

Abstract
Experimental work has elucidated molecular and cellular pathways of inflammation that promote atherosclerosis. Unraveling the roles of cytokines as inflammatory messengers provided a mechanism whereby risk factors for atherosclerosis can alter arterial biology, and produce a systemic milieu that favors atherothrombotic events. The discovery of the immune basis of allograft arteriosclerosis demonstrated that inflammation per se can drive arterial hyperplasia, even in the absence of traditional risk factors. Inflammation regulates aspects of plaque biology that trigger the thrombotic complications of atherosclerosis. Translation of these discoveries to humans has enabled both novel mechanistic insights and practical clinical advances.
For much of the last century, most considered atherosclerosis to be a cholesterol storage disease, characterized by the collection of cholesterol and thrombotic debris in the artery wall. The modern era of the cell biology of atherosclerosis in the 1960s and 1970s focused on the proliferation of smooth-muscle cells as the nidus for atherosclerotic plaques.1, 2 Over the last quarter century, the concept that inflammation plays a primordial role in atherogenesis has gained ascendency. Yet, as with many innovations in science and medicine, the roots of this seemingly modern concept stretch far back in time.
Inflammation: an enduring flame
Egyptian papyri from almost 5,000 years ago refer to heat and redness as concomitants of disease. In the 1st century, Aulus Cornelius Celsus defined the cardinal signs of inflammation: redness, swelling, heat, and pain. The advent of the microscope and aniline dyes laid the groundwork for the era of cellular pathology and the cell biology of inflammation. In the 19th century, keen observers described the diapedesis of leukocytes from the blood into tissues. Rudolf Virchow recognized the inflammatory nature of atherosclerotic plaques. “In some, particularly violent cases the softening manifests itself even in the arteries not as the consequence of a really fatty process, but as a direct product of inflammation.”3 Virchow also understood atherosclerosis as an active process of tissue reaction, rather than a mere encrustation of thrombus or deposition of fatty material, stating that “the frequency with which cells in a state of fatty degeneration are found in inflamed parts, affords sufficient proof, that in the course of inflammatory processes, which it is impossible we should ever regard as simply passive processes, such transformations must take place.” Virchow’s concept of atherogenesis, elements of which appear strikingly modern, unfortunately yielded to the view of atheroma as a primarily passive lipid collection for more than a century.
In the meantime, experimentalists laid the foundation for modern immunology. Paul Ehrlich studied antibodies and proposed the concept of complementarity of antigen and antibody, analogous to a key fitting into a lock. His innovative concepts formed the basis of the field of adaptive immunity. Ilya Mechnikov discovered phagocytosis at the end of the 19th century, providing the basis of the field we now call innate immunity.4 Ehrlich and Mechnikov shared the Nobel Prize in 1908 for their pioneering studies in immunity and host defenses. Yet, the application of these concepts to atherosclerosis lagged by almost a century.
Innate immunity in atherosclerosis
Since Virchow’s day, pathologists have recognized lipid-laden “foam cells” as a hallmark of atheromata. The advent of rigorous cell identification, enabled by the development of monoclonal antibodies, led to the confirmation that most foam cells arise from mononuclear phagocytes, although smooth-muscle cells and endothelial cells can also become engorged with lipids.5, 6 Still, most viewed macrophages as the graveyard of lipids in the plaque, rather than as active participants in atherogenesis. Death of foam cells would lead to formation of the “necrotic core” — conceived of as a depot of cellular debris and lipids. Indeed, some early schemes of the cell biology of atherosclerosis depicted atherogenesis as a bland process, without the participation of inflammatory cells.1, 2 The role of the mononuclear phagocyte as an effector emerged with the characterization of macrophage-derived mediators such as cytokines.7 The concept of dynamic interplay between mononuclear phagocytes and vascular cells during atherogenesis, once overlooked or doubted, has now become commonplace. (Figure 1)
Figure 1.
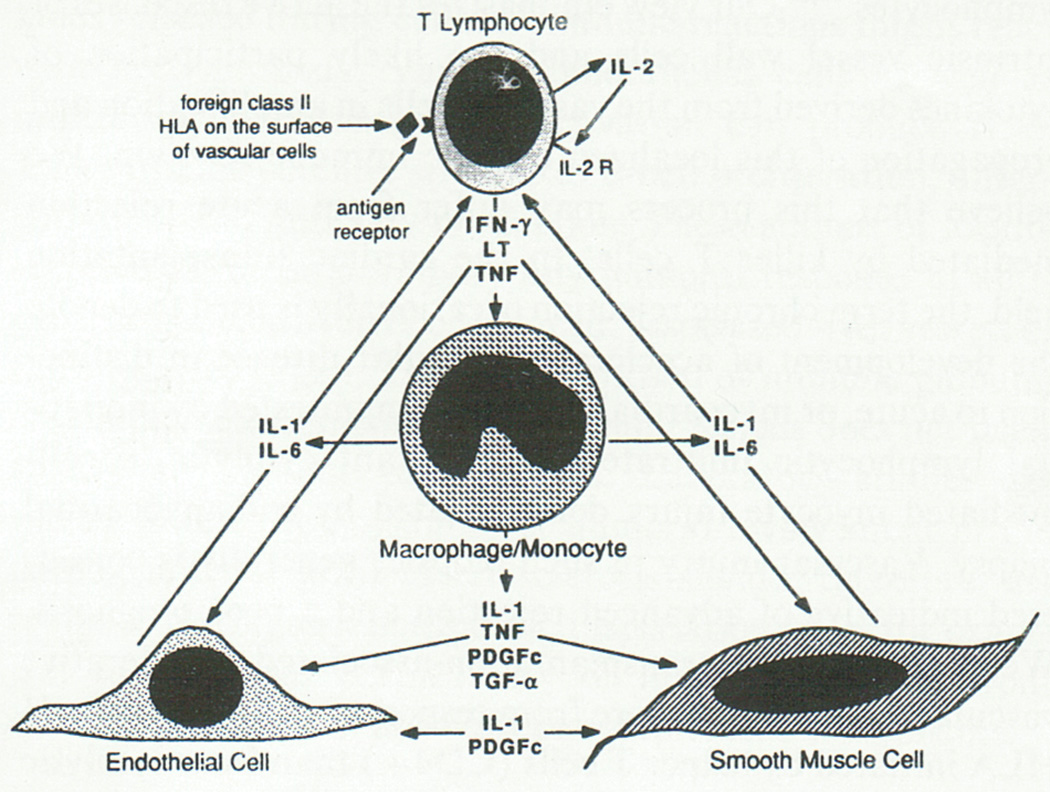
This drawing (made with the original MacPaint application by the author) presents an early depiction of the crosstalk between inflammatory cells and intrinsic factor wall cells mediated by cytokines. Work from around the world has now verified the principle points postulated in this primitive picture, a precursor of countless subsequent schemata. The T lymphocyte depicted at the apex of the diagram responds to antigenic stimulation by elaborating cytokines such as interferon gamma (IFN–γ), lymphotoxin (LT), and tumor necrosis factor-alpha (TNF–α). This sequence represents the adaptive immune response. These cytokines impinge upon the macrophage/monocyte depicted in the middle of the diagram that can elaborate mediators of innate immunity such as interleukin-1 and interleukin-6 in response, as well as a material then denoted as platelet-derived growth factor cross-reactive material (PDGFc), and transforming growth factor alpha (TGF–α) Monocytes/macrophages thus comprise a major cell type involved in innate immunity. These cytokines produced by mononuclear phagocytes in turn mediate paracrine signaling to endothelial cells (EC, lower left) or vascular smooth-muscle cells (SMC, lower right). From: Libby P, Salomon RN, Payne DD, Schoen FJ, Pober JS. Functions of vascular wall cells related to development of transplantation-associated coronary arteriosclerosis. Transplant Proc 1989;21(4):3677–3684.
Inflammatory cells function in atherogenesis
The discovery of adhesion molecules expressed by endothelial cells provided important insight into the initiation of atherosclerotic lesions.8 For example, vascular cell adhesion molecule-1 (VCAM-1), expressed by cytokine-stimulated endothelial cells, binds just those types of inflammatory cells that accumulate in the early atherosclerotic plaque: monocytes and T lymphocytes.9, 10 The characterization of leukocyte adhesion molecules provided a mechanism for the sticking of the mononuclear cells to intact endothelium in hypercholesterolemic rabbits observed by Poole and Florey in 1958.11 Once adherent to the endothelial surface due to the expression of inducible adhesion molecules, the mononuclear cells receive chemoattractant signals that beckon them to enter the intima. Chemoattractants such as monocyte chemoattractant protein-1 (MCP-1) contribute importantly to this process.12 A trio of chemokines induced by interferon-gamma (IFN-γ) also selectively recruit T lymphocytes to the nascent atherosclerotic plaque.13
Once resident in the arterial intima, monocytes mature into macrophages. In the plaque, these mononuclear phagocytes express scavenger receptors necessary for uptake of modified lipoproteins and hence, foam-cell formation. In a quest to identify the mechanisms that mediate macrophage maturation, we and others localized macrophage colony stimulating factor (M-CSF) in plaques.14, 15 In vitro studies showed that M-CSF could induce scavenger receptors and promote the proliferation of monocytes in early atherosclerotic lesions.14 Formation of the “necrotic core” may also reflect highly regulated functions of mononuclear phagocytes. We wrote in 1992:
“The death of lipid laden macrophages may not be a random event or simply caused by bursting like an over-inflated balloon due to lipid overload. Rather, this process may resemble apoptosis, a form of programmed cell death. The necrobiosis in the fatty core of lesions may be due in part to gradients of concentration of factors such as M-CSF required for survival of human monocytes. Overexpression of M-CSF may occur in the “shoulder” or leading edge of the evolving lesion, a region characterized by ongoing cellular activation. On the other hand, relative depletion of M-CSF in the central core of a plaque could favor necrobiosis.” 16
Macrophages also contribute to the thrombotic complications of atherosclerosis in pivotal ways. These phagocytes furnish the bulk of the enzymes that catabolize collagen, a key constituent of the plaque’s fibrous cap. We hypothesized in the early 1990s that overproduction of the interstitial collagenase members of the matrix metalloproteinase (MMP) family jeopardizes the biomechanical stability of the plaque’s protective fibrous cap, predisposing to plaque rupture. 17 We described overexpression of interstitial collagenases (MMP-1, MMP-8, and MMP-13) in human atheromata, and colocalized macrophages bearing these proteinases with degradation products of interstitial collagen within plaques.18 Our subsequent experiments elucidated the pro-inflammatory cytokines that can promote the expression of MMP interstitial collagenases by mononuclear phagocytes, setting the stage for plaque disruption and thrombosis. Our group subsequently substantiated the role of MMP collagenases in plaque collagen content using genetic or pharmacologic gain-of-function or loss-of-function experiments in mice.19–22
Macrophages and smooth-muscle cells within atherosclerotic plaques also overexpress the potent procoagulant tissue factor. We identified the inflammatory mediator CD40 ligand (CD154) as a disease-relevant activator of tissue factor expression by human macrophages.23 Thus, inflammation regulates both the thrombogenicity of the plaque and the integrity of the plaque’s protective fibrous cap. These findings provided firm evidence linking inflammation to the thrombotic complications of atherosclerosis. This thread of discoveries demonstrated that inflammatory mediators participate in all phases of atherogenesis — from lesion initiation through progression, and ultimately to the clinical complications of this disease.
Vessel wall cell–derived cytokines can provide kindling for the innate immune response in atherosclerosis
The discovery that vascular wall cells themselves can produce cytokines, protein mediators of inflammation and immunity, provided an important insight into the initiation of atherosclerosis. According to the original concept, cytokines functioned to signal between leukocytes, hence the name “interleukin”. Our work in the 1990s demonstrated that human endothelial cells and smooth-muscle cells not only responded to cytokines, but also could produce these pro-inflammatory mediators. 24, 25 (Figure 1) Products of oxidized lipoproteins, and angiotensin II — substances closely related to classical risk factors for atherosclerosis, such as hyperlipidemia and hypertension — could provoke vascular wall cells to produce cytokines. 26, 27 Thus, an early stimulus for recruitment of “professional” inflammatory cells to the lesion might arise from the production of cytokines by local vascular wall cells that elicit adhesion molecule and chemoattractant expression.
Before lesions take root, intrinsic vascular wall cells may sustain the initial assault from atherosclerotic risk factors, and respond by elaborating pro-inflammatory mediators that then recruit and activate “professional” inflammatory cells that amplify and sustain the inflammation in the nascent lesion. These findings stimulated the view of atherosclerosis as a dynamic and multilateral interchange between vascular wall cells and leukocytes, challenging the earlier notion of vascular cells as mere bystanders in arterial inflammation, and of foam cells as passive receptacles for lipid debris.
Inflammation and Death of Cells in Atheromata
As atherosclerotic lesions evolve, macrophage foam cells and smooth-muscle cells can undergo apoptosis. As noted above, macrophage death contributes to lipid core formation. Smooth-muscle cell apoptosis, a process we proposed as potentiated by inflammatory mediators, also likely contributes to lesion complication.28 We advanced in 1995 the explicit hypothesis that smooth-muscle cell death explains why regions of plaques that rupture and trigger thrombosis contain few of these cells.29, 30 Smooth-muscle cells produce most of the arterial interstitial collagen that lends strength to the fibrous cap. Thus, a paucity of smooth-muscle cells due to apoptosis could also contribute to a lack of collagen in inflamed plaques — a property associated with their propensity to rupture and provoke thrombosis. We also presented data in this early description of apoptosis in atheromata that pointed to what we termed “prolonged persistence” of cells undergoing apoptosis in these lesions. We stated: “Some apoptotic cells may not disappear from the atherosclerotic lesions, but accumulate in the fibrotic lesions in a ‘mummified’ state.”29 The elegant subsequent studies of Ira Tabas and colleagues have elaborated this concept of impaired clearance, or efferocytosis, of apoptotic cells in plaques. 31
Heterogeneity of mononuclear phagocytes in atherosclerosis
Recent work has shed new light on the long appreciated heterogeneity of monocyte/macrophage functions observed in atherosclerotic plaques. Our group provided early evidence in 1992 of heterogeneity of macrophage functions based on gene expression.32 The use of more recently recognized cell-surface markers has revealed a striking dichotomy in monocytes in atherosclerosis. Hypercholesterolemic mice have high levels of monocytes that exhibit particularly pro-inflammatory functions, delineated by high expression of the surface marker Ly6C, in the peripheral blood and spleen.33, 34 In severely hypercholesterolemic mice, extramedullary leukopoiesis in the spleen yields enhanced production of this pro-inflammatory subset of monocytes. These cells exit the spleen and can accumulate in atheromata, furnishing a substantial minority of the mononuclear phagocytes in experimental lesions. 35
The pro-inflammatory subset of monocytes may preferentially give rise to macrophages that exhibit a pro-inflammatory program known as “classical” activation. Some refer to macrophages exhibiting a primarily pro-inflammatory program as “M1 macrophages”. The simplistic dichotomization of macrophages into categories based on certain markers may have heuristic value, but applies more to mice than to humans, and glosses over overlapping patterns of activation that characterize complex chronic inflammatory processes in humans, such as atherosclerosis.36 Recent advances in understanding the heterogeneity of mononuclear phagocytes in atherosclerosis offer new mechanistic insight into this disease, and provide new avenues for manipulation of the immune response during atherogenesis. The extent to which these observations in mice with exaggerated levels of hypercholesterolemia apply to humans remains under investigation.
Innate immunity in atherogenesis: beyond phagocytes
Macrophages account for the vast majority of leukocytes found in atheromata. Other effector cells of innate immunity, while less numerous, may also contribute to the pathogenesis of atherosclerosis. Mast cells, long noted in the adventitia and postulated to contribute to vascular diseases, have recently emerged as participants in experimental atherogenesis. The morphologic findings of the 1950s have given way to pharmacologic and genetic interventions that disclose a pro-atherogenic role for mast cells in mouse atherosclerosis.37 Our group found that a genetically determined lack of mast cells ameliorates experimental atherosclerosis in mice.38 Adoptive transfer experiments have implicated mast cell–derived interleukin-6 and IFN-γ as pro-inflammatory mediators that contribute to lesion evolution. Eosinophils — and notably, immunoglobulin E activation — also can aggravate experimental atherosclerosis.39
Adaptive immunity in atherosclerosis
Macrophages comprise the vast majority of inflammatory cells in human and experimental atherosclerotic plaques. The cells of adaptive immunity — namely, T lymphocytes and B lymphocytes — also exist in atherosclerotic lesions, albeit in markedly lower numbers. Despite their minority status, lymphocytes — particularly T lymphocytes — appear to function decisively in the regulation of inflammation during atherogenesis. Armies have more foot soldiers than generals, and orchestras contain many instrumentalists but only one conductor; likewise, the less numerous T lymphocytes may regulate the innate inflammatory response in atherosclerosis, mediated by macrophages within plaques. A previous paper in this series by Hansson and Jonasson has covered in depth the discovery of cellular immunity in the atherosclerotic plaque, only briefly summarized here. 40 Immunolocalization first documented the presence of T lymphocytes in human atheromata. The expression of class II histocompatibility antigens by neighboring cells provided evidence for the functional significance of these T cells. T lymphocytes produce IFN-γ, the inducer of class II major histocompatibility complex antigens in smooth-muscle cells and macrophages. Adoptive transfer experiments proved a pathogenic role for CD4+ T lymphocytes in mouse atherosclerosis. 41 Further study disclosed an early predominance of IFN-γ–producing Th1 CD4+ cells in mouse atheromata. Subsequent studies have identified potential endogenous antigens, including low-density lipoprotein (LDL) and heat shock protein-60, which may stimulate adaptive immunity in atherosclerotic plaques. Like mononuclear phagocytes, T lymphocytes exhibit functional diversity. While Th1 CD4+ lymphocytes appear to accelerate atherogenesis, regulatory T cells, through the secretion of transforming growth factor beta (TGF-β) and in some cases IL-10, appear to limit atherosclerosis. Thus the balance between T-cell subsets may influence the formation and character of lesions. 42, 43
B cells also populate plaques. Humoral immunity, however, appears to mitigate atherogenesis. Thus B1 cells that give rise to natural antibody may protect against atherosclerosis. Splenectomy, an intervention that reduces B lymphocyte levels, can enhance atheroma formation in mice. 44 Vaccination with putative antigens, such as modified LDL, appears to protect against atherosclerosis. In contrast, B2 lymphocytes may aggravate atherogenesis. 45 Indeed, depletion of B cells with anti-CD20 antibody treatment limits lesion size in mice in an IL-17–dependent manner.46 Thus, the net influence of B cell functions in atherosclerosis remains unsettled.
Allograft vasculopathy: a special case of immune-mediated arteriosclerosis
Recipients of solid organ allografts develop a special type of arteriosclerosis known by many names, including allograft arteriopathy. This disease can develop rapidly — within a few months or a few years — even in the absence of traditional risk factors for atherosclerosis. Involvement of the donor arteries, with sparing of the host arteries, indicates that this accelerated arteriopathy does not result from a systemic change due to the transplanted state or from the medications used to control acute rejection.
We postulated in 1989 that an immune response directed against foreign class II histocompatibility antigens expressed by donor cells, stimulated a chronic cytokine-mediated immune response that led to this concentric fibroproliferative form of arteriosclerosis.47 (Figure 1) Our group demonstrated the expression of class II histocompatibility antigens on endothelial cells in the coronary arteries of cardiac allografts. 48 Further observations localized T lymphocytes in a sub-endothelial position in the intima, in a ring-like array. (Figure 2) We predicted a key role for IFN-γ derived from activated T cells in the induction of class II antigens and the initiation of the pathogenic cascade of allograft arteriopathy. Our group further postulated that the chronic immune response in this disease, akin to a delayed-type hypersensitivity reaction, differed from CD8 T cell–mediated cytolysis, the hallmark of parenchymal rejection. In 1997, our studies in genetically modified mice provided direct experimental support for these conjectures: IFN-γ deficiency prevented coronary arteriosclerosis but not myocardial rejection in cardiac allografts. 49 While other immune and non-immune mechanisms contribute to the pathogenesis of allograft arteriopathy, a combination of experimental and human observations establishes adaptive immunity as a key element in the development of this disease. This example illustrates indubitably that inflammatory mechanisms can produce arteriosclerosis in the absence of classical risk factors such as hyperlipidemia.
Figure 2.
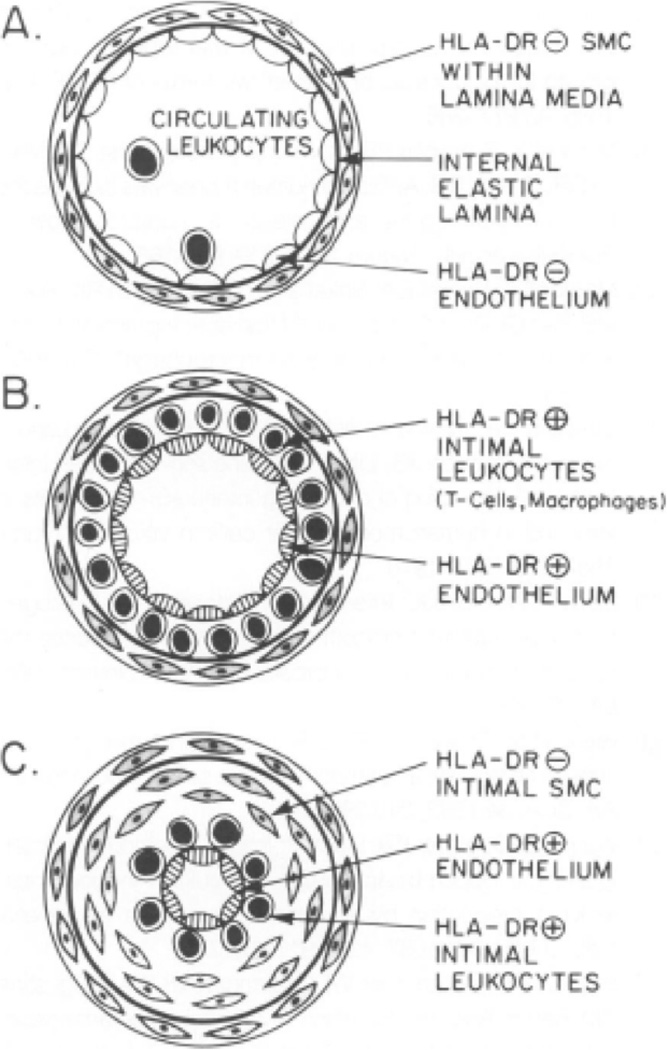
This illustration depicts a sequence of development of allograft arteriopathy proposed in 1991, based on morphologic evaluation of human lesions and in vitro immunologic experiments. A: Circulating T cells and monocytes contact human leukocyte antigen (HLA)-DR– endothelial cells (ECs). HLA-DR– medial smooth-muscle cells (SMCs) reside beneath the internal elastic lamina. B: In early allograft arteriopathy, HLA-DR+ ECs overlie T cells and macrophages within the intima. C: In advanced allograft arteriopathy, SMCs predominate within the deeper layers of the intima. HLA-DR+ ECs, T cells, and macrophages line the lumen. From Salomon RN, Hughes CCW, Schoen FJ, Payne DD, Pober JS, Libby P. Human coronary transplantation-associated arteriosclerosis: evidence for a chronic immune reaction to activated graft endothelial cells. Am J Pathol 1991;138(4):791–798.
Implications of inflammation in atherosclerosis for translation to human disease
The recognition of the operation of inflammation in atherogenesis spawned the application of biomarkers of inflammation to extend the experiments on animals and cultured cells, and observations on human specimens and intact subjects. Biomarkers of inflammation, such as C-reactive protein (CRP), rise in individuals with acute myocardial infarction.50, 51 Such observations, made many years ago, likely reflect the response to tissue injury. Evidence accumulated over the last 15 years demonstrates that subtle increases in biomarkers of inflammation (such as CRP) can augur prospective cardiovascular events in apparently well people.52 The fluctuations of CRP that predict enhanced cardiovascular risk occur within the range of this biomarker far below levels encountered in individuals with acute illnesses. The development of a high-sensitivity assay (denoted hsCRP) thus permitted accurate measurement of this biomarker as a tool to enhance risk stratification. In contrast to many other novel biomarkers, hsCRP adds to the traditional risk factors for atherosclerosis encompassed in the Framingham algorithm. The increase in the relative risk estimate of those with higher quantiles of hsCRP, adjusted for traditional risk factors, is modest — approximately 1.5 to 1.7. Yet after age and sex, the addition of traditional risk factors such as total cholesterol and systolic blood pressure yield about the same increment in risk prediction.53 Recently developed tools for gauging the clinical utility of novel biomarkers, such as the net reclassification index, show that hsCRP can correctly reclassify individuals — particularly in the group categorized as having intermediate risk according to traditional criteria. This intermediate risk group accounts for much of the burden of cardiovascular events. Some guidelines and recommendations from professional societies now incorporate hsCRP into risk-predicting algorithms. The Reynolds Risk Score, in particular, adds hsCRP and family history of premature coronary artery disease to traditional risk factors in a clinically useful manner.54
The successful application of biomarkers of inflammation to sharpen cardiovascular risk assessment, and its independence from traditional risk factors such as hypercholesterolemia, suggested that biomarkers of inflammation could identify individuals who might benefit from intervention, despite relatively low estimates of cardiovascular risk based on traditional risk factors. Statin drugs, for example, effectively lower LDL and reduce cardiovascular risk in broad categories of individuals, and also reduce inflammation as gauged by lowering of CRP. Within an individual, the statin-induced drop in LDL correlates very poorly with the fall in CRP. This observation, replicated in numerous large clinical trials, indicates that LDL and inflammation vary independently. Retrospective analysis of one large trial that treated individuals without known cardiovascular disease with a statin showed that the reduction of events in those with below-median LDL levels, but above-median CRP levels, resembled that achieved by treatment of those with LDL levels above the median. 55
These considerations inspired Dr. Paul M Ridker to design and conduct a large-scale clinical trial known as JUPITER (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). It enrolled more than 17,000 individuals without known cardiovascular disease with hsCRP greater than 2 mg/L and LDL cholesterol levels below 130 mg/dL. The study results showed a reduction greater than 40% in first-ever cardiovascular events due to statin treatment.56 The statin-treated group also had decreased all-cause mortality, even though the study was not powered for a mortality effect. Pre-specified analyses of JUPITER indicated that the clinical benefit derived both from LDL lowering and an anti-inflammatory effect reflected by reduction in CRP.57 These results in an apparently well population agreed with previous findings in clinical trials of patients who survived an acute coronary syndrome.
JUPITER was not designed to prove — nor could it prove — that a direct anti-inflammatory therapy reduces atherosclerotic events. Testing this hypothesis will require additional clinical trials, and several such investigations are underway or in the planning stage. The Canakinumab Anti-Thrombosis Outcome Study (CANTOS) will test whether administration of an antibody that neutralizes the pro-inflammatory cytokine interleukin-1-beta (IL-1β) can reduce cardiovascular events in survivors of myocardial infarction who have hsCRP levels persistently above 2 mg/dL, despite standard-of-care therapy including treatment with high-dose statins.58 The Cardiovascular Inflammation Reduction Trial (CIRT) will test whether treatment of a similar population with weekly low-dose methotrexate — a regimen used successfully in the management of rheumatoid arthritis — can reduce recurrent cardiovascular events. 59 In the realm of adaptive immunity, studies are exploring the feasibility of vaccination with oxidized LDL-derived antigens to elicit a humoral immune response that could protect against atherosclerotic events.60 Such studies should permit testing of the proposition that interfering with innate or adaptive immunity, and breaking the cycle of inflammation during atherosclerosis, can improve outcomes. Given the complexities and redundant mediators and signaling pathways involved in vascular inflammation, clinical validation of the “inflammation” hypothesis of atherogenesis may require testing a number of targets to find the “sweet spot” of an intervention that mitigates the disease without undue impairment of host defenses.
Conclusions
The concept of inflammation contributing to atherosclerosis, rooted in keen observations in the 19th century, has undergone a research renaissance in recent decades. New tools of biological and clinical research have established a modulatory role for inflammation and immunity in experimental atherosclerosis. The validation of these concepts in humans and the translation to clinical practice are works in progress. The new understanding of the participation of inflammation in atherosclerosis, and its complications, in no way challenges the importance of traditional risk factors — such as high LDL levels — as causal risk factors for this disease. (Figure 3) Indeed, inflammation provides a pathway that mechanistically links alterations in traditional risk factors and modifications in the biology of the artery wall that give rise to atherosclerosis and its complications. The coming years should prove fruitful in completing the canvas of the role of inflammation in atherosclerosis, and in translating these concepts to improve human health.
Figure 3.
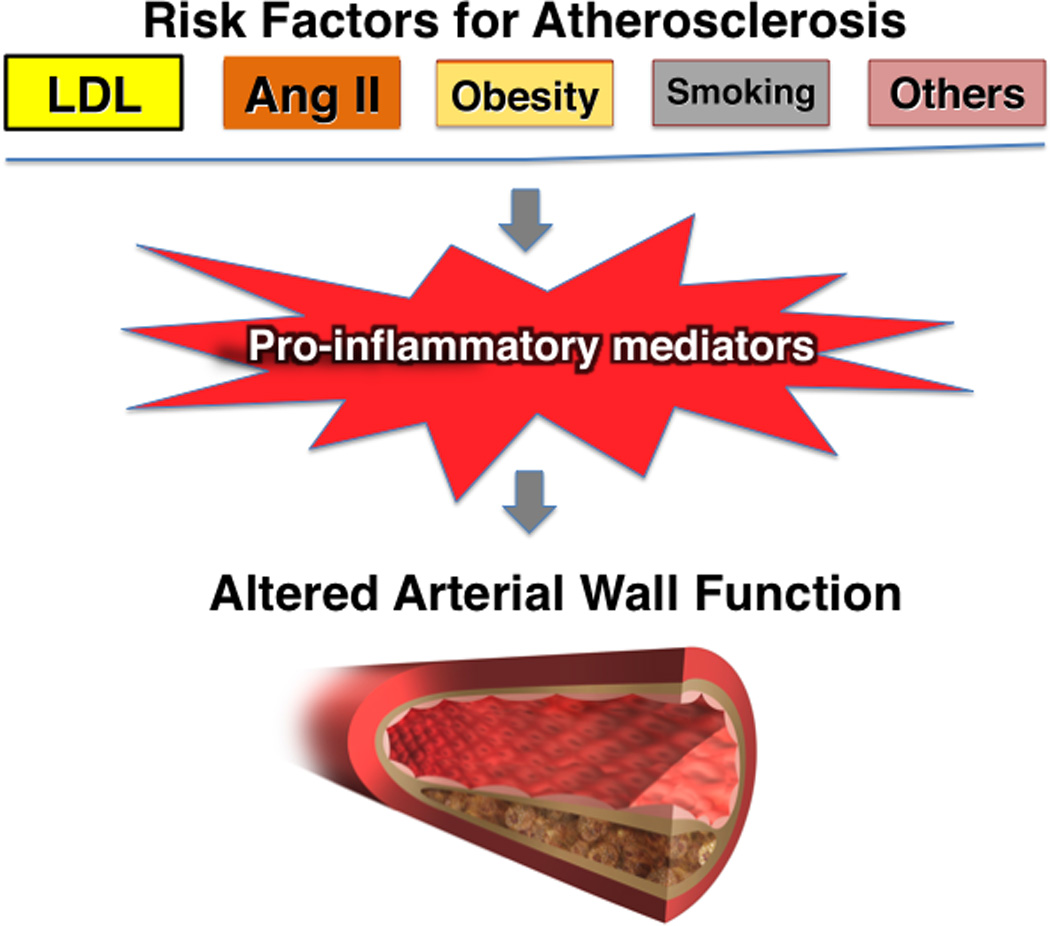
The relationship between traditional risk factors for atherosclerosis and inflammation. The concept of inflammation in atherosclerosis in no way diminishes the importance of the traditional risk factors for atherosclerosis depicted at the top of this diagram, including low-density lipoprotein (LDL) and angiotensin II (Ang II). Rather, the concept of inflammatory signaling and the participation of proinflammatory cytokines provides a mechanistic link between traditional risk factors and altered biological responses of the artery wall that drive atherosclerosis and its complications.
Acknowledgments
The author thanks the many trainees who have participated in this work through the decades, some of whose many contributions are cited in the References. I also thank the long-term professional colleagues who have contributed to the concepts of the participation of inflammation and immunity in vascular disease. Dr. Jordan S. Pober contributed decisively to early work on the pathogenesis of allograft arteriopathy. Dr. Göran K. Hansson participated pivotally in the evolution of these concepts over three decades in conversations, collaborative experiments, shared trainees, and joint writings. I thank him for his critical review of this manuscript. Dr. Paul M Ridker, my long–term colleague, spearheaded the clinical translation of inflammation biology to patient populations and clinical trials. I thank Ms. Sara Karwacki for expert editorial assistance, and Mr. David Lynn for outstanding technical transcription. Our work on inflammation and immunity in atherosclerosis has received support from the U.S. National Heart Lung and Blood Institute, the American Heart Association, the Donald W. Reynolds Foundation, and the Fondation Leducq.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ross R, Glomset JA. The pathogenesis of atherosclerosis I. New England Journal of Medicine. 1976;295(7):369–377. doi: 10.1056/NEJM197608122950707. [DOI] [PubMed] [Google Scholar]
- 2.Ross R, Glomset JA. The pathogenesis of atherosclerosis II. New England Journal of Medicine. 1976;295(8):420–425. doi: 10.1056/NEJM197608192950805. [DOI] [PubMed] [Google Scholar]
- 3.Virchow R. Cellular Pathology. London: John Churchill; 1858. [Google Scholar]
- 4.Karnovsky ML. Metchnikoff in Messina: a century of studies on phagocytosis. N Engl J Med. 1981;304(19):1178–1180. doi: 10.1056/NEJM198105073041922. [DOI] [PubMed] [Google Scholar]
- 5.Aqel NM, Ball RY, Waldmann H, Mitchinson MJ. Identification of macrophages and smooth muscle cells in human atherosclerosis using monoclonal antibodies. J Pathol. 1985;146:197–204. doi: 10.1002/path.1711460306. [DOI] [PubMed] [Google Scholar]
- 6.Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986;6:131–138. doi: 10.1161/01.atv.6.2.131. [DOI] [PubMed] [Google Scholar]
- 7.Libby P. Inflammatory and immune mechanisms in atherogenesis. In: Leaf A, Weber P, editors. Atheroclerosis Reviews. Vol 21. New York: Raven Press; 1990. pp. 79–89. [Google Scholar]
- 8.Bevilacqua MP, Pober JS, Mendrick DL, Cotran RS, Gimbrone Jr. MA. Identification of an inducible endothelial-leukocyte adhesion molecule. Proc. Natl. Acad. Sci. USA. 1987;84:9238–9242. doi: 10.1073/pnas.84.24.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cybulsky MI, Gimbrone Jr. MA. Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788–791. doi: 10.1126/science.1990440. [DOI] [PubMed] [Google Scholar]
- 10.Li H, Cybulsky MI, Gimbrone MA, Jr, Libby P. An atherogenic diet rapidly induces VCAM-1, a cytokine regulatable mononuclear leukocyte adhesion molecule, in rabbit endothelium. Arterioscler Thromb. 1993;13(2):197–204. doi: 10.1161/01.atv.13.2.197. [DOI] [PubMed] [Google Scholar]
- 11.Poole JCF, Florey HW. Changes in the endothelium of the aorta and the behavior of macrophages in experimental atheroma of rabbits. J Path Bact. 1958;75:245–253. doi: 10.1002/path.1700750202. [DOI] [PubMed] [Google Scholar]
- 12.Gu L, Okada Y, Clinton S, Gerard C, Sukhova G, Libby P, Rollins B. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low-density lipoprotein-deficient mice. Mol Cell. 1998;2:275–281. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- 13.Mach F, Sauty A, Iarossi AS, Sukhova GK, Neote K, Libby P, Luster AD. Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J Clin Invest. 1999;104:1041–1050. doi: 10.1172/JCI6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clinton S, Underwood R, Sherman M, Kufe D, Libby P. Macrophage-colony stimulating factor gene expression in vascular cells and in experimental and human atherosclerosis. Am J Pathol. 1992;140(2):301–316. [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenfeld M, Ylä-Herttuala S, Lipton B, Ord V, Witztum J, Steinberg D. Macrophage colony-stimulating factor mRNA and protein in atherosclerotic lesions of rabbits and humans. Am J Pathol. 1992;140(2):291–300. [PMC free article] [PubMed] [Google Scholar]
- 16.Libby P, Clinton SK. Cytokines as mediators of vascular pathology. Nouv Rev Fr Hematol. 1992;34(53):S47–S53. [PubMed] [Google Scholar]
- 17.Galis Z, Sukhova G, Lark M, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sukhova GK, Schonbeck U, Rabkin E, Schoen FJ, Poole AR, Billinghurst RC, Libby P. Evidence for increased collagenolysis by interstitial collagenases-1 and-3 in vulnerable human atheromatous plaques. Circulation. 1999;99(19):2503–2509. doi: 10.1161/01.cir.99.19.2503. [DOI] [PubMed] [Google Scholar]
- 19.Fukumoto Y, Deguchi JO, Libby P, Rabkin-Aikawa E, Sakata Y, Chin MT, Hill CC, Lawler PR, Varo N, Schoen FJ, Krane SM, Aikawa M. Genetically determined resistance to collagenase action augments interstitial collagen accumulation in atherosclerotic plaques. Circulation. 2004;110(14):1953–1959. doi: 10.1161/01.CIR.0000143174.41810.10. [DOI] [PubMed] [Google Scholar]
- 20.Deguchi JO, Aikawa E, Libby P, Vachon JR, Inada M, Krane SM, Whittaker P, Aikawa M. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation. 2005;112(17):2708–2715. doi: 10.1161/CIRCULATIONAHA.105.562041. [DOI] [PubMed] [Google Scholar]
- 21.Schneider F, Sukhova GK, Aikawa M, Canner J, Gerdes N, Tang SM, Shi GP, Apte SS, Libby P. Matrix-metalloproteinase-14 deficiency in bone-marrow-derived cells promotes collagen accumulation in mouse atherosclerotic plaques. Circulation. 2008;117(7):931–939. doi: 10.1161/CIRCULATIONAHA.107.707448. [DOI] [PubMed] [Google Scholar]
- 22.Quillard T, Tesmenitsky Y, Croce K, Travers R, Shvartz E, Koskinas KC, Sukhova G, Aikawa E, Aikawa M, Libby P. Selective inhibition of matrix metalloproteinase 13 (MMP-13) increases collagen content of established mouse atheromata. Arterioscler Thromb Vasc Biol. 2011;31(11):2464–2472. doi: 10.1161/ATVBAHA.111.231563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mach F, Schoenbeck U, Bonnefoy J-Y, Pober J, Libby P. Activation of monocyte/macrophage functions related to acute atheroma complication by ligation of CD40. Induction of collagenase, stromelysin, and tissue factor. Circulation. 1997;96:396–399. doi: 10.1161/01.cir.96.2.396. [DOI] [PubMed] [Google Scholar]
- 24.Libby P, Ordovàs JM, Auger KR, Robbins H, Birinyi LK, Dinarello CA. Endotoxin and tumor necrosis factor induce interleukin-1 gene expression in adult human vascular endothelial cells. Am. J. Path. 1986;124:179–186. [PMC free article] [PubMed] [Google Scholar]
- 25.Libby P, Ordovas JM, Birinyi LK, Auger KR, Dinarello. CA. Inducible interleukin-1 expression in human vascular smooth muscle cells. J. Clin. Invest. 1986;78:1432–1438. doi: 10.1172/JCI112732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lipton BA, Parthasarathy S, Ord VA, Clinton SK, Libby P, Rosenfeld ME. Components of the protein fraction of oxidized low density lipoprotein stimulate interleukin-1 alpha production by rabbit arterial macrophage-derived foam cells. Journal of Lipid Research. 1995;36(10):2232–2242. [PubMed] [Google Scholar]
- 27.Kranzhofer R, Schmidt J, Pfeiffer CA, Hagl S, Libby P, Kubler W. Angiotensin induces inflammatory activation of human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19(7):1623–1629. doi: 10.1161/01.atv.19.7.1623. [DOI] [PubMed] [Google Scholar]
- 28.Geng Y-J, Wu Q, Muszynski M, Hansson G, Libby P. Apoptosis of vascular smooth muscle cells induced by in vitro stimulation with interferon-gamma, tumor necrosis factor-alpha, and interleukin-1-beta. Arteriosclerosis, Thrombosis, and Vascular Biology. 1996;16:19–27. doi: 10.1161/01.atv.16.1.19. [DOI] [PubMed] [Google Scholar]
- 29.Geng Y-J, Libby P. Evidence for apoptosis in advanced human atheroma. Co-localization with interleukin-1 β-converting enzyme. Am J Pathol. 1995;147:251–266. [PMC free article] [PubMed] [Google Scholar]
- 30.Libby P. The molecular bases of the acute coronary syndromes. Circulation. 1995;91:2844–2850. doi: 10.1161/01.cir.91.11.2844. [DOI] [PubMed] [Google Scholar]
- 31.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10(1):36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salomon RN, Underwood R, Doyle MV, Wang A, Libby P. Increased apolipoprotein E and c-fms gene expression without elevated interleukin 1 or 6 levels indicate selective activation of macrophage functions in advanced human atheroma. Proc Natl Acad Sci U S A. 1992;89(7):2814–2818. doi: 10.1073/pnas.89.7.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117(1):195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117(1):185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo J-L, Iwamoto Y, Gorbatov R, Etzrodt M, Weber G, Ueno T, van Rooijen N, Mulligan-Kehoe MJ, Libby P, Nahrendorf M, Pittet MJ, Weissleder R, Swirski FK. Extramedullary hematopoiesis generates Ly-6Chigh monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125(2):364–374. doi: 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 37.Bot I, de Jager SC, Zernecke A, Lindstedt KA, van Berkel TJ, Weber C, Biessen EA. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation. 2007;115(19):2516–2525. doi: 10.1161/CIRCULATIONAHA.106.660472. [DOI] [PubMed] [Google Scholar]
- 38.Sun J, Sukhova GK, Wolters PJ, Yang M, Kitamoto S, Libby P, MacFarlane LA, Clair JM, Shi GP. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med. 2007;13(6):719–724. doi: 10.1038/nm1601. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Cheng X, Xiang M-X, Alanne-Kinnunen M, Wang J-A, Chen H, He A, Lin Y, Tang T-T, Tu X, Sjöberg S, Sukhova GK, Liao YH, Conrad DH, Yu L, Kawakami T, Kovanen PT, Libby P, Shi G-P. Immunoglobulin E promotes vascular wall cell inflammatory molecule expression, apoptosis, and atherogenesis. J Clin Invest. 2011;121(9):3564–3577. doi: 10.1172/JCI46028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hansson GK, Jonasson L. The discovery of cellular immunity in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2009;29(11):1714–1717. doi: 10.1161/ATVBAHA.108.179713. [DOI] [PubMed] [Google Scholar]
- 41.Zhou X, Nicoletti A, Elhage R, Hansson GK. Transfer of CD4(+) T cells aggravates atherosclerosis in immunodeficient apolipoprotein E knockout mice. Circulation. 2000;102(24):2919–2922. doi: 10.1161/01.cir.102.24.2919. [DOI] [PubMed] [Google Scholar]
- 42.Ait-Oufella H, Salomon BL, Potteaux S, Robertson A-KL, Gourdy P, Zoll J, Merval R, Esposito B, Cohen JL, Fisson S, Flavell RA, Hansson GK, Klatzmann D, Tedgui A, Mallat Z. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12:178–180. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 43.Cao Z, Wara AK, Icli B, Sun X, Packard RR, Esen F, Stapleton CJ, Subramaniam M, Kretschmer K, Apostolou I, von Boehmer H, Hansson GK, Spelsberg TC, Libby P, Feinberg MW. Kruppel-like factor KLF10 targets transforming growth factor-beta1 to regulate CD4(+)CD25(-) T cells and T regulatory cells. J Biol Chem. 2009;284(37):24914–24924. doi: 10.1074/jbc.M109.000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. 2002;109(6):745–753. doi: 10.1172/JCI07272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Binder CJ, Chou MY, Fogelstrand L, Hartvigsen K, Shaw PX, Boullier A, Witztum JL. Natural antibodies in murine atherosclerosis. Curr Drug Targets. 2008;9(3):190–195. doi: 10.2174/138945008783755520. [DOI] [PubMed] [Google Scholar]
- 46.Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, Taleb S, Van Vre E, Esposito B, Vilar J, Sirvent J, Van Snick J, Tedgui A, Tedder TF, Mallat Z. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. 2010;207(8):1579–1587. doi: 10.1084/jem.20100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Libby P, Salomon RN, Payne DD, Schoen FJ, Pober JS. Functions of vascular wall cells related to the development of transplantation-associated coronary arteriosclerosis. Transplant Proc. 1989;21:3677–3684. [PubMed] [Google Scholar]
- 48.Salomon RN, Hughes CCW, Schoen FJ, Payne DD, Pober JS, Libby P. Human coronary transplantation-associated arteriosclerosis: Evidence for a chronic immune reaction to activated graft endothelial cells. Am J Pathol. 1991;138(4):791–798. [PMC free article] [PubMed] [Google Scholar]
- 49.Nagano H, Mitchell RN, Taylor MK, Hasegawa S, Tilney NL, Libby P. Interferon-gamma deficiency prevents coronary arteriosclerosis but not myocardial rejection in transplanted mouse hearts. Journal of Clinical Investigation. 1997;100(3):550–557. doi: 10.1172/JCI119564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berk BC, Weintraub WS, Alexander RW. Elevation of C-reactive protein in "active" coronary artery disease. American Journal of Cardiology. 1990;65(3):168–172. doi: 10.1016/0002-9149(90)90079-g. [DOI] [PubMed] [Google Scholar]
- 51.Liuzzo G, Biasucci LM, Gallimore JR, Grillo RL, Rebuzzi AG, Pepys MB, Maseri A. The prognostic value of C-reactive protein and serum amyloid A protein in severe unstable angina. N Engl J Med. 1994;331(7):417–424. doi: 10.1056/NEJM199408183310701. [DOI] [PubMed] [Google Scholar]
- 52.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men [published erratum appears in N Engl J Med 1997 Jul 31;337(5):356] N Engl J Med. 1997;336(14):973–979. doi: 10.1056/NEJM199704033361401. [see comments] [DOI] [PubMed] [Google Scholar]
- 53.Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh J. Emerging Risk Factors Collaboration. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375(9709):132–140. doi: 10.1016/S0140-6736(09)61717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds Risk Score. Jama. 2007;297(6):611–619. doi: 10.1001/jama.297.6.611. [DOI] [PubMed] [Google Scholar]
- 55.Ridker PM, Rifai N, Clearfield M, Downs JR, Weis SE, Miles JS, Gotto AM., Jr Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med. 2001;344(26):1959–1965. doi: 10.1056/NEJM200106283442601. [DOI] [PubMed] [Google Scholar]
- 56.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, Macfadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ. Rosuvastatin to Prevent Vascular Events in Men and Women with Elevated C-Reactive Protein. N Engl J Med. 2008;359(21):2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 57.Ridker PM, Danielson E, Fonseca FAH, Genest J, Gotto AM, Jr, Kastelein JJP, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373(9670):1175–1182. doi: 10.1016/S0140-6736(09)60447-5. [DOI] [PubMed] [Google Scholar]
- 58.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011;162(4):597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 59.Ridker PM. Testing the inflammatory hypothesis of atherothrombosis: scientific rationale for the cardiovascular inflammation reduction trial (CIRT) J Thromb Haemost. 2009;7 Suppl 1:332–339. doi: 10.1111/j.1538-7836.2009.03404.x. [DOI] [PubMed] [Google Scholar]
- 60.Hansson GK, Nilsson J. Vaccination against atherosclerosis? Induction of atheroprotective immunity. Semin Immunopathol. 2009;31(1):95–101. doi: 10.1007/s00281-009-0151-x. [DOI] [PubMed] [Google Scholar]