

Abstract
Most applications of photoremovable protecting groups have used o-nitrobenzyl compounds and their (often commercially available) derivatives that, however, have several disadvantages. The focus of this review is on applications of the more recently developed title compounds, which are especially well suited for time-resolved biochemical and physiological investigations, because they release the caged substrates in high yield within a few nanoseconds or less. Together, these two chromophores cover the action spectrum for photorelease from >700 nm to 250 nm.
Introduction
o-Nitrobenzyl derivatives are by far the most commonly used photoremovable protecting groups (PPGs),1 despite their known disadvantages: following electronic excitation, the caged compounds are released on a time scale of microseconds at best,2 and o-nitroso aromatic ketones are produced as side products, which usually absorb more strongly than the parent PPG at the irradiation wavelengths and which may interfere with the effect of the released bioactive material under study, because of their toxic effect on biological tissues.3 With the need for higher time resolution in studies of the leading events in biological processes, there is a demand for clean, rapidly released triggers or initiators of these processes.
The technology to monitor events on the nanosecond to femtosecond time scales has become available and to exploit these methods the initiating processes must match or exceed the rates of detection. Recent advances in time-resolved Fourier transform infrared (TR FTIR) and attenuated total reflectance (TR ATF), for example, permit investigations of protein binding and enzyme catalysis at the nanosecond level.4 This review focuses on relatively recent additions to the PPG variety, p-hydroxyphenacyl (pHP)5 and coumarylmethyl derivatives,6 which offer an alternative to o-nitrobenzyl.
p-Hydroxyphenacyl photoremovables (pHP)
Anderson and Reese discovered the molecular reorganization that occurs upon irradiation of pHP chloride.7 The mechanism of the photochemical release of the leaving group (LG) from pHP diethyl phosphate (1) in aqueous solution is summarized in Scheme 1.8 Release of a typical caged substrate upon irradiation in neutral aqueous solution at λ = 250–350 nm cleanly and efficiently proceeds through its triplet excited state forming a putative spirodiketone 2, termed the Favorskii intermediate. The spirodiketone is subject to hydrolytic ring opening yielding p-hydroxyphenylacetic acid (3). The structural rearrangement closely follows the classical ground state Favorskii rearrangement of α-haloketones, hence the origin of its moniker as the “photo Favorskii rearrangement”. In contrast to the starting material, the photoproducts 3 (and traces of 4) are essentially transparent at wavelengths above 290 nm.
Scheme 1.

Mechanism for pHP photoisomerization and leaving group (LG) release.8
Leaving group effects
As a general rule, the most efficacious leaving groups are those with a pKa < 11. Alcohols and primary, secondary, and tertiary amines with pKa’s above the threshold of 11, with a few exceptions,9 have not been successfully released from pHP cages although little has been reported on these functional groups (vide infra).
Several studies have shown that the pHP triplet lifetimes, 3τ, and the rate constants for release, krel = Φdis/3τ, depend on the leaving group pKa.10–13 As shown in Table 1, for pKa’s that range from the reactive mesylate (−1.54) and tosylate (−0.43) to the less efficient phenols (8–11) and thiols (~8) give a range of rate constants krel that spans three orders of magnitude and quantum yields that vary by a factor of 20.
Table 1.
Representative pKa’s, disappearance quantum yields of pHP derivatives (Φdis), triplet state lifetimes (3τ), and rate constants for the release of the leaving groups (LG) calculated as krel = Φdis/3τ11–13
| LG | pKa12 (LG) | Φdis13 | 3τ/ns11 | log(krel/s−1) |
|---|---|---|---|---|
| Mesylate | −1.54 | 0.932 | 0.118 | 9.70 |
| Tosylate | −0.43 | 1.04 | 0.104 | 10.0 |
| Diethyl phosphate | 0.71 | 0.40 | 0.345 | 9.06 |
| p-CF3 benzoate | 3.69 | 0.201 | 0.625 | 8.51 |
| Formate | 3.75 | 0.94 | 0.667 | 9.15 |
| p-OCH3 benzoate | 4.09 | 0.288 | 0.833 | 8.54 |
| Benzoate | 4.21 | 0.316 | 1.16 | 8.44 |
| GABA | 4.76 | 0.21 | 0.345 | 8.78 |
| p-CN phenolate | 7.17 | 0.11 | 1.45 | 7.88 |
| Phenolate | 9.8 | 0.04 | 3.85 | 7.02 |
The dependence of the rate constants krel on the pKa’s of the LGs is demonstrated by a Brønsted linear free energy relationship, log(krel/s−1) = (9.57 ± 0.14) − (0.24 ± 0.03)pKa, i.e., βLG = −0.24 (Fig. 1). This correlation implies significant heterolytic bond breaking at the transition state for release.
Fig. 1.

Linear free energy relationship showing the dependence of the release rate constants krel on the pKa of the leaving group.
Laser flash transient absorption studies have detected the allyloxy–phenoxy biradical intermediate shown in Scheme 1, which intervenes between the excited triplet and the “Favorskii” intermediate.8 The biradical triplet results from the extrusion of both a proton and the leaving group anion from the triplet ketone 31. This indicates that the leaving group departs in its ground state since spin conservation requires only one triplet species be formed if the reaction is concerted. The simultaneous departure of both the proton and the leaving group is further supported by the observation of a solvent kinetic isotope effect (SKIE kH2O/kD2O = 2.17).8 The Brønsted βLG = −0.24 for the photochemical reaction finds precedence in gas-phase collision-induced dissociation (CID) fragmentation measurements of the conjugate base of the pHP derivatives in Table 1.12 A correlation coefficient of βLG = −0.19 ± 0.02 for the appearance energies (AEs) vs. gas-phase acidities of the leaving groups (−ΔHacid) reveals a similar extent (19% vs. 24%) of bond breaking at the transition state for the fragmentation process.
Chemical yield
An ideal PPG should give a quantitative yield of the unprotected functional group. Moreover, the irradiation dose required should be minimal to avoid unwanted photochemistry such as photoreactions of the products. Meeting these objectives avoids deleterious processes that compromise the efficacy of PPG-based methods. Poor chemical yields of released material, secondary photochemical reactions and over-irradiation leading to degradation of products and spectator components of the reaction mixture often result in complex mixtures of unwanted materials. For the p-hydroxyphenacyl derivatives, both criteria are met since the conversions are spectacularly clean and the quantum efficiencies approach unity.
Stability to thermal degradation
Esters, phenyl ethers and thio ethers of pHP are very stable in organic solvents. Most esters and all ethers are also stable in aqueous-based solvents at neutral pH and do not undergo hydrolysis at ambient temperatures. However, as the leaving group reactivity increases, e.g., sulfonate leaving groups, the caged compound’s stability decreases. Aqueous solutions of carboxylate esters may undergo slow hydrolysis at room temperature over a 24 h period forming the α-hydroxyketone. The rate of hydrolysis increases at higher pH (>9). No rearrangement products are detected, and thus no ground-state Favorskii rearrangement takes place under these basic conditions.14
Selected applications of pHP derivatives
The pHP PPG has been successfully deployed for time-resolved biochemical and physiological studies to delineate response kinetics,15 or to define spatial resolution in tracking signal transduction in neural networks.16 Several of these applications have been reviewed earlier.15 More recently, time-resolved absorption spectroscopy,8,14,17–20 Fourier transform IR (TR-FTIR)3,4,21 and resonance Raman (TR3),10,22,23 techniques have become available for better time-resolved applications in biological chemistry, such as examining substrate binding, enzyme conformational changes, dynamic inhibition and signal transduction kinetics. Several applications have incorporated an initiating pHP photo-activation step, again taking advantage of pHP’s rapid release rate.
The pHP phosphate derivatives are most frequently deployed for studies on enzyme catalysis, such as the photo-initiation step for kinetic studies,3,24 taking advantage of their very rapid and clean phosphate group release.3–5,10,18–24 For carboxylate release, such as the C-terminus of amino acids, peptides, and oligopeptides, the value has been in their ease of synthesis, clean, high yielding release, and biological compatibility. Bradykinin, L-glutamate, and GABA have been employed for several in vitro and in vivo studies, e.g., to activate the BK2 receptor, and as agonists or antagonists in mouse neuroreceptor stimulation studies of the auditory system.15,16 Assorted applications of pHP thio derivatives that take advantage of the high nucleophilicity of thiol for direct incorporation of the protecting group using pHP Br have been reported.25,26
The first examples of pHP phosphate release came from the investigations by Fendler et al.24 on ATPase hydrolysis of Na+, K+-ATP and at about the same time by Du et al.3,27 on pHP GTP as an initiator of the catalytic hydrolysis of GTP by Ras protein–GTPase. These two hallmark applications demonstrated the power of pHP as a phototrigger for the rapid release of the nucleotides ATP and GTP, in contrast to more commonly employed o-nitrobenzyl (oNB) and o-nitrophenylethyl (oNPE) PPGs. Fast cage release of the nucleotides was crucial for determining rate constants of the earliest events in a catalytic hydrolysis. In Du et al.’s work,27 kinetic isotope effects obtained from the rates of hydrolysis of 18O-labeled α-, β-, and γ-phosphates of GTP were determined from time-resolved FTIR analyses of the rates of bound and free phosphate. The changes in binding of GTP in the catalytic pocket during its hydrolysis of GDP were determined using 18O-induced spectral shifts from photolysis of the three pHP 18O-labelled GTP isomers. An increase in β-phosphate binding coupled with prior evidence of deprotonation of the γ-phosphate led the authors to conclude that hydrolysis occurs through a dissociative mechanism forming meta-phosphate intermediate and GDP. Rapid H2O addition to the meta-phosphate yields inorganic phosphate (H2PO42−, Pi), which is then released from the catalytic site in the rate-determining step. These studies relied heavily on the rapid release rate of pHP GTP to monitor the initial rates for binding at the catalytic site and to confirm its location (vide infra). Attempts to perform the same experiments using oNPE GTP were unsatisfactory and resulted in the early onset of extraneous IR signals masking the slower evolution of signals associated with the changes in binding from GTP to GDP.
In a follow-up communication,3 Du et al. documented the source of the difficulties encountered with the oNPE GTP study. The oNPE photolysis byproduct results from a photoredox reaction of the chromophore forming an α-nitrosoacetophenone. Nitroso ketones and aldehydes are the common byproducts for all o-NB-based caged compounds and are very susceptible to nucleophilic attack by amines and thiols. To counteract this effect, a common practice has been to add dithiothreitol (DTT) to the photolysis mixture in order to trap or disable any aryl-nitroso by products. Spurious signals that appeared early in the oNPE GTP photolysis were shown to arise from reactions of nitrosoacetophenone with the protein. In the absence of protein, the nitrosoacetophenone is readily detected.
The rate of oNPE cage release is also many orders of magnitude slower than that of the pHP analogs: krel = 1.0–10 s−1 (oNPE)2 vs. 108–109 s−1 (pHP).8,17,22 The vital time frame required for RasGTP catalysis by TR-FTIR was too fast for oNPE GTP initiation.3,4 The initial TR-FTIR fingerprints of the binding changes of the α- and β-phosphate groups on GTP were only observed with labelled pHP GTP as phototriggers.3,21,28,29
A similar comparison of TR-FTIR analyses of pHP ATP and oNB ATP-catalyzed hydrolysis was reported by Fendler et al.24 Here, the authors reached the same conclusions regarding the quality of data using the two methods for triggering the hydrolysis. Benzoin-protected ATP was also examined but did not provide useful results, primarily due to fluorescence and competition for incident radiation by the phenylbenzofuran photoproduct.
Gerwert et al.21 extended the TR-FTIR technique by developing it into a rapid high-throughput screening method as a competitive drug–protein binding assay. Using TR-FTIR difference spectra to tease out the changes in GTP–GDP binding in the presence and absence of small molecular weight pharmaceutical candidates, the degree of inhibition at key functional groups on the protein and/or induced changes in the protein’s conformation are measurable. The “on” and “off” signalling can be automated for high-throughput screening, a novel application of caged compounds with even broader applications for pharmaceutical drug development.30 This approach may be useful in assaying information about the active binding site of potential GTPase inhibitors.
Kötting et al.4,28 also determined the kinetic parameters and activation energies for GTP hydrolysis by GTPase in water and catalyzed by the Ras protein-GTPase in order to evaluate the extent of entropic vs. enthalpic influence of the protein on the catalytic cycle. Ras increased the rate of hydrolysis by 105 relative to H2O and 107 when Mg2+ was present in the aqueous solution. Temperature dependent rates showed that the ΔΔH0* of 5.2 kcal mol−1 for water vs. RasGTP was essentially the total source of the diminished activation barrier to the hydrolysis. The TΔΔS0* value was less than 0.5 kcal mol−1, essentially demonstrating that the protein was not providing enhanced ordering at the transition state in the hydrolysis process.
These values were determined using 18O frequency shifts at 1143 cm−1 for RasGTP and 1114 cm−1 for RasGDP to register the rate of formation of the RasGTP complex and its hydrolysis upon photolysis of pHP GTP. The frequency shifts for the α and γ labelled phosphates showed an upshift in comparing the GTPase activity in H2O vs. with protein-bound RasGTPase. The β-phosphate caused a downshift indicating a build-up of negative charge density at that position in accord with the earlier findings of Du et al.3 This was accounted for by the proximity of the Mg2+ ion and Lys16 near the β phosphate to encourage negative charge migration to the phosphate oxygen through electrostatic attraction (Fig. 2).
Fig. 2.

Mg2+ and Lys16 from Ras and the arginine finger from GAP draw negative charge towards the β-phosphate of GTP. The main catalytic effect of Ras and GAP is enthalpic and seems to originate from these electrostatic interactions. (Reprinted with permission by Chemical Physics (2004).28)
It should be pointed out that the synthesis of 18O-labeled phosphates of nucleotides like ATP and GTP is readily accomplished since the synthetic strategy for pHP caged phosphates simply involves the condensation of labelled pHP OPO(OH)2 with the mono- or diphosphorylated nucleoside bases (Scheme 2).3–5,15
Scheme 2.

Wittinghofer, Gerwert, Kötting, and coworkers have made important contributions to unveiling the specificity of Ras and Rap, two GTPase activating proteins (GAPs) that assist in catalytic hydrolysis of GTP.4,29,31,32 Subtle changes in the amino acid sequence and mutations can greatly affect the GTP and GDP binding and hydrolysis of GTP by RasGTPase altering the “on”–“off” signalling by the enzyme, which is considered one of the major determinants in uncontrolled cell growth.4
Regulation of phosphotyrosines, important in transmembrane signalling, is governed by the competitive rates of formation and hydrolysis of tyrosine phosphates. The analysis of the rates of phosphotyrosine formation by phosphotyrosine phosphatase (PTP) has been the target of several mechanistic investigations including small molecule inhibition. The hydrolysis of tyrosine phosphate involves the intervention of a neighbouring cysteine at the active site which attacks the phosphate forming a cysteine thiophosphate intermediate that is hydrolyzed to Pi. α-Haloketone derivatives including phenacyl halides act as small molecule inhibitors that block this pathway by reacting covalently with the cysteine thiol as “suicide inhibitors”.
Pei and coworkers33 showed that pHP Br inhibited PTP hydrolysis by reaction with the key cysteine (Cys 453) located at the enzyme active site. For PTP SHP1(ΔSHP2) the “suicide inhibition” was complete in a few minutes (kinact = 0.40 min−1) which could be replicated in human B cells. In this case, inhibition was shown to cause protein hyperphosphorylation. Unlike traditional “suicide inhibitors,” Pei’s work demonstrated that the pHP thio ether linkage could be severed from the enzyme photo-chemically at 350 nm. Up to 80% of the PTP SHP1(ΔSHP2) activity was restored with a 15 min exposure, providing a model for mechanistic investigations of “on”–“off” inhibition of enzyme catalysis and other fast kinetic studies at cysteine active sites.
Goeldner25 has further demonstrated that pHP thioethers undergo the same rearrangement as the ester counterparts to give 90% of the rearranged p-hydroxyphenylacetate products along with ~10% of p-hydroxyacetophenone, suggesting a small radical component for this reaction. In fact, the release of the thiol leads to disulfide dimers in ~70% yields with a quantum yield of 0.085 for 3′-thio-dTMP in Tris–HCl buffer (pH 7.2). No attempts were made in this study to suppress radical coupling reactions with citrate or other moderators. An interesting variant in the product mixture was 30% formation of p-hydroxyphenylacetate thioester 6. Its formation may be the result of nucleophilic addition by the released thiol (pKa = 8.4) to the spirodiketone 5, which then opens to 6 (Scheme 3).
Scheme 3.
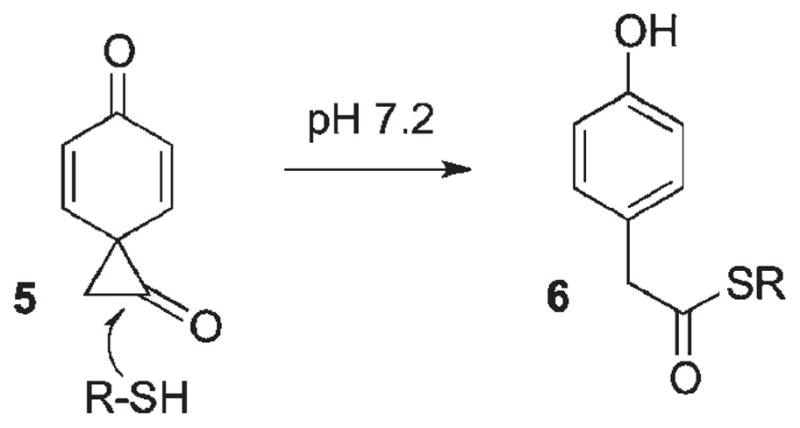
Three thiol derivatives (N-benzylcysteine, 3′-thio-deoxythymidine phosphate (3′-thio-dTMP, a substrate analog for thymidilate monophosphate kinase), and glutathione) were pHP “protected” by direct displacement on pHP Br with the thiols in 80–90% yields. The ease of thiol protection–deprotection by addition of pHP Br followed by photochemical deprotection has served as a basis for several studies on cysteine and thiophosphate substrates.
Bayley et al.26 compared the protection–deprotection effectiveness of oNB and pHP for thiophosphorylated tyrosine. Both protection sequences were accomplished in high yield (75% and 90%, respectively) by treatment of the thiophosphorylated tyrosyl peptide EPOYEEIPILG with oNB Br and pHP Br. Both were equally stable to storage as well as to reaction conditions without light (Scheme 4).
Scheme 4.

Irradiation at 312 nm released the thiophosphated peptide in 50–70% yields with quantum yields of 0.25–0.37 (oNB) and 0.56–0.65 (pHP). The chemical and quantum yields were pH dependent between pH 5.8 to 7.3, but at pH 8.2 the reactants and/or the products were not stable. Irradiation conditions at 312 nm resulted in deprotection from pHP ten times faster than from oNB due primarily to a combination of higher efficiency and larger absorptivity for pHP at this excitation wavelength. Both deprotection reactions provided the oligopeptide regaining 50–70% of its original binding capacity as determined by radio-labelled binding studies with recombinant SH2 domain.
Bayley et al.26 extended this approach to the protection–deprotection of thiophosphorylated threonine, Thr-197, in the catalytic site of a cell-signalling protein kinase A found in the catalytic Cα subunit by phosphorylation. The phosphothreonine unit plays a key role in stabilizing the active conformation of the Cα catalytic subunit. The thiophosphorylated threonine was effectively protected by direct reaction with pHP Br to yield pHP-PST197 Cα. It is noteworthy that the thiophosphorylated threonine (pKa = 5.8) was selectively protected with pHP Br in the presence of potentially competing cysteine residues. Both the binding capability and the specific activity of the caged enzyme were dramatically reduced, to 2.4% (binding assay measured on TNB-thio agarose beads) and by a 17-fold decrease in 32P kinase activity. Photolysis at 312 nm at pH 7.3 restored 85–90% of the binding activity and the specific activity was nearly recovered (15-fold increase) with a quantum yield of 0.21. The role of the pHP group in this instance is to block or diminish H-bonding and electrostatic attractions between the threonine-197 and Arg-165 with Lys-189, the amino acids that stabilize the closed form of the Cα subunit.
Protecting groups have a long history of application, which takes advantage of the ability to control the exposure of reactive functional groups to reactive environments during multistep syntheses of peptides, nucleotides and polymers.34 While photoremovable caged compounds have long been considered for applications in these areas, related applications on solid surfaces35 are receiving more attention. Three attractive characteristics for photochemical approaches toward systematic exposure of functional groups are the location and the density of the exposed functionalities. Added to this is the temporal control which can be important for investigations of signal processing. These attributes have encouraged an explosive array of applications36 and surface-bound caged compounds have been exploited for controlled exposure of groups such as carboxylic acids, amines, thiols, and many other reactive functional groups bound to glass surfaces, beads, polymeric supports, and other solid surfaces.
Photoremovable protecting groups applied to regulation of PCR and gene expression have been explored with mixed results. Many attempts to control polynucleotide selectivity and activity using PPGs have also been attempted.37 However, a recent study by Pickens and Gee9 has opened a potentially useful new application using pHP protection of the 3′-hydroxy group on thymidine that, as its corresponding phosphoramidite, could be employed in PCR. In this study, the 3′-hydroxy was protected as its pHP ether. Photolysis of pHP 7 released thymidine quantitatively in 15 s (Scheme 5). The authors suggested that the protected phosphoramidite might serve as a photoreversible blocker for selective PCR modification of the nucleotide array (Fig. 3).
Scheme 5.

Synthesis and release of pHP caged thymidine 7.
Fig. 3.

Caging methodologies to regulate oligo synthesis. (With permission of Tetrahedron Lett.)
(Coumarin-4-yl)methyl [coumarylmethyl] photoremovable protecting groups
The development of coumarylmethyl cages as a new class of photoremovable groups commenced with the discovery by Givens and Matuszewski of the photoreactivity of a coumarinylmethyl group in releasing phosphate ester 8 (Scheme 6).6
Scheme 6.

Schmidt and coworkers38 have probed the mechanism of release from several coumarylmethyl cages CM–LG using quantitative quantum yield and time-resolved measurements of the photochemistry and fluorescence of coumarylmethyl esters and the corresponding alcohols (vide infra, Scheme 14 and Table 3). Heterolytic bond cleavage, kcl, proceeds from the relaxed excited singlet state 1CM–LG* in competition with fluorescence and nonradiative decay processes, kf and knr, forming a singlet ion pair 1[CM+…LG−] in the initial reaction step (Fig. 4). Escape from the solvent cage first affords the solvent-separated ions CM+ and LG−. The coumarylmethyl cation CM+ reacts with water to yield the product alcohol CM–OH and H+ in addition to the leaving group anion LG−.
Scheme 14.

Table 3.
Effect of pKa on quantum yields and cleavage rate constants kcl38
| LG | pKa | Φ/10−3 | k/109 s−1 |
|---|---|---|---|
| C6H13CO2− | 4.89 | 4.3 | 0.32 |
| p-CH3O–C6H4CO2− | 4.41 | 4.5 | 0.20 |
| C6H5CO2− | 3.99 | 5.2 | 0.18 |
| p-CN–C6H4CO2− | 3.54 | 6.4 | 4.0 |
| −OP(O)(OEt)2 | 0.71 | 37 | 6.6 |
Fig. 4.

Mechanism of the photocleavage of coumarylmethyl cages. Adapted from ref. 38.
The cleavage rate constants, kcl, were estimated from measurements of the fluorescence quantum yield, Φfl, and the fluorescence lifetime, τfl, of the alcohols CM–OH and the fluorescence and photochemical quantum yields, Φfl and Φr, of the coumarylmethyl esters CM–LG. The resulting rate constants kcl range from 0.1 to 50 × 109 s−1 and obey a linear free energy relationship with the acidity constants pKa of the LG acids, i.e., those releasing the strongest acid are fastest. A similar relationship was found for the heterolytic cleavage from the triplet state of pHP esters (Fig. 1). The ratio of escape to recombination from the ion pair could not be determined directly, but the measurements indicated that the factors that accelerate the heterolytic bond cleavage retard the ion recombination reaction.
Coumarin-based PPGs have gained considerable attention, especially for applications in biological chemistry,39 primarily due to its longer-wavelength absorption, extending farther into the visible region (400–500 nm) and the fast release rate from its excited singlet. These advantages have been offset by coumarin’s marginal quantum yields, low aqueous solubility and the occasional inconvenience of a strong fluorescence and competitive absorptivity from the coumarin photolysis byproducts. However, many successful efforts have been reported where clever modifications to the coumarin group have resulted in somewhat higher quantum yields and better aqueous solubility. These, together with adaptations that expand the leaving group range (including alcohols and amines), have raised researchers’ awareness to this relatively new class of PPGs.
The first generation of coumarylmethyl cages possessing an –OH or –OMe substituent at C7 were characterized by very fast release rates and great hydrolytic stability. However, they had a drawback of poor solubility and relatively low quantum yields for all leaving groups except phosphates. 6-Bromo-derivatives where designed to lower the pKa of the 7-hydroxy group by two units to effect a complete deprotonation at physiological pH, thereby enhancing water solubility and causing a bathochromic shift of 60 nm (Table 2). The downside of this modification was the increased susceptibility of some of the bromo-substituted caged compounds to hydrolyze in the dark. Further development in this area resulted in emergence of a second-generation of coumarylmethyl PPGs bearing an amino group at C7, which significantly improved the spectroscopic and photochemical properties of the cage, moving the absorption maxima to 350–400 nm and recording the highest quantum yields among the analogues. Solubility of the aminocoumarylmethyl cages could be improved by appending polar groups such as carboxylates to the aniline moiety. Strong fluorophoric properties, which are even more enhanced in the polyaromatic analogs, lend a convenient tool for monitoring of the reaction course.
Table 2.
Typical coumarin chromophores
| λmax | Structure | Abbreviation | Solventa | Ref. |
|---|---|---|---|---|
| 320–330 nm |
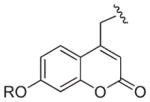
|
HCM (R = H) | C | 40 |
| D | 41 | |||
| 7-MCM, Bpm (R = Me) | C | 40,42 | ||
| F | 43 | |||
| A,D | 38,41,44 | |||
| CMCM (R = CH2CO2H) | D | 45 | ||
| 330–380 nm |
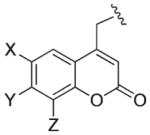
|
DMCM (X = Y = OMe, Z = H) | F | 43 |
| A | 38,46 | |||
| BCMCM (X = Y = OCH2CO2H, Z = H) | D | 45 | ||
| B | 47 | |||
| 7,8-BCMCM (X = H, Y = Z = OCH2CO2H) | G | 48 | ||
| BHCM, Bhc (X = Br, Y = OH, Z = H) | E | 49,50 | ||
| G | 51 | |||
| F | 43 | |||
| BMCM (X = Br, Y = OMe, Z = H) | E | 49 | ||
| BBHCM (X = Br, Y = OH, Z = N(CH2CO2But)2) (X = Br, Y = n-C17H35CO2, Z = H) | G | 51 | ||
| B | 52 | |||
| 350–400 nm |
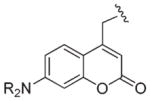
|
ACM (R = H) | D | 41 |
| DMACM (R = Me) | B | 53 | ||
| A | 38,44 | |||
| DEACM (R = Et) | H,B | 54–57 | ||
| D | 58 | |||
| F | 43 | |||
| A | 38,46 | |||
| BCMACM (R = CH2CO2H) | B | 59,60 | ||
| D | ||||
| BBCMACM (R = CH2CO2But) | G | 61 | ||
| 340–360 nm |
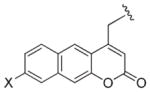
|
Obb (X = H) Bbl (X = MeO) |
C | 42,62 |
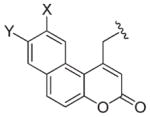
|
Obc (X = OH, Y = H) Obm (X = OMe, Y = H) Bba (X = H, Y = OMe) |
C | 42,62,63 |
Solvents: HEPES–MeOH (A); HEPES buffer (B); EtOH (C); PB or PBS buffer (D); KMOPS buffer (E); KMOPS–MeOH (F); HEPES–MeCN (G); water (H).
Applications of coumarin caged compounds
Phosphates
Photolysis of 4-methoxycoumarylmethyl phosphate was the first example that employed a coumarylmethyl PPG6 and current literature reveals that coumarinylmethyl phosphates continue to be the most frequently encountered application of the coumarin cage. Most caged phosphates have good stability to hydrolysis (although some analogues may have limited water solubility), adequate quantum efficiencies (Φ = 0.03–0.2), and are easily synthesized, which, taken together with very fast release kinetics, make coumarin-caged phosphates very attractive candidates for biological applications. Depending on specific needs, properties of caged phosphate can be tuned by modifying substituents on the coumarin core. Studies by Lima et al. also emphasize the importance of pH control in photolysis of ionizable 4-methylcoumarin-caged phosphates, which show significant dependence of quantum yields on pH variations.64 A review by Furuta provides useful guidelines for choosing a suitable cage.65 The most recent applications of caged phosphates in biological studies are described below.
Caged peptide 9 was used as a modular domain-binding peptide in the investigation of PI3K kinase-mediated phosphorylation of proteins. Irradiation of 9 in K-MOPS buffer solution quantitatively produced phosphopeptide 10 and hydroxymethylcoumarin 11 with a quantum yield of 0.12 (Scheme 7).66 Peptide 10 was also successfully released in living cells upon exposure at 365 nm without causing any toxicity.
Scheme 7.

Schultz and coworkers demonstrated that 7-diethylaminocoumarin-4-ylmethyl-caged bioinactive phosphatidylinositol 3,4,5-triphosphate (PI(3,4,5)P3) can be effectively transported through the plasma membrane and then uncaged to release PI(3,4,5)P3, inducing membrane ruffling and pH-domain translocation in the presence of the PI3-kinase inhibitor wortmannin (Fig. 5).67 The cell entry by PI(3,4,5)P3 was conveniently monitored by fluorescence microscopy. Biological response was observed within 1 min following irradiation by a short pulse of a 375 nm laser, and was superior to that of the membrane-permeable uncaged analogue. It should be mentioned that synthesis of PI(3,4,5)P3 suffered from low yield (2.5% overall) due to poor stability of the coumarylmethyl group under alkylation conditions.
Fig. 5.

Mechanism of action of PI(3,4,5)P3: (a) cell entry; (b) enzymatic removal of Bt and AM protecting groups produces cgPI(3,4,5)P3; (c) light-induced removal of coumarin protecting group; (d) PI(3,4,5)P3 induces translocation of EGFP-Grp1-PH domains to the plasma membrane.67
Due to very fast release rates (2 × 108 s−1), high photoefficiencies and hydrolytic stability, the coumarin-4-ylmethyl cage is ideal for studies of biological processes involving release of nucleotides and nucleosides. Furthermore, the intense absorptivity of 7-aminocoumarins at longer wavelengths (350–400 nm), far beyond that of other photoremovable groups, permits efficient photorelease nicely avoiding biologically lethal wavelengths. Thus, Bendig and Giese suggested 7-diethylaminocoumarin-4-ylmethyl cytidine 5′-diphosphate (CDP) 12 (λmax = 392 nm; ε = 16 200 M−1 cm−1) as a suitable model for studies of a long-range radical transfer mechanism in E. coli ribonucleotide reductase (RNR) (Scheme 8).57 An important advantage of this model over o-nitrobenzyl and other analogues is that photo-activation of CDP occurs extremely fast, which permits analysis of conformational changes preceding the radical transfer, and at wavelengths where the molar absorptivity of RNR is very low.57
Scheme 8.

The release of ATP in cell cultures and in acutely isolated brain slices was tested to probe the ability to evoke Ca2+ ion waves53 (Scheme 9). The release of nucleoside triphosphate from caged precursor 13 in HEPES solution was twice as efficient as that of the nitrobenzyl-caged analog at 365 nm and ten times more efficient at 405 nm. To measure the response in cells, cytosolic Ca2+ concentrations were recorded from confiuent astroglial cultures stained with the Ca2+ indicator fluo-3. The experiments confirmed biological inertness of the caged ATP 13. In contrast, irradiation of the latter with an argon laser at 364 nm caused a notable increase of fluorescence in cells close to the UV spot. The response was successfully elicited three times, provided the cells were allowed to recover after each dose of radiation. The Ca2+ response showed the typical time course of an ATP-triggered signal: a fast increase in cytosolic Ca2+ concentration and a subsequent slow decay back to the initial concentration.53
Scheme 9.

Light-controlled, in vitro enzymatic polymerization of nucleic acids through the release of ATP from a coumarin-4-ylmethyl-caged precursor was demonstrated by Baptista (Scheme 9)54 by irradiating a ribonucleotide mixture (CTP, UTP, and GTP) and caged ATP 13. A control sample (no light) showed no reaction, whereas formation of the transcription product was formed on irradiation, reaching a maximum at 25 μM of ATP followed by a decrease in RNA formation. The decrease in RNA was explained by inhibition of transcription due to accumulation of byproduct 15.54
The release rate of the cyclic nucleotide depends on its structure and the solvent polarity.68 High electron donor ability of the purine base compared with diethyl phosphate leads to more stable intermediate tight ion pairs resulting in greater quantum yields. Furthermore, the higher quantum yields for the axial vs. equatorial isomers of caged cyclic phosphates (0.13–0.21 for 16 vs. 0.07–0.09 for 17, Scheme 10) were attributed to increased steric interaction and larger partial charge transfer between the purine base and coumarylmethyl cation for the axial vs. equatorial isomer.
Scheme 10.

The effect of substituents on coumarin PPGs was systematically investigated for a series of caged cAMPs (Scheme 11).44 In addition to influencing the absorption maxima (Table 2), changing donor substituents at the 6- and 7-position of coumarin also significantly affects the quantum yields. The highest quantum yields are obtained for 7-dialkylamino derivatives (0.21–0.28), which were rationalized by more efficient stabilization of the coumarylmethyl carbocation through the electron-donating amino substituents and, as a result, more efficient ion pair escape (Scheme 11).44,68
Scheme 11.

Iwamura and coworkers were the first to probe photorelease of cAMP from a coumarin cage in studies of biological responses within living cells.69,70 A test experiment using zebra fish melanophores demonstrated that inactive, caged cAMP (18, X = Br, Y = AcO, Scheme 11) successfully penetrated through the plasma membrane into the melanophore and released a sufficient amount of cAMP upon irradiation at 340–365 nm to induce motile response of melanin granules. This has been revisited for coumaryl PPGs possessing a polar aminobis(methylcarboxylate) group (19, Scheme 12), which significantly improved water solubility of the caged nucleoside monophosphates.47,60 Furthermore, the fluorescence quantum yields of 19a,b were an order of magnitude lower than that of the corresponding coumarin-4-ylmethanol 20, which permitted convenient monitoring of the reaction by fluorescence spectroscopy. Upon irradiation of HEK293 cells transfected with DNA encoding CNGA2 channels, loaded with 19b, an increase in both the intracellular fluorescence and the cAMP-induced current resulting from activation of olfactory neurons (CNGA2) was observed.47 From these studies, coumarylmethyl PPGs can be used as an analytical method for quantitative dose–response relationships for cellular reactions triggered by cAMP.
Scheme 12.

In addition to significant advances achieved in caging of DNA and RNA through the phosphate backbone using methods described above, complementary approaches for base-caged nucleosides have also been developed to enable control of Watson–Crick pairing and double strand formation. Bromocoumarylmethyl-caged deoxycytidine 21 and deoxyadenosines 22 used by Furuta et al. proved most efficient compared to o-nitro-benzyl caged analogues in the release of nucleosides upon irradiation at 350 nm (Scheme 13).49
Scheme 13.

The coumarylmethyl chromophore has been increasingly examined for 2-photon excitation efficacy using near IR (>700 nm) lasers.71 Thus, it was also shown that the two-photon uncaging action cross-section of 22a (0.35 GM at 740 nm) is in the practical range for biological applications. The O-caged deoxyguanosines 23 reported by Heckel showed significantly lower quantum yields at 365 nm as compared to P-caged cyclic nucleotides or the 2-(o-nitrophenyl)propyl analogue 24; however, the high extinction coefficient of 23 makes photochemical effectiveness for release nearly two-fold better than that for 24.72 Furthermore, uncaging of nucleobase in 23 at 405 nm proceeded 80 times faster than the corresponding oNE PPG in 24, permitting wavelength-selective photorelease.
Release of carboxylate: carboxylic acids and amino acids
The photoefficiency of the 7-alkoxycoumarin-caged carboxylates, especially aliphatic acids, is generally rather poor,65 primarily attributed to the higher pKa of a carboxylic acid as compared with a phosphate leaving group. Thus, Schmidt et al. demonstrated a strong decrease of the rate constants of heterolytic cleavage, kcl, with increasing pKa of the released acid (Fig. 4, Scheme 14, Table 3).38
The release of 2,4-D represents a practical, agricultural application of coumarins as PPGs. The UV-Vis photochemistry of 2,4-D caged by 7-substituted coumarylmethyl protecting groups was measured in several aqueous organic solvents to explore the effects of substituents and the pH on the solubility and the conversion at different wavelengths (Scheme 15).73 All compounds in the tested series showed rather low efficiencies; the highest quantum yield was observed for 7-hydroxy-derivative 26 (0.018 and 0.012 at 310 and 350 nm, respectively).
Scheme 15.

Similarly, a comparison of quantum yields of three GABA conjugates 25a–c at different wavelengths performed by Costa et al. revealed rather low photoefficiency in the UV range optimal for most biological applications (≥350 nm) (Scheme 16).42,68
Scheme 16.

In contrast to poor quantum yields observed in the previous examples employing 7-alkoxycoumaryl group, photorelease of amino acids linked through the carboxylate moiety to 7-aminocoumarylmethyl chromophore was much more efficient (Scheme 17).46,55,56,74–76 Quantum yields for caged GABA, glycine and glutamate derivatives range from 10 to 20% (Table 4).46,56 It was proposed that the photocleavage product yield can be attenuated by ion-pair internal return of the released group in competition with escape from the solvent cage.46 Stability of the carboxylate-caged amino acids toward hydrolysis may become an issue and thus should always be examined prior to photolysis to ensure stability of the caged compound in aqueous buffer solution. In fact, it was found that phenylalanine caged with the water-soluble bis(carboxymethyl)amino derivative (26c) was unstable in HEPES buffer at pH 7.2.74 Spontaneous hydrolysis was also observed for glutamate when caged with 6-bromo-7-hydroxycoumarin-4-ylmethyl.66
Scheme 17.

Table 4.
Photorelease of coumarylmethyl-caged neurotransmitters
| Y | X | LG | λ/nm (pH) | Φ | Ref. | |
|---|---|---|---|---|---|---|
| 26a | N(CH2CO2H)2 | H | GABA | 380 (7.2) | 0.2 | 2 |
| 26b | N(CH2CO2H)2 | H | Glu | 380 (7.2) | 0.1 | 2 |
| 26c | N(CH2CO2H)2 | H | Phe | — (7.2) | — | 71 |
| 26d | NEt2 | H | Gly | 400 (7.4) | 0.12 | 55 |
| 26e | NEt2 | H | Glu | 400 (7.4) | 0.11 | 28 |
| 26f | NEt2 | H | GABA | 400 (7.4) | 0.14 | 29 |
| 26g | NEt2 | a | GABA | 400 (7.4) | 0.1 | 55 |
X = –CONH–CH2–CO2Et.
Coumarin-caged neurotransmitters are ideal models because they can be efficiently photolyzed by visible and near UV light that is much less harmful to cells, thereby opening biological studies to both in vivo and in vitro activation of agonists and antagonists at various neurotransmitter receptors. On the other hand, the photolysis byproducts do not appear to inhibit or activate these same receptors. The experimental design in these studies vary from more sophisticated laser-flash, time-resolved procedures to the more readily available, inexpensive light sources such as Rapp flash lamps.
Hess and coworkers employed photorelease of caged GABA, glycine, and glutamate as a tool for studying the mechanism of action of neurotransmitter receptors using HEK 293 cells transfected with cDNA encoding the corresponding receptors. Flash lamp photolysis (385–450 nm) uncaged glycine56 and glutamate55 from precursors 26d,e in HEK extracellular buffer. The efficiency of photorelease was assessed by the whole-cell current evoked by the released amino acid.55,56 Both caged glycine (26d) and glutamate (26e) were inert toward the corresponding receptors. The analogous GABA derivative 26f, however, inhibited GABAA receptors.75,76 This problem was addressed through the installation of an additional substituent at 4-methylene group (26g, X = CONHCH2CO2Et, Scheme 17) of the coumarin phototrigger which helped eliminate unwanted activity of the caged precursor.76
Carbamate-caged amino acids
To address the hydrolysis issue, amino acids caged at the N-terminus via a carbamate linkage were employed. Thus, carbamate-caged blockers for glutamate transporter, L-threo-β-benzyloxyaspartate (L-TBOA) and its more potent m-(trifluoromethyl)benzamide analog (L-TFB–TBOA) were found to be more stable in aqueous solutions than the corresponding carboxylates (Scheme 18).45 Furthermore, hydrophilic coumarin-4-ylmethylcarbamate cage 27b,c (R2 = OCH2COOH) photolyzed efficiently with quantum yields 0.03 and 0.02 (for 27b and 27c, respectively), which is comparable to that of the complementary carboxylate-caged analogs. Tests were conducted using 27a–c on biological activity by the glutamate uptake inhibition assay using excitatory amino acid transporter (EAAT2) stably expressed on MDCK cells. It was shown that the activity of carbamate- and carboxylate-protected blockers was efficiently masked by the coumarin cage. The release of the more potent L-TFB-TBOA upon photolysis provided a sufficiently effective concentration of the blocker for applications in biological preparations. It was also confirmed by uptake assay that the blocker activity of the released L-TFB-TBOA was not impeded by the photolysis byproducts.45
Scheme 18.

Comparison of irradiation doses required to release amino acids caged through different linkages revealed the following relative photosensitivity order: anhydride > ester > carbamate > carbonate, with carbamates being slightly more resistant to irradiation than carboxylates (Scheme 19).40 It should be mentioned that the rate-limiting step in all these cases except carboxylate 31 is decarboxylation of the released carbamic and carbonic acid, which depends on the nature of the released group,77,78 and varies with pH. The rates of decarboxylation reactions are usually quite slow, k−CO2 = 10−3 s−1,79–81 and subject to both acid and base catalysis.
Scheme 19.

Schmidt’s calculated shifts in the λmax of 7-amino-substituted coumarin-caged GABA 32 suggested a notable bathochromic shift as compared with the 7-hydroxy- and 7-methoxy derivatives.41 Indeed, the absorption maximum of caged GABA 32 was shifted by 22–24 nm and readily released amino acid at 300–400 nm with a quantum yield of 0.04 (Scheme 20). Photolysis of caged GABA 32 on auditory neurons in the lateral superior olive in brainstem slices of mice triggered membrane current mediated by specific activation of the GABAA receptor by whole-cell patch clamp recordings of individual neurons.41 It was shown that GABA derivative 32 had no intrinsic biological activity and good stability in dark solutions, while another caged neurotransmitter, 6-bromo-7-hydroxycoumarin-4-ylmethoxycarbonyl dopamine, was found to be more susceptible to hydrolysis in aqueous solutions, which, however, was slow enough on the timescale of the physiological experiments to allow useful studies.51
Scheme 20.

Applications of photoremovable groups in protein chemistry face a number of limitations, including synthetic challenges associated with chemo- and site-specific installation of PPGs, difficulty in transporting caged proteins into a cell, poor tissue penetration and damaging effects by UV light causing unwanted side reactions, such as photooxidation, and incomplete uncaging in biological environment. Often, small PPG chromophores fail to efficiently suppress the activity of large biological entities resulting in notable residual activity.82 Described below are several recent reports of the successful use of coumarin PPGs in caging strategies for peptides and other large biomolecules.
The coumarylmethyl cage was successfully used to mask the activity of oligopeptides Aβ1–24 and Aβ27–42 (Scheme 21).61 An elegant approach termed “click peptide” involved an O–N intramolecular acyl migration triggered by cleavage of the PPG. Screening of different photoremovable groups, including 6-nitroveratryl, p-dimethylaminophenacyl and various 7-aminocoumarylmethyl analogs revealed that only the bis(carboxymethyl) aminocoumarylmethyl derivative possessed the necessarily important features of sufficient hydrophilicity and stability to hydrolysis. Importantly, the protected oligopeptide precursor 33 was significantly more water soluble than the target peptide Aβ1–42 (35), and did not self-assemble, because the O-acyl (instead of N-acyl) fragments in 33 suppressed the conformational transition into a β-sheet analogous to 35. Irradiation of 33 at 355 nm quantitatively removed the protecting group within 2 min; subsequent incubation of 34 for 30 min at 37 °C enabled smooth acyl migration to produce 35 (Scheme 21).61
Scheme 21.

Another example showcasing the application of a photoremovable coumarin group in synthesis of peptidoconjugates was demonstrated by Nagamune et al. (Scheme 22).83 Temporal and spatial control of peptide release was achieved upon irradiation of a peptide conjugate possessing a photocleavable backbone. Solutions of conjugates 36 and 38 possessing a bifunctional bromocoumarin carbamate linker were photolyzed at 350 nm, releasing peptides 37 and 39 with quantum yields of 0.16 and 0.26, respectively. A similar approach was employed for site-specific caging and photorelease of plasmid DNA.84
Scheme 22.

Coumarin-containing lipids are another example for successful use of photocleavable fragments implanted in the backbone of a complex biological molecule (Scheme 23).52 Light-mediated release of the liposomal content via lipid cleavage represents an attractive alternative for controlled drug release. A hydrophilic head of the lipid (an amino acid) was separated from the hydrophobic tail by a bifunctional coumarin phototrigger carbamate link at one end and a carboxylate group at C7 as the tether (40). The hydrophobic head was cleaved upon irradiation, which released the amino acid from the coumarylmethyl stearate 41. Liposomes that incorporated 20% of 40 were compared in size before and after irradiation and found to decrease in diameter by 50% as a result of the liposome reorganization and leakage.52
Scheme 23.

A caging strategy for masking the biological activity of a complex, heavily functionalized molecule such as Isotaxel was demonstrated by Kiso et al. (Scheme 24).85 Isotaxel, an O-acyl isoform of the anticancer agent Paclitaxel, was modified to suppress the pharmacophore activity through caging the benzylamine moiety with a coumaryl PPG. The resulting photoresponsive prodrug, called Phototaxel (42), was selectively activated by visible light (430 nm) releasing Isotaxel (43), which in turn was converted into Paclitaxel (44) by a spontaneous intra-molecular O–N-acyl migration (Scheme 24).85 The yield of released Paclitaxel was found to be 69%; partial loss of material was attributed to formation of unidentified side photoproducts. To improve water solubility of the Phototaxel prodrug, the N-ethyl groups of the coumarin cage were replaced with N-(2-(dimethylamino)ethyl)acetamide groups (Scheme 24).86 N- and O-protected Paclitaxel prodrugs 45 and 46 with carbamate and carbonate-linked coumarins were synthesized. Both caged compounds showed excellent water solubility; however, carbonate 46 was not stable to aqueous solutions. Notably, direct continuous irradiation (365 nm) by a UV ray lamp released Paclitaxel from 45 very effectively, whereas pulsed laser activation (355 nm) resulted in substantial decomposition.86
Scheme 24.

Effective release by irradiation at long wavelengths and excellent quantum yields for the 7-aminocoumarin derivatives make them the protecting group of choice vis-à-vis other PPGs for wavelength-selective uncaging.87,88 Thus, del Campo et al. used a pair of wavelength-based nearly orthogonal photoactivated protecting groups, a 7-diethylaminocoumarin (λmax = 390–400 nm) and a nitroveratryloxycarbonyl (λmax = 356 nm), each individually attached through an amino group tethered to a silica surface (Scheme 25).87 Sequential spatially controlled wavelength exposures, first with 412 nm and then 345 nm light, selectively deprotected coumarin and nitroveratryloxycarbonyl groups creating patterns which were visualized by in situ subsequent coupling of the uncaged amino groups with different fluorescent dyes. Reversing the exposure sequence to first 345 nm light followed by 412 nm resulted in simultaneous cleavage of both chromophores at 345 nm due to the overlap and similar extinction coefficients of both PPGs at this wavelength.
Scheme 25.

The study further expanded the controllable variables from spatial and time-resolution to include wavelength selectivity by employing a variety of caging groups that are activated over a range of excitation wavelengths. A clear objective of this study was to identify truly orthogonal chromophores for activation of functional groups, an objective of many synthetic applications in protecting group chemistry. Seven caging chromophores, ranging from pHP (λmax 270 nm) to coumaryl (λmax 350 nm), were attached to a carboxylate group poised on an alkyl tether bound to a glass surface by a Si(OMe)3 linkage. Systematic variation of the exposure wavelength to the coated silica plate released the functional carboxylate groups. The chromophores gave an effective wavelength range between 250 and 450 nm. Analysis of the seven chromophores further demonstrated that pairwise, triplet and quartet sets of orthogonal protecting groups from the variety of caging chromophore combinations could be generated on a single platform. This approach holds promise for new applications of sensors that offer more precise control for molecular detection and for communication purposes.
A photoremovable coumarin protection was employed by Wylie and Shoichet in a clever design of three-dimensional pattern-writing medium (Scheme 26).89 The matrix, composed of modified aragose 47 possessing an exposed amino group protected with 6-bromo-7-hydroxycoumarin, was chosen for its high efficiency in two-photon activation. Confocal microscopy using a Ti-sapphire laser for 3D multiphoton uncaging cleaved the coumarin protecting group liberating the primary amine moieties in the gel.
Scheme 26.

Thiols
Analogously, unmasking sulfide groups upon UV light exposure in thiolated aragose gels 48 provided a convenient, chemospecific 3D pattern at the reaction site in preparation for selective immobilization of biologically relevant substrates (Scheme 26).90 This approach preserves the mechanical properties of the patterned material, enabling the creation of complex 3D biochemical sites buried within hydrogels.89,90
Photolabile coumarin thiocarbonate protecting groups for thiols were developed by Hagen et al.48,74 Two complementary PPGs, 7-aminocoumarin 49 (λmax 450 nm) and 7,8-biscarboxy-methoxycoumarin 50 (λmax 324 nm), were employed to mask two different cysteine residues in resact, a sea urchin peptide hormone guanylate cyclase plasma membrane (Scheme 27).48 Notably, both PPGs were stable to TFA, which makes them orthogonal to many conventional peptide protecting groups. Irradiation of the protected resact with 402 nm light led to selective cleavage of 49, releasing 50-OH and two resact isomers, the result of an S-to-S acyl shift of the remaining 50 group, thereby freeing a Cys1 side chain. Subsequent irradiation of the product mixture at 325 nm quantitatively removed 50 from the peptide. Although careful tuning of substituents on the PPG allowed clean wavelength-selective photochemistry, the observed acyl migration within resact rendered thiocarbonate PPGs inappropriate for selective protection of cysteine residues in a peptide.48
Scheme 27.

Alcohols and phenols
Furuta et al. compared several carbonate-tethered coumarin phototriggers for their efficient release of alcohols (Scheme 28).43 All coumarin PPGs tested were found to be at least an order of magnitude more efficient than o-nitrobenzyl. Within the coumarins, the 7-methoxy- and 6-bromo-7-hydroxy-analogs 51a and 51b provided the highest quantum yields, and 51b was the most efficient among all the tested cages. A few biologically relevant alcohols caged with 51b showed moderate to good resistance to hydrolysis, and all caged compounds efficiently released substrates upon photolysis at 350 nm.43
Scheme 28.

Alkylation of the phenolic moiety of rosamine (52) with coumarin-4-ylmethyl chloride followed by base-promoted spirocyclization destroyed conjugation in the xanthene ring and completely shut off the fluorescence of 53 (Scheme 29).91 Brief exposure of 53 to visible light induced a large fluorescence enhancement typical for rosamine dye (577 nm), indicating efficient probe release and restored conjugation in xanthene. Notably, the fluorescence spike in this case was much more pronounced indicating very efficient photorelease, when compared with release of rhodamine from the o-nitrobenzyl cage.92
Scheme 29.

Caged capsaicin 54, possessing the {7-[bis(carboxymethyl) amino]coumarin-4-yl}methoxycarbonyl PPG, was employed to study capsaicin’s effect on HEK 293 cells transfected with TRPV1 channels – cation channels that mediate pain perception in nociceptive somatosensory neurons (Scheme 30).93 Preliminary tests showed that carbonate 54 had no residual activity at the micromolar level, was sufficiently resistant to hydrolysis in the dark, and efficiently released capsaicin 55 at 365 or 405 nm.51,93 Isothermal titration calorimetry showed that the modified capsaicin 54 did not measurably translocate across a phospholipid membrane and thus can be selectively applied to one side of the plasma membrane. The effect of intracellular and extracellular photorelease of capsaicin from membrane-impermeant 54 was investigated. It was found that extracellular release of 55 triggered large whole-cell current and strong desensitization, whereas the analogous response was rather modest from intracellular release of capsaicin.93
Scheme 30.

Carbonyl compounds
The first example of utilizing photo activation of coumarylmethyl-caged carbonyl compounds in a biological system was reported by Hagen et al. (Scheme 31).94 The rate of the primary reaction (56 → 57) was found to be (1.2 ± 0.5) × 108 s−1 and the yield of progesterone (57) release was about 30%. The quantum yield was low but acceptable for the application, due to high absorptivities that ensure high photosensitivity. Caged 56 was also sensitive to 2PE; exposure of 56 to femtosecond pulses at 755 nm caused significant release of progesterone. However, the efficiency was ca. 5 times lower than that observed for an analogous glutamate derivative.66
Scheme 31.

Conclusions
The coumarylmethyl and pHP photoremovable protecting groups are being developed as photoinitiators for mechanistic studies in biological transformations that require nanosecond release rates. They are synthetically accessible and can be installed readily on most substrates. Recent advances in time resolved spectroscopy encourage yet further development of these new PPGs.
Acknowledgments
We thank the NIH (RO1GM72910) for support.
Biographies

Richard S. Givens (1940) is an Emeritus Professor (2010) of Chemistry at the University of Kansas. He earned his BA (Chemistry, H-G. Gilde) from Marietta College and PhD (H. E. Zimmerman) from the University of Wisconsin in 1966. He was an NIH postdoctoral associate (G. A. Russell) at Iowa State. He has authored or coauthored more than 120 scientific publications and 5 patents on physical organic chemistry, photochemistry, and their applications in biology and is co-editor of Dynamic Studies in Biology: Phototriggers, Photoswitches and Caged Biomolecules (Wiley-VCH, 2005).

Marina Rubina received her BS degree in chemistry and chemical education from Syktyvkar State University (Russia) in 1996. She was a research associate at the Moscow State University (Russia) before earning her PhD degree with Vladimir Gevorgyan (2004) at the University of Illinois at Chicago. She was then a postdoctoral associate with Michael Rubin and Richard Givens at the University of Kansas. Currently, she is Laboratory Director in the Department of Chemistry and a Research Associate at the Center for Environmentally Beneficial Catalysis at the University of Kansas.

Jakob Wirz (1942) is Emeritus Professor of Chemistry. He studied at the ETH Zürich with Prof. E. Heilbronner and, after postdoctoral studies in London with Professors G. Porter and D. Barton, habilitated at the University of Basel, where he led a research group until 2007. He co-authored the book Photochemistry of Organic Compounds (Wiley, 2009) with Professor P. Klán and published over 170 papers focusing on photochemistry and spectroscopy, laser flash photolysis, reaction mechanisms, and photochemical protecting groups. He continues to serve as Deputy editor-in-chief for Photochemical & Photobiological Sciences.
Footnotes
This paper is part of a themed issue on photoremovable protecting groups: development and applications.
Contributor Information
Richard S. Givens, Email: givensr@ku.edu.
Marina Rubina, Email: mrubin3@ku.edu.
Jakob Wirz, Email: J.Wirz@unibas.ch.
Notes and references
- 1.(a) Pelliccioli AP, Wirz J. Photoremovable protecting groups: reaction mechanisms and applications. Photochem Photobiol Sci. 2002;1:441. doi: 10.1039/b200777k. [DOI] [PubMed] [Google Scholar]; (b) Bochet CG. Photolabile protecting groups and linkers. J Chem Soc, Perkin Trans. 2002;1:125. [Google Scholar]; (c) Goeldner M, Givens R, editors. Dynamic Studies in Biology: Phototriggers, Photoswitches and Caged Biomolecules. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 2.(a) Il’ichev YV, Schwörer MA, Wirz J. Photochemical reaction mechanisms of 2-nitrobenzyl compounds: methyl ethers and caged ATP. J Am Chem Soc. 2004;126:4581. doi: 10.1021/ja039071z. [DOI] [PubMed] [Google Scholar]; (b) Hellrung B, Kamdzhilov Y, Schwörer M, Wirz J. Photorelease of alcohols from 2-nitrobenzyl ethers proceeds via hemiacetals and may be further retarded by buffers intercepting the primary aci-nitro intermediates. J Am Chem Soc. 2005;127:8934. doi: 10.1021/ja052300s. [DOI] [PubMed] [Google Scholar]
- 3.Du X, Frei H, Kim S-H. Comparison of nitrophenylethyl and hydroxyphenacyl caging groups. Biopolymers. 2001;62:147. doi: 10.1002/bip.1007. [DOI] [PubMed] [Google Scholar]
- 4.Kötting C, Güldenhaupt J, Gerwert K. Time-resolved FTIR spectroscopy for monitoring protein dynamics exemplified by functional studies of as protein bound to a lipid bilayer. Chem Phys. 2011 doi: 10.1016/j.chemphys.2011.08.007. [DOI] [Google Scholar]
- 5.(a) Givens RS, Park CH. p-Hydroxyphenacyl ATP: a new photo-trigger. Tetrahedron Lett. 1996;37:6259. [Google Scholar]; (b) Park CH, Givens RS. New photoactivated protecting groups. 6. p-Hydroxyphenacyl: a phototrigger for chemical and biochemical probes. J Am Chem Soc. 1997;119:2453. [Google Scholar]
- 6.Givens RS, Matuszewski B. Photochemistry of phosphate esters: an efficient method for the generation of electrophiles. J Am Chem Soc. 1984;106:6860. [Google Scholar]
- 7.Anderson JC, Reese CB. A photo-induced rearrangement involving aryl participation. Tetrahedron Lett. 1962;3:1. [Google Scholar]
- 8.Givens RS, Heger D, Hellrung B, Kamdzhilov Y, Mac M, Conrad PG, Lee E, Cope JI, Mata-Segreda JF, Schowen RL, Wirz J. The photo-Favorskii reaction of p-hydroxyphenacyl compounds is initiated by water-assisted, adiabatic extrusion of a triplet biradical. J Am Chem Soc. 2008;130:3307. doi: 10.1021/ja7109579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pickens CJ, Gee KR. Photolabile thymidine cleavable with a 532 nanometer laser. Tetrahedron Lett. 2011;52:4989. [Google Scholar]
- 10.Ma C, Kwok WM, Chan WS, Du Y, Kan JTW, Toy PH, Phillips DL. Ultrafast time-resolved transient absorption and resonance Raman spectroscopy study of the photodeprotection and rearrangement reactions of p-hydroxyphenacyl caged phosphates. J Am Chem Soc. 2006;128:2558. doi: 10.1021/ja0532032. [DOI] [PubMed] [Google Scholar]
- 11.Heger D, Wirz J. unpublished results. [Google Scholar]
- 12.Remes M, Roithova J, Schroeder D, Cope ED, Perera C, Senadheera SN, Stensrud K, Ma C-C, Givens RS. Gas-phase fragmentation of deprotonated p-hydroxyphenacyl derivatives. J Org Chem. 2011;76:2180. doi: 10.1021/jo1025223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For quantum yields, see ref. 5, 8, 10 and 15, and Cope E. PhD thesis. University of Kansas; 2007.
- 14.Zhang K, Corrie JET, Munasinghe VRN, Wan P. Mechanism of photosolvolytic rearrangement of p-hydroxyphenacyl esters: evidence for excited-state intramolecular proton transfer as the primary photochemical step. J Am Chem Soc. 1999;121:5625.We have also tested our pHP derivatives for stability in base (pH > 9) in control experiments and found no rearrangement products.
- 15.(a) Givens RS, Weber JFW, Jung AH, Park C-H. New photo-protecting groups: desyl and p-hydroxyphenacyl phosphate and carboxylate esters. In: Marriott G, editor. Methods in Enzymology. Vol. 291. Academic Press; New York: 1998. p. 1. [DOI] [PubMed] [Google Scholar]; (b) Givens RS, Yousef AL. p-Hydroxyphenacyl: a photoremovable protecting group for caging bio-active substrates. :55. ref. 1c. [Google Scholar]
- 16.See: Givens RS, Weber JFW, Conrad PG, II, Orosz G, Donahue SL, Thayer SA. New phototriggers 9: p-hydroxyphenacyl as a C-terminal photoremovable protecting group for oligopeptides. J Am Chem Soc. 2000;122:2687.Conrad PG, II, Givens RS, Weber JF, Kandler K. New phototriggers:1 extending the p-hydroxyphenacyl π–π* absorption range. Org Lett. 2000;2:1545. doi: 10.1021/ol005856n.for early examples of amino acid neurotransmitter and oligopeptide applications
- 17.Givens RS, Stensrud K, Conrad PG, II, Yousef AL, Perera C, Senadheera SN, Heger D, Wirz J. p-Hydroxyphenacyl photoremovable protecting groups: robust photochemistry despite substituent diversity. Can J Chem. 2011;89:364. doi: 10.1139/V10-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conrad PG, II, Givens RS, Hellrung B, Rajesh CS, Ramseier M, Wirz J. p-Hydroxyphenacyl phototriggers: the reactive excited state of phosphate photorelease. J Am Chem Soc. 2000;122:9346. [Google Scholar]
- 19.Givens RS, Conrad PG, II, Yousef AL, Lee J-I. Photoremovable protecting groups. In: Horspool WM, editor. CRC Handbook of Organic Photochemistry and Photobiology. 2. ch 69 2003. [Google Scholar]
- 20.Givens RS, Lee J-I. The p-hydroxyphenacyl photoremovable protecting group. J Photoscience. 2003;10:37. [Google Scholar]
- 21.Kötting C, Surveyzdis Y, Bojja RS, Metzler-Nolte N, Gerwert K. Label-free screening of drug–protein interactions by trFTIR spectroscopic assays exemplified by Ras interactions. Appl Spectrosc. 2010;64:967. doi: 10.1366/000370210792434341. [DOI] [PubMed] [Google Scholar]
- 22.(a) Ma C, Kwok WM, Chan WS, Zuo P, Kan JTW, Toy PH, Phillips DL. Ultrafast time-resolved study of photophysical processes involved in the photodeprotection of p-hydroxyphenacyl caged phototrigger compounds. J Am Chem Soc. 2005;127:1463. doi: 10.1021/ja0458524. [DOI] [PubMed] [Google Scholar]; (b) Chen X, Ma C, Kwok WM, Guan X, Du Y, Phillips DL. A theoretical investigation of p-hydroxyphenacyl caged phototrigger compounds: an examination of the excited state photochemistry of p-hydroxyphenacyl acetate. J Phys Chem A. 2006;110:12406. doi: 10.1021/jp064490e. [DOI] [PubMed] [Google Scholar]; (c) Ma C, Chan WS, Kwok WM, Zuo P, Phillips DL. Time-resolved resonance Raman study of the triplet state of the p-hydroxyphenacyl acetate model phototrigger compound. J Phys Chem B. 2004;108:9264. doi: 10.1021/jo049331a. [DOI] [PubMed] [Google Scholar]; (d) Ma C, Zuo P, Kwok WM, Chan WS, Kan JTW, Toy PH, Phillips DL. Time-resolved resonance Raman study of the triplet states of p-hydroxyacetophenone and the p-hydroxyphenacyl diethyl phosphate phototrigger compound. J Org Chem. 2004;69:6641. doi: 10.1021/jo049331a. [DOI] [PubMed] [Google Scholar]
- 23.Chen X, Ma C, Kwok WM, Guan X, Du Y, Phillips DL. A theoretical investigation of p-hydroxyphenacyl caged phototrigger compounds: how water induces the photodeprotection and subsequent rearrangement reactions. J Phys Chem B. 2007;111:11832. doi: 10.1021/jp073529s. [DOI] [PubMed] [Google Scholar]
- 24.Geibel S, Barth A, Amslinger S, Jung AH, Burzik C, Clarke RJ, Givens RS, Fendler K. P3-[2-(4-hydroxyphenyl)-2-oxo]ethyl ATP for the rapid activation of the Na+, K+-ATPase. Biophys J. 2000;79:1346. doi: 10.1016/S0006-3495(00)76387-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Specht A, Loudwig S, Peng L, Goeldner M. p-Hydroxyphenacyl bromide as photoreversible thiol label: a potential phototrigger for thiol-containing biomolecules. Tetrahedron Lett. 2002;43:8947. [Google Scholar]
- 26.(a) Zou K, Miller WT, Givens RS, Bayley H. Caged thiophosphotyrosine peptides. Angew Chem, Int Ed. 2001;40:3049. doi: 10.1002/1521-3773(20010817)40:16<3049::AID-ANIE3049>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]; (b) Zou K, Cheley S, Givens RS, Bayley H. Catalytic subunit of protein kinase A caged at the activating phosphothreonine. J Am Chem Soc. 2002;124:8220. doi: 10.1021/ja020405e. [DOI] [PubMed] [Google Scholar]
- 27.Du X, Frei H, Kim S-H. The mechanism of GTP hydrolysis by Ras probed by Fourier transform infrared spectroscopy. J Biol Chem. 2000;275:8492. doi: 10.1074/jbc.275.12.8492. [DOI] [PubMed] [Google Scholar]
- 28.Kötting C, Gerwert K. Time-resolved FTIR studies provide activation free energy, activation enthalpy and activation entropy for GTPase reactions. Chem Phys. 2004;307:227. [Google Scholar]
- 29.Sot B, Kötting C, Deaconescu D, Suveyzdis Y, Gerwert K, Wittinghofer A. Unravelling the mechanism of dual-specificity GAPs. EMBO J. 2010;29:1205. doi: 10.1038/emboj.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conrad PGC, Chavli RV, Givens RS. Caged substrates applied to high content screening: an introduction with an eye to the future. In: Giuliano KA, Haskins J, Taylor DL, editors. High Content Screening: A Powerful Approach in Systems Cell Biology and Drug Discovery, Methods in Molecular Biology. Humana Press Inc; Totowa, NJ, United States: 2007. p. 253. [DOI] [PubMed] [Google Scholar]
- 31.(a) Kötting C, Kallenbach A, Suveyzdis Y, Eichholz C, Gerwert K. Surface change of Ras enabling effector binding monitored in real time at atomic resolution. ChemBioChem. 2007;8:781. doi: 10.1002/cbic.200600552. [DOI] [PubMed] [Google Scholar]; (b) Warscheid B, Brucker S, Kallenbach A, Meyer HE, Gerwert K, Kötting C. Systematic approach to group-specific isotopic labeling of proteins for vibrational spectroscopy. Vib Spectrosc. 2008;48:28. [Google Scholar]
- 32.Brucker S, Gerwert K, Kötting C. Try 39 of ran preserves the Ran. GTP gradientby inhibiting GTP hydrolysis. J Mol Biol. 2010;401:1–6. doi: 10.1016/j.jmb.2010.05.068. [DOI] [PubMed] [Google Scholar]
- 33.Arabaci G, Guo X-C, Beebe KD, Coggeshall KM, Pei D. r-Haloacetophenone derivatives as photoreversible covalent inhibitors of protein tyrosine phosphatases. J Am Chem Soc. 1999;121:5085. [Google Scholar]
- 34.Greene TW, Wutts PGM. Greene’s Protective Groups in Organic Synthesis. 4. Wiley-Interscience, J. Wiley and Sons; 2007. [Google Scholar]
- 35.Alvarez M, Alonso JM, Filevich O, Bhagawati M, Etchenique R, Piehler J, del Campo A. Modulating surface density of proteins via caged surfaces and controlled light exposure. Langmuir. 2011;27:2789. doi: 10.1021/la104511x. [DOI] [PubMed] [Google Scholar]
- 36.Ellis-Davies GCR. Caged compounds: photorelease technology for control of cellular chemistry and physiology. Nat Methods. 2007;4:619. doi: 10.1038/nmeth1072.Dorman KG, Prestwich GD. Using photolabile ligands in drug discovery and development. Trends Biotechnol. 2000;18:54. doi: 10.1016/s0167-7799(99)01402-x.Kikuchi Y, Nakanishi J, Shimizu T, Nakayama H, Inoue S, Yamaguchi K, Iwai H, Yoshida Y, Horiike Y, Takarada T, Maeda M. Arraying heterotypic single cells on photoactivatable cell-culturing substrates. Langmuir. 2008;24:13084. doi: 10.1021/la8024414.and references therein; Pirrung MC, Rana VS. Photoremovable protecting groups in DNA synthesis and microarray fabrication. :341. ref. 1c.
- 37.See, for example: (Casey JP, Blidner RA, Monroe WT. Caged siRNAs for spatiotemporal control of gene silencing. Mol Pharmacol. 2009;6:669. doi: 10.1021/mp900082q.Lee H-M, Larson DR, Lawrence DS. Illuminating the chemistry of life: design, synthesis, and applications of “caged” and related photoresponsive compounds. ACS Chem Biol. 2009;4:409. doi: 10.1021/cb900036s.Mayer G, Heckel A. Biologically active molecules with a “light switch”. Angew Chem, Int Ed. 2006;45:4900. doi: 10.1002/anie.200600387.Young DD, Deiters A. Photochemical control of biological processes. Org Biomol Chem. 2007;5:999. doi: 10.1039/b616410m.
- 38.Schmidt R, Geissler D, Hagen V, Bendig J. Mechanism of photo-cleavage of (coumarin-4-yl)methyl esters. J Phys Chem A. 2007;111:5768. doi: 10.1021/jp071521c. [DOI] [PubMed] [Google Scholar]
- 39.Schultz C. Molecular tools for cell and systems biology. HFSP J. 2007;1:230. doi: 10.2976/1.2812442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fonseca ASC, Gonçalves MST, Costa SPG. Photocleavage studies of fluorescent amino acid conjugates bearing different types of linkages. Tetrahedron. 2007;63:1353. [Google Scholar]
- 41.Cürten B, Kullmann PHM, Bier ME, Kandler K, Schmidt BF. Synthesis, photophysical, photochemical and biological properties of caged GABA, 4-[[(2H-1-benzopyran-2-one-7-amino-4-methoxy) carbonyl] amino] butanoic acid. Photochem Photobiol. 2005;81:641. doi: 10.1562/2004-07-08-RA-226. [DOI] [PubMed] [Google Scholar]
- 42.Fernandes MJG, Gonçalves MST, Costa SPG. Comparative study of polyaromatic and polyheteroaromatic fluorescent photocleavable protecting groups. Tetrahedron. 2008;64:3032. [Google Scholar]
- 43.Suzuki AZ, Watanabe T, Kawamoto M, Nishiyama K, Yamashita H, Ishii M, Iwamura M, Furuta T. Coumarin-4-ylmethoxycarbonyls as phototriggers for alcohols and phenols. Org Lett. 2003;5:4867. doi: 10.1021/ol0359362. [DOI] [PubMed] [Google Scholar]
- 44.Eckardt T, Hagen V, Schade B, Schmidt R, Schweitzer C, Bendig J. Deactivation behavior and excited-state properties of (coumarin-4-yl)methyl derivatives. 2. Photocleavage of selected (coumarin-4-yl)methyl-caged adenosine cyclic 3′,5′-monophosphates with Fluorescence enhancement. J Org Chem. 2002;67:703. doi: 10.1021/jo010692p. [DOI] [PubMed] [Google Scholar]
- 45.Takaoka K, Tatsu Y, Yumoto N, Nakajima T, Shimamoto K. Synthesis of carbamate-type caged derivatives of a novel glutamate transporter blocker. Bioorg Med Chem Lett. 2004;12:3687. doi: 10.1016/j.bmc.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 46.Senda N, Momotake A, Arai T. Synthesis and photocleavage of 7-[{bis(carboxymethyl)amino}coumarin-4-yl]methyl-caged neurotransmitters. Bull Chem Soc Jpn. 2007;80:2384. [Google Scholar]
- 47.Hagen V, Frings S, Bendig J, Lorenz D, Wiesner B, Kaupp UB. Fluorescence spectroscopic quantification of the release of cyclic nucleotides from photocleavable [bis(carboxymethoxy)coumarin-4-yl]methyl esters inside cells. Angew Chem, Int Ed. 2002;41:3625. doi: 10.1002/1521-3773(20021004)41:19<3625::AID-ANIE3625>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 48.Kotzur N, Briand B, Beyermann M, Hagen V. Wavelength-selective photoactivatable protecting groups for thiols. J Am Chem Soc. 2009;131:16927. doi: 10.1021/ja907287n. [DOI] [PubMed] [Google Scholar]
- 49.Furuta T, Watanabe T, Tanabe S, Sakyo J, Matsuba C. Phototriggers for nucleobases with improved photochemical properties. Org Lett. 2007;9:4717. doi: 10.1021/ol702106t. [DOI] [PubMed] [Google Scholar]
- 50.Lu M, Fedoryak OD, Moister BR, Dore TM. Bhc-diol as a photolabile protecting group for aldehydes and ketones. Org Lett. 2003;5:2119. doi: 10.1021/ol034536b. [DOI] [PubMed] [Google Scholar]
- 51.Hagen V, Kilic F, Schaal J, Dekowski B, Schmidt R, Kotzur N. [8-[Bis(carboxymethyl)aminomethyl]-6-bromo-7-hydroxycoumarin-4-yl] methyl moieties as photoremovable protecting groups for compounds with COOH, NH2, OH, and C=O functions. J Org Chem. 2010;75:2790. doi: 10.1021/jo100368w. [DOI] [PubMed] [Google Scholar]
- 52.Subramaniam R, Xiao Y, Li Y, Qian SY, Sun W, Mallik S. Light-mediated and H-bond facilitated liposomal release: the role of lipid head groups in release efficiency. Tetrahedron Lett. 2010;51:529. [Google Scholar]
- 53.Geissler D, Kresse W, Wiesner B, Bendig J, Kettenmann H, Hagen V. DMACM-caged adenosine nucleotides: ultrafast phototriggers for ATP, ADP and AMP activated by long-wavelength irradiation. Chem-BioChem. 2003;4:162. doi: 10.1002/cbic.200390027. [DOI] [PubMed] [Google Scholar]
- 54.Pinheiro AV, Baptista P, Lima JC. Light activation of transcription: photocaging of nucleotides for control over RNA polymerization. Nucleic Acids Res. 2008;36:e90. doi: 10.1093/nar/gkn415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shembekar VR, Chen Y, Carpenter BK, Hess GP. A protecting group for carboxylic acids that can be photolysed by visible light. Biochemistry. 2005;44:7107. doi: 10.1021/bi047665o. [DOI] [PubMed] [Google Scholar]
- 56.Shembekar VR, Chen Y, Carpenter BK, Hess GP. Coumarin-caged glycine that can be photolyzed within 3 microseconds by visible light. Biochemistry. 2007;46:5479. doi: 10.1021/bi700280e. [DOI] [PubMed] [Google Scholar]
- 57.Schönleber RO, Bendig J, Hagen V, Giese B. Rapid photolytic release of cytidine 5′-diphosphate from a coumarin derivative: a new tool for the investigation of ribonucleotide reductases. Bioorg Med Chem. 2002;10:97. doi: 10.1016/s0968-0896(01)00254-1. [DOI] [PubMed] [Google Scholar]
- 58.Skwarczynski M, Noguchi M, Hirota S, Sohma Y, Kimura T, Hayashi Y, Kiso Y. Development of first photoresponsive prodrug of paclitaxel. Bioorg Med Chem Lett. 2006;16:4492. doi: 10.1016/j.bmcl.2006.06.030. [DOI] [PubMed] [Google Scholar]
- 59.Senda N, Momotake A, Arai T. Synthesis and photocleavage of 7-[{bis(carboxymethyl)amino}coumarin-4-yl]methyl-caged neurotransmitters. Bull Chem Soc Jpn. 2007;80:2384. [Google Scholar]
- 60.Hagen V, Dekowski B, Nache V, Schmidt R, Geißler D, Lorenz D, Eichhorst J, Keller S, Kaneko H, Benndorf K, Wiesner B. Coumarinylmethyl esters for ultrafast release of high concentrations of cyclic nucleotides upon one- and two-photon photolysis. Angew Chem, Int Ed. 2005;44:7887. doi: 10.1002/anie.200502411. [DOI] [PubMed] [Google Scholar]
- 61.Taniguchi A, Skwarczynski M, Sohma Y, Okada T, Ikeda K, Prakash H, Mukai H, Hayashi Y, Kimura T, Hirota S, Matsuzaki K, Kiso Y. Controlled production of amyloid β peptide from a phototriggered, water-soluble precursor “click peptide”. ChemBioChem. 2008;9:3055. doi: 10.1002/cbic.200800503. [DOI] [PubMed] [Google Scholar]
- 62.Piloto AM, Rovira D, Costa SPG, Gonçalves MST. Oxobenzo[f]benzopyrans as new fluorescent photolabile protecting groups for the carboxylic function. Tetrahedron. 2006;62:11955. [Google Scholar]
- 63.Fernandes MJG, Gonçalves MST, Costa SPG. Neurotransmitter amino acid – oxobenzo[f]benzopyran conjugates: synthesis and photorelease studies. Tetrahedron. 2008;64:11175. [Google Scholar]
- 64.Pinheiro AV, Parola AJ, Baptista PV, Lima JC. pH effect on the photochemistry of 4-methylcoumarin phosphate esters: caged-phosphate case study. J Phys Chem A. 2010;114:12795. doi: 10.1021/jp103045u. [DOI] [PubMed] [Google Scholar]
- 65.Furuta T. Coumarin-4-ylmethyl phototriggers. :29. ref. 1c. [Google Scholar]
- 66.Kawakami T, Cheng H, Hashiro S, Nomura Y, Tsukiji S, Furuta T, Nagamune T. A caged phosphopeptide-based approach for photochemical activation of kinases in living cells. ChemBioChem. 2008;9:1583. doi: 10.1002/cbic.200800116. [DOI] [PubMed] [Google Scholar]
- 67.Mentel M, Laketa V, Subramanian D, Gillandt H, Schultz C. Photoactivatable and cell-membrane-permeable phosphatidylinositol 3,4,5-trisphosphate. Angew Chem, Int Ed. 2011;50:3811. doi: 10.1002/anie.201007796. [DOI] [PubMed] [Google Scholar]
- 68.Schade B, Hagen V, Schmidt R, Herbrich R, Krause E, Eckardt T, Bendig J. Deactivation Behavior and excited-state properties of (coumarin-4-yl)methyl derivatives. 1. Photocleavage of (7-methoxycoumarin-4-yl)methyl-caged acids with Fluorescence enhancement. J Org Chem. 1999;64:9109. doi: 10.1021/jo010692p. [DOI] [PubMed] [Google Scholar]
- 69.Furuta T, Torigai H, Sugimoto M, Iwamura M. Photochemical properties of new photolabile cAMP derivatives in a physiological saline solution. J Org Chem. 1995;60:3953. [Google Scholar]
- 70.Furuta T, Takeuchi H, Isozaki M, Takahashi Y, Kanehara M, Sugimoto M, Watanabe T, Noguchi K, Dore TM, Kurahashi T, Iwamura M, Tsien RY. Bhc-cNMPs as either water-soluble or membrane-permeant photo-releasable cyclic nucleotides for both one and two-photon excitation. ChemBioChem. 2004;5:1119. doi: 10.1002/cbic.200300814. [DOI] [PubMed] [Google Scholar]
- 71.Warther D, Gug S, Specht A, Bolze F, Nicoud J-F, Mourot A, Goeldner M. Two-photon uncaging: new prospects in neuroscience and cellular biology. Bioorg Med Chem. 2010;18:7753. doi: 10.1016/j.bmc.2010.04.084. [DOI] [PubMed] [Google Scholar]
- 72.Menge C, Heckel A. Coumarin-caged dG for improved wavelength-selective uncaging of DNA. Org Lett. 2011;13:4620. doi: 10.1021/ol201842x. [DOI] [PubMed] [Google Scholar]
- 73.Atta S, Jana A, Ananthakirshnan R, Dhuleep PSN. Fluorescent caged compounds of 2,4-dichlorophenoxyacetic acid (2,4-D): photo-release technology for controlled release of 2,4-D. J Agric Food Chem. 2010;58:11844. doi: 10.1021/jf1027763. [DOI] [PubMed] [Google Scholar]
- 74.Hagen V, Dekowski B, Kotzur N, Lechler R, Wiesner B, Briand B, Beyermann M. {7-[bis(carboxymethyl)amino]coumarin-4-yl}methoxycarbonyl derivatives for photorelease of carboxylic acids, alcohols/ phenols, thioalcohols/thiophenols, and amines. Chem–Eur J. 2008;14:1621. doi: 10.1002/chem.200701142. [DOI] [PubMed] [Google Scholar]
- 75.Shembekar VR, Carpenter BK, Ramachandran L, Hess GP. Development of photolabile protecting groups that rapidly release bio-active compounds on photolysis with visible light. Polym Prepr. 2004;45:8893. [Google Scholar]
- 76.Fan L, Lewis RW, Hess GP, Ganem B. A new synthesis of caged GABA compounds for studying GABAA receptors. Bioorg Med Chem Lett. 2009;19:3932. doi: 10.1016/j.bmcl.2009.03.065. [DOI] [PubMed] [Google Scholar]
- 77.Johnson SL, Morrison DL. Kinetics and mechanism of decarboxylation of N-arylcarbamates. Evidence for kinetically important zwitterionic carbamic acid species of short lifetime. J Am Chem Soc. 1972;94:1323. doi: 10.1021/ja00759a045. [DOI] [PubMed] [Google Scholar]
- 78.Papageorgiou G, Barth A, Corrie JET. Flash photolytic release of alcohols from photolabile carbamates or carbonates is rate-limited by decarboxylation of the photoproduct. Photochem Photobiol Sci. 2005;4:216. doi: 10.1039/b417153e. [DOI] [PubMed] [Google Scholar]
- 79.Rossi FM, Kao JPY. Nmoc-DBHQ: A new caged molecule for modulating sarcoplasmic/endoplasmic reticulum Ca2+ ATPase activity with light flashes. J Biol Chem. 1997;272:3266. doi: 10.1074/jbc.272.6.3266. [DOI] [PubMed] [Google Scholar]
- 80.Pocker Y, Davison BL, Deits TL. Decarboxylation of monosubstituted derivatives of carbonic acid. Comparative studies of water- and acid-catalyzed decarboxylation of sodium alkyl carbonates in water and water-d2. J Am Chem Soc. 1978;100:3564. [Google Scholar]
- 81.Rossi FM, Margulis M, Tang C-M, Kao JPY. N-Nmoc-glutamate: A new caged glutamate with high chemical stability and low pre-photolysis activity. J Biol Chem. 1997;272:32933. doi: 10.1074/jbc.272.52.32933. [DOI] [PubMed] [Google Scholar]
- 82.Loudwig S, Bayley H. Light-activated proteins: an overview. :253–304. ref. 1c. [Google Scholar]
- 83.Katayama K, Tsukiji S, Furuta T, Nagamune T. A bromocoumarin-based linker for synthesis of photocleavable peptidoconjugates with high photosensitivity. Chem Commun. 2008:5399. doi: 10.1039/b812058g. [DOI] [PubMed] [Google Scholar]
- 84.Yamaguchi S, Chen Y, Nakajima S, Furuta T, Nagamune T. Light-activated gene expression from site-specific caged DNA with a biotinylated photolabile protection group. Chem Commun. 2010;46:2244. doi: 10.1039/b922502a. [DOI] [PubMed] [Google Scholar]
- 85.Skwarczynski M, Noguchi M, Hirota S, Sohma Y, Kimura T, Hayashi Y, Kiso Y. Development of first photoresponsive prodrug of paclitaxel. Bioorg Med Chem Lett. 2006;16:4492. doi: 10.1016/j.bmcl.2006.06.030. [DOI] [PubMed] [Google Scholar]
- 86.Noguchi M, Skwarczynski M, Prakash H, Hirota S, Kimura T, Hayashi Y, Kiso Y. Development of novel water-soluble photocleavable protective group and its application for design of photoresponsive paclitaxel prodrugs. Bioorg Med Chem. 2008;16:5389. doi: 10.1016/j.bmc.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 87.San Miguel V, Bochet CG, del Campo A. Wavelength-selective caged surfaces: how many functional levels are possible? J Am Chem Soc. 2011;133:5380. doi: 10.1021/ja110572j. [DOI] [PubMed] [Google Scholar]
- 88.Stegmaier P, Alonso JM, del Campo A. Photoresponsive surfaces with two independent wavelength-selective functional levels. Langmuir. 2008;24:11872. doi: 10.1021/la802052u. [DOI] [PubMed] [Google Scholar]
- 89.Wylie RG, Shoichet MS. Two-photon micropatterning of amines within an agarose hydrogel. J Mater Chem. 2008;18:2716. [Google Scholar]
- 90.Woznick JH, Shoichet MS. Three-dimensional chemical patterning of transparent hydrogels. Chem Mater. 2008;20:55. [Google Scholar]
- 91.Lin W, Long L, Tan W, Chen B, Yuan L. Coumarin-caged rosamine probes based on a unique intramolecular carbon–carbon spirocyclization. Chem–Eur J. 2010;16:3914. doi: 10.1002/chem.201000015. [DOI] [PubMed] [Google Scholar]
- 92.Ottl J, Gabriel D, Marriott G. Preparation and photoactivation of caged fluorophores and caged proteins using a new class of heterobifunctional, photocleavable cross-linking reagents. Bioconjugate Chem. 1998;9:143. doi: 10.1021/bc970147o. [DOI] [PubMed] [Google Scholar]
- 93.Gilbert D, Funk K, Dekowski B, Lechler R, Keller S, Möhrlen F, Frings S, Hagen V. Caged capsaicins: new tools for the examination of TRPV1 channels in somatosensory neurons. ChemBioChem. 2007;8:89. doi: 10.1002/cbic.200600437. [DOI] [PubMed] [Google Scholar]
- 94.Kilic F, Kashikar ND, Schmidt R, Alvarez L, Dai L, Weyand I, Wiesner B, Goodwin N, Hagen V, Kaupp UB. Caged progesterone: a new tool for studying rapid nongenomic actions of progesterone. J Am Chem Soc. 2009;131:4027. doi: 10.1021/ja808334f. [DOI] [PubMed] [Google Scholar]