

Abstract
Folate deficiency and hypomethylation have been implicated in a number of age-related neurodegenerative disorders including dementia and Parkinson's disease (PD). Levodopa (l-dopa) therapy in PD patients has been shown to cause an increase in plasma total homocysteine as well as depleting cellular concentrations of the methyl donor, S-adenosylmethionine (SAM), and increasing the demethylated product S-adenosylhomocysteine (SAH). Modulation of the cellular SAM/SAH ratio can influence activity of methyltransferase enzymes, including leucine carboxyl methyltransferase that specifically methylates Ser/Thr protein phosphatase 2A (PP2A), a major Tau phosphatase. Here we show in human SH-SY5Y cells, in dopaminergic neurons, and in wild-type mice that l-dopa results in a reduced SAM/SAH ratio that is associated with hypomethylation of PP2A and increased phosphorylation of Tau (p-Tau) at the Alzheimer's disease-like PHF-1 phospho-epitope. The effect of l-dopa on PP2A and p-Tau was exacerbated in cells exposed to folate deficiency. In the folate-deficient mouse model, l-dopa resulted in a marked depletion of SAM and an increase in SAH in various brain regions with parallel downregulation of PP2A methylation and increased Tau phosphorylation. l-Dopa also enhanced demethylated PP2A amounts in the liver. These findings reveal a novel mechanism involving methylation-dependent pathways in l-dopa induces PP2A hypomethylation and increases Tau phosphorylation, which may be potentially detrimental to neuronal cells.
Introduction
Administration of levodopa (l-dopa) to Parkinson's disease (PD) patients has proven to be an effective treatment for replenishing depleted levels of dopamine (DA) that are characteristic of this neurodegenerative movement disorder (Cotzias et al., 1969; Poewe et al., 2010). Combining l-dopa with a peripheral decarboxylase inhibitor (Rinne et al., 1972), such as carbidopa or benserazide, can reduce the metabolism of l-dopa in peripheral tissues and maximize the amount that enters into the CNS. More recent pharmacotherapies include the addition of catechol-O-methyltransferase (COMT) inhibitors, which prolong the half-life of l-dopa and DA resulting in an enhanced effect (Ericsson, 1971; Roberts et al., 1993). This combination therapy can reduce l-dopa-related side effects such as nausea and vomiting. Despite the success of l-dopa combination therapies, which provide almost immediate symptomatic relief, these treatments do not halt the underlying neurodegenerative process. The effectiveness of therapy wanes with time and is associated with dyskinesias and “on–off” phenomenon (Nutt, 2001; Stacy, 2009). Additionally, l-dopa is associated with adverse metabolic effects in relation to methylation and homocysteine metabolism. Several investigators have shown that PD patients treated with l-dopa have increased levels of plasma total homocysteine (tHcy) (Irizarry et al., 2005; Müller and Kuhn, 2009; Zoccolella et al., 2009). Elevated tHcy is a well established independent risk factor for vascular disease (McCully, 2007; Zhou and Austin, 2009) and has more recently been associated with depression (Bottiglieri, 2005) and increased risk for Alzheimer's disease (AD) (Selhub et al., 2010; Zhuo et al., 2011). Increased tHcy may occur as a result of COMT-dependent methylation of l-dopa to 3-O-methyl-dopa (3-OMD). In this reaction, the methyl group originates from S-adenosylmethionine (SAM) producing S-adenosylhomocysteine (SAH), which is then converted to Hcy (Fig. 1). l-Dopa administration has been shown to decrease SAM and increase SAH levels in rat tissues (Wagner et al., 1984; Miller et al., 1997). This may affect other methylation-dependent pathways involving DNA, proteins, and other small molecular weight compounds, thereby compromising normal cell function (Cantoni, 1987). The amount of folate present in the diet has been shown to influence tissue concentrations of SAM and SAH. We have recently demonstrated that C57BL/6 mice reared on a folate-deficient diet have a reduced SAM/SAH ratio in brain tissue that is associated with hypomethylation of the catalytic C subunit of Ser/Thr protein phosphatase 2A (PP2A), a major brain Tau phosphatase. This resulted in altered PP2A substrate specificity and subsequent accumulation of phosphorylated-Tau (p-Tau) in several brain regions (Sontag et al., 2008). Downregulation of PP2A methylation has been reported in autopsy AD brain tissue, and correlates with enhanced neuronal p-Tau levels (Sontag et al., 2004). Accumulation of p-Tau species is a significant early event involved in the neuropathology of dementia and other neurodegenerative disorders (Iqbal et al., 2010). In view of these findings, we have undertaken a study to determine the interaction between l-dopa and folate deficiency and the effect on methylation cycle metabolites, PP2A methylation, and p-Tau in cultured cells and regional mouse brain tissue.
Figure 1.

Schematic model showing the metabolism of l-dopa and its potential effect on PP2A methylation and Tau phosphorylation. Exogenously administered l-dopa is either decarboxylated to form DA or may be methylated in a COMT-dependent reaction that uses SAM as the methyl donor to form 3-OMD. The by-product of COMT activity is SAH, which is converted to Hcy by the reversible reaction catalyzed by SAH hydrolase. Folate deficiency promotes accumulation of Hcy, which may be converted back to SAH, a potent inhibitor of methyltransferases. Methylation of PP2A C subunit by the methyltransferase LCMT1 promotes assembly and stabilization of PP2A/Bα heterotrimers that dephosphorylate Tau on Ser/Thr residues. Conversely, GSK-3β and other kinases (data not shown here) can phosphorylate Tau on Ser/Thr residues. Met, methionine; MTHF, methyltetrahydrofolate; THF, tetrahydrofolate, SAHH, SAH hydrolase; MTR, methionine synthase; GSK3β, glycogen synthase kinase 3 β.
Materials and Methods
Materials.
Unless indicated, all chemicals and drugs used in this study were obtained from Sigma-Aldrich.
Cell culture and treatment.
Human SH-SY5Y neuroblastoma cells (American Type Culture Collection) were maintained in RPMI-1640 medium (Invitrogen) containing 2.5 mm HEPES, pH 7.4, 10% fetal bovine serum (FBS; HyClone and ThermoFisher Scientific) and 10 μg/ml gentamycin (Invitrogen). Exponentially growing SH-SY5Y cells were trypsinized and plated in normal cell culture medium. After 24 h, cells were washed with PBS and serum starved by overnight incubation in RPMI-1640 medium supplemented with 1% dialyzed FBS (Invitrogen) before treatment.
Mature human dopaminergic neurons were obtained after culturing human-induced pluripotent cell-derived neural progenitor cells for 14 d using the ReproNeuro DA kit and following exactly the manufacturer's instructions (ReproCELL). A subset of cells was assessed for expression of β-III-tubulin and tyrosine hydroxylase (TH) before treatment. When indicated, human SH-SY5Y cells and dopaminergic neurons were incubated for 2 h in regular or folate-free RPMI-1640 media supplemented with 2% dialyzed FBS (Invitrogen), as reported earlier for N2a cells (Sontag et al., 2008). Cells were treated in the same medium with each drug or vehicle alone for the indicated period of time just before harvesting for Western blot analysis. When indicated, cell morphology and viability were assessed as described previously (Sontag et al., 2008).
Confocal microscopy.
A subset of human dopaminergic cells was stained following a standard protocol (Nunbhakdi-Craig et al., 2007). Briefly, cells cultured on glass coverslips were fixed for 20 min with 4% paraformaldehyde, permeabilized for 5 min with 0.1% Triton X-100, incubated for 1 h in blocking buffer, and stained for 1 h with anti-β-III-tubulin (Sigma) followed by incubation for 1 h with Alexa Fluor 488-conjugated goat antibodies (Invitrogen). The samples were mounted with Fluoromount (Fisher Scientific) and examined on an Olympus FV1000 CLSM confocal microscope using a 40× objective. Captured images (z-stacks) were transferred to Adobe Photoshop/Illustrator CS5 for printing (Adobe Systems).
Animals.
All experiments with mice were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at Baylor Research Institute. Male C57BJ/6 mice were maintained in a temperature-controlled animal facility on a 12 h light/dark cycle and were allowed access to food and water ad libitum. At 4 weeks of age, mice were placed on an amino acid-defined diet and divided into three groups (n = 12 per group) containing various amounts of folate as follows: normal folate (NF; 6.7 mg/kg), low folate (LF; 0.2 mg/kg), or folate deficient (FD; 0 mg/kg). All diets contained succinylsulfathiazole (10 mg/kg) to inhibit gastrointestinal bacterial growth and prevent absorption of folate from this source. After 8 weeks on the diet, mice in each diet group received either two intraperitoneal injections of either saline or a combination of l-dopa (100 mg/kg) and benserazide (10 mg/kg). The second injection was administered 45 min after the first. All mice were killed 30 min after the second injection by CO2 asphyxiation. Blood was obtained by cardiac puncture and brain tissue rapidly removed for regional dissection of left and right hemispheres. Liver tissue was also obtained. Tissues were stored at −80°C until time of analysis with the regions from the left hemisphere used for metabolite analysis and the regions from the right hemisphere used for Western blot analysis.
Metabolite analysis.
Brain and liver tissue were deproteinized with 5 and 10 volumes, respectively, of perchloric acid (PCA; 0.1 m) containing diethylenetriaminepentaacetic acid (1 mg/ml) and dithioerythritol (0.1 mg/ml). After centrifugation, 10 μl of PCA extract was injected into an HPLC system coupled to UV detection for the analysis of SAM and SAH as previously described (Bottiglieri, 1990). l-Dopa, DA, and their methylated metabolites, 3-OMD and 3-methoxytyramine (3-MT) were measured by HPLC with colorimetric electrochemical detection as previously described (Ogburn et al., 2006). Plasma tHcy was determined by HPLC with fluorescence detection (Ubbink et al., 1991). Plasma folate was quantified with a Simultrac-SNB RIA kit (MP Biomedicals).
Determination of protein expression and PP2A methylation levels.
Total brain homogenates were prepared from each brain region exactly as described previously (Sontag et al., 2008). Total cell homogenates were prepared in buffer (Tris 25 mm, pH 7.4, 150 mm NaCl, 2 mm EDTA, 25 m sodium fluoride, 1 mm sodium orthovanadate, 2 mm dithiothreitol, 2 μm okadaic acid (OA), 5 mm PMSF, and 1% NP-40) containing a mixture of protease inhibitors (Roche) (Sontag et al., 2007). Detergent-soluble and -insoluble extracts were obtained after further centrifugation for 45 min at 4°C at 30,000 × g in a Jouan Refrigerated Benchtop Centrifuge (Jouan). Pellets were resuspended in the same buffer and further sonicated to disrupt protein aggregates before analysis. Equivalent amounts of proteins (∼30 μg) from cell extracts or equivalent aliquots (5 μl) of brain homogenates were analyzed on 4–12% Bis-Tris gels using the NU-PAGE system (Invitrogen) followed by Western blotting and densitometry exactly as described previously (Sontag et al., 2007, 2008). Monoclonal methylation- or demethylation-specific (Millipore) and methylation-independent (BD Biosciences) anti-C antibodies were used to quantify PP2A methylation levels by Western blotting and densitometry (Sontag et al., 2004, 2007, 2008). In some experiments, equivalent aliquots of cell or brain tissue homogenates were incubated for 30 min at 37°C in the absence or presence of 0.2 N sodium hydroxide (NaOH). This alkaline treatment results in complete demethylation of PP2A at Leu-309 and unmasking of the epitope recognized by antibodies directed against the C-terminal 299–309 region of PP2A C subunit. Thus, it can allow for detection of total C expression levels with anti-demethyl C antibodies, and for verification of the specificity of methyl-dependent anti-C antibodies (Sontag et al., 2004). Tau phosphorylation was determined exactly as described previously (Sontag et al., 2007, 2008) using monoclonal “PHF-1” recognizing Tau phosphorylated at the Ser396/Ser404 epitope (a gift from Dr. Peter Davies, Albert Einstein College of Medicine, New York) and rabbit anti-Tau antibodies (rPeptide) to detect total Tau. The expression of TH was assessed using a rabbit antibody raised against the recombinant human protein (a gift from Dr. Philip Dickson, University of Newcastle, New South Wales, Australia). Anti-actin or β-tubulin (Sigma-Aldrich) antibodies were used to normalize for protein loading.
Statistics.
Data were analyzed using one-way ANOVA with Dunnett's multiple-comparison t test. Differences with p values <0.05 were considered statistically significant.
Results
l-Dopa induces the accumulation of demethylated PP2A and p-Tau in human SH-SY5Y neuroblastoma cells and dopaminergic neurons
Incubation of human SH-SY5Y neuroblastoma cells for 2 h with l-dopa induced a dose-dependent decrease in both soluble and insoluble methylated PP2A C subunit levels and concomitant accumulation of demethylated PP2A enzymes (Fig. 2A,B). Time course experiments showed maximal effects on endogenous PP2A methylation after 2 h incubation with 50–100 μm l-dopa. The reduction of methylated C levels induced by 50 μm l-dopa was also similar to that observed with 100 nm of OA (Fig. 2C), a phosphatase inhibitor known to induce PP2A demethylation (Favre et al., 1997). Of note, the signal obtained with the anti-methyl C antibody was lost after alkaline treatment of cell extracts, which induces complete PP2A demethylation (Sontag et al., 2004), thereby confirming the antibody's specificity. SH-SY5Y cells are a widely used cell culture model for PD studies, and it is well established that long-term exposure of these cells to l-dopa is associated with oxidative stress and cellular toxicity, ultimately resulting in cell death (Martin et al., 2005). Accordingly, we observed considerable morphological changes and cell death after incubation of cells for 16 h with 50 μm l-dopa. In contrast, under our experimental conditions based on short exposure of SH-SY5Y cells to the drug, we did not observe any loss of cell viability (data not shown), as reported previously (Martin et al., 2005). Moreover, incubation of SH-SY5Y cells for 2 h with 50 μm l-dopa in the presence of N-acetylcysteine (NAC) and ascorbic acid, two antioxidants known to protect against l-dopa-mediated oxidative stress (Weingarten et al., 2001), did not prevent l-dopa-induced decrease in endogenous PP2A methylation (Fig. 2D).
Figure 2.
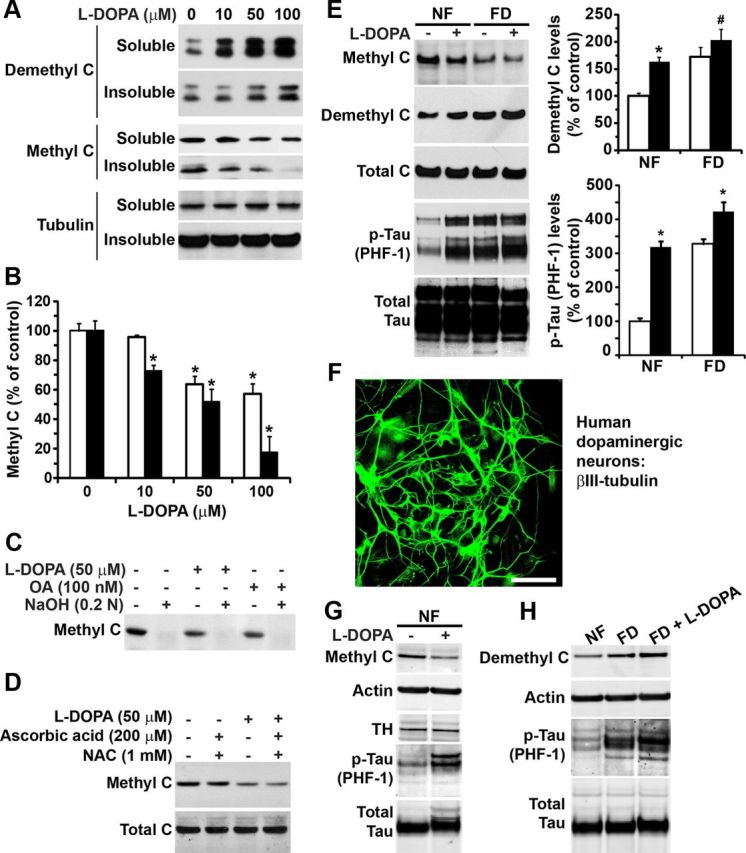
l-Dopa induces the accumulation of demethylated PP2A and p-Tau in human SH-SY5Y neuroblastoma cells and dopaminergic neurons. A–D, Serum-starved SH-SY5Y cells were incubated for 2 h with the indicated concentrations of drugs or vehicle alone in the same medium. A, Representative immunoblot of demethylated and methylated C expression levels in detergent-soluble and -insoluble cell extracts. Note that it has been reported that the PP2A C subunit can migrate as a doublet or a single band by gel electrophoresis; whether one or two bands are seen can vary for the same sample from gel to gel, and the underlying reasons remain unknown (Ogris et al., 1997; Nunbhakdi-Craig et al., 2007). B, Methylated C levels in soluble (white bars) and insoluble (black bars) cell extracts (n = 4, mean ± SD; *p < 0.001, l-dopa vs vehicle). C, Like OA, l-dopa induces a decrease in the amounts of methylated C in total cell homogenates. The specificity of the anti-methyl C antibody was verified by treating aliquots of total cell extracts with NaOH, which induces PP2A demethylation. D, The addition of antioxidants, including ascorbic acid and NAC, does not prevent l-dopa-induced decrease in methylated C levels. E, SH-SY5Y cells were incubated for 2 h in NF or FD medium in the absence (−) or presence (+) of 50 μm l-dopa before harvesting. Representative immunoblots of total cell extracts are shown. The levels of demethylated C and Tau phosphorylated at the PHF-1 epitope were quantified in l-dopa-treated (black bars) and untreated (white bars) cells (n = 4, mean ± SD; *p < 0.001, #p < 0.01, l-dopa vs vehicle). F, Human dopaminergic neurons stained with anti-β-III-tubulin, a neuronal marker. Scale bar, 40 μm. G, Representative immunoblots of total cell extracts from human dopaminergic neurons incubated for 2 h in NF medium in the absence (−) or presence (+) of 100 μm l-dopa. Note that cells are also positive for the presence of TH. H, Representative immunoblots of total cell extracts from human dopaminergic neurons incubated for 2 h in NF, FD, or FD medium containing 100 μm l-dopa.
Next, we further investigated the effect of l-dopa in folate-deprived SH-SY5Y cells. We have previously shown that PP2A methylation is decreased in N2a cells that have been incubated for 2–4 h in FD medium (Sontag et al., 2008). Likewise, a decrease in methylated and parallel increase in demethylated C subunit levels were observed after incubation of SH-SY5Y cells for 2 h in FD medium (Fig. 2E). Interestingly, we found that the combination of l-dopa and folate starvation resulted in enhanced accumulation of demethylated C levels relative to control cells exposed to l-dopa in NF medium. We have previously reported that reduced PP2A methylation in cultured N2a cells and in vivo is associated with increased phosphorylation of endogenous Tau at several epitopes (Sontag et al., 2007, 2008). Accordingly, decreased PP2A methylation also coincided with enhanced p-Tau at the PHF-1 (phospho-Ser396/Ser404) epitope in SH-SY5Y cells incubated in FD medium or treated with l-dopa in NF medium). The greatest increase in demethylated C and p-Tau levels was observed after exposure of SH-SY5Y cells to l-dopa in FD medium, indicating that l-dopa augments the effect of folate deficiency. Significantly, we found similar effects in human dopaminergic neurons (Fig. 2F). Treatment of neurons with l-dopa induced a 40–49% decrease in methylated PP2A and concomitant 168–179% increase in endogenous p-Tau (PHF-1) levels (Fig. 2G). Relative to controls in NF medium, incubation of neurons for 2 h in FD medium induced a 171–177% and 201–209% increase in endogenous demethylated PP2A and p-Tau amounts, respectively (Fig. 2H). As observed in SH-SY5Y cells, these effects were further accentuated in dopaminergic neurons treated with l-dopa in FD medium. Folate starvation in the presence of l-dopa resulted in a 200–210% increase in demethylated PP2A levels, and 235–250% increase in p-Tau levels in the dopaminergic neurons, relative to untreated controls maintained in NF medium.
l-Dopa exacerbates the effects of folate deficiency on methylation cycle metabolites and PP2A methylation state in mouse tissues
Our findings in cell culture systems prompted us to investigate the effect of l-dopa in a mouse model where we could determine organ-specific changes in methylation cycle metabolites and PP2A methylation following l-dopa treatment and folate deficiency. We have previously demonstrated that mice reared on FD diets have increased levels of plasma tHcy, which is indicative of reduced functional activity of methionine synthase, a key regulatory enzyme in folate metabolism and the methylation cycle (Sontag et al., 2008). Here, we found that administration of l-dopa led to a significant fivefold increase in plasma tHcy concentrations when compared with saline-treated mice (Table 1). This l-dopa-induced increase in plasma tHcy was significantly greater in mice that had been reared on either LF or FD diets (Table 1). Importantly, l-dopa administration had no effect on plasma folate levels, although the expected decrease in plasma folate concentration was similar in mice reared on either LF or FD diets between saline and l-dopa-treated mice (Table 1). Of particular interest is the effect of l-dopa and folate deficiency on hepatic concentrations of SAM and SAH. l-Dopa administration significantly decreased SAM in liver tissue in mice reared on an NF diet (Fig. 3A). This effect of l-dopa was markedly exacerbated in mice reared on an LF diet. Mice reared on FD diets had a similar low level of SAM; however, l-dopa did not have any further effect. The concentration of SAH in liver tissue was significantly higher in mice reared on an FD diet. In these mice, l-dopa led to an even greater increase in SAH (Fig. 3B). As a result, the hepatic SAM/SAH ratio was reduced when l-dopa was administered to mice on either an NF or FD diet (Fig. 3C). These changes in SAM and SAH were associated with a significant increase in the percentage of demethylated PP2A (Fig. 3D,E). Relative to mice on control diets, demethylated C levels were increased in mice on LF and FD diets. Remarkably, l-dopa administered to mice on an NF diet increased the amounts of demethylated C, which were significantly greater than given to mice on an FD diet (Fig. 3D,E).
Table 1.
Plasma folate and tHCY in l-dopa treated mice
| NF |
LF |
FD |
||||
|---|---|---|---|---|---|---|
| Saline | l-dopa | Saline | l-dopa | Saline | l-dopa | |
| Folate (nmol/L) | 138 ± 28 | 130 ± 18 | 20.3 ± 2.9* | 24.9 ± 4.6* | 5.2 ± 1.1* | 4.8 ± 0.7* |
| tHcy (μmol/L) | 8.8 ± 1.7 | 47.8 ± 17.7^ | 13.5 ± 2.4* | 63.8 ± 16.3*^ | 86.5 ± 15.0* | 131.5 ± 12.4*^ |
LF and FD diets are effective in lowering folate concentrations in blood, which for each diet were similar between saline and l-dopa-treated mice. l-Dopa treatment induced a marked increase in plasma tHcy levels, an effect that was exacerbated by LF and FD diets. Values represent mean ± SD for six mice in each group.
*Compared to NF diet in the same treatment group (ANOVA with Dunnett's multiple-comparison t test; p < 0.05).
^Compared to saline treated in the same diet group (t test; p < 0.05).
Figure 3.

Folate deficiency and l-dopa treatment lowers SAM and increases SAH levels in liver tissue and methylation of PP2A. A, Liver SAM levels. B, Liver SAH levels. C, Liver SAM/SAH ratio. D, Western blot analysis of demethylated PP2A. E, Open bars, saline treated (n = 6, mean ± SD); gray bars, l-dopa treated (n = 6, mean ± SD). *Compared with normal folate diet in the same treatment group (ANOVA with Dunnett's multiple-comparison t test, p < 0.05); ^Compared with saline treated in the same diet group (t test, p < 0.05).
Exogenously administered l-dopa is rapidly absorbed and decarboxylated to DA. Coadministration of benserazide, an inhibitor of l-amino acid decarboxylase, prevents metabolism to DA in peripheral tissues. However, l-dopa is also methylated to 3-OMD, a reaction that requires SAM and produces SAH. We have shown that l-dopa concentrations increase in mouse brain tissue following its administration with a concomitant increase in DA that varies depending on the distribution of dopaminergic innervation in various regions (Table 2). As expected, the highest levels of DA were found in the striatum, a dopaminergic neuron-rich region, followed by midbrain, frontal cortex, and cerebellum. In addition, methylation of l-dopa and DA led to significant increases in 3-OMD and 3-MT concentrations, respectively, in all brain regions, although the methylated monoamines were significantly less in mice reared on an FD diet compared with mice on an NF diet (Table 2). SAM is the methyl donor for these reactions involving COMT and it produces SAH (Fig. 1). Methylation of l-dopa and DA by COMT accounts for the marked decrease in SAM levels and increase in SAH levels in all brain regions following the exogenous administration of l-dopa (Fig. 4, top and middle). Of particular significance is that the l-dopa-induced decrease in SAM and increase in SAH were exacerbated in most brain regions by LF and FD diets. We have previously shown decreased SAM and increased SAH levels in mice reared on LF and FD diets, compared with mice on an NF diet, and consequently the SAM/SAH ratio is markedly reduced (Sontag et al., 2008). Here, we found that administration of l-dopa in mice on either LF or FD diets resulted in a greater reduction in the SAM/SAH ratio in several brain regions compared with saline-treated mice on corresponding diets (Fig. 4, bottom). This effect was less pronounced in the striatum with respect to other regions since the SAM/SAH ratio is relatively low, due to lower levels of SAM and higher levels of SAH, in mice on an NF diet.
Table 2.
The effect of l-dopa on regional brain concentrations of dopamine and related metabolites
| Diet |
l-dopa |
DA |
3-OMD |
3-MT |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Saline | l-dopa | Saline | l-dopa | Saline | l-dopa | Saline | l-dopa | ||
| Striatum | NF | nd | 12 ± 6 | 41 ± 14 | 160 ± 27* | nd | 43.2 ± 11.0 | 2.78 ± 0.16 | 2.63 ± 0.75 |
| LF | nd | 24 ± 10 | 66 ± 22 | 205 ± 49* | nd | 37.4 ± 12.5 | 5.13 ± 1.33 | 1.90 ± 0.41* | |
| FD | nd | 19 ± 6 | 66 ± 21 | 234 ± 25*^ | nd | 25.2 ± 3.5^ | 4.05 ± 1.50 | 1.50 ± 0.32*^ | |
| Frontal cortex | NF | nd | 177 ± 29 | 0.79 ± 0.27 | 16.7 ± 1.9* | nd | 64.4 ± 5.5 | 0.10 ± 0.06 | 0.92 ± 0.18* |
| LF | nd | 206 ± 25 | 0.84 ± 0.21 | 15.8 ± 1.2* | nd | 58.8 ± 9.2 | 0.13 ± 0.03 | 0.66 ± 0.09* | |
| FD | nd | 159 ± 24 | 1.01 ± 0.64 | 15.5 ± 2.5* | nd | 32.9 ± 2.0^ | 0.14 ± 0.08 | 0.51 ± 0.16*^ | |
| Midbrain | NF | nd | 106 ± 17 | 6.5 ± 1.8 | 51.5 ± 5.9* | nd | 58.3 ± 5.6 | 0.74 ± 0.20 | 1.50 ± 0.28* |
| LF | nd | 110 ± 11 | 3.7 ± 1.1 | 55.4 ± 3.8* | nd | 53.5 ± 8.8 | 0.61 ± 0.21 | 1.13 ± 0.12* | |
| FD | nd | 90 ± 26 | 3.8 ± 1.1 | 52.3 ± 6.0* | nd | 29.2 ± 1.8^ | 0.45 ± 0.14 | 0.98 ± 0.20*^ | |
| Cerebellum | NF | nd | 227 ± 33 | 0.16 ± 0.05 | 11.8 ± 3.9* | nd | 70.9 ± 7.1 | nd | 0.61 ± 0.11 |
| LF | nd | 268 ± 42 | 0.13 ± 0.06 | 12.6 ± 2.8* | nd | 64.3 ± 12.3 | nd | 0.43 ± 0.08 | |
| FD | nd | 193 ± 49 | 0.14 ± 0.06 | 13.9 ± 2.6* | nd | 33.1 ± 3.0^ | nd | 0.33 ± 1.10 | |
Exogenous l-dopa is decarboxylated to DA. In the striatum, a dopaminergic neuron-rich region, this conversion is more pronounced resulting in lower levels of l-dopa and higher levels of DA compared to other brain regions. COMT dependent O-methylation of l-dopa to 3-OMD and DA to 3-MT occurs in all regions. Significant lower levels of 3-OMD and 3-MT are found in l-dopa-treated mice fed an FD diet due to inhibition of COMT as a result of elevated SAH levels and a reduced SAM/SAH ratio (Fig. 4). Values represent mean ± SD for six mice in each group. nd, none detected. Symbols are the same as in Table 1.
Figure 4.

The effect of l-dopa and folate deficiency on SAM and SAH concentrations in regional brain areas. Bar shading: Open, NF diet; light gray, LF diet; dark gray, FD diet; no hatch, saline treated (n = 6, mean ± SD); hatch, l-dopa treated (n = 6, mean ± SD). *Compared with normal folate diet in the same treatment group (ANOVA with Dunnett's multiple-comparison t test, p < 0.05); ^Compared with saline treated in the same diet group (t test, p < 0.05).
l-Dopa-induced decrease in PP2A methylation is associated with an increase in p-Tau in regional mouse brain tissue
We have shown that PP2A methylation levels are reduced in brain tissue from mice reared on LF and FD diets (Sontag et al., 2008). Here we demonstrate that PP2A-methylated C subunit levels are significantly decreased following administration of l-dopa with respect to saline-treated mice fed an NF diet. Moreover, decreased PP2A methylation is significantly greater when l-dopa was given to mice fed either an LF diet (cortex and cerebellum only) or an FD diet (all brain regions) compared with l-dopa-treated mice fed an NF diet (Fig. 5A,B). Reductions in PP2A methylation were associated with a concomitant increase in each brain region of p-Tau at the PHF-1 epitope (Fig. 6A,B). We observed that l-dopa increased Tau phosphorylation in mice fed an NF diet, and this effect was augmented in mice on an LF or FD diet.
Figure 5.

The effect of l-dopa and folate deficiency on PP2A methylation in regional brain areas. A, Representative immunoblots of demethylated and total C subunit expression levels in cortical and striatal brain tissues. B, PP2A methylation levels were determined after densitometric analysis of immunoblots. Bar shading and symbols as in Figure 4.
Figure 6.

The effect of l-dopa and folate deficiency on p-Tau (PHF-1) in regional brain areas. A, Western blot analysis of p-Tau (PHF-1). B, p-Tau levels were determined after densitometric analysis of immunoblots. Bar shading and symbols as in Figure 4.
Discussion
Substantial evidence implicates folate deficiency and hypomethylation in the etiology and pathogenesis of neurodegenerative disorders including AD (Mattson and Shea, 2003). In support of this are reports that have identified a central role for folate- and methylation-dependent pathways in processing both Tau and amyloid precursor proteins that are well established neuropathological hallmarks of AD (Sontag et al., 2007, 2008; Fuso and Scarpa, 2011). SAM is the sole methyl-group donor and a critical metabolite that regulates the activity of numerous methyltransferase reactions (Chiang et al., 1998), including PP2A methylation (Janssens et al., 2008). Intracellular levels of SAM are maintained by a highly regulated methylation cycle that is dependent on folate and vitamin B12. These act as cofactors for methionine synthase, a key enzyme for the conversion of homocysteine to methionine (Fig. 1). In addition, drugs that interfere with folate metabolism or the methylation cycle can affect cellular SAM levels. One such compound is l-dopa, which is O-methylated by SAM in a COMT-dependent reaction. Here we report for the first time the combined effect of folate deficiency and l-dopa on methylation cycle metabolites, PP2A methylation, and p-Tau levels in cultured cells and in a mouse model. We first show in SH-SY5Y cells that l-dopa dose dependently induces a decrease in both soluble and insoluble methylated PP2A levels. Carboxyl methylation of PP2A catalytic subunit on Leu-309 by SAM-dependent leucine carboxyl methyltransferase (LCMT1) enhances the biogenesis and stability of PP2A enzymes containing the Bα regulatory subunit (PP2A/Bα), the primary PP2A isoforms that dephosphorylate Tau (Sontag et al., 1996, 1999, 2008; Xu et al., 2008). Methylated PP2A/Bα enzymes fractionate in both soluble and insoluble (containing microtubule-associated forms) fractions from neuronal cells (Sontag et al., 1995, 2007). This has implications for PP2A-dependent regulation of Tau and microtubules, as methylated PP2A/Bα enzymes associate with microtubules (Sontag et al., 1995) and regulate microtubule dynamics (Nunbhakdi-Craig et al., 2007) and the ability of Tau to bind to and stabilize microtubules (Sontag et al., 1996). Importantly, PP2A/Bα can only bind to and dephosphorylate Tau when both proteins are not bound to microtubules (Sontag et al., 1999). PP2A methylation was markedly decreased by l-dopa in both SH-SY5Y cells and human dopaminergic neurons. The extent of PP2A demethylation correlated with an increase in p-Tau (PHF-1) levels. We observed a synergistic effect of l-dopa and folate deficiency, implying that under these conditions the methylation cycle is severely compromised. l-dopa and folate deficiency can perturb the methylation cycle in several ways: (1) impaired conversion of Hcy to methionine leading to reduced synthesis of SAM; (2) accumulation of Hcy may be metabolized to SAH, a potent competitive inhibitor of methyltransferase enzymes (Cantoni GL, 1987; Chiang et al., 1998); (3) methylation of l-dopa to 3-OMD will deplete intracellular pools of SAM as well as increasing SAH; and (4) l-dopa competes with methionine for transport across cell membranes through the large neutral amino acid transporter and limits SAM synthesis (Baldessarini and Karobath, 1972).
In support of our in vitro findings, exogenous administration of l-dopa increased plasma tHcy in mice fed an NF diet, an effect that was exacerbated in mice fed either an LF or FD diet. While plasma tHcy is a sensitive marker of folate deficiency (Stabler, 2000; Selhub et al., 2010), it may also reflect the activity of COMT-dependent methylation of l-dopa. This is confirmed by the l-dopa-induced decrease in SAM, increase in SAH, and corresponding decrease in the SAM/SAH ratio in liver tissue that we and other investigators have observed (Wagner et al., 1984, Miller et al., 1997). LCMT1 activity is affected by reduced availability of SAM and competitive inhibition from increased SAH, which is reflected by increased levels of demethylated PP2A in liver tissue. These findings have significant implications for patients with PD and those with other neurodegenerative disorders who receive l-dopa therapy, as well as children diagnosed with l-dopa responsive dystonia due to inherited disorders of tetrahydrobiopterin synthesis and TH deficiency (Swoboda, 2006). In the later group, l-dopa treatment may commence early in life and continue indefinitely. Increased levels of plasma tHcy following l-dopa treatment in PD have been reported by several investigators (Yasui et al., 2000; Irizarry et al., 2005; Müller and Kuhn, 2009; Yuan et al., 2009). Furthermore, l-dopa treatment in PD is associated with decreased SAM (Cheng et al., 1997; De Bonis et al., 2010) and increased SAH concentrations in erythrocytes (De Bonis et al., 2010). CSF tHcy became significantly higher following l-dopa treatment in 18 patients with PD compared with pretreatment levels (Isobe et al., 2010). Likewise, l-dopa therapy in children was associated with a significant decrease in the concentration of SAM in CSF that was inversely correlated with elevated concentrations of CSF 3-OMD (Surtees and Hyland, 1990). These observations clearly establish that l-dopa treatment has significant effects on methylation cycle metabolites.
Our findings confirm that l-dopa has a marked effect on methylation cycle metabolites in brain tissue. Decarboxylation of l-dopa to DA occurs predominately in the striatum; however, O-methylation of l-dopa to 3-OMD, as well as methylation of DA to 3-MT, occurs in all brain regions resulting in a considerable decrease in the concentration of SAM. In some brain regions (cortex, midbrain, and cerebellum), l-dopa-induced decrease in SAM was aggravated by folate deficiency. Significantly, in all regions of the brain studied, accumulation of the demethylated metabolite SAH was highest in mice treated with l-dopa and exposed to FD diets. Folate deficiency restricts metabolism of Hcy causing intracellular SAH accumulation, thereby compounding the effect of l-dopa. SAH is a competitive inhibitor of methyltransferases, including COMT, and this is evident from the significantly lower levels of 3-OMD and 3-MT in all brain regions following l-dopa in mice reared on an FD diet. Furthermore, we demonstrated that l-dopa significantly decreased PP2A methylation in mice fed an NF diet. We attribute this to a decrease in the SAM/SAH ratio and downregulation of LCMT1, an effect that was greatest in mice fed an LF or FD diet. In support of this, we have previously shown that folate deficiency in mice results in a reduced SAM/SAH ratio and downregulation of LCMT1 in brain tissue (Sontag et al., 2008). l-Dopa-induced loss of methylated PP2A was associated with increased p-Tau (PHF-1) levels in all brain regions studied. Methotrexate, a folate antagonist, promotes PP2A demethylation and Tau hyperphosphorylation in rat primary cortical neurons (Yoon et al., 2007), an effect probably mediated through inhibition of folate metabolism and altered SAM and Hcy metabolism. Enhanced Tau phosphorylation is potentially neurotoxic to cells by promoting microtubule destabilization and favoring Tau mislocalization and aggregation (Iqbal et al., 2010). Our findings suggest a novel mechanism in which l-dopa increases phosphorylation of Tau protein, which has major implications for patients with PD or other neurodegenerative diseases. In this context, COMT inhibitors may be protective against this effect. In one study, tolcapone, a centrally acting COMT inhibitor, was shown to lower plasma tHcy and SAH levels in PD patients treated with l-dopa and a peripheral decarboxylase inhibitor (Müller and Kuhn, 2006). COMT inhibition in l-dopa-treated PD patients was proposed to potentially reduce Hcy-mediated neuronal degeneration, risk of onset of dementia, vascular disease, and polyneuropathy, which are commonly prevalent in PD. An earlier study found that rats given a centrally acting COMT inhibitor before l-dopa attenuated or prevented entirely the l-dopa-induced decrease in SAM and increase in SAH in both peripheral and regional brain tissue (Miller et al., 1997). Moreover, folate intake has been recently reported to normalize plasma tHcy levels in hyperhomocysteinemic PD patients (Belcastro et al., 2010). Altogether, these data reinforce the importance of monitoring methylation pathways during l-dopa treatment.
In conclusion, our studies have demonstrated a novel mechanism involving methylation–dependent pathways that can lead to deregulation of PP2A and accumulation of p-Tau with potential detrimental effects in neuronal cells. This effect is exacerbated when folate metabolism is compromised, which not only occurs through dietary insufficiency as shown in this study but also from the use of antifolate drugs or the existence of common polymorphisms related to the folate-methylation cycle. This dietary–genetic–drug interaction is particularly relevant in PD patients since hyperphosphorylation of Tau is an early event in dementia (Hanger et al., 2009) and there is increasing evidence for a contribution of Tau in PD pathogenesis (Lei et al., 2010). For instance, p-Tau accumulates in PD Lewy bodies, which are cytoplasmic inclusions enriched in α-synuclein. Tau also enhances α-synuclein aggregation and toxicity in cellular models of synucleinopathy (Badiola et al., 2011). Indeed, there is ample experimental proof for synergistic effects of α-synuclein and Tau protein interactions in neurodegenerative disorders (Jellinger, 2011). Notably, PP2A has been shown to dephosphorylate hyperphosphorylated aggregates of α-synuclein in a mouse model (Lee et al., 2011). Thus, deregulation of PP2A by long-term use of l-dopa may promote an increase in both hyperphosphorylated Tau and α-synuclein. It remains to be seen if therapies that enhance PP2A methylation are effective in reducing toxicity associated with these neurodegenerative proteinopathies.
Footnotes
This work was supported by National Institutes of Health Grant AG18883 (E.S.); startup funds from the University of Newcastle, Australia (E.S.); and a project grant from the National Health and Medical Research Council of Australia (E.S. and J.-M.S.).We thank Dr. Peter Davies (Albert Einstein College of Medicine, NY) for the gift of PHF-1 antibodies and Dr. Philip Dickson (University of Newcastle, Australia) for the gift of anti-TH antibodies.
T.B. reports having been the chairman of the Advisory Board for Methylation Sciences Inc., holding stock options in Methylation Sciences Inc., being a scientific consultant to Pamlab LLC, and having received research funding from Pamlab LLC, which is a distributor of B vitamins as a medical food.
References
- Badiola N, de Oliveira RM, Herrera F, Guardia-Laguarta C, Gonçalves SA, Pera M, Suárez-Calvet M, Clarimon J, Outeiro TF, Lleó A. Tau enhances α-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS One. 2011;6:e26609. doi: 10.1371/journal.pone.0026609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldessarini RJ, Karobath M. Effects of L-DOPA and L-3-O-methyl-DOPA on uptake of (3H)L-methionine by synaptosomes. Neuropharmacology. 1972;11:715–720. doi: 10.1016/0028-3908(72)90080-9. [DOI] [PubMed] [Google Scholar]
- Belcastro V, Pierguidi L, Castrioto A, Menichetti C, Gorgone G, Ientile R, Pisani F, Rossi A, Calabresi P, Tambasco N. Hyperhomocysteinemia recurrence in levodopa-treated Parkinson's disease patients. Eur J Neurol. 2010;17:661–665. doi: 10.1111/j.1468-1331.2009.02894.x. [DOI] [PubMed] [Google Scholar]
- Bottiglieri T. Isocratic high performance liquid chromatographic analysis of S-adenosylmethionine and S-adenosylhomocysteine in animal tissues: the effect of exposure to nitrous oxide. Biomed Chromatogr. 1990;4:239–241. doi: 10.1002/bmc.1130040606. [DOI] [PubMed] [Google Scholar]
- Bottiglieri T. Homocysteine and folate metabolism in depression. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1103–1112. doi: 10.1016/j.pnpbp.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Cantoni GL. The role of S-adenosylhomocysteine in the biological utilization of S-adenosylmethionine. Prog Clin Biol Res. 1987;198:47–65. [PubMed] [Google Scholar]
- Cheng H, Gomes-Trolin C, Aquilonius SM, Steinberg A, Löfberg C, Ekblom J, Oreland L. Levels of L-methionine S-adenosyltransferase activity in erythrocytes and concentrations of S-adenosylmethionine and S-adenosylhomocysteine in whole blood of patients with Parkinson's disease. Exp Neurol. 1997;145:580–585. doi: 10.1006/exnr.1997.6466. [DOI] [PubMed] [Google Scholar]
- Chiang PK. Biological effects of inhibitors of S-adenosylhomocysteine hydrolase. Pharmacol Ther. 1998;77:115–134. doi: 10.1016/s0163-7258(97)00089-2. [DOI] [PubMed] [Google Scholar]
- Cotzias GC, Papavasiliou PS, Gellene R. Modification of Parkinsonism–chronic treatment with L-dopa. N Engl J Med. 1969;280:337–345. doi: 10.1056/NEJM196902132800701. [DOI] [PubMed] [Google Scholar]
- De Bonis ML, Tessitore A, Pellecchia MT, Longo K, Salvatore A, Russo A, Ingrosso D, Zappia V, Barone P, Galletti P, Tedeschi G. Impaired transmethylation potential in Parkinson's disease patients treated with L-Dopa. Neurosci Lett. 2010;468:287–291. doi: 10.1016/j.neulet.2009.11.014. [DOI] [PubMed] [Google Scholar]
- Ericsson AD. Potentiation of the L-Dopa effect in man by the use of catechol-O-methyltransferase inhibitors. J Neurol Sci. 1971;14:193–197. doi: 10.1016/0022-510x(71)90088-8. [DOI] [PubMed] [Google Scholar]
- Favre B, Turowski P, Hemmings BA. Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin-A, okadaic acid, and tautomycin. J Biol Chem. 1997;272:13856–13863. doi: 10.1074/jbc.272.21.13856. [DOI] [PubMed] [Google Scholar]
- Fuso A, Scarpa S. One-carbon metabolism and Alzheimer's disease: is it all a methylation matter? Neurobiol Aging. 2011;32:1192–1195. doi: 10.1016/j.neurobiolaging.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010;7:656–664. doi: 10.2174/156720510793611592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry MC, Gurol ME, Raju S, Diaz-Arrastia R, Locascio JJ, Tennis M, Hyman BT, Growdon JH, Greenberg SM, Bottiglieri T. Association of homocysteine with plasma amyloid beta protein in aging and neurodegenerative disease. Neurology. 2005;65:1402–1408. doi: 10.1212/01.wnl.0000183063.99107.5c. [DOI] [PubMed] [Google Scholar]
- Isobe C, Abe T, Terayama Y. L-dopa therapy increase homocysteine concentration in cerebrospinal fluid from patients with Parkinson's disease. J Clin Neurosci. 2010;17:717–721. doi: 10.1016/j.jocn.2009.09.034. [DOI] [PubMed] [Google Scholar]
- Janssens V, Longin S, Goris J. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail) Trends Biochem Sci. 2008;33:113–121. doi: 10.1016/j.tibs.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Interaction between α-Synuclein and Other Proteins in Neurodegenerative Disorders. ScientificWorldJournal. 2011;11:1893–1907. doi: 10.1100/2011/371893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Chen W, Junn E, Im JY, Grosso H, Sonsalla PK, Feng X, Ray N, Fernandez JR, Chao Y, Masliah E, Voronkov M, Braithwaite SP, Stock JB, Mouradian MM. Enhanced phosphatase activity attenuates α-synucleinopathy in a mouse model. J Neurosci. 2011;31:6963–6971. doi: 10.1523/JNEUROSCI.6513-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei P, Ayton S, Finkelstein DI, Adlard PA, Masters CL, Bush AI. Tau protein: relevance to Parkinson's disease. Int J Biochem Cell Biol. 2010;42:1775–1778. doi: 10.1016/j.biocel.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Martin TM, Benghuzzi H, Tucci M. The effect of conventional and sustained delivery of levodopa on SH-SY5Y neuroblastoma cells. Biomed Sci Instrum. 2005;41:382–387. [PubMed] [Google Scholar]
- Mattson MP, Shea TB. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003;26:137–146. doi: 10.1016/S0166-2236(03)00032-8. [DOI] [PubMed] [Google Scholar]
- McCully KS. Homocysteine, vitamins, and vascular disease prevention. Am J Clin Nutr. 2007;86:1563S–1568S. doi: 10.1093/ajcn/86.5.1563S. [DOI] [PubMed] [Google Scholar]
- Miller JW, Shukitt-Hale B, Villalobos-Molina R, Nadeau MR, Selhub J, Joseph JA. Effect of L-Dopa and the catechol-O-methyltransferase inhibitor Ro 41–0960 on sulfur amino acid metabolites in rats. Clin Neuropharmacol. 1997;20:55–66. doi: 10.1097/00002826-199702000-00007. [DOI] [PubMed] [Google Scholar]
- Müller T, Kuhn W. Tolcapone decreases plasma levels of S-adenosyl-L-homocysteine and homocysteine in treated Parkinson's disease patients. Eur J Clin Pharmacol. 2006;62:447–450. doi: 10.1007/s00228-006-0132-0. [DOI] [PubMed] [Google Scholar]
- Müller T, Kuhn W. Homocysteine levels after acute levodopa intake in patients with Parkinson's disease. Mov Disord. 2009;24:1339–1343. doi: 10.1002/mds.22607. [DOI] [PubMed] [Google Scholar]
- Nunbhakdi-Craig V, Schuechner S, Sontag JM, Montgomery L, Pallas DC, Juno C, Mudrak I, Ogris E, Sontag E. Expression of protein phosphatase 2A mutants and silencing of the regulatory Balpha subunit induce a selective loss of acetylated and detyrosinated microtubules. J Neurochem. 2007;101:959–971. doi: 10.1111/j.1471-4159.2007.04503.x. [DOI] [PubMed] [Google Scholar]
- Nutt JG. Motor fluctuations and dyskinesia in Parkinson's disease. Parkinsonism Relat Disord. 2001;8:101–108. doi: 10.1016/s1353-8020(01)00024-4. [DOI] [PubMed] [Google Scholar]
- Ogburn KD, Bottiglieri T, Wang Z, Figueiredo-Pereira ME. Prostaglandin J2 reduces catechol-O-methyltransferase activity and enhances dopamine toxicity in neuronal cells. Neurobiol Dis. 2006;22:294–301. doi: 10.1016/j.nbd.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Ogris E, Gibson DM, Pallas DC. Protein phosphatase 2A subunit assembly: the catalytic subunit carboxy terminus is important for binding cellular B subunit but not polyomavirus middle tumor antigen. Oncogene. 1997;15:911–917. doi: 10.1038/sj.onc.1201259. [DOI] [PubMed] [Google Scholar]
- Poewe W, Antonini A, Zijlmans JC, Burkhard PR, Vingerhoets F. Levodopa in the treatment of Parkinson's disease: an old drug still going strong. Clin Interv Aging. 2010;5:229–238. doi: 10.2147/cia.s6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne UK, Sonninen V, Sirtola T. Treatment of Parkinson's disease with L-DOPA and decarboxylase inhibitor. Z Neurol. 1972;202:1–20. doi: 10.1007/BF00316422. [DOI] [PubMed] [Google Scholar]
- Roberts JW, Cora-Locatelli G, Bravi D, Amantea MA, Mouradian MM, Chase TN. Catechol-O-methyltransferase inhibitor tolcapone prolongs levodopa/carbidopa action in parkinsonian patients. Neurology. 1993;43:2685–2688. doi: 10.1212/wnl.43.12.2685. [DOI] [PubMed] [Google Scholar]
- Selhub J, Troen A, Rosenberg IH. B vitamins and the aging brain. Nutr Rev. 2010;68(Suppl 2):S112–S118. doi: 10.1111/j.1753-4887.2010.00346.x. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Bloom GS, Mumby MC. A novel pool of protein phosphatase 2A is associated with microtubules and is regulated during the cell cycle. J Cell Biol. 1995;128:1131–1144. doi: 10.1083/jcb.128.6.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Lee G, Bloom GS, Mumby MC. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron. 1996;17:1201–1207. doi: 10.1016/s0896-6273(00)80250-0. [DOI] [PubMed] [Google Scholar]
- Sontag E, Hladik C, Montgomery L, Luangpirom A, Mudrak I, Ogris E, White CL., 3rd Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J Neuropathol Exp Neurol. 2004;63:1080–1091. doi: 10.1093/jnen/63.10.1080. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Lee G, Brandt R, Kamibayashi C, Kuret J, White CL, 3rd, Mumby MC, Bloom GS. Molecular interactions among protein phosphatase 2A, tau, and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J Biol Chem. 1999;274:25490–25498. doi: 10.1074/jbc.274.36.25490. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Sontag JM, Diaz-Arrastia R, Ogris E, Dayal S, Lentz SR, Arning E, Bottiglieri T. Protein phosphatase 2A methyltransferase links homocysteine metabolism with Tau and amyloid precursor protein regulation. J Neurosci. 2007;27:2751–2759. doi: 10.1523/JNEUROSCI.3316-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag JM, Nunbhakdi-Craig V, Montgomery L, Arning E, Bottiglieri T, Sontag E. Folate deficiency induces in vitro and mouse brain region-specific downregulation of leucine carboxyl methyltransferase-1 and protein phosphatase 2A B(alpha) subunit expression that correlate with enhanced Tau phosphorylation. J Neurosci. 2008;28:11477–11487. doi: 10.1523/JNEUROSCI.2816-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabler SP. Using homocysteine and related metabolites to diagnose vitamin deficiency states. Biofactors. 2000;11:51–52. doi: 10.1002/biof.5520110114. [DOI] [PubMed] [Google Scholar]
- Stacy M. Medical treatment of Parkinson disease. Neurol Clin. 2009;27:605–631. v. doi: 10.1016/j.ncl.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Surtees R, Hyland K. L-3,4-dihydroxyphenylalanine (levodopa) lowers central nervous system S-adenosylmethionine concentrations in humans. J Neurol Neurosurg Psychiatry. 1990;53:569–572. doi: 10.1136/jnnp.53.7.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swoboda KJ. Inherited disorders of amine biosynthesis. Future Neurol. 2006;1:605–614. [Google Scholar]
- Ubbink JB, Hayward Vermaak WJ, Bissbort S. Rapid high-performance liquid chromatographic assay for total homocysteine levels in human serum. J Chromatogr. 1991;565:441–446. doi: 10.1016/0378-4347(91)80407-4. [DOI] [PubMed] [Google Scholar]
- Wagner J, Danzin C, Huot-Olivier S, Claverie N, Palfreyman MG. High-performance liquid chromatographic analysis of S-adenosylmethionine and its metabolites in rat tissues: interrelationship with changes in biogenic catechol levels following treatment with L-dopa. J Chromatogr. 1984;290:247–262. doi: 10.1016/s0021-9673(01)93580-0. [DOI] [PubMed] [Google Scholar]
- Weingarten P, Bermak J, Zhou QY. Evidence for non-oxidative dopamine cytotoxicity: potent activation of NF-kappa B and lack of protection by anti-oxidants. J Neurochem. 2001 Mar;76:1794–1804. doi: 10.1046/j.1471-4159.2001.00190.x. [DOI] [PubMed] [Google Scholar]
- Xu Y, Chen Y, Zhang P, Jeffrey PD, Shi Y. Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol Cell. 2008;31:873–885. doi: 10.1016/j.molcel.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui K, Kowa H, Nakaso K, Takeshima T, Nakashima K. Plasma homocysteine and MTHFR C677T genotype in levodopa-treated patients with PD. Neurology. 2000;55:437–440. doi: 10.1212/wnl.55.3.437. [DOI] [PubMed] [Google Scholar]
- Yoon SY, Choi HI, Choi JE, Sul CA, Choi JM, Kim DH. Methotrexate decreases PP2A methylation and increases Tau phosphorylation in neuron. Biochem Biophys Res Commun. 2007;363:811–816. doi: 10.1016/j.bbrc.2007.09.060. [DOI] [PubMed] [Google Scholar]
- Yuan RY, Sheu JJ, Yu JM, Hu CJ, Tseng IJ, Ho CS, Yeh CY, Hung YL, Chiang TR. Methylenetetrahydrofolate reductase polymorphisms and plasma homocysteine in levodopa-treated and non-treated Parkinson's disease patients. J Neurol Sci. 2009;287:64–68. doi: 10.1016/j.jns.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Zhou J, Austin RC. Contributions of hyperhomocysteinemia to atherosclerosis: Causal relationship and potential mechanisms. Biofactors. 2009;35:120–129. doi: 10.1002/biof.17. [DOI] [PubMed] [Google Scholar]
- Zhuo JM, Wang H, Praticò D. Is hyperhomocysteinemia an Alzheimer's disease (AD) risk factor, an AD marker, or neither? Trends Pharmacol Sci. 2011;32:562–571. doi: 10.1016/j.tips.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoccolella S, dell'Aquila C, Abruzzese G, Antonini A, Bonuccelli U, Canesi M, Cristina S, Marchese R, Pacchetti C, Zagaglia R, Logroscino G, Defazio G, Lamberti P, Livrea P. Hyperhomocysteinemia in levodopa-treated patients with Parkinson's disease dementia. Mov Disord. 2009;24:1028–1033. doi: 10.1002/mds.22511. [DOI] [PubMed] [Google Scholar]