

Introduction
Electromechanical dyssynchrony can markedly worsen heart failure (HF) morbidity and mortality, independent of traditional risk factors.1-4 Depending on the metric used, current estimates of the prevalence of dyssynchrony vary from 25 to 30% in patients with HF (based on QRS widening) up to 60% based on tissue Doppler or magnetic resonance imaging measures of dyssynchronous contraction of the left ventricle.5, 6 Cardiac resynchronization therapy (CRT) or biventricular pacing has emerged as a promising option to treat patients with HF and dyssynchronous contraction.7-9 The past few decades have seen the rise of pharmacotherapy, primarily through agents that antagonize the effect of excessive concentrations of circulating neurohormones, yet, HF-related morbidity and mortality remain high.3-6 Biventricular stimulation has been demonstrated to improve contractile performance in patients with mechanical dyssynchrony acutely and chronically, while also prolonging long-term survival -something not yet achieved by drug therapy.10 While the clinical and mechanical effectiveness of CRT are well described, 30% of patients do not benefit from CRT and clinical criteria to identify CRT non-responders remain elusive8, 11, 12 Currently, the most widely used predictor of reverse remodeling is the presence of marked mechanical dyssynchrony before CRT, as indexed by the width of the QRS.13 Mechanical dyssynchrony seems important, yet imaging-based measures have not predicted response well14 and even improvement in dyssynchrony after initiation of CRT only weakly predicts chronic response.15 Limited understanding of the molecular mechanisms underlying reverse cardiac remodeling induced by CRT has hampered the selection of potential responders. In this review, we focus on the electrophysiological aspects and molecular networks underlying the benefits of CRT. We will review how CRT homogenizes regional differences in stress kinase signaling and electrical remodeling, and then review its global effect on myocyte function, and its broader impact on the cardiac ventricular transcriptome. A comprehensive understanding of the molecular features of dyssynchronous contraction in the failing heart (DHF) and its reversibility by biventricular pacing promises to identify sets of biological marker for the selection of patients who will benefit most from CRT and, in a more general sense, to advance our knowledge of HF-associated pathophysiological processes.
Beyond the mechano-energetics
Until recently, the prevailing view of CRT efficacy is that it reduces mechanical inefficiency from discoordinate contraction, allowing more blood to be ejected at a lower energy cost. Multisite ventricular pacing had been proposed for ventricular arrhythmia termination and ultimately for improvement in hemodynamic performance in patients with heart failure.16 CRT was developed in the mid-1990s after investigators found that biventricular (or left ventricular only) preexcitation could restore mechanical synchrony and improve acute left ventricular mechanics,17 energetic efficiency,18 and regional metabolism.19 Subsequent large-scale clinical trials demonstrated that CRT can acutely and chronically enhance cardiac work and systolic performance in selected patients.7, 20, 21 Using a canine model of dyssynchronous ventricular contraction, our group has corroborated the mechanical benefits of CRT. Dogs treated with biventricular rapid pacing after an initial period of dyssynchronous HF of 3 weeks, had a slight, but significant improvement in ejection fraction and stroke volume, whereas both ejection fraction and stroke volume continued to decline in dogs with continued heart failure with dyssynchronous contraction (DHF) (Fig. 1A).22 In this model, resynchronization of left ventricular contraction was confirmed by MRI circumferential uniformity ratio estimate (CURE) index or standard tissue Doppler parameters (Fig 1B). The observed improvement of myocardial function in both animal models and HF patients raised the question of whether wall motion is all there is to left ventricular preexcitation, or, whether effective CRT might also reverse cellular remodeling. In fact, another mechanical circulatory support mechanism, left ventricular assist devices (LVADs) are known to induce myocardial changes at the cellular and structural level such that a small number of patients may recover sufficient cardiac function that permits device removal.23-25 Hints at changes in cellular signaling pathways by CRT first came from human studies of responders versus “non-responders”, where myocardial gene expression changes of calcium handling proteins, beta-receptors, and natriuretic peptides were reversed preferentially in responders.26-28 Patients with effective CRT display chronic enhancement of circulating apelin, a secreted hormone that can block adverse remodeling and has positive inotropic effects.29 Circulating biomarkers of extracellular matrix remodeling also accompanies successful CRT, including decreases in tenascin-C, and matrix metalloproteinase 9 (MMP-9).30 Chronic CRT also has anti-inflammatory effects and reduces monocyte chemoattractant protein-1, interleukin-8, and interleukin-6 levels.31 While these studies do not identify the underlying mechanisms by which CRT exerts its beneficial effect, they may suggest biomarkers for therapies that both enhance systolic function and survival in HF patients.
Figure 1.
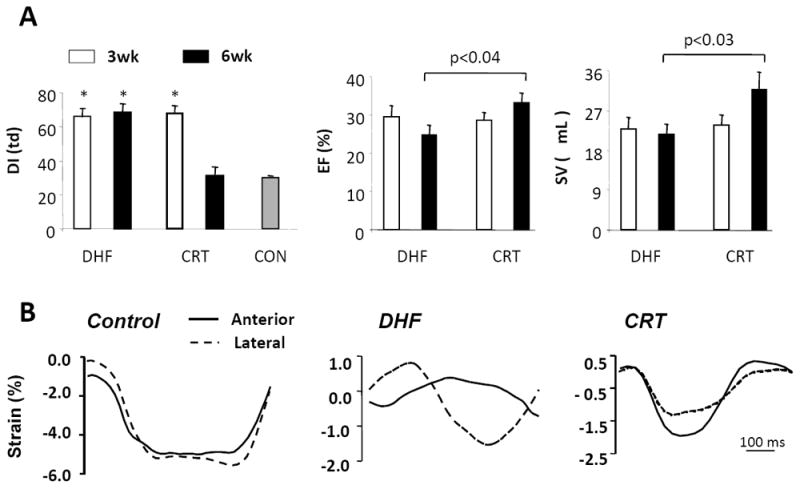
Development of a canine model of cardiac dyssynchrony and resynchronization. In this model, both dyssynchronous heart failure (DHF) and resynchronized heart failure [cardiac resynchronization therapy (CRT)] groups are exposed first to tachypacing for 3 weeks (in the presence of a preestablished left bundle branch block). This protocol is continued for an additional 3 weeks in DHF hearts, but is switched to rapid biventricular pacing in CRT hearts. A: Left ventricular dyssynchrony (DI, left) is assessed by the variance in timing delay in systolic motion using tissue Doppler. This is similar in both groups at 3 weeks and declines to control levels (synchrony) only in the CRT group. Slight improvements in ejection fraction (EF, middle) and stroke volume (SV, right) are noted in the CRT group, but not in the DHF group at 6 weeks. B: Myocardial strain patterns (tissue Doppler images) obtained in the anterior and lateral wall (tissue Doppler) from a normal control, DHF heart, and CRT heart. With DHF, major disparities in the timing of shortening and reciprocal shortening/stretch in each region are ameliorated by CRT. (adapted from Chakir et al. 22).
Regional Molecular Changes in HF and CRT
1. Stress response-kinases
Initial molecular insights into DHF were provided in a report by our group32 examining the regional effects of DHF on molecular signaling. This study revealed the selective down-regulation of Ca2+ handling proteins and connexin 43, and up-regulation of mitogen-activated protein kinase in the lateral wall only, referred to as molecular polarization. This regional molecular change was not observed in synchronous HF. In a more recent study by Chakir et al.33 the lateral wall of DHF ventricles exhibited an increase in p38 MAPK and Ca2+-calmodulin kinase II (CaMKII) activation and increased TNF-α expression which were both reversed by CRT (Fig. 2). These localized differences in stress kinase activation were consistent with disparities in regional workload in DHF and its equalization by CRT (Fig. 1). The changes in stress-response kinases are potentially important given the impact of these proteins on muscle function, survival, and fibrosis. P38 MAPK stimulates fibrosis and apoptosis and is associated with contractile failure.34, 35 CaMKII is an important mediator of β-adrenergic related toxicity leading to apoptosis,36 cardiac hypertrophy,37, 38 and the generation of cardiac arrhythmias.39, 40 TNF-α stimulates fibrosis and apoptosis, and overexpression induces dilated cardiomyopathy.41 Expression TNF-α can be triggered by abnormal mechanical loading,42 and its decline solely in the lateral wall of CRT hearts supports this mechanism. This change is also supported by recent human data reporting lower levels of left ventricular TNF-α after 6 months of CRT.43
Figure 2.
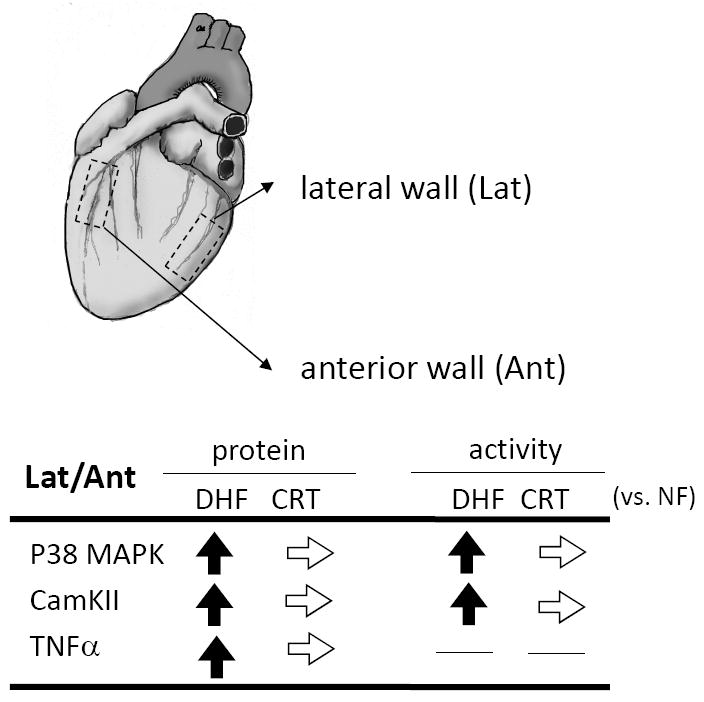
CRT reverses regional disparities in activation of stress response kinases. Summarized data for activity and expression of p38 MAPK, CaMKII, and TNF-α show relative increases in lateral wall in DHF hearts that are reversed by CRT. (adapted from Chakir et al.33).
2. Electrophysiological changes in DHF and CRT
In addition to molecular polarization, DHF is characterized by regional heterogeneities in cellular and tissue electrophysiological properties. The hallmark signature of cells and tissues isolated from failing hearts, independent of the etiology, is action potential (AP) prolongation.44-50 AP prolongation in DHF is most prominent in cells isolated from the late-activated lateral LV wall, and is an index of the exaggeration of the physiological heterogeneity of electrical properties in the failing heart.51 CRT significantly shortens the AP in lateral myocytes and thus reduces LV regional heterogeneity in action potential duration (APD) (Fig. 3). Regional alterations in ionic currents underlie the AP remodeling; however, the molecular mechanisms of regional ionic current remodeling in DHF and CRT are controversial. A prominent increase of TNF-α and CaMKII in the lateral wall might play a role in regional AP remodeling. TNF-α decreases the transient outward potassium current (Ito) and prolongs the APD in rat ventricular myocytes.52 Recently, Xie et al. suggested that increased oxidative stress in HF activates CaMKII and triggeres ventricular arrhythmias.53 CaMKII modulates Ca2+ currents, sarcoplasmic reticulum (SR) function,54, 55 and increases persistent Na+ current,56, 57 resulting in prolongation of APD.58 It is possible and indeed likely, that other regional alterations in Ca2+ handling or increased persistent Na+ current contribute to regional differences in the APD and AP profile in DHF, and the regionally-specific effects of bi-ventricular pacing on this phenotype.
Figure 3.
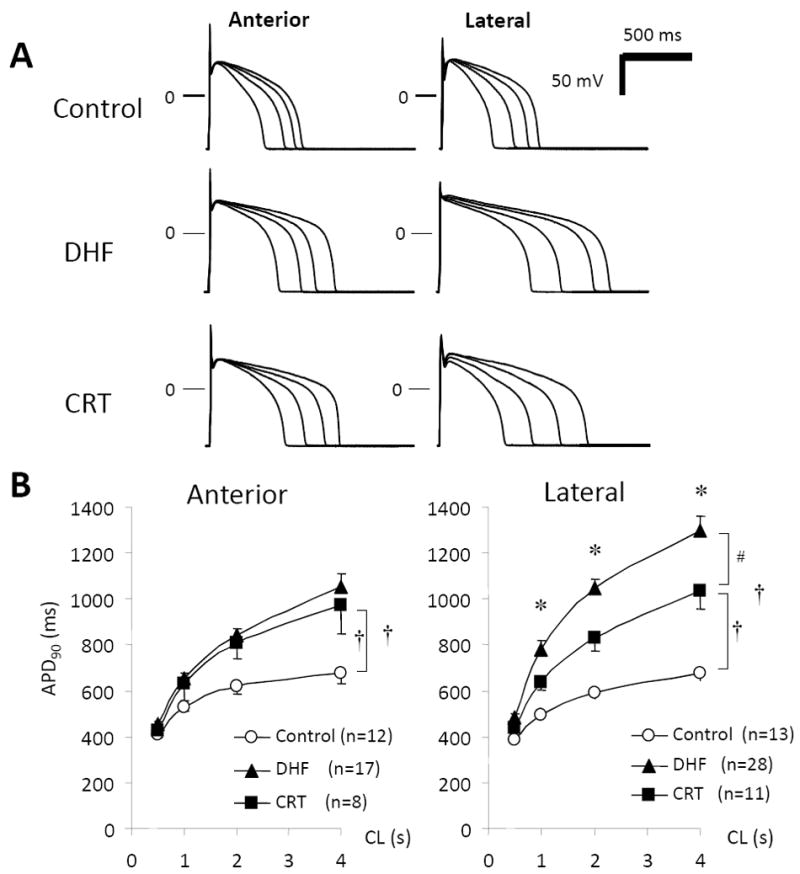
APD in LV myocytes from control, DHF, and CRT hearts. A, Representative superimposed APs recorded at pacing cycle lengths (CL) of 0.5, 1.0, 2.0, and 4.0 seconds, B, Relationship between pacing CL and APD at 90% recovery (APD90) from anterior and lateral myocytes in each group. †P<0.05 vs. control; #P<0.05 vs. DHF; *P<0.05 vs. anterior (adapted from Aiba et al.51).
3. Effects of DHF and CRT on the regional left ventricular transcriptome
In previous studies, we demonstrated that CRT can reverse the regional heterogeneities of electrical remodeling and stress-response kinases. However, this may be just the tip of iceberg, as most analyses were focused on individual proteins, and most likely missed a broader impact of dyssynchrony and CRT on regional molecular expression patterns. To test this hypothesis, we used a global gene expression profiling approach in the aforementioned canine model of dyssynchronous HF and CRT which allowed us to examine mRNA expression in anterior and lateral left ventricular myocardium. As a result of this unbiased and global assessment of transcriptional activity, we identified over six-times as many genes to be differentially expressed between NF and DHF hearts in anterior compared to lateral LV myocardium of the same hearts (2173 vs. 346 transcripts, respectively; false discovery rate, FDR<5%).59 We found prominent down regulation of metabolic pathways (oxidative phosphorylation, fatty-acid, amino acid and glucose metabolism), while various cell signaling pathways were up regulated (MAPK, JAK-STAT, TGF-β) in the anterior LV wall of dyssynchronous failing heart. The greater down-regulation of metabolic transcripts in anterior compared with lateral LV regions is also in good agreement with human studies: using gated PET with 18F-fluorodeoxyglucose (FDG) and 99mTc-sestamibi SPECT to noninvasively measure myocardial glucose metabolism and myocardial perfusion, respectively, Nowak and colleagues found that glucose metabolism is reduced more than perfusion in the anteroseptal compared with LV lateral wall in patients with DCM and left bundle branch block.19
Importantly, the disparity in the number of regulated transcripts between the early- and late-activated LV regions gave rise to an increased regional heterogeneity of gene expression within the dyssynchronously contracting LV myocardium. These dyssynchrony-induced regional gene expression changes were reversed by CRT to levels comparable to NF hearts (Fig. 4). Experimentally, this has been shown to couple with rebalancing of glucose metabolism19 and myocardial blood flow60 (rising in the anterior and declining in lateral walls), and such findings are consistent with CRT-associated increases in transcripts levels encoding oxidative phosphorylation and various metabolic pathways in anterior samples. Our results indicate that by re-coordinating contraction, regional heterogeneity of gene expression can be essentially returned to normal, even in a failing heart, on a genome-wide level.
Figure 4.
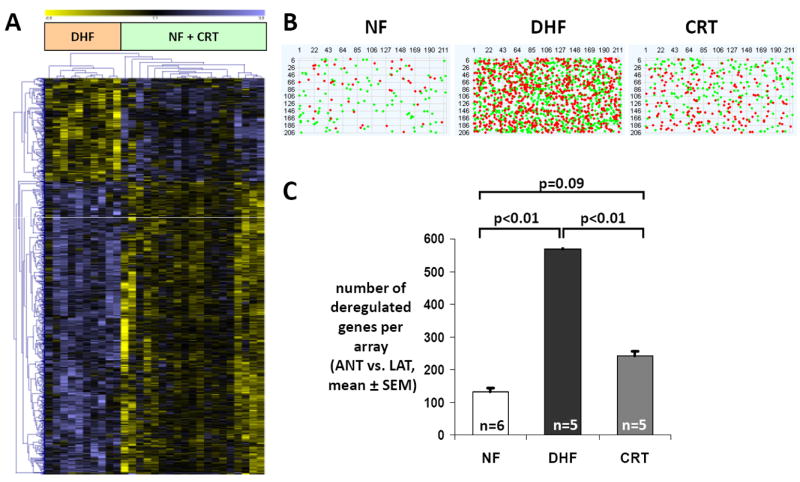
Dyssynchrony leads to increased regional heterogeneity in gene expression within the LV that is partially reduced with CRT. A. Unsupervised clustering of differentially expressed transcripts identified by multiclass Significance Analysis of Microarrays (SAM, false discovery rate <5%) using Euclidean distance for 1-color microarray data shows that transcript expression from CRT hearts clusters with NF hearts rather than with DHF samples. Each row represents data for 1 gene. The gene expression level is color coded with yellow and blue representing low and high expression, respectively. The difference in gene expression between the anterior and lateral wall from the same heart was compared for NF, DHF, and CRT. B. Pseudoimages of representative microarrays from NF, DHF, and CRT hearts with 211 columns and 206 rows (44K array). RNA from the anterior and lateral regions was labeled with Cy3 and Cy5 and hybridized in a 2-color design onto 1 array. Red and green dots represent statistically significant transcripts between anterior and lateral wall, respectively. C. Bar plot of the number of deregulated genes comparing the anterior and lateral regions in NF, DHF, and CRT hearts. In DHF, the number of differentially expressed transcripts between anterior and lateral wall increases 4-fold, whereas it is greatly reduced by CRT (modified from Barth et al.59).
Global effects of CRT on myocyte function
In addition to reversing regional molecular polarization, CRT globally corrects electrophysiological abnormalities and improves β-adrenergic responsiveness and mitochondrial energetic efficiency. All these changes may play an important role in the ability of CRT to enhance the systolic work performance of the failing heart acutely and chronically, while also improving long-term survival.
1. CRT reverses K+ channel remodeling and reduces afterdepolarizations
Downregulation of K+ currents is the most consistent ionic current change in animal models 47, 49, 50, 61 and human HF.45 K+ current downregulation may promote ventricular tachycardia (VT)/ventricular fibrillation (VF)50 either by direct prolongation of AP44 in the voltage range at which ICa,L reactivation occurs, predisposing to the development of early afterdepolarizations (EADs)62 or by heterogeneously reducing repolarization reserve and promoting functional reentry.63 CRT dramatically reduces the frequency of EADs in cells isolated from both the anterior and lateral LV (Fig. 5A&B).
Figure 5.
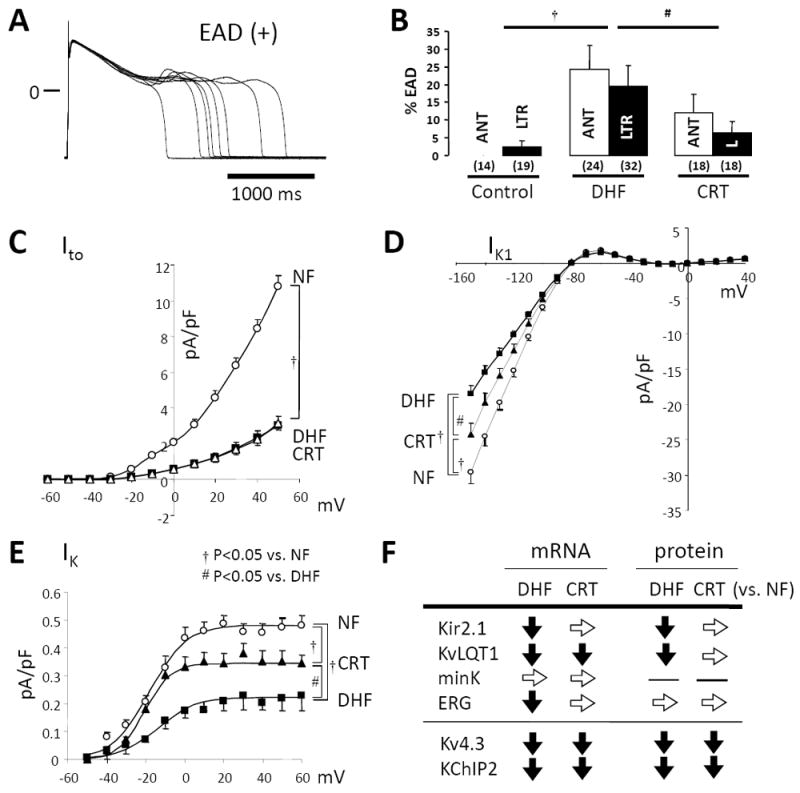
CRT reverses K+ channel remodeling and reduces EADs. A, Representative superimposed APs recorded in myocytes isolated from the lateral wall of DHF hearts with EADs. B, Bar plot of frequency of EADs (%EADs indicates fraction of APs with EADs). C-E, DHF significantly reduces the inward rectifier IK1, the delayed rectifier (IK) and transient outward K+ currents (Ito) in both anterior and lateral cells. CRT partially restores the DHF-induced reduction of IK1 and IK, but not Ito in both anterior and lateral cells. F. Changes in steady-state K+ channel mRNA subunit and protein expression. ANT or A indicates anterior; LTR or L, lateral (modified from Aiba et al.51).
We recently reported on these modifications in more detail, revealing that DHF significantly reduced the inward rectifier (IK1), delayed rectifier (IK), and transient outward potassium currents (Ito) in both anterior and lateral myocytes.51 CRT restored DHF-induced K+ current reductions throughout the ventricle, with the exception of Ito (Fig. 5C-E). In fact, K+ channels are the most diverse class of ion channels. The detailed changes in K+ channels vary with the model of HF or with species.64 Ito is unique among regulated K+ currents in HF, since it is downregulated uniformly in HF,45-47, 50, 65 yet not reversed by CRT (Fig. 5C).51 In parallel, Kv4.3 and KChIP2 mRNA and protein expression are downregulated in DHF without restoration by CRT (Fig. 5F).51
IK1 (Kir 2 family of genes) maintains the resting membrane potential and contributes to terminal repolarization. Reduced inward IK1 density in HF46, 49, 66, 67 may contribute to prolongation of APD and enhanced susceptibility to spontaneous depolarizations including delayed afterdepolarizations (DADs).67, 68 CRT, even in the setting of continued HF, partially restores IK1 density (Fig. 5D),51 decreases membrane resistance, and, in the setting of improved Ca2+ handling in CRT (see below), may reduce the frequency of arrhythmogenic DADs. Kir2.1 mRNA and protein levels are partially restored by CRT in the canine model (Fig. 5F).51
IK plays a prominent role in the late phase of repolarization,69 therefore changes in either the slow (IKs) or fast (IKr) activating components of this current could contribute significantly to AP prolongation in HF. CRT partially restores DHF-induced downregulation of IK density in both anterior and lateral LV myocytes without a significant change in mRNA or protein levels of KvLQT1- or mink-subunits for IKs, whereas mRNA level of ERG, a subunit of IKr, was restored by CRT in both the anterior or lateral LV wall (Fig. 5E&F).51
2. Ca2+ handling in DHF and CRT
HF is associated with major changes in Ca2+ handling, which underlies the observed reduction in force of contraction of the failing heart. Consistent with this heart failure phenotype, myocytes isolated from DHF hearts, showed calcium transients with a markedly reduced peak amplitude and slowed kinetics51, 70 in both anterior and lateral wall (Fig. 6A). CRT dramatically restored both the amplitude and kinetics of the Ca2+-transient. This result is striking, recalling that our CRT model involves 6 weeks of tachypacing, and CRT and DHF hearts have a similar degree of left ventricular dilation and elevation of end-diastolic pressure (LVEDP).
Figure 6.
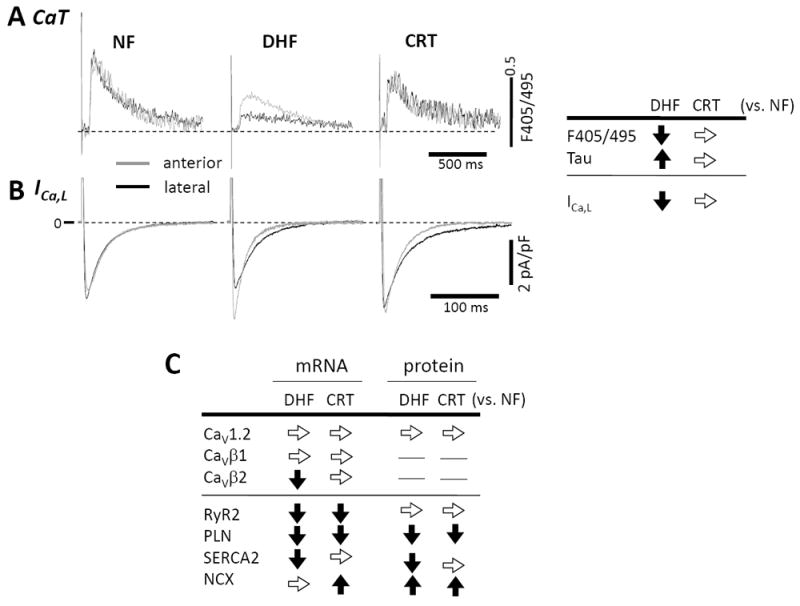
CRT restores DHF-induced reduction in Ca2+ currents (ICa,L) and transients (CaT). A. Compared with the control, DHF significantly reduced the CaT amplitude and slowed the rate of decay of the transient in both anterior and lateral myocytes. CRT partially restored the CaT amplitude of lateral cells and hastened the slowed decay of the CaT in anterior cells. B. In DHF, the peak ICa,L density was smaller and the decay was slower in the lateral myocytes, whereas the peak current density was larger and decay was faster in the anterior myocytes. The reduced current density is observed over a wide range of activation voltages. CRT restored the peak ICa,L density in lateral cells, but the decay was still slow (modified from Aiba et al.51).
On a beat-by-beat basis, the Ca2+ transient is elicited by the influx of a small amount of Ca2+ through L-type Ca2+ currents (ICa,L) and the subsequent large-scale Ca2+ release from the SR through the ryanodine receptor (RyR2). During diastole, cytosolic Ca2+ is taken up into the SR by the phospholamban (PLN)-regulated SR Ca2+-ATPase (SERCA2A). In DHF, we found that the reduction of ICa,L and Ca2+ transients is more pronounced in the lateral wall vs. the anterior wall. Importantly, CRT restored the DHF-induced reduction of peak ICa,L density, thus eliminating the anterior-lateral ICa,L density gradient (Fig. 6B).51 However, no significant differences in CaV1.2 (CaVα1C) mRNA and protein or CaVβ1 subunit mRNA expression were found among control, DHF, and CRT hearts (Fig. 6C).51 Yet, CaVβ2 mRNA was decreased significantly in DHF, but not in CRT myocytes when compared to non-failing myocardium.51
We further tested for the molecular basis for changes in Ca2+ transient. We found that mRNA and protein levels of SERCA2A, PLN and RyR2 were downregulated and Na+/Ca2+ exchanger (NCX1) up regulated without a change in CRT (Fig. 6C). There were also no regional differences in mRNA and protein expression in any of these mediators of Ca2+ handling in DHF and CRT. These results suggest that the differences of Ca2+ handling function in DHF and its restoration by CRT are post-translational.
3. Rest and β-adrenergic-stimulated myocyte contractility
The first hint of a positive impact of CRT on cardiac β-signaling was provided by clinical studies demonstrating reduced muscle sympathetic nerve activity in patients with severe HF and dyssynchrony,71 and CRT-mediated enhanced neural norepinephrine reuptake and retention.72 To more directly study myocyte β-adrenergic signaling, our group measured sarcomere shortening after administration of isoproterenol.22 Consistent with many models of HF, DHF myocytes displayed a highly blunted contractility at rest and during stimulation with isoproterenol compared with myocytes from non-failing hearts. Both basal and isoproterenol-stimulated cell shortening were markedly improved by CRT throughout the ventricles, and their recovery was well correlated with an increase in the amplitude and hastening of the kinetics of the Ca2+ transients (Fig. 7A).22
Figure 7.
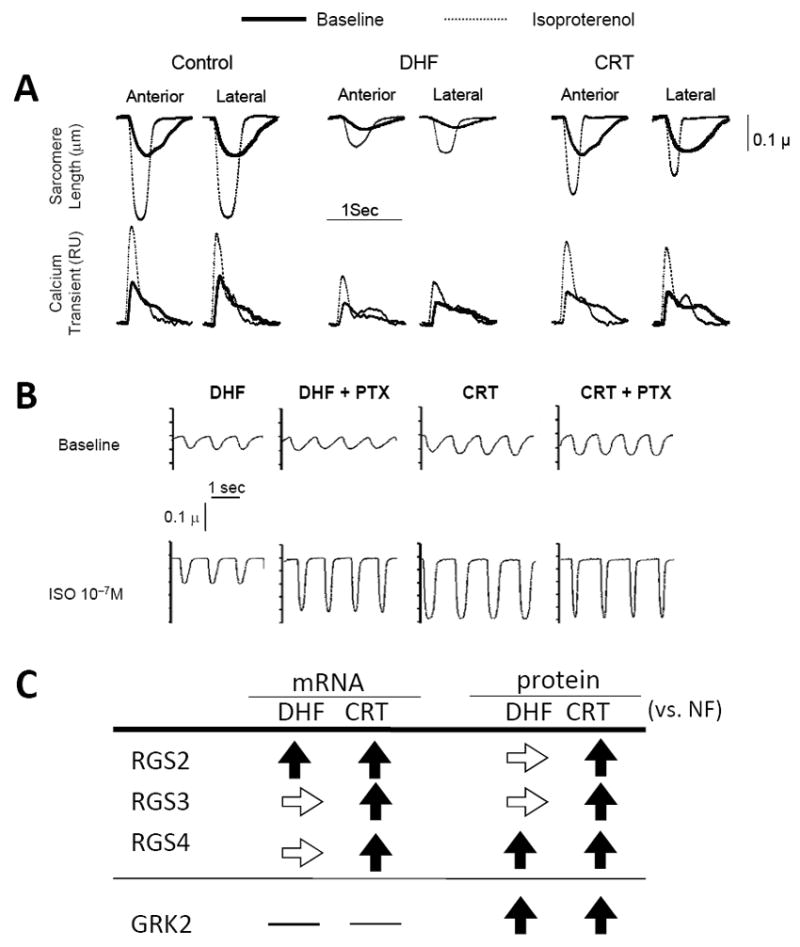
CRT improves rest and beta-adrenergic responsiveness in isolated myocytes from both early and late activated regions. A. Example time tracings of sarcomere length (top traces) and whole-cell calcium transients (Fura2- AM; bottom traces) for myocytes isolated from anterior versus lateral walls from control, DHF, and CRT left ventricles. Baseline and results with isoproterenol (ISO) stimulation are shown. Compared to controls, DHF cells displayed marked depression of resting function and calcium transients, and the ISO response in both behaviors was also very blunted. Rest and beta-adrenergic stimulated shortening and calcium transients were both strikingly improved by CRT. B. DHF results in enhanced inhibitory G-protein (Gi) coupling in DHF myocytes that is suppressed by CRT. In DHF, pretreatment with the Gi inhibitor pertussis toxin (PTX) enhanced ISO-stimulated contraction, whereas CRT myocytes had enhanced shortening without ISO and showed no further increase with addition of PTX. C. Summarized data for up- and down-regulation of the regulator of G-protein signaling (RGS) proteins by CRT. →, no change; ↓, decreased; and ↑, increased (adapted from Chakir et al. 22).
We examined the underlying mechanisms for enhanced β-adrenergic responsiveness by CRT. Both β1- and β2-receptor gene expression and receptor number were depressed by DHF, and CRT enhanced β1- but not β2-receptor number, as in humans.73 Functional analysis of adenylyl cyclase activity revealed that it was also depressed by DHF, and CRT augmented cAMP production. Among the most striking changes, however, was inhibitory G-protein (Gi) coupled signaling. As shown in Fig. 7B, myocytes from DHF hearts showed marked potentiation of the isoproterenol response if the myocytes were first incubated with pertussis toxin, which inhibits Gi. In contrast, CRT myocytes displayed enhanced responses at baseline and showed no effect with pertussis toxin, as if Gi already was inhibited by CRT. Consistent with human heart failure, Giα was up-regulated in both DHF and CRT animals and can therefore by itself not account for the enhanced β-adrenergic responsiveness with CRT. However, we found selective up-regulation of proteins called regulators of G-protein signaling (RGS) (Fig. 7C). RGS proteins negatively regulate G-coupled signaling by acting as selective GTPase accelerators, removing GTP from the activated α-subunit, and allowing the trimeric G-protein complex to reform suppressing G-protein coupled signaling. RGS3, a protein known to suppress Gi, was selectively up-regulated in human CRT responders as well as canine models of resynchronization. Moreover CRT appeared to improve contraction through RGS-mediated enhancement in coupling of β2-adrenergic receptors to stimulatory G-proteins (Gαs).74 Activation of the β2- Gαs axis may represent a general strategy how to improve functional reserve in patients with HF and dyssynchronous contraction, perhaps even those who do not respond to CRT.
4. Cell survival signaling
As reported both in humans43 and in our canine model,33 DHF hearts display an increase in apoptosis. In the canine pacing tachycardia model, this was supported by TUNEL staining, caspase-3 activity, and nuclear poly ADP-ribose polymerase cleavage. Importantly, apoptosis was suppressed by CRT. One of the most striking changes was a marked decline in Akt phosphorylation/activity with DHF that was also reversed by CRT (Fig. 8). Akt is generally considered a pro-survival kinase, and Akt phosphorylation of the pro-apoptotic protein BAD results in the interaction of BAD with the chaperone 14-3-3, reducing apoptosis. In the canine model, we observed reduced BAD phosphorylation (and 14-3-3 interaction) with DHF, a finding that was reversed with CRT. The anti-apoptotic impact of CRT appears global in nature, as molecular changes in BAD and 14-3-3 were observed in both anterior and lateral left ventricular myocardium. There are many other factors that regulate cell survival signaling that may also be modified by CRT. The mechanism by which the loss of dyssynchrony reactivates Akt to modify its downstream protein targets such as BAD remains unknown. It is unlikely that improved left ventricular function which is modest in this model with CRT accounts for the changes in Akt signaling. However, activation of secreted factors coupled to the abnormal cell-mechanical loading may prove an important pathway, and this is currently being explored.
Figure 8.
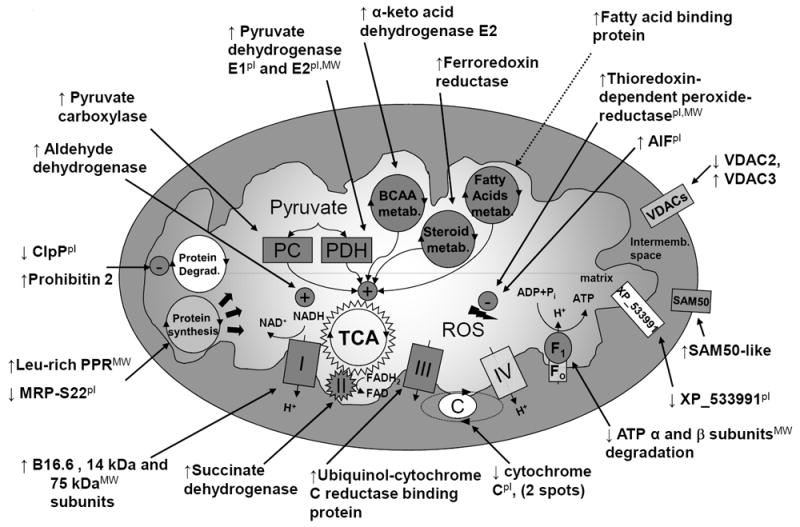
Schema of mitochondrial protein changes with CRT. The pI indicates observed pI differs >1 pH unit from predicted one; MW, observed MW differs more than 10 kDa from predicted one. FAD indicates flavin-adenine dinucleotide; FADH2, flavin-adenine dinucleotide, reduced form; NAD+, oxidized nicotinamide-adenine dinucleotide; Pi, inorganic phosphate; and TCA, tricarboxylic acid (adapted from Agnetti et al.75).
5. CRT and mitochondria
Given the central role of mitochondria in apoptosis pathways, the improved survival signaling prompted us to test the hypothesis that mitochondrial function was favorably altered by CRT. Agnetti et al.75 compared changes in mitochondrial protein expression and post-translational modification between DHF and CRT animals in lateral left ventricular myocardium, demonstrating salutary effects on mitochondrial respiration and efficiency of oxidative phosphorylation with CRT. Using optimized 2D electrophoresis of the mitochondrial sub-proteome, ≈1200 protein spots were resolved revealing 31 quantitative protein changes between DHF and CRT. Most changes were in proteins of the respiratory chain, including all of the complexes of oxidative phosphorylation (except complex IV; Fig. 8), consistent with CRT modulating ATP production.75 CRT also increased the metabolic pathways supplying the substrates (pyruvate carboxylase and pyruvate dehydrogenase, E1 and E2 subunits) and key enzymes (aldehyde dehydrogenase, α-keto acid dehydrogenase E2, and ferredoxin reductase) fuelling the Krebs cycle. Importantly, mitochondrial oxidative efficiency (ADP/O2) was depressed by DHF and enhanced by CRT. CRT also reduced oxidative stress, potentially by enhanced mitochondrial reactive oxygen species (ROS) -scavenging proteins. These mitochondrial changes have not been reported with other HF therapies, and may represent a selective response to CRT.
Conclusion
We and others have found profound basic cellular and molecular changes in DHF, many of which are not observed in HF with synchronous ventricular contraction. Remarkably, CRT specifically targets and reverses many of these changes. Some of the observed cellular and molecular alterations appear global in nature, though some cascades exhibit regional specificity, e.g. activation of stress kinases in the late activating lateral wall and transcriptional changes in the anterior wall. The improvement in cell survival, and increase in potassium currents, myocyte contractility and β-AR responsiveness occur throughout the ventricle. These results suggest that CRT can influence the failing heart in a way that enhances both regional mechanical work and global pump function and may be the basis for improved long-term survival with CRT. The relative contribution of global vs. regional cellular and molecular signaling in DHF and CRT remain incompletely understood. We hypothesize that the beneficial effects of CRT on global myocardial function result from restoration of the normal sequence of excitation and contraction, involving local, region-specific myocardial changes in neurohumoral activation, and mechanical forces.13 Ongoing studies are attempting to further investigate this interesting question.
The characteristic molecular and electrophysiological alterations of DHF also promise to identify a molecular signature through which CRT ameliorates the heart failure phenotype. As noted, HF and depressed ejection fraction do not necessarily predict the response to specific pharmacologic therapies, and variations in the underlying genetics as well as molecular and cellular biology are increasingly thought to be key determining factors.76, 77 We do not yet know whether underlying cellular and/or molecular signaling responses to dyssynchrony vary among individuals with DHF, but we speculate that a lack of depressed Akt activation, IK signaling or up-regulated Gi coupling in a given DHF patient might diminish the effectiveness of CRT. The level of molecular heterogeneity may prove to be a useful marker that dyssynchrony has adversely impacted the ventricle function beyond HF-specific molecular changes.
Acknowledgments
Funding Sources This work was supported by National Heart, Lung, and Blood Institute (NHLBI) grants P01 HL77189-01 (G.F.T.) and RO1 HL72488 (G.F.T.). G.F.T. is the Michel Mirowski M.D. Professor of Cardiology.
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Bader H, Garrigue S, Lafitte S, Reuter S, Jais P, Haissaguerre M, Bonnet J, Clementy J, Roudaut R. Intra-left ventricular electromechanical asynchrony. A new independent predictor of severe cardiac events in heart failure patients. J Am Coll Cardiol. 2004;43:248–256. doi: 10.1016/j.jacc.2003.08.038. [DOI] [PubMed] [Google Scholar]
- 2.Cho GY, Song JK, Park WJ, Han SW, Choi SH, Doo YC, Oh DJ, Lee Y. Mechanical dyssynchrony assessed by tissue Doppler imaging is a powerful predictor of mortality in congestive heart failure with normal QRS duration. J Am Coll Cardiol. 2005;46:2237–2243. doi: 10.1016/j.jacc.2004.11.074. [DOI] [PubMed] [Google Scholar]
- 3.Iuliano S, Fisher SG, Karasik PE, Fletcher RD, Singh SN. QRS duration and mortality in patients with congestive heart failure. Am Heart J. 2002;143:1085–1091. doi: 10.1067/mhj.2002.122516. [DOI] [PubMed] [Google Scholar]
- 4.Kashani A, Barold SS. Significance of QRS complex duration in patients with heart failure. J Am Coll Cardiol. 2005;46:2183–2192. doi: 10.1016/j.jacc.2005.01.071. [DOI] [PubMed] [Google Scholar]
- 5.Chalil S, Stegemann B, Muhyaldeen S, Khadjooi K, Smith RE, Jordan PJ, Leyva F. Intraventricular dyssynchrony predicts mortality and morbidity after cardiac resynchronization therapy: a study using cardiovascular magnetic resonance tissue synchronization imaging. J Am Coll Cardiol. 2007;50:243–252. doi: 10.1016/j.jacc.2007.03.035. [DOI] [PubMed] [Google Scholar]
- 6.Yu CM, Lin H, Zhang Q, Sanderson JE. High prevalence of left ventricular systolic and diastolic asynchrony in patients with congestive heart failure and normal QRS duration. Heart. 2003;89:54–60. doi: 10.1136/heart.89.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bristow MR, Saxon LA, Boehmer J, Krueger S, Kass DA, De Marco T, Carson P, DiCarlo L, DeMets D, White BG, DeVries DW, Feldman AM. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med. 2004;350:2140–2150. doi: 10.1056/NEJMoa032423. [DOI] [PubMed] [Google Scholar]
- 8.Cleland JG, Daubert JC, Erdmann E, Freemantle N, Gras D, Kappenberger L, Tavazzi L. The effect of cardiac resynchronization on morbidity and mortality in heart failure. N Engl J Med. 2005;352:1539–1549. doi: 10.1056/NEJMoa050496. [DOI] [PubMed] [Google Scholar]
- 9.Vardas PE, Auricchio A, Blanc JJ, Daubert JC, Drexler H, Ector H, Gasparini M, Linde C, Morgado FB, Oto A, Sutton R, Trusz-Gluza M, Vahanian A, Camm J, De Caterina R, Dean V, Dickstein K, Funck-Brentano C, Filippatos G, Hellemans I, Kristensen SD, McGregor K, Sechtem U, Silber S, Tendera M, Widimsky P, Zamorano JL, Priori SG, Blomstrom-Lundqvist C, Brignole M, Terradellas JB, Castellano P, Cleland J, Farre J, Fromer M, Le Heuzey JY, Lip GY, Merino JL, Montenero AS, Ritter P, Schlij MJ, Stellbrink C. Guidelines in cardiac pacing and resynchronization therapy. Kardiol Pol. 2007;65:1449–1487. discussion 1488-1449. [PubMed] [Google Scholar]
- 10.Leclercq C, Singh JP. Cardiac resynchronization therapy: from treatment to prevention. Eur Heart J. 2011;32:1580–1582. doi: 10.1093/eurheartj/ehr016. [DOI] [PubMed] [Google Scholar]
- 11.Abraham WT, Fisher WG, Smith AL, Delurgio DB, Leon AR, Loh E, Kocovic DZ, Packer M, Clavell AL, Hayes DL, Ellestad M, Trupp RJ, Underwood J, Pickering F, Truex C, McAtee P, Messenger J. Cardiac resynchronization in chronic heart failure. N Engl J Med. 2002;346:1845–1853. doi: 10.1056/NEJMoa013168. [DOI] [PubMed] [Google Scholar]
- 12.Linde C, Leclercq C, Rex S, Garrigue S, Lavergne T, Cazeau S, McKenna W, Fitzgerald M, Deharo JC, Alonso C, Walker S, Braunschweig F, Bailleul C, Daubert JC. Long-term benefits of biventricular pacing in congestive heart failure: results from the MUltisite STimulation in cardiomyopathy (MUSTIC) study. J Am Coll Cardiol. 2002;40:111–118. doi: 10.1016/s0735-1097(02)01932-0. [DOI] [PubMed] [Google Scholar]
- 13.Kass DA. Pathobiology of cardiac dyssynchrony and resynchronization. Heart Rhythm. 2009;6:1660–1665. doi: 10.1016/j.hrthm.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung ES, Leon AR, Tavazzi L, Sun JP, Nihoyannopoulos P, Merlino J, Abraham WT, Ghio S, Leclercq C, Bax JJ, Yu CM, Gorcsan J, 3rd, St John Sutton M, De Sutter J, Murillo J. Results of the Predictors of Response to CRT (PROSPECT) trial. Circulation. 2008;117:2608–2616. doi: 10.1161/CIRCULATIONAHA.107.743120. [DOI] [PubMed] [Google Scholar]
- 15.Bleeker GB, Mollema SA, Holman ER, Van de Veire N, Ypenburg C, Boersma E, van der Wall EE, Schalij MJ, Bax JJ. Left ventricular resynchronization is mandatory for response to cardiac resynchronization therapy: analysis in patients with echocardiographic evidence of left ventricular dyssynchrony at baseline. Circulation. 2007;116:1440–1448. doi: 10.1161/CIRCULATIONAHA.106.677005. [DOI] [PubMed] [Google Scholar]
- 16.Thomas AC, Mower MM. Multiple chambered pacing for treatment of congestive heart failure. Pacing Clin Electrophysiol. 1995;18:749–750. doi: 10.1111/j.1540-8159.1995.tb04676.x. [DOI] [PubMed] [Google Scholar]
- 17.Kass DA, Chen CH, Curry C, Talbot M, Berger R, Fetics B, Nevo E. Improved left ventricular mechanics from acute VDD pacing in patients with dilated cardiomyopathy and ventricular conduction delay. Circulation. 1999;99:1567–1573. doi: 10.1161/01.cir.99.12.1567. [DOI] [PubMed] [Google Scholar]
- 18.Nelson GS, Berger RD, Fetics BJ, Talbot M, Spinelli JC, Hare JM, Kass DA. Left ventricular or biventricular pacing improves cardiac function at diminished energy cost in patients with dilated cardiomyopathy and left bundle-branch block. Circulation. 2000;102:3053–3059. doi: 10.1161/01.cir.102.25.3053. [DOI] [PubMed] [Google Scholar]
- 19.Nowak B, Sinha AM, Schaefer WM, Koch KC, Kaiser HJ, Hanrath P, Buell U, Stellbrink C. Cardiac resynchronization therapy homogenizes myocardial glucose metabolism and perfusion in dilated cardiomyopathy and left bundle branch block. J Am Coll Cardiol. 2003;41:1523–1528. doi: 10.1016/s0735-1097(03)00257-2. [DOI] [PubMed] [Google Scholar]
- 20.Abraham WT. Cardiac resynchronization therapy. Prog Cardiovasc Dis. 2006;48:232–238. doi: 10.1016/j.pcad.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Cleland JG, Daubert JC, Erdmann E, Freemantle N, Gras D, Kappenberger L, Tavazzi L. Longer-term effects of cardiac resynchronization therapy on mortality in heart failure [the CArdiac REsynchronization-Heart Failure (CARE-HF) trial extension phase] Eur Heart J. 2006;27:1928–1932. doi: 10.1093/eurheartj/ehl099. [DOI] [PubMed] [Google Scholar]
- 22.Chakir K, Daya SK, Aiba T, Tunin RS, Dimaano VL, Abraham TP, Jaques-Robinson KM, Lai EW, Pacak K, Zhu WZ, Xiao RP, Tomaselli GF, Kass DA. Mechanisms of enhanced beta-adrenergic reserve from cardiac resynchronization therapy. Circulation. 2009;119:1231–1240. doi: 10.1161/CIRCULATIONAHA.108.774752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burkhoff D, Holmes JW, Madigan J, Barbone A, Oz MC. Left ventricular assist device-induced reverse ventricular remodeling. Prog Cardiovasc Dis. 2000;43:19–26. doi: 10.1053/pcad.2000.7190. [DOI] [PubMed] [Google Scholar]
- 24.Burkhoff D, Klotz S, Mancini DM. LVAD-induced reverse remodeling: basic and clinical implications for myocardial recovery. J Card Fail. 2006;12:227–239. doi: 10.1016/j.cardfail.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 25.Thohan V, Stetson SJ, Nagueh SF, Rivas-Gotz C, Koerner MM, Lafuente JA, Loebe M, Noon GP, Torre-Amione G. Cellular and hemodynamics responses of failing myocardium to continuous flow mechanical circulatory support using the DeBakey-Noon left ventricular assist device: a comparative analysis with pulsatile-type devices. J Heart Lung Transplant. 2005;24:566–575. doi: 10.1016/j.healun.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 26.Iyengar S, Haas G, Lamba S, Orsinelli DA, Babu GJ, Ferketich AK, Yamokoski L, Periasamy M, Abraham WT. Effect of cardiac resynchronization therapy on myocardial gene expression in patients with nonischemic dilated cardiomyopathy. J Card Fail. 2007;13:304–311. doi: 10.1016/j.cardfail.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Mullens W, Bartunek J, Wilson Tang WH, Delrue L, Herbots L, Willems R, De Bruyne B, Goethals M, Verstreken S, Vanderheyden M. Early and late effects of cardiac resynchronization therapy on force-frequency relation and contractility regulating gene expression in heart failure patients. Heart Rhythm. 2008;5:52–59. doi: 10.1016/j.hrthm.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 28.Vanderheyden M, Mullens W, Delrue L, Goethals M, de Bruyne B, Wijns W, Geelen P, Verstreken S, Wellens F, Bartunek J. Myocardial gene expression in heart failure patients treated with cardiac resynchronization therapy responders versus nonresponders. J Am Coll Cardiol. 2008;51:129–136. doi: 10.1016/j.jacc.2007.07.087. [DOI] [PubMed] [Google Scholar]
- 29.Francia P, Salvati A, Balla C, De Paolis P, Pagannone E, Borro M, Gentile G, Simmaco M, De Biase L, Volpe M. Cardiac resynchronization therapy increases plasma levels of the endogenous inotrope apelin. Eur J Heart Fail. 2007;9:306–309. doi: 10.1016/j.ejheart.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Hessel MH, Bleeker GB, Bax JJ, Henneman MM, den Adel B, Klok M, Schalij MJ, Atsma DE, van der Laarse A. Reverse ventricular remodelling after cardiac resynchronization therapy is associated with a reduction in serum tenascin-C and plasma matrix metalloproteinase-9 levels. Eur J Heart Fail. 2007;9:1058–1063. doi: 10.1016/j.ejheart.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Lappegard KT, Bjornstad H. Anti-inflammatory effect of cardiac resynchronization therapy. Pacing Clin Electrophysiol. 2006;29:753–758. doi: 10.1111/j.1540-8159.2006.00430.x. [DOI] [PubMed] [Google Scholar]
- 32.Spragg DD, Leclercq C, Loghmani M, Faris OP, Tunin RS, DiSilvestre D, McVeigh ER, Tomaselli GF, Kass DA. Regional alterations in protein expression in the dyssynchronous failing heart. Circulation. 2003;108:929–932. doi: 10.1161/01.CIR.0000088782.99568.CA. [DOI] [PubMed] [Google Scholar]
- 33.Chakir K, Daya SK, Tunin RS, Helm RH, Byrne MJ, Dimaano VL, Lardo AC, Abraham TP, Tomaselli GF, Kass DA. Reversal of global apoptosis and regional stress kinase activation by cardiac resynchronization. Circulation. 2008;117:1369–1377. doi: 10.1161/CIRCULATIONAHA.107.706291. [DOI] [PubMed] [Google Scholar]
- 34.Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, Saffitz J, Chien K, Xiao RP, Kass DA, Wang Y. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A. 2001;98:12283–12288. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liao P, Wang SQ, Wang S, Zheng M, Zhang SJ, Cheng H, Wang Y, Xiao RP. p38 Mitogen-activated protein kinase mediates a negative inotropic effect in cardiac myocytes. Circ Res. 2002;90:190–196. doi: 10.1161/hh0202.104220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, Brown JH, Devic E, Kobilka BK, Cheng H, Xiao RP. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest. 2003;111:617–625. doi: 10.1172/JCI16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116:1853–1864. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116:675–682. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grueter CE, Colbran RJ, Anderson ME. CaMKII, an emerging molecular driver for calcium homeostasis, arrhythmias, and cardiac dysfunction. J Mol Med (Berl) 2007;85:5–14. doi: 10.1007/s00109-006-0125-6. [DOI] [PubMed] [Google Scholar]
- 40.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 41.Feldman AM, Combes A, Wagner D, Kadakomi T, Kubota T, Li YY, McTiernan C. The role of tumor necrosis factor in the pathophysiology of heart failure. J Am Coll Cardiol. 2000;35:537–544. doi: 10.1016/s0735-1097(99)00600-2. [DOI] [PubMed] [Google Scholar]
- 42.Kapadia SR, Oral H, Lee J, Nakano M, Taffet GE, Mann DL. Hemodynamic regulation of tumor necrosis factor-alpha gene and protein expression in adult feline myocardium. Circ Res. 1997;81:187–195. doi: 10.1161/01.res.81.2.187. [DOI] [PubMed] [Google Scholar]
- 43.D’Ascia C, Cittadini A, Monti MG, Riccio G, Sacca L. Effects of biventricular pacing on interstitial remodelling, tumor necrosis factor-alpha expression, and apoptotic death in failing human myocardium. Eur Heart J. 2006;27:201–206. doi: 10.1093/eurheartj/ehi579. [DOI] [PubMed] [Google Scholar]
- 44.Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res. 2003;93:638–645. doi: 10.1161/01.RES.0000092248.59479.AE. [DOI] [PubMed] [Google Scholar]
- 45.Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73:379–385. doi: 10.1161/01.res.73.2.379. [DOI] [PubMed] [Google Scholar]
- 46.Kaab S, Dixon J, Duc J, Ashen D, Nabauer M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998;98:1383–1393. doi: 10.1161/01.cir.98.14.1383. [DOI] [PubMed] [Google Scholar]
- 47.Kaab S, Nuss HB, Chiamvimonvat N, O’Rourke B, Pak PH, Kass DA, Marban E, Tomaselli GF. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ Res. 1996;78:262–273. doi: 10.1161/01.res.78.2.262. [DOI] [PubMed] [Google Scholar]
- 48.Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87:425–456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- 49.Rose J, Armoundas AA, Tian Y, DiSilvestre D, Burysek M, Halperin V, O’Rourke B, Kass DA, Marban E, Tomaselli GF. Molecular correlates of altered expression of potassium currents in failing rabbit myocardium. Am J Physiol Heart Circ Physiol. 2005;288:H2077–2087. doi: 10.1152/ajpheart.00526.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsuji Y, Zicha S, Qi XY, Kodama I, Nattel S. Potassium channel subunit remodeling in rabbits exposed to long-term bradycardia or tachycardia: discrete arrhythmogenic consequences related to differential delayed-rectifier changes. Circulation. 2006;113:345–355. doi: 10.1161/CIRCULATIONAHA.105.552968. [DOI] [PubMed] [Google Scholar]
- 51.Aiba T, Hesketh GG, Barth AS, Liu T, Daya S, Chakir K, Dimaano VL, Abraham TP, O’Rourke B, Akar FG, Kass DA, Tomaselli GF. Electrophysiological consequences of dyssynchronous heart failure and its restoration by resynchronization therapy. Circulation. 2009;119:1220–1230. doi: 10.1161/CIRCULATIONAHA.108.794834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernandez-Velasco M, Ruiz-Hurtado G, Hurtado O, Moro MA, Delgado C. TNF-alpha downregulates transient outward potassium current in rat ventricular myocytes through iNOS overexpression and oxidant species generation. Am J Physiol Heart Circ Physiol. 2007;293:H238–245. doi: 10.1152/ajpheart.01122.2006. [DOI] [PubMed] [Google Scholar]
- 53.Xie LH, Chen F, Karagueuzian HS, Weiss JN. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res. 2009;104:79–86. doi: 10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kohlhaas M, Zhang T, Seidler T, Zibrova D, Dybkova N, Steen A, Wagner S, Chen L, Brown JH, Bers DM, Maier LS. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ Res. 2006;98:235–244. doi: 10.1161/01.RES.0000200739.90811.9f. [DOI] [PubMed] [Google Scholar]
- 55.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–911. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 56.Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN, Undrovinas A. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am J Physiol Heart Circ Physiol. 2008;294:H1597–1608. doi: 10.1152/ajpheart.00484.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–1293. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 59.Barth AS, Aiba T, Halperin V, DiSilvestre D, Chakir K, Colantuoni C, Tunin RS, Dimaano VL, Yu W, Abraham TP, Kass DA, Tomaselli GF. Cardiac resynchronization therapy corrects dyssynchrony-induced regional gene expression changes on a genomic level. Circ Cardiovasc Genet. 2009;2:371–378. doi: 10.1161/CIRCGENETICS.108.832345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valzania C, Gadler F, Winter R, Braunschweig F, Brodin LA, Gudmundsson P, Boriani G, Eriksson MJ. Effects of cardiac resynchronization therapy on coronary blood flow: evaluation by transthoracic Doppler echocardiography. Eur J Heart Fail. 2008;10:514–520. doi: 10.1016/j.ejheart.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 61.Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004;95:754–763. doi: 10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- 62.Aiba T, Shimizu W, Inagaki M, Noda T, Miyoshi S, Ding WG, Zankov DP, Toyoda F, Matsuura H, Horie M, Sunagawa K. Cellular and ionic mechanism for drug-induced long QT syndrome and effectiveness of verapamil. J Am Coll Cardiol. 2005;45:300–307. doi: 10.1016/j.jacc.2004.09.069. [DOI] [PubMed] [Google Scholar]
- 63.Roden DM, Spooner PM. Inherited long QT syndromes: a paradigm for understanding arrhythmogenesis. J Cardiovasc Electrophysiol. 1999;10:1664–1683. doi: 10.1111/j.1540-8167.1999.tb00231.x. [DOI] [PubMed] [Google Scholar]
- 64.Aiba T, Tomaselli GF. Electrical remodeling in the failing heart. Curr Opin Cardiol. 2010;25:29–36. doi: 10.1097/HCO.0b013e328333d3d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nabauer M, Beuckelmann DJ, Erdmann E. Characteristics of transient outward current in human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73:386–394. doi: 10.1161/01.res.73.2.386. [DOI] [PubMed] [Google Scholar]
- 66.Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. 2002;283:H1031–1041. doi: 10.1152/ajpheart.00105.2002. [DOI] [PubMed] [Google Scholar]
- 67.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 68.Nuss HB, Kaab S, Kass DA, Tomaselli GF, Marban E. Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am J Physiol. 1999;277:H80–91. doi: 10.1152/ajpheart.1999.277.1.H80. [DOI] [PubMed] [Google Scholar]
- 69.Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. A weaker IKs contributes to the longer action potential of the M cell. Circ Res. 1995;76:351–365. doi: 10.1161/01.res.76.3.351. [DOI] [PubMed] [Google Scholar]
- 70.Nishijima Y, Sridhar A, Viatchenko-Karpinski S, Shaw C, Bonagura JD, Abraham WT, Joshi MS, Bauer JA, Hamlin RL, Gyorke S, Feldman DS, Carnes CA. Chronic cardiac resynchronization therapy and reverse ventricular remodeling in a model of nonischemic cardiomyopathy. Life Sci. 2007;81:1152–1159. doi: 10.1016/j.lfs.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Najem B, Unger P, Preumont N, Jansens JL, Houssiere A, Pathak A, Xhaet O, Gabriel L, Friart A, De Roy L, Vandenbossche JL, van de Borne P. Sympathetic control after cardiac resynchronization therapy: responders versus nonresponders. Am J Physiol Heart Circ Physiol. 2006;291:H2647–2652. doi: 10.1152/ajpheart.00373.2006. [DOI] [PubMed] [Google Scholar]
- 72.Erol-Yilmaz A, Verberne HJ, Schrama TA, Hrudova J, De Winter RJ, Van Eck-Smit BL, De Bruin R, Bax JJ, Schalij MJ, Wilde AA, Tukkie R. Cardiac resynchronization induces favorable neurohumoral changes. Pacing Clin Electrophysiol. 2005;28:304–310. doi: 10.1111/j.1540-8159.2005.09508.x. [DOI] [PubMed] [Google Scholar]
- 73.Vanderheyden M, Mullens W, Delrue L, Goethals M, Verstreken S, Wijns W, de Bruyne B, Bartunek J. Endomyocardial upregulation of beta1 adrenoreceptor gene expression and myocardial contractile reserve following cardiac resynchronization therapy. J Card Fail. 2008;14:172–178. doi: 10.1016/j.cardfail.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 74.Chakir K, Depry C, Dimaano VL, Zhu WZ, Vanderheyden M, Bartunek J, Abraham TP, Tomaselli GF, Liu SB, Xiang YK, Zhang M, Takimoto E, Dulin N, Xiao RP, Zhang J, Kass DA. Galphas-biased beta2-adrenergic receptor signaling from restoring synchronous contraction in the failing heart. Sci Transl Med. 2011;3:100ra88. doi: 10.1126/scitranslmed.3001909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Agnetti G, Kaludercic N, Kane LA, Elliott ST, Guo Y, Chakir K, Samantapudi D, Paolocci N, Tomaselli GF, Kass DA, Van Eyk JE. Modulation of mitochondrial proteome and improved mitochondrial function by biventricular pacing of dyssynchronous failing hearts. Circ Cardiovasc Genet. 2010;3:78–87. doi: 10.1161/CIRCGENETICS.109.871236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liggett SB, Cresci S, Kelly RJ, Syed FM, Matkovich SJ, Hahn HS, Diwan A, Martini JS, Sparks L, Parekh RR, Spertus JA, Koch WJ, Kardia SL, Dorn GW., 2nd A GRK5 polymorphism that inhibits beta-adrenergic receptor signaling is protective in heart failure. Nat Med. 2008;14:510–517. doi: 10.1038/nm1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mialet Perez J, Rathz DA, Petrashevskaya NN, Hahn HS, Wagoner LE, Schwartz A, Dorn GW, Liggett SB. Beta 1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat Med. 2003;9:1300–1305. doi: 10.1038/nm930. [DOI] [PubMed] [Google Scholar]