, deoxynucleotide triphosphates (dNTPs), M-MLV reverse transcriptase (Invitrogen, San Diego, CA), and RNAase inhibitors (Roche Applied Science, Indianapolis, IN). cDNA (6 μL) was subjected to PCR using specific primers sets (10 μM): CDK6 forward 5’-CTTTGCCTAGTTCATCGATATC-3’, reverse 5’-CCGAGTAGTGCATCGCGATCTAA-3’; GAPDH forward 5’-TGAACGGGAAGCTCACTGG-3’, reverse 5’-TCCACCACCCTGTTGCTGTA-3’. The cycling conditions were 27 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, extension at 72 °C for 30 s, followed by a final extension at 72 °C for 10 minutes. The resulting PCR products were fractionated by electrophoresis in a 1% agarose gel along with 1-kb plus DNA ladder and further visualized by a UV transilluminator.
2.11. Chromatin immunoprecipitation assay
MCF-7 cells were seeded in 150 mm plates and treated with 200 μM I3C, 1 μM 1-benzyl-I3C compounds or with the DMSO vehicle control (two plates for each treatment) for 48 hours. Media were replaced every 24 hours. The cells were then incubated with formaldehyde at a final concentration of 1% for 10 minutes on a shaking platform followed by glycine to a final concentration of 0.125 M for 5 minutes. The cells were washed twice with ice-cold PBS, collected in PBS containing in 5 mM EDTA, and then pelleted. The cell pellets were resuspended in 500 μL of cell lysis buffer (50 mM HEPES pH 7.5, 140 mM NaCl, 1% Triton X-100, 0.1% NaDeoxycholate) containing protease inhibitors and incubated on ice for 20 minutes. The samples were centrifuged at 4 °C for 15 minutes at 1,000 rpm, and subjected to sonication by a Branson Sonifier Cell Disruptor 200 (Branson Sonic Power. Co., Danbury, Connecticut). After another centrifugation at 12,000 rpm for 15 minutes, the supernatant fractions were collected into a new tube and the protein concentrations quantified using the Lowry Method (Bio-Rad Laboratories, Hercules, CA). Immunoprecipitations were carried out using 2 mg of protein, and 1% of the amount (20 μg) was saved at -80 °C as “input”. Samples were pre-cleared with 40 μl of a 1:1 slurry of protein G-Sepharose beads (GE Health BioSciences AB, Sweden) at 4 °C for 30 minutes, and followed by brief centrifugation to remove pre-cleared beads. An amount of 0.5 μg of mouse anti-Sp1 antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was added to samples for overnight incubation at 4 °C on a rocking platform. To each sample, 40 μL of protein G-Sepharose beads were added, and the slurries were incubated at 4 °C for 1 hour on a rocking platform. After the incubation, the beads were pelleted by brief centrifugation at 4,000 rpm for 5 minutes, and washed twice with cell lysis buffer and twice with wash buffer (10 mM Tris pH 8.0, 250 mM LiCl, 0.5% NP-40, 0.5% NaDeoxycholate, and 1 mM EDTA). To each of the samples, 150 μL of elution buffer (50 mM Tris pH 8.0, 1% SDS, 10 mM EDTA) were added, as well as the pre-saved 1% input samples. All the samples were incubated at 65 °C for 10 minutes. The samples were centrifuged briefly, and the resulting pellet was washed three times with cell lysis buffer and twice with wash buffer. The immunoprecipitation samples were centrifuged briefly, and the elution process was repeated and the supernatants were pooled. To the pooled eluted material and the input sample, NaCl was added to a final concentration of 0.3 M, and the samples were incubated at 65 °C overnight. The DNA was then purified using a PCR purification kit (Qiagen, Valencia, CA) following the recommended protocol. For PCR, 2 μl of the isolated chromatin was used and the cycling conditions were an initial incubation at 95 °C for 3 minutes, then 35 cycles at 95 °C for 1 minutes, 58 °C for 1 minute, 72 °C for 1 minute, and 10 minutes final extension at 72 °C. The primer set targeting the Sp1 binding site on CDK6 promoter used was: forward 5’-GTCCTTCCGCTCCAGCTT -3’ and reverse 5’-CACGTCAATGTCACGGCTTA -3’; the control primer set was (CDK6 +1474/ +1573): forward 5’ - GAAAAGCATGGGCAGAACAT -3’ and reverse 5’- TCTCTGGGCGGACATACTCT -3’.
2.12. Growth of human breast cancer cell xenografts in athymic mice
MCF-7 breast cancer cells (5 × 106), which are estrogen receptor-alpha positive, were mixed with equal volume of matrigel (BD Biosciences, CA) and inoculated subcutaneously into right and left flanks of athymic nude mice. These mice received 1 mg/ml 17β-estradiol (Sigma, MO) in Dulbecco’s phosphate buffered saline (DPBS) (Mediatech, CA) every day to promote growth of the xenografts. Once the tumors reached a palpable size, the mice were given daily subcutaneous injections of I3C (300 mg/kg body mass) or 1-benzyl-I3C (30 mg/kg body mass). The stock solutions of I3C (300 mM) or 1-benzyl-I3C (30 mM) were dissolved initially in DMSO and then diluted in appropriate volume of DPBS. The indoles were injected subcutaneously in order to reduce the potential for formation of I3C condensation products, such as the natural dimer DIM, that normally occur in the acid conditions of the stomach. In this regard, it is important to point out that the structure of 1-benzyl-I3C prevents its conversion into a DIM-like compound. Mice were also palpated and tumor size measured weekly with calipers. Tumor volume was calculated using the following equation V = a × b2 /2, where a is the width and b is the length of the tumors. Xenografted tumors were excised at the termination of the experiment and photographed. A similar xenograft experiment in athymic nude mice was performed using MDA-MB-231 breast cancer cells, which are estrogen receptor-alpha negative. For these xenograft studies, the athymic mice were not injected with 17-beta-estradiol and five weeks was needed to assess tumor volume because of the increased lag time needed for formation of palpable tumors in the MDA-MB-231 cell xenografts compared to the MCF-7 cell xenografts.
3. Results
3.1. Synthesis of 1-benzyl-I3C and increased potency on the DNA synthesis of MCF-7 human breast cancer cells compared to I3C
Our previous structure-activity analysis of I3C revealed that substituents linked to position 1 nitrogen in the indole ring that inhibit dehydration and the formation of the reactive indolenine increases potency (which stabilizes the compound and prevents formation of oligomeric self condensation products), whereas, the C-3 hydroxy methyl substituent on the indole ring is required for maintaining biological activity [38]. Furthermore, increasing the number of carbons in the N-alkoxy derivatives caused a significant increase in potency suggesting that the hydrophobic properties of the substituents at this indole ring position are critical to increasing potency of the resulting molecules. Based on these experimental results, we synthesized a novel I3C derivative, 1-benzy-indole-3-carbinol (1-benzyl-I3C), that was designed to be stable, contains increased hydrophobic character of the substituent, and maintains the C-3 hydroxy methyl substituent. The synthesis of 1-Benzyl-indole-3-carbinol is detailed in the methods section and involves the sequential benzylation of indole, then formylation to the aldehyde, and finally reduction to the carbinol (see figure 1).
The stability of 10-5 M solutions of I3C and 1-benzyl-I3C was compared at pH 2, pH 4 and pH 5.5. At pH 2 both I3C and 1-benzyl-I3C decompose in ethanol-water solution in several minutes; however, at higher pH 1-benzyl-I3C is significantly more stable than I3C. At pH 4 in ethanol-citrate phosphate buffer solution, 1-benzyl-I3C requires at least 12 hours before decomposing, whereas, I3C totally disappears after 2.5 hrs. At pH 5.5 in ethanol-citrate phosphate buffer solution, I3C totally decomposes in 3 days, whereas, significant amounts of 1-benzyl-I3C remains even after 10 days incubation (data not shown). The higher stability of 1-benzyl-I3C in comparison with I3C may be dependent on the steric effect of bulky 1-benzyl substituent, which hampers oligomerization of the 1-benzyl-indol-3-yl methyl cation formed under protonation. Thus, the biological effects of 1-benyzl-I3C are not dependent on its conversation into a DIM-like derivative.
To compare the efficacies of the anti-proliferative effects of 1-benzyl-I3C and the I3C parental compound, MCF-7 human breast cancer cells were treated with increasing concentrations of indole for 72 hours and DNA synthesis monitored by [3H]thymidine incorporation. As shown in Fig 2A, both 1-benzyl-I3C and I3C strongly inhibited DNA synthesis to approximately the same extent, however, significantly lower concentrations of 1-benzyl-I3C induced the maximal growth arrest. The dose-dependent effects of 1-benzyl-I3C and I3C on DNA synthesis were examined in multiple independent experiments, and as shown in figure 2B, 1-benzyl-I3C was approximately 1000-fold more potent than I3C at inhibiting DNA synthesis of MCF-7 human breast cancer cells. The IC50 values, the concentration required to inhibit DNA incorporation by 50%, were 0.05 μM 1-benzyl-I3C and 0.52 μM for I3C. This result suggests the 1-benzyl-I3C is the most potent anti-proliferative I3C derivative characterized to date.
Fig. 2.
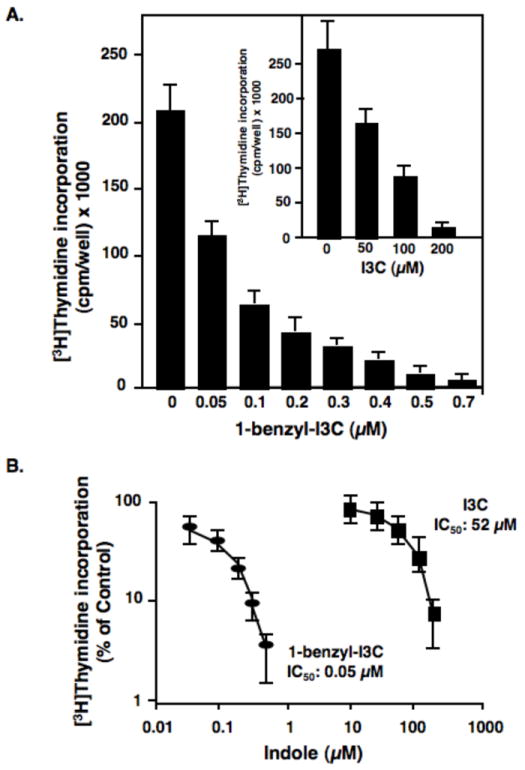
Dose-dependent effects of 1-benzyl-I3C and I3C on the DNA synthesis of human MCF-7 breast cancer cells. (A) Triplicate samples of asynchronously growing human MCF-7 breast cancer cells were treated with increasing concentrations of 1-benzyl-I3C, I3C or the vehicle control (DMSO) for 72 hours. During the last three hours, cells were pulsed with 3 μCi [3H]thymidine (84 Ci/mmol), and the incorporation into DNA was determined by acid precipitation as described in the Materials and Methods section. Triplicates were averaged and expressed as counts per minute per well. (B) To determine the IC50 values of the I3C and 1-benzyl-I3C inhibition of DNA synthesis of MCF-7 cells, the results of the [3H]thymidine incorporation data are plotted as the percentage of vehicle control treated cells for each indole. Data are the mean of three experiments; bars, +/-SE.
3.2. 1-Benzyl-I3C arrests the growth and induces a G1-cell cycle arrest of both estrogen responsive and non-responsive human breast cancer cells
A key property of I3C is the ability to arrest the growth of both estrogen responsive and non-responsive human breast cancer cell lines [10, 11], and we therefore tested whether 1-benzyl-I3C has a similar anti-proliferative property at more potent efficacies. The dose-dependent effects of 1-benzyl-I3C on the rates of [3H]thymidine incorporation at 72 hours of indole treatment of estrogen responsive MCF-7 human breast cancer cells (which express estrogen receptor-alpha) were compared to estrogen independent and highly invasive MDA-MB-231 human breast cancer cells (which do not express estrogen receptor-alpha). As shown in Fig. 3A, 1-benzyl-I3C was effective in suppressing DNA synthesis of both the MCF-7 and MDA-MB-231 human breast cancer cell lines, and a greater than 90 percent inhibition of DNA synthesis was observed at approximately 0.2 μM 1-benzyl-I3C. We previously showed that I3C displays the same level of growth inhibition in both cell lines at approximately 200 μM indole [11] showing an approximately 1000-fold increased potency for 1-benzyl-I3C when compared to I3C. Unless otherwise indicated, 0.2 μM 1-benzyl-I3C was employed to assess anti-proliferative effects of this synthetic indole in human breast cancer cells.
Fig. 3.
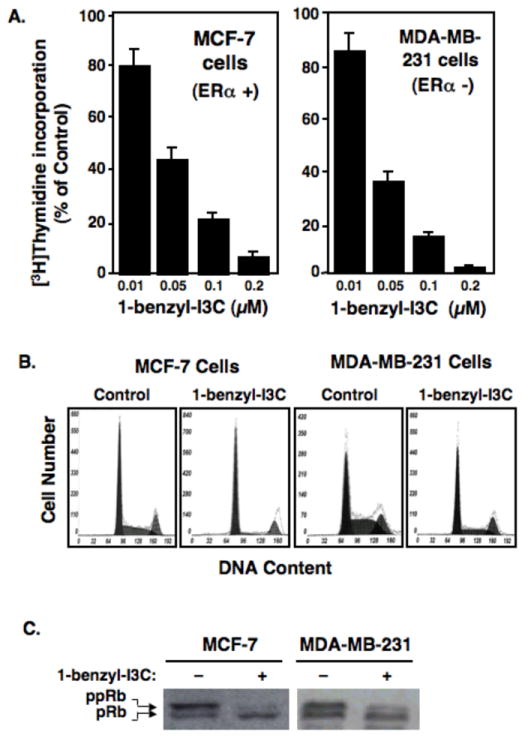
1-benzyl-I3C inhibits DNA synthesis and induces a G1 cell cycle arrest of estrogen-responsive and estrogen-independent unresponsive human breast cancer cell lines. (A) Estrogen responsive MCF-7 cells and estrogen independent MDA-MB-231 cells were treated for 72 hours with the indicated concentrations of 1-benzyl-I3C and during the last three hours, cells were pulsed labeled with 3 μCi [3H]thymidine and the incorporation into DNA and percent incorporation calculated as described in the figure 2 legend. Data represent mean of three independent experiments. (B) MCF-7 cells and MBA-MD-231 cells were treated for 72 hours with either 0.2 μM 1-benzyl-I3C or the DMSO vehicle control, and effects on the cell cycle monitored by flow cytometry for DNA content of propidium iodide stained nuclei. (C) MCF-7 and MDA-MB-231 cells were treated with or without 0.2 μM 1-benzyl-I3C for 72 hours, and western blots of electrophoretically fractionated total cell extracts analyzed for total phosphorylated-Rb (pRb) and hyperphosphorylated-Rb (ppRB) using the corresponding Rb-specific antibodies.
To examine the cell cycle profile of the growth arrest, MCF-7 or MDA-MB-231 human breast cancer cells were treated for 72 hours with 0.2 μM 1-benzyl-I3C or with the DMSO vehicle control, and the nuclear DNA content analyzed by flow cytometry analysis of propidium iodide stained nuclei. As shown in figure 3B, typical of proliferating cells, in the absence of indole treatment both cell populations were in all phases of the cell cycle, although the highly invasive MDA-MB-231 cells displayed a higher level of S phase cells. Consistent with the [3H]thymidine incorporation results, 1-benzyl-I3C induced a significant enhancement in the number of cells displaying a G1 phase level of DNA content and reduction in S phase DNA content compared to the vehicle controls in both cell lines. Similar to that observed for the inhibition of [3H]thymidine incorporation, the half maximal stimulation in G1 phase arrested cells and reduction in S phase cells occurred at approximately 0.05 μM 1-benzyl-I3C (data not shown). The 1-benzyl-I3C mediated G1 cell cycle arrest resulted in a qualitatively similar flow cytometry profiles as I3C [11], but at a significantly lower indole concentration.
The retinoblastoma protein (Rb) becomes hyperphosphorylated by the G1-acting cyclin dependent kinases (CDK) during progression through the cell cycle. The effects of 0.2 μM 1-benzyl-I3C on production of the hyperphosphorylated Rb protein (ppRb) was examined in both MDA-MB-231 and MCF-7 breast cancer cells treated for 72 hours with or without this highly potent I3C derivative. As shown in figure 3C, western blots revealed that 1-benzyl-I3C strongly down-regulated production of the hyperphosphorylated Rb in both cell lines at a concentration of 0.2 μM 1-benzyl-I3C that is approximately 3 orders of magnitude less than what is needed for a similar maximal effect by 200 μM of the I3C parent compound [11].
3.3. Effects of 1-benzyl-I3C on expression of G1 cell cycle components
I3C regulates the expression and activity of a select subset of G1-acting cell cycle components, and therefore we examined the cell cycle effects of 1-benzyl-I3C to determine if this novel derivative elicits an I3C-type response in human breast cancer cells. MCF-7 cells were treated with 0.2 μM 1-benzyl-I3C for the indicated durations, which is approximately 1000-fold lower concentration than what we previously reported for I3C [11]. As shown in Figure 4 (western blots), similar to our previous results with the I3C parent compound [10, 11, 28], 1-benzyl-I3C markedly reduced the level of CDK6 and increase p21 and p27 protein levels in MCF-7 breast cancer cells. These effects were observed as early as 24 hours of treatments and reaches maximal response by 72 hours. However, 1-benzyl-I3C displayed a somewhat expanded effect on cell cycle components compared to what we have previously reported for I3C [10] in that the level of cyclin D was more effectively down-regulated, and the level of the CDK inhibitor p15 was increased by treatment with the indole derivative. Similar to I3C, the cellular levels of Cyclin E, CDK2, CDK4 remained relatively unaltered. The protein levels of the cell cycle components examined by western blots in 1-benzyl-I3C treated and untreated cells were quantified by densitometry. At each time point, the level of each cell cycle protein in 1-benzyl-I3C treated cells was determined relative to the level detected in untreated control cells. As shown in Figure 4 (lower panel), treatment with 1-benzyl-I3C stimulates an approximate 50% increase in the levels of p21 and p27 proteins during the 72-hour time, and strongly down-regulated the levels of CDK4, Cyclin D1 and CDK6 protein.
Fig. 4.
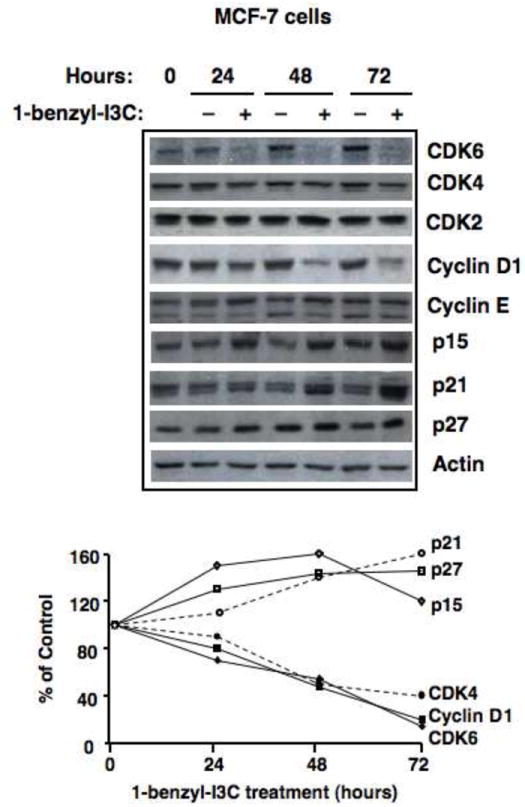
Time course of effects of 1-benzyl-I3C on expression of G1 cell cycle components. Upper panel: Human MCF-7 breast cancer cells were treated with 0.2 μM 1-benzyl-I3C or the DMSO vehicle control for 0, 24, 48 or 72 hours followed by western blot analysis of the indicated G1 cell cycle components. Actin protein levels were used a gel loading control. Lower panel: The level of observed cell cycle protein in 1-benzyl-I3C treated cells was compared to control cells and quantified at each time point by densitometric analysis of the western blot.
3.4. Effects of 1-benzyl-I3C and I3C on the endogenous interactions of Sp1 with the CDK6 promoter
One of the hallmark responses to I3C in human breast cancer cells is the down-regulation of CDK6 expression by disrupting the interactions of the Sp1 transcription factor with an Ets-Sp1 composite element in the CDK6 promoter [28]. To assess whether 1-benzyl-I3C maintains this I3C-specific property but at an increased efficacy, the effects of 200 μM I3C or 1 μM 1-benzyl-I3C and on CDK6 transcript levels and on endogenous interactions of Sp1 with the CDK6 promoter were examined in human breast cancer cells. A slightly higher concentration of 1-benzyl-I3C compared to the cell cycle studies was used to ensure maximal effects on transcription factor interactions with the CDK6 promoter. As shown in Figure 5, RT-PCR analysis revealed that treatment with either 1-benzyl-I3C or I3C strongly down-regulated CDK6 transcripts within 48 hours of indole treatment. Chromatin immunoprecipitation (ChIP) was used to compare the effects of 1-benzyl-I3C and I3C on the endogenous interactions of the Sp1 transcription factor with the composite Ets-Sp1 element that is located at -680 to -638 bp within the CDK6 promoter. An analysis of endogenous Sp1 interactions with the CDK6 promoter had not been previously attempted. Treatment with either indole strongly disrupted the endogenous interactions of Sp1 with the CDK6 promoter (Fig. 5). Under these conditions, either I3C or 1-benzyl-I3C had no effect on endogenous Sp1 interactions with CDK4 promoter, which helps confirm that this indole response is promoter specific (data not shown). Thus, 1-benzyl-I3C regulates the same anti-proliferative pathways targeting the CDK6 promoter as its I3C parental compound.
Fig. 5.
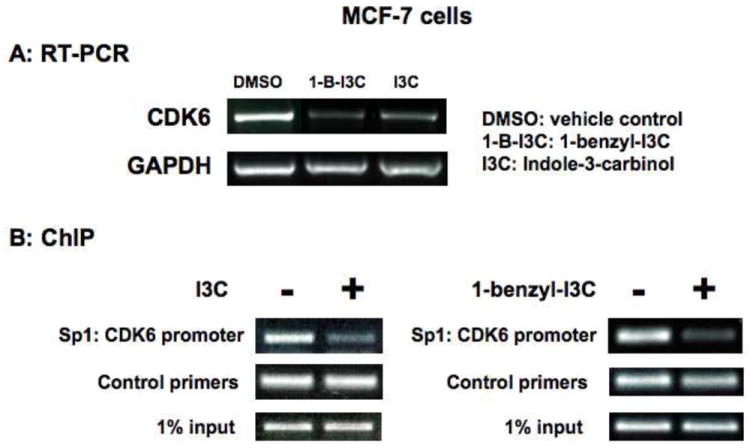
Effects of I3C and 1-benzyl-I3C on CDK6 transcript expression and on endogenous interactions of Sp1 with the CDK6 promoter. (A) MCF-7 cells were treated with 1 μM 1-benzyl-I3C (1-B-I3C), 200 μM I3C or the DMSO vehicle control for 48 hours, total RNA isolated and the levels of CDK6 transcripts determined by RT-PCR analysis using CDK6-specific oligonucleotide primers. The level of the constitutively expressed GAPDH was used as a positive control for each condition. (B) Chromatin immunoprecipitation was used to evaluate the effects of 1-benzyl-I3C and I3C on endogenous interactions of the Sp1 transcription factor with the Ets-Sp1 composite element in the CDK6 promoter. Chromatin was isolated from MCF-7 cells treated with or without either 200 μM I3C or 1 μM 1-benzyl-I3C for 48 hours. Sp1 was immunoprecipitated from total cell extracts using Sepharose G bound to anti-Sp1 antibody and DNA released from Sp1 was amplified using the oligonucleotide primers described in the methods and materials section. Input samples represent total genomic DNA from each treatment (loading control).
3.5. Effects of 1-benzyl-I3C on the expression and enzymatic activities of the G1-acting cyclin dependent kinases
To further characterize the 1-benzyl-I3C induced G1 cell cycle arrest of human breast cancer cells, the kinase activity and protein levels of all three G1 CDKs (CDK2, CDK4 and CDK6) were examined with an in vitro kinase activity assay of immunoprecipitated protein using GST-Rb as a substrate. MCF-7 breast cancer cells were treated with or without 0.2 μM 1-benzyl-I3C for 72 hours, and CDK immunoprecipitations were either analyzed for kinase activity or for the level of individual immunoprecipitated CDK proteins by western blots. As shown in figure 6A, the total enzymatic activities of all three G1 cyclin/CDK protein complexes were significantly reduced with 1-benzyl-I3C treatment. The loss of CDK6 kinase activity was significantly reduced in proportion to the down-regulation of CDK6 protein demonstrating that 1-benzyl-I3C did not alter CDK6 enzymatic specific activity. Thus, similar to I3C, the loss of CDK6 protein accounts for the loss of total cellular CDK6 kinase activity. In contrast, the primary effect of 1-benzyl-I3C on both CDK4 and CDK2 is to significantly reduce their enzyme specific activities, which is likely due to the significant increases in level of the p15, p21 and p27 CDK inhibitors. In this regard, the loss of CDK4 specific enzymatic activity is an enhanced anti-proliferative property of 1-benzyl-I3C compared to the parental I3C compound [28]. Figure 6B, quantifies the 1-benzyl-I3C inhibitory effects on CDK6, CDK4 and CDK2 enzymatic activities over a 72 hour time course and shows that the G1 CDK kinase activities are essentially ablated by 72 hours indole treatment, which likely accounts for the near complete block in cell cycle progression by this time point.
Fig. 6.
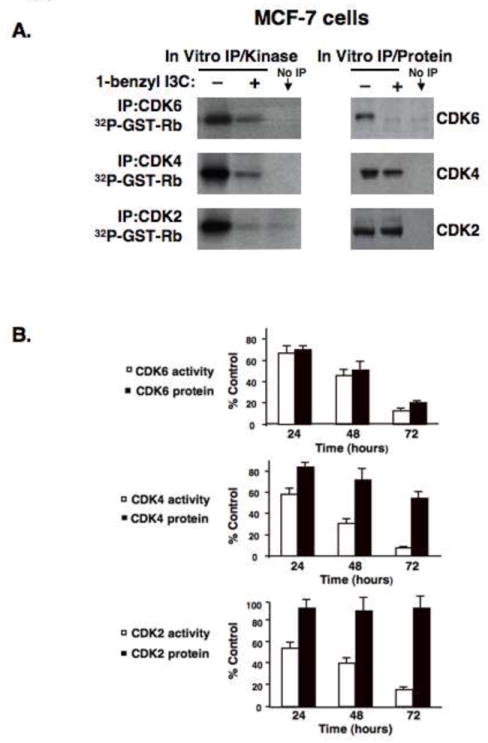
Effects of 1-benzyl-I3C on the specific enzymatic activities of the G1 acting Cyclin Dependent Kinases. (A) MCF-7 cells were treated with either 0.2 μM 1-benzyl-I3C or the DMSO vehicle control for 72 hours followed by immunoprecipitations of CDK2, CDK4, or CDK6 as described in Materials and Methods section. One aliquot of each immunoprecipitation was assayed in vitro for protein kinase activity using GST-Rb as a substrate (left panels), and the other aliquot was used to assess total CDK protein levels in each immunoprecipitation by western blot analysis (right panels). The No IP lane represents an immunoprecipitation with a non-specific IgG. (B) MCF-7 were treated with 0.2 μM 1-benzyl-I3C for indicated times, and at each time point, the enzymatic activities and protein levels of immunoprecipitated CDKs were performed as described above. The amount of immunoprecipitated CDK protein was quantified by densitometry, and the level of [32P]GST-Rb generated from the corresponding kinase assays was determined by phosphoimager analysis as described in Materials and Methods. The values are expressed as percentage of growing control cells not with the vehicle control DMSO at each time point, which was calculated by dividing the values of 1-benzyl-I3C treated cells with the values of vehicle control treated cells. Error bars represent standard error of the mean three independent experiments.
3.6. 1-Benzyl-I3C cooperates with tamoxifen to arrest the growth of estrogen responsive human breast cancer cells
We previously demonstrated that a combination of I3C and tamoxifen more effectively inhibits the proliferation of estrogen responsive human cancer cells compared to the effects of either compound alone [31]. These results suggest that one of the potential therapeutic uses of I3C-based compounds, such as 1-benzyl-I3C, is a combined treatment with suboptimal concentrations of the anti-estrogen tamoxifen in order to reduce the adverse side effects of tamoxifen. Therefore, to determine whether 1-benzyl-I3C at a concentration that elicits its half-maximal response has a similar cooperative effect with tamoxifen as the I3C parental compound, MCF-7 cells were treated with combinations of 1 μM tamoxifen and/or 0.05 μM 1-benzyl-I3C for 48 hours. Cells were also treated with a combination of 1 μM tamoxifen and/or 100 μM I3C DNA. DNA synthesis was monitored by incorporation of [3H]thymidine during the last three hours of treatment, and the number of cells arrested in the G1 phase of the cell cycle determined by flow cytometry of propidium stained nuclei. As shown in figure 7 (upper panel), similar to the cooperative effects of I3C and tamoxifen, a combination of a suboptimal concentration of 1-benzyl-I3C and tamoxifen more effectively inhibit the DNA synthesis of MCF-7 human breast cancer cells compared to the effect of each reagent added individually. Flow cytometry analysis revealed that similar to I3C, a combination of 1-benzyl-I3C and tamoxifen more effectively induced the number of MCF-7 cells arrested in the G1 phase of cell cycle (Figure 7, lower panel) compared to the effects of either compound alone. These results show that at significantly lower concentrations compared to I3C, 1-benzyl-I3C displays the I3C property of cooperating with tamoxifen to more effectively arrest the growth of estrogen responsive human breast cancer cells.
Fig. 7.
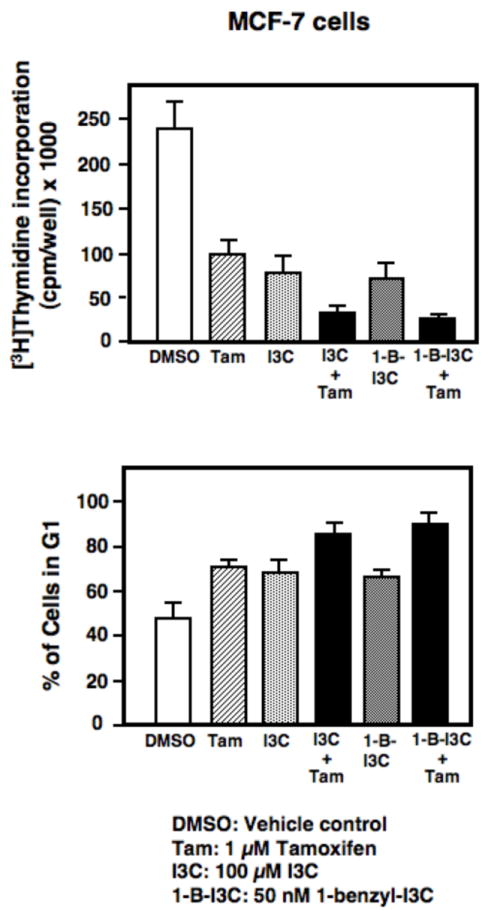
Cooperative effects of 1-benzyl-I3C and I3C with tamoxifen on the inhibition of DNA synthesis and stimulation in G1 phase arrested cells in MCF-7 cells. MCF-7 cells were treated with the indicated combinations of 100 μM I3C, 0.5 μM 1-benzyl-I3C (1-B-I3C) and/or 1 μM tamoxifen for 72 hours. The effects on DNA synthesis was monitored by pulsed labeling with 3 μCi [3H]thymidine during the last three hours of incubation (upper panel). The effects on the number of cells arrested in the G1 phase of the cell cycle was monitored by flow cytometry for DNA content of propidium iodide stained nuclei (lower panel). The reported data represent the mean of three independent experiments.
3.7. 1-Benzyl-I3C down-regulates the level of estrogen receptor-alpha
The underlying mechanism for the ability of I3C to cooperate with tamoxifen to growth arrest of MCF-7 cells is that this indole down-regulates expression of estrogen receptor-alpha (ER-alpha), whereas tamoxifen antagonizes ER-alpha activity by acting as a selective estrogen receptor modulator [9, 27, 31]. To determine whether 1-benzyl-I3C has a similar effect on ER-alpha production, MCF-7 human breast cancer cells were treated with a range of concentrations of either I3C and 1-benzyl-I3C and the level of total ER-alpha protein monitored by western blots of total cell extracts. As shown, Figure 8, both 1-benzyl-I3C and I3C strongly down-regulated ER-alpha levels, however, the concentrations of 1-benzyl-I3C required for this response was significantly lower compared to the concentrations required for the I3C parental compound. Actin was used as a constitutive gel loading control.
Fig. 8.
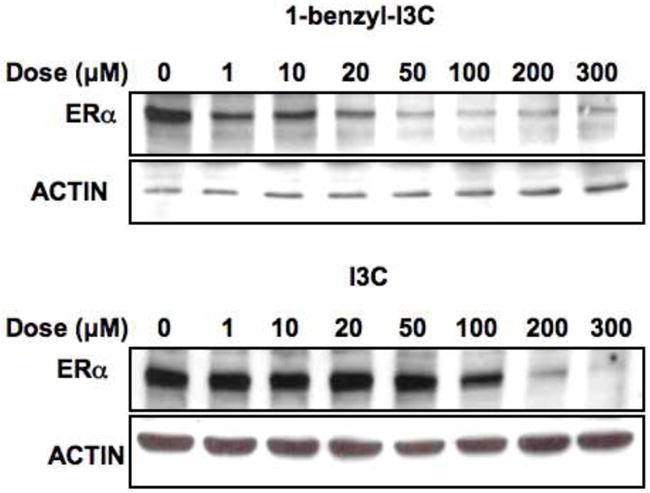
Dose-dependent effects of 1-benzyl-I3C and I3C on expression of estrogen receptor-alpha protein. MCF-7 cells were treated with indicated concentrations of 1-benzyl-I3C or I3C for 24 hours, and the level of estrogen receptor-alpha (ERα) determined by western blot analysis of electrophoretically fractionated total cell extracts. Western blot analysis of actin levels was used as a gel loading control.
3.8. In vivo effects of 1-benzyl-I3C and I3C on the growth of human breast cell derived tumors in athymic mice
Because the in vivo hormonal milieu is considerably different than the defined culture conditions used for cell proliferation assays, it was important to examine the in vivo bioactivity of 1-benzyl-I3C on tumors derived from xenografts of human MCF-7 human breast cancer cells in athymic mice. MCF-7 cells express estrogen receptor-alpha and are highly estrogen responsive. The athymic mice were injected with 17-beta-estradiol everyday in order to boost growth of the MCF-7 cell-derived tumors and to mitigate any potential effects of the indoles on estrogen levels or metabolism. MCF-7 cell xenografts were grown to an average size of 35 mm3, and then the mice were injected with 300 mg/Kg body mass of I3C, a ten-fold lower concentration of 1-benzyl-I3C or with the DMSO vehicle control. 1-Benzyl-I3C was employed at a higher concentration compared to that used in comparison to I3C in the cell line studies to account for possible sequestration in the subcutaneous as well as mammary adipose tissue. As shown in Figure 9, both I3C and 1-benzyl-I3C were effective in inhibiting growth of MCF-7 cell xenografts at approximately equivalent rates, although a significantly lower concentration of 1-benzyl-I3C was used. Both indole compounds suppressed xenograft growth within three weeks of treatment, whereas, in vehicle control treated animals the xenografts showed robust growth. In vehicle control treated cells, the MCF-7 cell derived tumors displayed intense gross tumor vascularization, whereas, in sharp contrast, in I3C and 1-benzyl-I3C treated animals the xenografts appeared white and much smaller in size (Figure 9, lower panel). The vehicle control treated xenografts felt hard, which is consistent with intense growth of cells, whereas, the texture of the residual tumors from I3C and 1-benzyl-I3C treated mice was of a pliable nature, consistent with reduced cell density in the xenografts. Another interesting gross observation was that the vehicle control treated host mouse mammary fat pad showed intense vascularization, whereas this process was strongly inhibited by I3C and 1-benzyl-I3C (data not shown). A similar xenograft experiment was carried out using the estrogen receptor-alpha negative MDA-MB-231 human breast cancer cells. Preliminary studies show that compared to the calculated volumes of the vehicle control xenografts, I3C and 1-benzyl-I3C inhibited tumor growth by approximately 75% (data not shown). Thus, 1-benzyl-I3C caused a marked in vivo growth arrest of human breast cancer cell xenografts, suggesting that this compound represents a significantly more effective I3C-derived compound compared to its I3C parental compound.
Fig. 9.
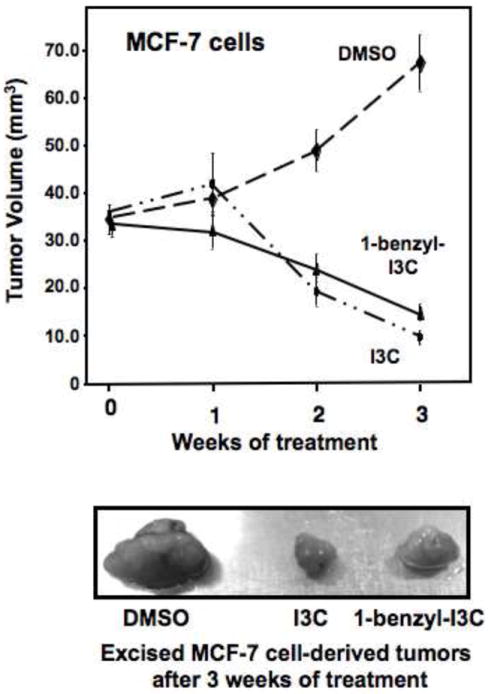
In vivo effects of 1-benzyl-I3C and I3C on the growth of human MCF-7 breast cancer cells derived tumors from xenografts in athymic mice. MCF-7 cell xenografts were grown to a tumor size of 35 mm3 in athymic mice, and then the animals were injected with 30 mg/kg body mass 1-benzyl-I3C, 300 mg/kg body mass I3C or with the DMSO vehicle control as described in the Methods and Materials section. Tumor size was monitored throughout a three-week time course, and the residual tumors were excised and photographically visualized (lower panel).
4. Discussion
In vivo and cell line studies have shown that the natural phytochemical I3C has potent anti-proliferative properties [6-10] and have implicated this indole as a potential therapeutic for treatment of human reproductive cancers. A key limitation for the use of I3C is the need for relatively high concentrations and the conversation of a portion of this compound intracellularly into self condensation products such as DIM [35], which can also inhibit cell proliferation [6, 7, 9, 10]. Attempts by other laboratories to develop more potent synthetic I3C-based compounds have been only partially successful because of the generally low enhancement of potency of the biological activity with most compounds limited to 5-10 fold increased in potency [36], with only one compound showing as much as a 100-fold increased potency for apoptosis [21]. In addition, there was a limited assessment of anti-proliferative mechanisms of the I3C derivatives to show that they displayed I3C-specific responses. Also, for certain synthetic I3C derivatives, the final structures were altered so drastically that the final compounds closely resembles self-condensation products of I3C [37], suggesting that the observed biological activities were not related to the I3C parental compound. In this regard, several recent studies have generated more potent derivatives of DIM, which is an I3C dimerization product, that showed enhanced potencies for cytoxicity and induction of apoptosis of cancer cells [42, 43], and for activation of the Nur77 and PPAR-gamma orphan nuclear receptors [7, 44]. In a systematic structure-activity analysis of I3C, we previously reported that addition of substituents to the nitrogen at position 1 in the indole ring with the more hydrophobic alkyl groups showing the highest potency [38]. Based on this analysis, a novel I3C derivative, 1-benzyl-I3C was synthesized that is designed to display a significantly increased hydrophobic character. In addition, 1-benzyl-I3C is significantly more stable than I3C, a property that is likely due to the steric effect of the bulky 1-benzyl substituent, which hampers oligomerization of the 1-benzyl-indol-3-yl methyl cation.
The metabolism of 1-benzyl-I3C is not known; however, based on the structure, the formulation in vitro of compounds hydroxylated or epoxydated in the phenyl ring is not possible. We cannot exclude metabolic degradation in vivo at some extent leading to an N-debenzylated product or to a compound hydroxyalted or somehow oxidated in the benzyl moiety. We have shown that 1-benzyl-I3C is a rather stable compound and is not totally degraded even after 10 days in ethanol-citrate phosphate buffer solution at pH 5.5. This result directly implies that the condensation of 1-benzyl-I3C into dibenzyl-DIM in vitro (pH neutral or somewhat higher) occurs only a very low rate and therefore the biological effects of 1-benzyl-I3C are not dependent on its conversation into a DIM-like derivative.
Our results show that 1-benzyl-I3C is the most potent synthetic derivative of I3C to date, with an approximately 1000-fold increased potency of the cell cycle arrest of both human estrogen responsive and estrogen independent human breast cancer cells. 1-Benzyl-I3C has an IC50 value in the nano molar range as compared to micro molar amounts of I3C, and thereby overcomes the problem of relatively high concentrations of I3C that is required to suppress the growth of human breast cancer cells, and implicates the feasible development of 1-benzyl-I3C as a therapeutic option against human breast cancer, and likely other types of cancers.
Importantly, 1-benzyl-I3C maintained the hallmark I3C anti-proliferative responses in estrogen-sensitive and independent human breast cancer cells, in particular the G1 cell cycle arrest and control of G1 acting cell cycle components, suggesting that addition of the 1-benzyl moiety does not qualitatively disrupt the biological properties of I3C. More specifically, treatment with this I3C derivative caused a robust inhibition of CDK2 specific enzymatic activity, and down-regulated CDK6 transcript and protein levels that accounts for a loss of total cellular CDK6 activity. Inhibition of G1-acting CDKs leads to a decrease in the level of endogenous and in vitro Rb phosphorylation, which prevents transcription of S-phase genes and DNA synthesis. We previously showed that I3C that disrupts Sp1 interactions with the CDK6 promoter [28], although an analysis of endogenous interactions were not evaluated. Furthermore, chromosome immunoprecipitation assays revealed that treatment with either 1-benzyl-I3C or the I3C parental compound disrupted the endogenous interactions of the Sp1 transcription factor with its Sp1-Ets composite element in the CDK6 promoter. DIM, the major bioactive condensation product of I3C, has no effect on CDK6 expression, thus showing that 1-benzyl-I3C maintains its I3C specific properties at a significantly increased potency.
In human breast cancer cells, 1-benzyl-I3C markedly reduced CDK4 kinase activity compared to the relatively moderate effects of the I3C parental compound. This significant effect of 1-benzyl-I3C is likely due to an increase in the level of p15, a potent CDK4 inhibitor. In addition, 1-benzyl-I3C stimulated the levels of the p21 and p27 CDK inhibitors. Thus, 1-benzyl-I3C has a more potent and somewhat expanded efficacy of its cell cycle effect compared to the I3C parental compound. Thus, the cumulative inhibition of cellular kinase activities of all three G1 acting cyclin/CDK protein complexes may account for the ~1000 fold more potency of 1-benzyl-I3C towards the suppression of human breast cancer cell growth. It also worth mentioning that the increased hydrophobic nature of 1-benzyl-I3C compared to I3C may potentially play a role in its enhanced potency through an improved stability and/or augmented cellular uptake. We recently established that the direct I3C inhibition of elastase enzymatic activity disrupts cyclin E protein processing and is required for the G1 cell cycle arrest [30]. We are currently exploring whether the enhanced bioactivity of 1-benzyl-I3C is due, in part, to its increased affinity for elastase, and perhaps other indole target proteins in human breast cancer cells.
The fact that 1-benzyl-I3C can effectively inhibit the growth of hormone responsive and hormone unresponsive breast cancer cells suggests that this novel I3C derivative can potentially be used for treating a wide range of breast cancer types, which has been a fundamental problem with the available therapeutic compounds. Furthermore, similar to what we have shown for the I3C parental compound, 1-benzyl-I3C inhibits expression of estrogen receptor-alpha, which mediates the mitogenic properties of estrogens, and acts with tamoxifen to inhibit the proliferation of steroid responsive MCF-7 human breast cancer cells to a greater extent than either compound alone. This result implicates the use of 1-benzyl-I3C in combination with anti-estrogen therapies that would allow lower doses and thereby reduced side effects or decreased resistance to the anti-estrogens.
1-Benzyl-I3C elicited a potent anti-proliferative response of human breast cancer cell derived tumors in athymic mice, demonstrating that this synthetic indole is bioactive in the in vivo environment. Treatment with either 1-benzyl-I3C or I3C caused comparable inhibitory effects on growth rates of the xenografts as well as on gross blood vessel density, although significantly reduced amounts of the I3C derivative were utilized compared to I3C. Estrogen stimulation is critical for MCF-7 xenograft growth [45], and the experiments required implantation of estrogen capsules for the generation of palpable tumor prior to the indole treatments. In addition to direct effects of estrogens on the cell cycle progression, estrogens have been shown to directly affect tumoral angiogenesis [46]. We propose that the indole inhibition of MCF-7 tumor growth is likely due to a combination of the direct cell cycle arrest of MCF-7 cells and by inhibiting estrogen dependent extratumoral vascularization. Given that the only treatment options for the treatment of breast cancer is hormone therapy, chemotherapy, radiation therapy, immune therapy and/or the surgical removal of the breast, the cellular and in vivo anti-proliferative properties of 1-benzyl-I3C and the low effective concentration makes this novel I3C derivative a promising candidate for its future development as a therapeutic agent for human reproductive cancers.
Acknowledgments
We greatly appreciate the comments and suggestions by the entire Firestone, Bjeldanes and Preobrazhenskaya laboratories during the course of our work.
This study was supported by National Institutes of Health Public Service grant CA102360 awarded from the National Cancer Institute. S.N. Sundar was supported by a postdoctoral fellowship from the Susan G. Komen Breast Cancer Fund, and C. N. Marconett was supported by a dissertation fellowship from the California Breast Cancer Research Program (#13GB-1801).
Abbreviations
- 1-benzyl-I3C
1-benzyl-indole-3-carbinol
- CDK
cyclin dependent kinase
- ChIP
chromatin immunoprecipitation
- DIM
3,3’-diindolylmethane
- ER-alpha
estrogen receptor-alpha
- I3C
indole-3-carbinol
- Rb
retinoblastoma protein
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van Poppel G, Verhoeven DT, Verhagen H, Goldbohm RA. Brassica vegetables and cancer prevention. Epidemiology and mechanisms. Adv Exp Med Biol. 1999;472:159–168. doi: 10.1007/978-1-4757-3230-6_14. [DOI] [PubMed] [Google Scholar]
- 2.Terry P, Wolk A, Persson I, Magnusson C. Brassica vegetables and breast cancer risk. JAMA. 2001;285:2975–2977. doi: 10.1001/jama.285.23.2975. [DOI] [PubMed] [Google Scholar]
- 3.Lopez-Otin C, Diamandis EP. Breast and prostate cancer: an analysis of common epidemiological, genetic, and biochemical features. Endocr Rev. 1998;19:365–396. doi: 10.1210/edrv.19.4.0337. [DOI] [PubMed] [Google Scholar]
- 4.Higdon JV, Delage B, Williams DE, Dashwood RH. Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol Res. 2007;55:224–236. doi: 10.1016/j.phrs.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moiseeva EP, Manson MM. Dietary chemopreventive phytochemicals: too little or too much? Cancer Prev Res. 2009;2:611–616. doi: 10.1158/1940-6207.CAPR-08-0102. [DOI] [PubMed] [Google Scholar]
- 6.Aggarwal BB, Ichikawa H. Molecular targets and anti-cancer potential of indole-3-carbinol and its derivatives. Cell Cycle. 2005;4:1201–1215. doi: 10.4161/cc.4.9.1993. [DOI] [PubMed] [Google Scholar]
- 7.Safe S, Papineni S, Chintharlapalli S. Cancer chemotherapy with indole-3-carbinol, bis(3’-indolyl)methane and synthetic analogs. Cancer Lett. 2008;269:326–338. doi: 10.1016/j.canlet.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim YS, Milner JA. Targets for indole-3-carbinol in cancer prevention. Journal of Nutritional Biochemistry. 2005;16:65–73. doi: 10.1016/j.jnutbio.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Firestone GL, Sundar SN. Minireview: Modulation of Hormone Receptor Signaling by Dietary Anticancer Indoles. Molecular Endocrinology. 2009;23:1940–1947. doi: 10.1210/me.2009-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Firestone GL, Bjeldanes LF. Indole-3-Carbinol (I3C) and 3-3’diindolylmethane (DIM) anti-proliferative signaling pathways control cell cycle gene transcription in human breast cancer cells by regulating promoter-Sp1 tanscription factor interactions. J Nutrition. 2003;133:2448S–2455S. doi: 10.1093/jn/133.7.2448S. [DOI] [PubMed] [Google Scholar]
- 11.Cover CM, Hsieh SJ, Tran SH, Hallden G, Kim GS, Bjeldanes LF, Firestone GL. Indole-3-carbinol inhibits the expression of cyclin-dependent kinase-6 and induces a G1 cell cycle arrest of human breast cancer cells independent of estrogen receptor signaling. J Biol Chem. 1998;273:3838–3847. doi: 10.1074/jbc.273.7.3838. [DOI] [PubMed] [Google Scholar]
- 12.Brew CT, Aronchik I, Hsu JC, Sheen JH, Dickson RB, Bjeldanes LF, Firestone GL. Indole-3-carbinol activates the ATM signaling pathway independent of DNA damage to stabilize p53 and induce G1 arrest of human mammary epithelial cells. Int J Cancer. 2006;118:857–868. doi: 10.1002/ijc.21445. [DOI] [PubMed] [Google Scholar]
- 13.Garcia HH, Brar GA, Nguyen DH, Bjeldanes LF, Firestone GL. Indole-3-carbinol (I3C) inhibits cyclin-dependent kinase-2 function in human breast cancer cells by regulating the size distribution, associated cyclin E forms, and subcellular localization of the CDK2 protein complex. J Biol Chem. 2005;280:8756–8764. doi: 10.1074/jbc.M407957200. [DOI] [PubMed] [Google Scholar]
- 14.Moiseeva EP, Heukers R, Manson MM. EGFR and Src are involved in indole-3-carbinol-induced death and cell cycle arrest of human breast cancer cells. Carcinogenesis. 2007;28:435–445. doi: 10.1093/carcin/bgl171. [DOI] [PubMed] [Google Scholar]
- 15.Rahman KM, Sarkar FH, Banerjee S, Wang Z, Liao DJ, Hong X, Sarkar FH. Therapeutic intervention of experimental breast cancer bone metastasis by indole-3-carbinol in SCID-human mouse model. Mol Cancer Ther. 2006;5:2747–2756. doi: 10.1158/1535-7163.MCT-06-0221. [DOI] [PubMed] [Google Scholar]
- 16.Chinni SR, Li Y, Upadhyay S, Koppolu PK, Sarkar FH. Indole-3-carbinol (I3C) induced cell growth inhibition, G1 cell cycle arrest and apoptosis in prostate cancer cells. Oncogene. 2001;20:2927–2936. doi: 10.1038/sj.onc.1204365. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Hsu JC, Kinseth MA, Bjeldanes LF, Firestone GL. Indole-3-carbinol (I3C) induces a G1 cell cycle arrest of human LNCaP prostate cancer cells and inhibits expression of prostate specific antigen. Cancer. 2003;98:2511–2520. doi: 10.1002/cncr.11844. [DOI] [PubMed] [Google Scholar]
- 18.Hsu JC, Dev A, Wing A, Brew CT, Bjeldanes LF. Firestone, Indole-3-carbinol mediated cell cycle arrest of LNCaP human prostate cancer cells requires the induced production of activated p53 tumor suppressor protein. Biochem Pharmacol. 2006;72:1714–1723. doi: 10.1016/j.bcp.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 19.Garikapaty VP, Ashok BT, Tadi K, Mittelman A, Tiwari RK. 3,3’-Diindolylmethane downregulates pro-survival pathway in hormone independent prostate cancer. Biochem Biophys Res Commun. 2006;340:718–725. doi: 10.1016/j.bbrc.2005.12.059. [DOI] [PubMed] [Google Scholar]
- 20.Souli E, Machluf M, Morgenstern A, Sabo E, Yannai S. Indole-3-carbinol (I3C) exhibits inhibitory and preventive effects on prostate tumors in mice. Food Chem Toxicol. 2008;46:863–870. doi: 10.1016/j.fct.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 21.Weng JR, Tsai CH, Kulp SK, Wang D, Lin CH, Yang HC, Ma Y, Sargeant A, Chiu CF, Tsai MH, Chen CS. A potent indole-3-carbinol derived antitumor agent with pleiotropic effects on multiple signaling pathways in prostate cancer cells. Cancer Research. 2007;67:7815–7824. doi: 10.1158/0008-5472.CAN-07-0794. [DOI] [PubMed] [Google Scholar]
- 22.Meng Q, Qi M, Chen DZ, Yuan R, Goldberg ID, Rosen EM, Auborn K, Fan S. Suppression of breast cancer invasion and migration by indole-3-carbinol: associated with up-regulation of BRCA1 and E-cadherin/catenin complexes. J Mol Med. 2000;78:155–165. doi: 10.1007/s001090000088. [DOI] [PubMed] [Google Scholar]
- 23.Brew CT, Aronchik I, Kosco K, McCammon J, Bjeldanes LF, Firestone GL. Indole-3-carbinol inhibits MDA-MB-231 breast cancer cell motility and induces stress fibers and focal adhesion formation by activation of Rho kinase activity. Int J Cancer. 2009;124:2294–2302. doi: 10.1002/ijc.24210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kojima T, Tanaka T, Mori H. Chemoprevention of spontaneous endometrial cancer in female Donryu rats by dietary indole-3-carbinol. Cancer Research. 1994;54:1446–1449. [PubMed] [Google Scholar]
- 25.Sharma S, Stutzman JD, Kelloff GJ, Steele VE. Screening of potential chemopreventive agents using biochemical markers of carcinogenesis. Cancer Research. 1994;54:5848–5855. [PubMed] [Google Scholar]
- 26.Bradlow HL. Review. Indole-3-carbinol as a chemoprotective agent in breast and prostate cancer. In Vivo. 2008;22:441–445. [PubMed] [Google Scholar]
- 27.Sundar SN, Kerekatte V, Equinozio CN, Doan VB, Bjeldanes LF, Firestone GL. Indole-3-carbinol selectively uncouples expression and activity of estrogen receptor subtypes in human breast cancer cells. Mol Endocrinol. 2006;20:3070–3082. doi: 10.1210/me.2005-0263. [DOI] [PubMed] [Google Scholar]
- 28.Cram EJ, Liu BD, Bjeldanes LF, Firestone GL. Indole-3-carbinol inhibits CDK6 expression in human MCF-7 breast cancer cells by disrupting Sp1 transcription factor interactions with a composite element in the CDK6 gene promoter. J Biol Chem. 2001;276:22332–22340. doi: 10.1074/jbc.M010539200. [DOI] [PubMed] [Google Scholar]
- 29.Moiseeva EP, Heukers R. Indole-3-carbinol-induced modulation of NF-kappaB signalling is breast cancer cell-specific and does not correlate with cell death. Breast Cancer Res Treat. 2008;109:451–462. doi: 10.1007/s10549-007-9669-6. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen HH, Aronchik I, Brar GA, Nguyen DH, Bjeldanes LF, Firestone GL. The dietary phytochemical indole-3-carbinol is a natural elastase enzymatic inhibitor that disrupts cyclin E protein processing. Proc Natl Acad Sci U S A. 2008;105:19750–19755. doi: 10.1073/pnas.0806581105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cover CM, Hsieh SJ, Cram EJ, Hong C, Riby JE, Bjeldanes LF, Firestone GL. Indole-3-carbinol and tamoxifen cooperate to arrest the cell cycle of MCF-7 human breast cancer cells. Cancer Research. 1999;59:1244–1251. [PubMed] [Google Scholar]
- 32.Grose KR, Bjeldanes LF. Oligomerization of indole-3-carbinol in aqueous acid. Chem Res Toxicol. 1992;5:188–193. doi: 10.1021/tx00026a007. [DOI] [PubMed] [Google Scholar]
- 33.De Kruif CA, Marsman JW, Venekamp JC, Falke HE, Noordhoek J, Blaauboer BJ, Wortelboer HM. Structure elucidation of acid reaction products of indole-3-carbinol: detection in vivo and enzyme induction in vitro. Chem Biol Interact. 1991;80:303–315. doi: 10.1016/0009-2797(91)90090-t. [DOI] [PubMed] [Google Scholar]
- 34.Riby JE, Feng C, Chang YC, Schaldach CM, Firestone GL, Bjeldanes LF. The major cyclic trimeric product of indole-3-carbinol is a strong agonist of the estrogen receptor signaling pathway. Biochemistry. 2000;39:910–918. doi: 10.1021/bi9919706. [DOI] [PubMed] [Google Scholar]
- 35.Staub RE, Feng C, Onisko B, Bailey GS, Firestone GL, Bjeldanes LF. Fate of indole-3-carbinol in cultured human breast tumor cells. Chem Res Toxicol. 2002;15:101–109. doi: 10.1021/tx010056m. [DOI] [PubMed] [Google Scholar]
- 36.Brandi G, Paiardini M, Cervasi B, Fiorucci C, Filippone P, De Marco C, Zaffaroni N, Magnani M. A new indole-3-carbinol tetrameric derivative inhibits cyclin-dependent kinase 6 expression, and induces G1 cell cycle arrest in both estrogen-dependent and estrogen-independent breast cancer cell lines. Cancer Research. 2003;63:4028–4036. [PubMed] [Google Scholar]
- 37.Chao WR, Yean D, Amin K, Green C, Jong L. Computer-Aided Rational Drug Design: A Novel Agent (SR13668) Designed to Mimic the Unique Anticancer Mechanisms of Dietary Indole-3-Carbinol to Block Akt Signaling. J Med Chem. 2007;50:3412–3415. doi: 10.1021/jm070040e. [DOI] [PubMed] [Google Scholar]
- 38.Jump SM, Kung J, Staub R, Kinseth MA, Cram EJ, Yudina LN, Preobrazhenskaya MN, Bjeldanes LF, Firestone GL. N-Alkoxy derivatization of indole-3-carbinol increases the efficacy of the G1 cell cycle arrest and of I3C-specific regulation of cell cycle gene transcription and activity in human breast cancer cells. Biochem Pharmacol. 2008;75:713–724. doi: 10.1016/j.bcp.2007.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heaney H, Ley SV. 1-benzylindole. Organic Syntheses. 1973;53:1858. [Google Scholar]
- 40.James PN, Snyder HR. Indole-3-aldehyde. Organic Syntheses. 1959;39:30–33. [Google Scholar]
- 41.Mukhanov VI, Kaganskii MM, Sorokin AA, Antonyan SG, Tananova GV, Mikhailevskaya LL, Kinzirskii AS, Preobrazhenskaya MN. Search for new drugs. Pharm Chem J. 1994;28:446–451. [Google Scholar]
- 42.Abdelrahim M, Newman K, Vanderlaag K, Samudio I, Safe S. Safe, 3,3’-diindolylmethane (DIM) and its derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5. Carcinogenesis. 2006;27:717–728. doi: 10.1093/carcin/bgi270. [DOI] [PubMed] [Google Scholar]
- 43.Maciejewska D, Rasztawicka M, Wolska I, Anuszewska E, Gruber B. Novel 3,3’-diindolylmethane derivatives: synthesis and cytotoxicity, structural characterization in solid state. Eur J Med Chem. 2009;44:4136–4147. doi: 10.1016/j.ejmech.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 44.Cho SD, Yoon K, Chintharlapalli S, Abdelrahim M, Lei P, Hamilton S, Khan S, Ramaiah SK, Safe S. Nur77 agonists induce proapoptotic genes and responses in colon cancer cells through nuclear receptor-dependent and nuclear receptor-independent pathways. Cancer Research. 2007;67:674–683. doi: 10.1158/0008-5472.CAN-06-2907. [DOI] [PubMed] [Google Scholar]
- 45.Huseby RA, Maloney TM, McGrath CM. Evidence for a direct growth-stimulating effect of estradiol on human MCF-7 cells in vivo. Cancer Research. 1984;44:2654–2659. [PubMed] [Google Scholar]
- 46.Lindner DJ, Borden EC. Effects of tamoxifen and interferon-beta or the combination on tumor-induced angiogenesis. Int J Cancer. 1997;71:456–461. doi: 10.1002/(sici)1097-0215(19970502)71:3<456::aid-ijc25>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]