

Abstract
Proper folding of secreted and transmembrane proteins made in the rough endoplasmic reticulum (ER) requires oxygen for disulfide bond formation. Accordingly, ischemia can impair ER protein folding and initiate the ER stress response, which we previously showed is activated in the ischemic heart and in culture cardiac myocytes subjected to simulated ischemia. ER stress and ischemia activate the transcription factor, activating transcription factor 6 (ATF6), which induces numerous genes, many of which have not been identified, or examined in the heart. Using an ATF6 transgenic mouse model, we previously showed that ATF6 protected the heart from ischemic damage; however, the mechanism of this protection remains to be determined. In this study, we showed that, in the mouse heart, and in cultured cardiac myocytes, ATF6 induced the protein disulfide isomerase associated 6 (PDIA6) gene, which encodes an ER enzyme that catalyzes protein disulfide bond formation. Moreover, in cultured cardiac myocytes, ER stress-mediated PDIA6 promoter activation was ATF6-dependent, and required an ER stress response element (ERSE) and a nearby CCAAT box element. Electromobility shift assays and chromatin immunoprecipitation showed that ATF6 bound to the ERSE in the PDIA6 promoter, in vitro, and in the mouse heart, in vivo. Gain- and loss-of-function studies showed that PDIA6 protected cardiac myocytes against simulated ischemia/reperfusion-induced death in a manner that was dependent on the catalytic activity of PDIA6. Thus, by facilitating disulfide bond formation, and enhanced ER protein folding, PDIA6 may contribute to the protective effects of ATF6 in the ischemic mouse heart.
1. Introduction
Perturbation of protein synthesis in the endoplasmic reticulum (ER) by impaired glycosylation, decreased oxygen-mediated disulfide bond formation, or depletion of ER calcium results in the accumulation of terminally misfolded proteins [1-3]. Misfolded proteins trigger the ER stress response, also known as the unfolded protein response [3]. During the ER stress response, several transcription factors contribute to the induction of a gene program that encodes proteins designed to restore ER protein folding; however, if this initial attempt to restore protein folding is insufficient, continued ER stress can lead to apoptosis. One of the transcription factors activated during ER stress is activating transcription factor 6 (ATF6) [4, 5]. ATF6 binds to specific elements in certain ER stress response genes, including ER stress response elements, or ERSEs, through which the transcription of a subset of genes is induced [6-8], which increases expression of proteins, some of which are targeted to the ER where they enhance the folding of nascent proteins in this organelle. The genes induced by ATF6 during the initial, or acute phase of the ER stress response are thought to be protective, since they encode proteins that are mostly oriented toward restoration of ER protein folding [8, 9]. However, if this initial phase does not effectively restore ER protein folding, continued ER stress activates apoptosis [10].
To examine the function of the ATF6 branch of the ER stress response in the heart, we previously developed a line of transgenic (TG) mice that expresses a conditionally activated form of ATF6 in the myocardium. We have shown that in these TG mice, when ATF6 is activated, the hearts are resistant to ischemia/reperfusion injury [9]. A microarray analysis of ATF6 TG mouse hearts showed that 381 genes were induced by ATF6; however, many of those genes were not previously known to be ATF6-regulated, and/or have not been previously studied in the heart [8]. Many of the ATF6-inducible genes in the heart have consensus, or near-consensus ERSEs in their regulatory regions, suggesting mechanisms by which ATF6 might induce them [11]. One of the genes induced in the array was protein disulfide isomerase associated 6, or PDIA6, which has not been studied in the heart. PDIA6 was of interest, not only because of it was ATF6-inducible, but also because it encodes a protein with a predicted ER retention sequence, KDEL, and its predicted molecular mass matched that of a previously uncharacterized ER stress-inducible protein observed in previous studies [12].
PDIA6 belongs to the protein disulfide isomerase (PDI) family of proteins; some members of this family are encoded by ER stress response genes. The PDIs are a specialized family of ER-resident chaperones that facilitate the formation and isomerization of disulfide bonds in the ER lumen [13]. Disulfide bond formation is one of the rate-limiting steps in ER protein folding, and it is dependent on molecular oxygen [14]. Accordingly, hypoxia-mediated activation of the ER stress response during myocardial ischemia may up-regulate genes, such as PDIA6, that increase ER-protein folding and disulfide bond formation [15, 16]. This increased ER-protein folding capacity may aid in the survival of cardiac myocytes and limit ischemic damage [17-19]. Consistent with this hypothesis are previous studies showing that the ER stress response is activated in the cultured cardiac myocytes by simulated ischemia (sI), and in the border zone of infarcted mouse hearts, in vivo [12, 15]. Moreover, PDIs have been linked to cardioprotection [18, 20]. In our efforts to determine the mechanism by which the ATF6 branch of the ER stress response protects the heart from ischemic damage, in the present study, we identified PDIA6 as an ATF6-inducible, cardioprotective protein. While much is known about other members of the PDI family of proteins, PDIA6 is one of the least studied; moreover, to the best of our knowledge, it has not been studied in the heart. Accordingly, the present study was undertaken to determine the mechanism by which PDIA6 is induced in cardiac myocytes, and to examine whether PDIA6 gain- and loss-of-function affect cardiac myocyte survival during ischemic stress.
2. Materials and Methods
2.1 Cultured Cardiac Myocytes
Neonatal rat ventricular myocytes (NRVMCs) were isolated as described [21] from 1-2-day-old Sprague-Dawley rat hearts digested with collagenase, and purified by passage through a Percoll gradient, as described [22].
2.2 Immunoblotting
Immunoblotting was carried out as previously described [23] using antibodies raised against KDEL (cat# SPA-827, Stressgen, Ann Arbor, MI), PDIA6 (cat# ab37756, Abcam, Cambridge, MA) or GAPDH (cat# RDI-TRK5G4-6C5, Fitzgerald Industries International, Concord, MA) at dilutions of 1:1000, 1:1000, and 1:1.5×105, respectively.
2.3 Immunopurification/Depletion
Ten μg of KDEL antibody (cat# SPA-827, Stressgen, Ann Arbor, MI) were combined with 40μl of a 50% slurry of protein A sepharose beads (Pierce Classic IP Kit, cat# 26146, Thermo Scientific, Rockford, IL) and conjugated to the beads using 10μl of disuccinimidylsuberate (DSS) (cat# S1855, Sigma-Aldrich, St Louis, MO) at 13 mg/ml in DMSO for 1hr at 22°C. The KDEL antibody conjugated beads were then used as per manufacturer’s instructions (Pierce Classic IP Kit, cat# 26146, Thermo Scientific, Rockford, IL).
2.4 ATF6-MER TG Mice
The generation of ATF6-MER (mutant mouse estrogen receptor) transgenic (TG) mice featuring cardiomyocyte-specific transgene expression was described previously [9]. Non-transgenic (NTG) and ATF6-MER transgenic (TG) mice were treated with vehicle or tamoxifen, which activates ATF6 only in the TG mouse hearts. Tamoxifen (cat# T5648, Sigma-Aldrich, St Louis, MO) was suspended at 10mg/mL in 100μL of 95% ethanol and 900μL of sunflower oil and sonicated until clarified. Animals were injected intra-peritoneal (IP) with 20 mg/kg tamoxifen, or with vehicle, once daily for 5 days. After 5 days, RNA was extracted from mouse heart ventricles, as described [9].
2.5 Tandem mass spectrometry coupled to liquid chromatography (LC-MS/MS)
Protein extracts were subjected to SDS-PAGE and then silver stained; bands of interest were then excised and then digested with trypsin (10μg/mL) at 37°C overnight. The digest was then subjected to LC-MS/MS analysis, essentially as described in Shevchenko et al 1996 [24], using a linear quadrupole ion trap ThermoFinnigan LTQ mass spectrometer (San Jose, CA) equipped with a Michrom Paradigm MS4 HPLC, a SpectraSystems AS3000 autosampler, and a nanoelectrospray source.
Mass spectrometric data were acquired by the Arizona Proteomics Consortium supported by NIEHS grant ES06694 to the SWEHSC, NIH/NCI grant CA023074 to the AZCC and by the BIO5 Institute of the University of Arizona.
2.6 Adenovirus Constructs
Recombinant adenovirus (AdV) encoding only GFP (AdV-Con), GFP and constitutively active ATF6α (GenBank™ accession number NM_001107196) (AdV-ATF6) were generated, as described [12], using the AdEasy System (cat# 240009, Aligent Technologies, La Jolla, CA) [25]. PDIA6 (GenBank™ accession number NM_001004442) was cloned from cDNA isolated from NRVMCs using primers in Table s1, cloning. PCR-based mutagenesis (QuikChange site-directed mutagenesis kit, cat# 200518, Stratagene, Santa Clara, CA) was used, as per the manufacturer’s instructions, the generate a catalytically inactive mutant of PDIA6 (PDIA6 CD) where the cysteine at positions 60, 63, 195, and 198 in PDIA6 were mutated to alanine. These forms of PDIA6 were then used to generate recombinant AdV, as described above.
2.7 MicroRNA Constructs
Recombinant adenovirus encoding either miRNA targeted to ATF6α (miATF6), PDIA6 (miPDIA6) or a negative control (miCon) were created using the Gateway System (cat# 11828-029, Life Technologies, Inc., Carlsbad, CA). The generation of the adenovirus encoding miRNA targeted to ATF6 was previously described [15]. Two hairpin sequences to either ATF6α or PDIA6 were generated using Life Technologies’s online miRNA designer and ATF6α cDNA (GenBank™ accession number NM_001107196) or PDIA6 cDNA (GenBank™ accession number NM_001004442) (Table s2). The negative control sequence (GTCTCCACGCGCATTACATTT) was provided by Life Technologies, and is not targeted toward any known gene.
2.8 Virus Generation
The recombinant adenovirus discussed in sections 2.6 and 2.7 were created using the AdEasy system, as described [26].
2.9 Simulated Ischemia/Simulated Reoxygenation
NRVMCs were maintained for 16 hours in 2% FBS-supplemented medium, then subjected to simulated ischemia (sI) or simulated ischemia followed by reperfusion (sI/R), essentially as described [26]. Briefly, for sI, the medium was replaced with glucose-free Dulbecco’s modified Eagle’s medium/F-12 containing 2% dialyzed fetal bovine serum, and cultures were placed in a gas-tight chamber outfitted with a BioSpherix PROOX model 110 controller, which was used to set the [O2] to 0.1%. For sI/R, following sI, the medium was replaced with glucose-containing Dulbecco’s modified Eagle’s medium/F-12 supplemented with 2% fetal bovine serum albumin, and cultures were placed in an incubator at ~20 21% O2.
2.10 Quantitative real-time PCR (qRT-PCR)
RNA was extracted from NRVMCs or heart tissue using Quick-RNA MiniPrep Kit, as per the manufacturer’s instructions (cat# R1055, Zymo Research, Irvine, CA). cDNA was generated using Superscript III, as per the manufacturer’s instructions (cat# 18080-300, Life Technologies, Carlsbad, CA). qRT-PCR was performed as described in Martindale et al 2006 [9] using the Biopioneer 2x qPCR Master mix (cat# QPCR-10, Biopioneer, San Diego, CA) and the primers in Table s1, qRT-PCR.
2.11 Luciferase Reporter Constructs
The rat 5’-flanking sequence of the PDIA6 gene (GenBank™ accession number NM_001004442) from nt -296 to +18 were cloned into the pGL2 luciferase reporter vector (cat# E1631, Promega, Madison, WI). Using PCR-based mutagenesis, as per manufacturer’s instructions (QuikChange site-directed mutagenesis kit, cat# 200518, Stratagene, Santa Clara, CA), the CCAAT box at -142, and/or the endoplasmic reticulum stress response element (ERSE) at -109 were changed from ATTGG and CCAAT-N9-CCACG to TCCAG and GATCT-N9-AACAT, respectively.
2.12 Luciferase Reporter Assay
NRVMCs were transfected, as described [26], with 15μg of a plasmid encoding the luciferase reporter constructs generated above. The cultures were then plated and after various treatments, the cell extracts were assayed for luciferase activity, as described in Craig et al 2000 [26].
2.13 Electro-Mobility Shift Assay
Double-stranded synthetic oligonucleotides (Table s3) were labeled with 32P-dCTP (cat# BLU513Z250UC, Perkin-Elmer, Waltham, MA) using Klenow fragment, as per manufacturer’s instructions (Klenow Fragment, cat# EP0051, Thermo-Fisher, Glen Burnie, MD). The double-stranded synthetic oligonucleotides were then used as 32P-labeled probes, as described in the figure legends. Nuclear extracts were isolated from NRVMCs, as described in Dignam et al 1983 [27]. Nuclear extracts served as the source of nuclear proteins (e.g. NF-Y, YY1, and TFII-I) that are needed to observe ATF6 binding to ER stress response elements [28]. Binding assays were carried out as described in Doroudgar et al 2009 [15].
2.14 In Vivo Quantitative ChIP
Hearts from NTG and TG mice treated with tamoxifen were flash-frozen, and 25μg of tissue were processed, as per manufacturer’s instructions (ChampionChIP One-Day kit, cat# 334471, SA Biosciences, Frederick, MD). Approximately 200μg of samples were used for pre-clearing, then 30μl of the subsequent supernatant (pre-cleared fraction) were used in the immunoprecipitation, using either 10μg of FLAG antibody (cat# F1804, Sigma-Aldrich, St Louis, MO), or non-immune IgG beads, which served as the non-immune control, as per manufacturer’s instructions (ChampionChIP One-Day kit, cat# 334471, SA Biosciences, Fredrick, MD).
Primer sets (Table s1, ChIP) that flank the ER stress response elements in the promoter regions of genes-of-interest were designed, or in the case of GAPDH, which does not contain ER stress response elements, primers were designed to overlap a randomly selected portion of the promoter.
2.15 Live/Dead Assay
Assessment of cell death in NRVMCs was performed using Hoescht (cat# H21486; Life Technologies, Inc., Carlsbad, CA) and propidium iodide (cat# P1304MP, Life Technologies, Inc., Carlsbad, CA), as described [11].
2.16 Animals
Approximately 18 adult male C57/BL6 mice (6 NTG and 12 ATF6-MER TG mice), and 100 1-4 day-old Harlan Sprague-Dawley rats were used in this study. All procedures involving animals were carried out in accordance with the San Diego State University Institutional Animal Care and Use Committee.
2.17 Statistical Analyses
Data are reported as mean ± SEM and analyzed via Student’s-t test, or 1-way ANOVA with Newman–Keuls post-hoc analysis, when appropriate, using Graphpad Prism version 4. Unless otherwise stated in the figure legends, *, δ, ψ = p ≤ 0.05, **, δδ = p ≤ 0.01 and ***, δδδ= p ≤ 0.001 different from all other values.
3. Results
3.1 Identification of PDIA6 in cardiac myocytes
We previously showed that, in cardiac myocytes, ER stresses, such as simulated ischemia (sI), or simulated ischemia/reperfusion (sI/R), increased the expression of numerous ER proteins, including some that assist in ER protein folding, such as chaperones and protein disulfide isomerases [8, 9, 11, 12]. Many of these proteins, such as glucose-regulated protein-78 (GRP78) and glucose-regulated protein-94 (GRP94) [29], have a C-terminal KDEL motif through which they bind to the KDEL-receptor, which facilitates their retrograde transport from the Golgi to the ER, and thus their retention in the ER [30, 31]. An antibody raised against KDEL (anti-KDEL) is often used to assess expression of such proteins in response to ER stress [32]. When anti-KDEL was used in immunoblots of extracts of cultured cardiac myocytes that had been treated with tunicamycin (TM), or subjected to sI, or sI/R, the expression of two major KDEL-cross-reactive proteins, known to be GRP78 and GRP94, was increased, as expected (Fig. 1A, GRP94 and GRP78). A previously unidentified anti-KDEL cross-reactive protein of about 50 kD was also induced (Fig. 1A, arrow), suggesting that it has a function in ER-stressed cardiac myocytes.
Fig 1. Identification of PDIA6 as a 50kD anti-KDEL cross-reactive protein.
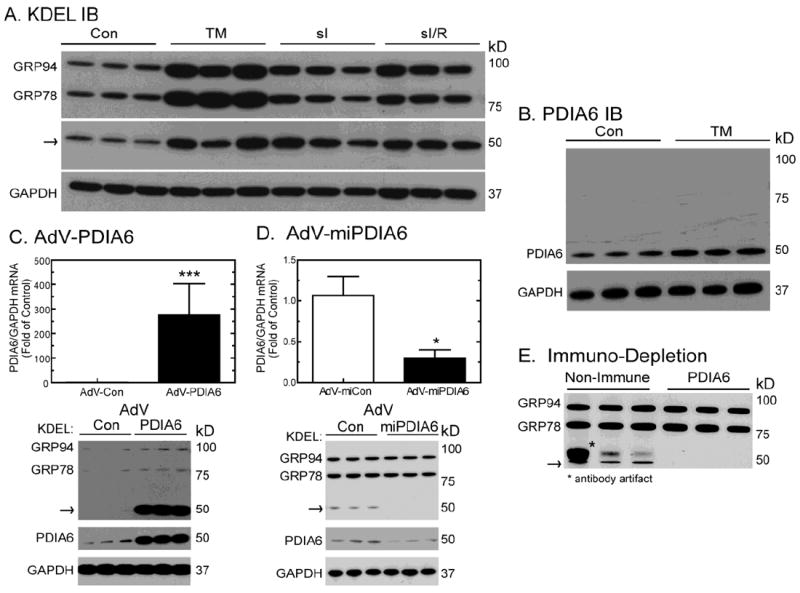
Panels A and B- NRVMCs were treated with or without 10μg/ml of TM for 24hrs, or subjected to 20hrs of simulated ischemia (sI), or 20hrs of simulated ischemia followed by 24hrs of simulated reperfusion (sI/R). Culture extracts were then analyzed by immunoblotting using anti-KDEL, anti-PDIA6, and anti-GAPDH antibodies. The arrow shows the location of the 50 kD anti-KDEL cross-reactive protein. n = 3 cultures per treatment
Panel C- NRVMCs were infected with either a control adenovirus (AdV-Con), or an adenovirus encoding PDIA6 (AdV-PDIA6). Forty-eight hours later, culture extracts were analyzed by qRT-PCR to determine the levels of PDIA6 and GAPDH mRNAs (top). Values are mean PDIA6/GAPDH mRNA ± SE (n=3). Cultures were also analyzed by immunoblotting with anti-KDEL or anti-PDIA6 antibodies (bottom), n=3 cultures per treatment.
Panel D- NRVCMs were infected with an adenovirus encoding a control microRNA (AdV-miCon), or a microRNA designed to reduce the expression of PDIA6 (AdV-miPDIA6). Forty-eight hours later, culture extracts were analyzed by qRT-PCR to determine the levels of PDIA6 and GAPDH mRNAs (top). Values are mean PDIA6/GAPDH mRNA ± SE (n=3). Cultures were also analyzed by immunoblotting with anti-KDEL or anti-PDIA6 antibodies (bottom), n=3 cultures per treatment.
Panel E- NRVMCs were treated with or without 10μg/ml TM for 24hrs, and lysates were incubated with either a non-immune antibody, or with anti-PDIA6, followed by immune complex removal, then analysis of the supernatants by immunoblotting with anti-KDEL, n=3 cultures per treatment. The arrow shows the location of the 50 kD anti-KDEL cross-reactive protein.
In order to determine the function of the 50 kD anti-KDEL cross-reactive protein in cardiac myocytes, it was necessary to determine its identity. Accordingly, proteins in cardiac myocyte extracts with C-terminal KDEL motifs were isolated by anti-KDEL affinity purification; the 50 kD anti-KDEL cross-reactive material was analyzed by LC-MS/MS and shown to contain protein disulfide isomerase-associated 6 (PDIA6), a member of the protein disulfide isomerase (PDI) family of thiol oxidoreductases. In order to verify this initial identification, immunoblots were carried out using a PDIA6-specific antibody (anti-PDIA6), and the results were compared to those obtained with anti-KDEL. In comparison to the 50 kD anti-KDEL cross-reactive protein, there was an increase in the 50 kD anti-PDIA6 cross-reactive band in response to TM (Fig. 1B). Moreover, when cultured cardiac myocytes were infected with an adenovirus (AdV) encoding native PDIA6 (AdV-PDIA6), there was a 275-fold increase in PDIA6 mRNA (Fig. 1C, top), as expected, as well as coordinate increases in the levels of 50 kD anti-KDEL, and anti-PDIA6 cross-reactive material (Fig. 1C, bottom arrow and PDIA6). Conversely, when cardiac myocytes were infected with an AdV encoding a microRNA targeted to endogenous PDIA6 mRNA (AdV-miPDIA6), there was a 70% decrease in PDIA6 mRNA (Fig. 1C, top), as well as coordinate decreases in the levels of the 50 kD anti-KDEL and anti-PDIA6 cross-reactive material (Fig. 1D, bottom arrow and PDIA6). Furthermore, when anti-PDIA6 was used to deplete cardiacmyocyte lysates, the 50 kD anti-KDEL cross-reactive material decreased (Fig. 1E, arrow). Taken together the results Figure 1 and the LC-MS/MS protein sequencing demonstrates that the 50 kD anti-KDEL cross-reactive protein was PDIA6.
3.2 Effects of ER stress on PDIA6 expression
To characterize the mechanism of PDIA6 induction in cardiac myocytes, the effects of the ER stress-activated transcription factor, ATF6, were examined. When cardiac myocytes were infected with a recombinant AdV encoding activated ATF6 (AdV-ATF6) [33], ATF6 mRNA was increased about 50-fold (Fig. 2A). In cultures infected with AdV-ATF6, PDIA6 mRNA and protein increased by 8- and 13-fold, respectively (Fig. 2B and 2C). To explore the effect of endogenous ATF6 on PDIA6 induction, a recombinant AdV encoding a microRNA (miRNA) targeted to rat ATF6 (AdV-miATF6) was used [15]. Cultures infected with AdV-miATF6 exhibited a 70% reduction in the quantity of ATF6 mRNA (Fig. 3A), thus validating the utility of this reagent for knocking down endogenous ATF6 in cultured cardiac myocytes. TM-mediated induction of PDIA6 mRNA and protein was reduced by about 50% and 80%, respectively (Fig. 3B, bar 4; Fig. 3C, bar 4). Taken together, the results shown in Figures 2 and 3 demonstrate that in cultured cardiacmyocytes, ER stress-mediated PDIA6 induction was partly dependent upon transcriptional induction by ATF6. Since ER stressors, such as TM, activate ATF6, which increases transcription of certain ER stress response genes, the effects of ER stress and ATF6 on PDIA6 promoter activation were examined.
Fig 2. Effect of AdV-ATF6 on PDIA6 expression.
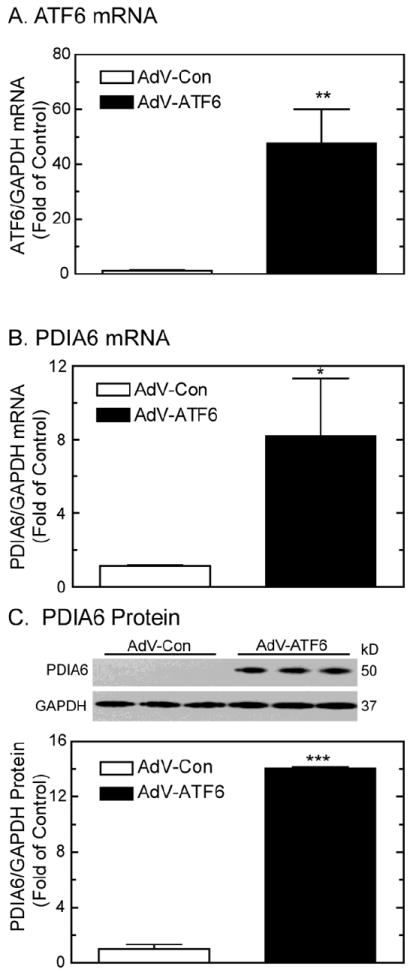
Panel A- NRVMCs were infected with AdV-Con or AdV-ATF6. Twenty-four hours later, cultures were analyzed for ATF6 and GAPDH mRNAs. Values are mean ATF6/GAPDH mRNA ± SE, n=3 cultures per treatment.
Panel B- NRVMCs were infected with AdV-Con or AdV-ATF6. Forty-eight hours later, culture extracts were analyzed for PDIA6 and GAPDH mRNA. Values are mean PDIA6/GAPDH mRNA ± SE, n=3 cultures per treatment.
Panel C- NRVMCs were treated as described for Panel A, except extracts were analyzed for PDIA6 and GAPDH protein by immunoblotting. Relative blot intensities of PDIA6/GAPDH expressed as fold of control (bottom). Values are mean relative PDIA6/GAPDH ± SE, n=3 cultures per treatment.
Fig 3. Effect of knocking down endogenous ATF6 on PDIA6 expression.
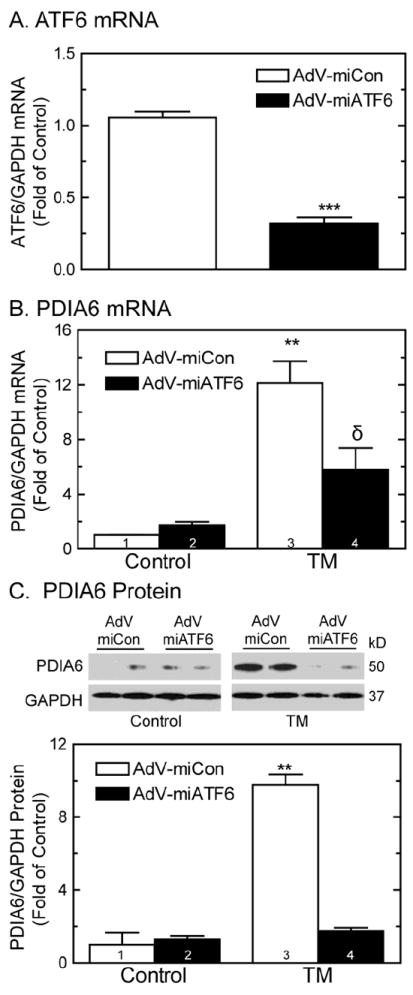
Panel A- NRVMCs were infected with adenovirus encoding AdV-miCon, or AdV-miATF6. Twenty-four hours later, cultures were analyzed for ATF6 and GAPDH mRNAs. Values are mean ATF6/GAPDH mRNA ± SE, n=3 cultures per treatment.
Panel B- NRVMCs were infected with AdV-miCon, or AdV-miATF6. Twenty-four hours later, cultures were treated with or without 10μg/ml of TM for 24hrs, then analyzed for PDIA6 and GAPDH mRNA. Values are mean PDIA6/GAPDH mRNA ± SE, n=3 cultures per treatment.
Panel C- NRVMCs were infected with AdV-miCon, or AdV-miATF6. Twenty-four hours later, cultures were treated with or without 10μg/ml of TM for 24hrs, and then analyzed for PDIA6 and GAPDH by immunoblotting. Image quantification of the blots shown at the top of Panel C are plotted at the bottom. Values are PDIA6/GAPDH ± SE, n=3 cultures per treatment.
3.3 Regulation of PDIA6 promoter activation and ER stress response element identification
A construct composed of the nucleotides -296 to +18 of the PDIA6 promoter driving firefly luciferase (Fig. 4A) was transfected into cultured cardiac myocytes, which were then treated with TM, sI, or AdV-ATF6. These treatments increased PDIA6 promoter activity by ~2- to 6-fold (Fig. 4B, bars 1-3; Fig. 4C, bars 1, 2). A search for ER stress response elements (ERSEs) revealed an region spanning nts -109 to -90 of the PDIA5 5’-flanking sequence that comprised a CCAAT box element, followed by 9 nts then a CCACG sequence, which fulfills the requirements of an ERSE [4, 28]. Transcriptional induction via ERSEs by ATF6 requires that the nuclear protein, NF-Y, binds to the CCAAT box portion of the ERSE, and that ATF6 binds to NF-Y and to the CCACG portion of the ERSE [28]. Thus, while NF-Y can bind directly to ERSEs via CCAAT sequences, ATF6 binding to ERSEs requires that ATF6 also binds to NF-Y. The search also revealed a CCAAT box about 30 nt 5’ of the ERSE, between nts -142 to -137, which was not part of a consensus ERSE. NF-Y binding to such isolated CCAAT boxes can increase transcription of some ER stress response genes [34, 35]. Since isolated CCAAT boxes and ERSEs can mediated transcriptional induction upon ER stress, the effects of mutating these two elements on PDIA6 promoter activity were examined. Mutating the isolated CCAAT box alone (Fig. 4A, M1) slightly increased promoter activation in response to TM or sI (Fig. 4B, bars 5, 6), while it slightly decreased promoter activation in response to ATF6 (Fig. 4C, bar 4). However, mutations to the ERSE that are predicted to disrupt NF-Y and ATF6 binding (Fig. 4A, M2) decreased promoter activation by 3- to 4-fold in response to all of the treatments (Fig. 4B, bars 8, 9, Fig. 4B, bars 5, 6). When the isolated CCAAT box and ERSE were both mutated (Fig. 4A, M1+M2), promoter activation was completely lost (Fig. 4B, bars 10-12; Fig. 4C, bars 7, 8). Thus, while the ERSE seemed to be the dominant element, the isolated CCAAT box exerted inhibitory effects in the native promoter, but in the absence of the ERSE, it appeared to be capable of mediating some promoter activation, albeit relatively small.
Fig 4. Effect of TM, sI, or AdV-ATF6 on the PDIA6 Promoter.
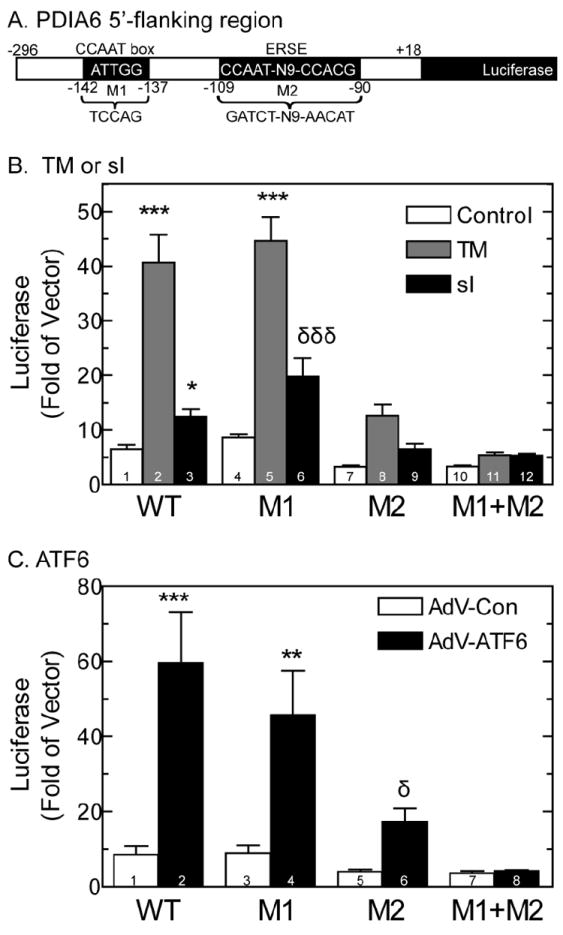
Panel A- The promoter and 5’-flanking region of the rat PDIA6 gene from -296 to +18 is shown, as are the putative CCAAT box and ERSE at -142 to-137 and 109 to -90, respectively. M1 and M2 denote mutations 1 and 2 that were made in the PDIA6 5’-flanking sequence. The sequences of the native CCAAT box and ERSE are shown, and the mutations are shown below. These mutations were prepared in a manner predicted to disrupt NF-Y and ATF6 binding to these elements.
Panel B- NRVMCs were transfected with either an empty vector control, or with wild type or mutated PDIA6-luciferase constructs, as shown. After 24h, cells were subjected to 20hrs of sI, or treated with 10μg/ml of TM for 24hrs, and then analyzed for reporter activity. Values are the mean luciferase activity expressed as fold of empty vector control ± SE, n=3 cultures per treatment.
Panel C- NRVMCs were transfected as described in Panel B, and then infected with either AdV-Con, or AdV-ATF6. Twenty four hours later, cell extracts were analyzed for reporter activity. Values are the mean luciferase expressed as fold empty vector control ± SE, n=3 cultures per treatment.
To examine the mechanisms by which these elements regulate the PDIA6 promoter, electromobility shift assays (EMSAs) were performed using oligonucleotide probes that mimicked the region of the promoter containing either the isolated CCAAT box, or the ERSE (Fig. 5A, CCAAT box probe, ERSE probe). Since the binding of ATF6 to ERSEs requires NF-Y, nuclear extracts of untreated cardiac myocytes were used in the EMSAs as a source of NF-Y, as previously described [28, 36-38]. Incubation of nuclear extract with either the ERSE or CCAAT box probe resulted in formation of a complex (Fig. 5B, lanes 2 and 7), which is due to NF-Y binding to the CCAAT box region of each probe. Addition of ATF6 decreased the mobility of the ERSE probe (Fig. 5B, lane 3), and the addition of ATF6 and an ATF6 antibody further decreased the mobility of ERSE probe (Fig. 5B, lane 4), which verified ATF6 binding [39]. In contrast, neither ATF6, nor ATF6 antibody affected the mobility of the CCAAT box probe (Fig. 5B, lanes 8 and 9), demonstrating that ATF6 did not bind to the isolated CCAAT box. The reduction of the intensity of the complex observed when ATF6 was added to the CCAAT box probe (Fig. 5B, lanes 8 and 9) is most likely due to the titration of NF-Y off the probe by ATF6, which can bind to NF-Y in the absence of DNA [28].
Fig 5. Electromobility shift assays.
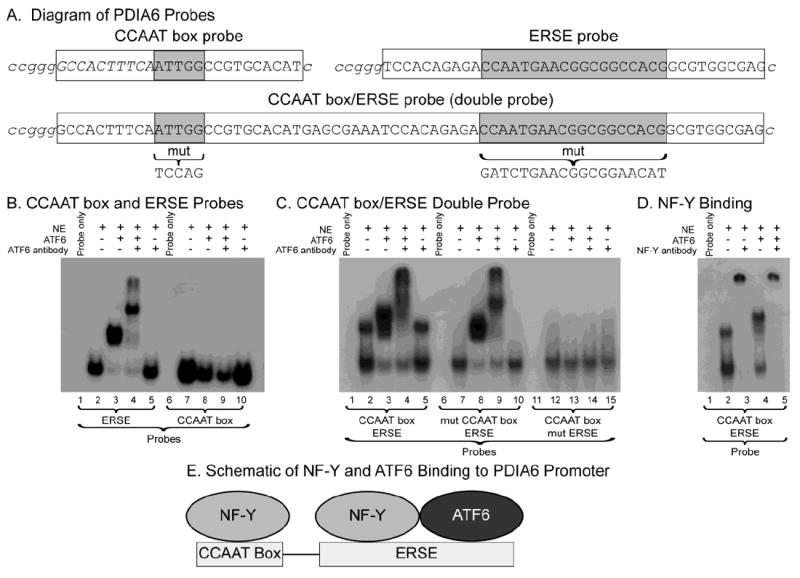
Panel A- Shown are diagrams of the probes containing the isolated PDIA6 ERSE, PDIA6 CCAAT box and a “double” probe containing both elements. 32P-labled overhangs are shown in italics and the relevant elements are highlighted in gray.
Panel B- ERSE and CCAAT box Probes: EMSA reactions were carried out as described in the methods using radiolabled ERSE or CCAAT box probes, as shown. Reactions analyzed in lanes 1 and 6 contained only the probes, while those in lanes 2-5 and 7-10 contained nuclear extract from NRVMCs, recombinant ATF6(116-373) obtained from in vitro transcription/translation reactions, and/or anti-ATF6, as shown.
Panel C- CCAAT box/ERSE Double Probe: Binding reactions were carried out using either wild type probe (lanes 1-5), probe with the isolated CCAAT box mutated (lanes 6-10), or probe with the ERSE mutated (lanes 11-15). All other additions were made as described in the methods and in the legend to Panel A.
Panel D- Effect of NF-Y Antibody: Binding reactions were carried out using the CCAAT box/ERSE double probe and the other additions, as shown.
Panel E- Diagram of NF-Y and ATF6 binding to the CCAAT box and ERSE in the PDIA6 promoter.
Since the promoter/luciferase experiments suggested that the isolated CCAAT box and the ERSE might both contribute to regulating the PDIA6 promoter, EMSAs were carried out using a probe that had both the isolated CCAAT box and the ERSE in their native relative positioning (Fig. 5A, CCAAT box/ERSE double probe). Incubation of nuclear extract with the double probe resulted in two complexes (Fig. 5C, lane 2). Mutations to either element in the double probe disrupted the slower-migrating complex, but not the faster-migrating complex (Fig. 5C, lanes 7 and 12), indicating that the slower complex was due to NF-Y binding to both sites, while the faster complex was due to NF-Y binding to either one of the sites. Addition of an NF-Y antibody disrupted both complexes (Fig. 5D, lanes 2 and 3) further demonstrating that it was responsible for the formation of both complexes. Addition of ATF6 to the double probe resulted in the formation of a new, slower migrating complex, (Fig. 5C, lane 3) that was sensitive to addition of ATF6 antibody (Fig. 5C, lane 4), indicating that the slowly migrating complex in Figure 5C, lane 3 was the result of ATF6 binding. The complexes formed using the double probe in which the CCAAT box was mutated (Fig. 5C, lanes 6-10) resembled those formed using the ERSE probe (Fig. 5B, lanes 1-5), indicating that NF-Y and ATF6 bound to the ERSE in the double probe. Moreover, the complex formed using the double probe in which the ERSE was mutated (Fig. 5C, lanes 11-15) resembled that formed using the CCAAT box probe (Fig. 5B, lanes 6-10), indicating that NF-Y, but not ATF6, bound to the isolated CCAAT box in the double probe.
Taken together, these results showed that NF-Y bound to the isolated and ERSE-associated CCAAT box elements in the double probe, while ATF6 bound only to the ERSE (Fig. 5E). Moreover, with the results shown in Figure 4, the results in Figure 5 suggest that, in the native promoter, binding of NF-Y to the CCAAT box element may reduce promoter activity, perhaps because ATF6 may be titrated off the ERSE by the NF-Y on the isolated CCAAT box element, thus reducing the amount of ATF6 available to bind to the ERSE. However, since some promoter activity remained after mutation of the ERSE, and since this remaining activity was extinguished by mutations to both the ERSE and the CCAAT box, it is apparent that the CCAAT box alone confers some PDIA6 promoter activation in response to ER stress.
3.4 ATF6 regulates PDIA6 expression, in vivo
Since ATF6 was required for maximal PDIA6 promoter activation, and since it bound to oligonucleotides mimicking the ERSE in the promoter, we determined whether ATF6 bound to the PDIA6 promoter, in vivo, using ATF6 transgenic (TG) mouse hearts. In a model in which a FLAG tagged tamoxifen-inducible form of ATF6 was expressed (ATF6 TG), ER stress response genes are activated in response to tamoxifen treatment [9]. To determine whether PDIA6 was induced by ATF6 in this mouse model, non-transgenic (NTG) and ATF6 TG mice were treated with, or without tamoxifen, then RNA was isolated from their hearts and was examined for PDIA6 expression. PDIA6 mRNA levels increased by about 5-fold in tamoxifen-treated ATF6 TG mice, but were unaffected in the other groups (Fig. 6A), thus verifying that ATF6 induced PDIA6 in mouse hearts, in vivo. Chromatin immunoprecipitation (ChIP) showed that the PDIA6 promoter was enriched in tamoxifen-treated ATF6 TG mouse hearts, but not in NTG mouse hearts when DNA bound to ATF6 was isolated (Fig. 6B; PDIA6). ChIP analyses of the same samples for the ER stress response gene, heme-oxygenase 1 (HO-1) served as a negative control (Fig. 6B; HO-1), since its promoter lacks ERSEs, or any other known ATF6 binding sites [8, 40]. These results demonstrate that activated ATF6 bound directly to the PDIA6 promoter in the mouse heart and regulated its activity, in vivo.
Fig 6. PDIA6 expression in ATF6 TG mouse hearts and ATF6 binding to the PDIA6 promoter in mouse hearts, in vivo.
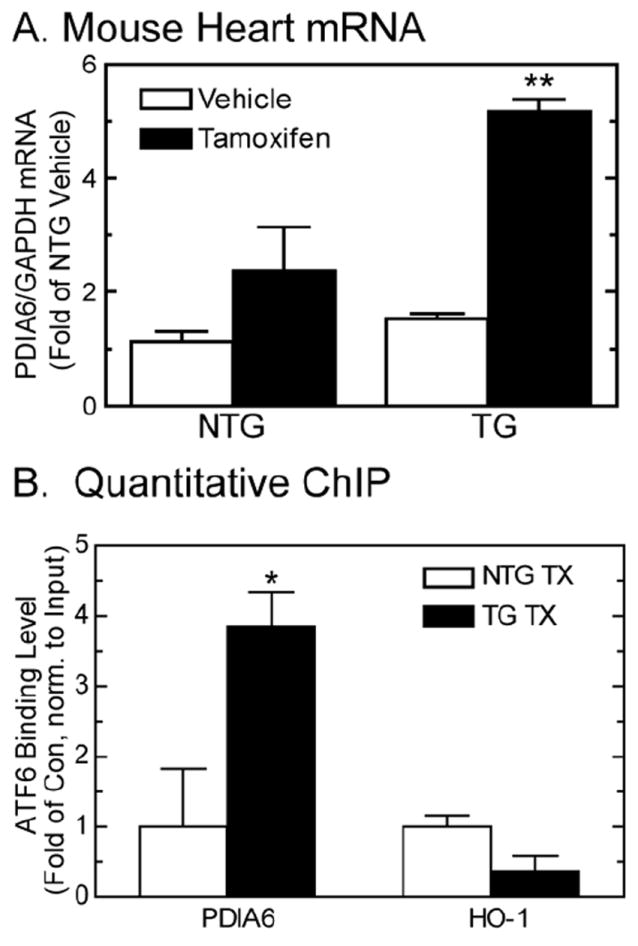
Panel A- NTG and ATF6 TG mice were treated with or without tamoxifen for 5 days, after which hearts were extracted and analyzed for PDIA6 and GAPDH mRNA by qRT-PCR. Values are mean PDIA6/GAPDH mRNA ± SE, n=3 mice per treatment.
Panel B- Quantitative ChIP analysis was carried out to determine the ability of ATF6 to bind to the PDIA6 and hemoxygenase-1 (HO-1) promoters in the mouse heart, in vivo. Chromatin was isolated from NTG and ATF6 TG mouse hearts, and then subjected to FLAG IP. Isolated chromatin was then examined by quantitative PCR using primers targeted to the mouse PDIA6 and mouse HO-1 promoters. Shown is the quantitation of the PCR products obtained, n=3 mice per treatment.
3.5 PDIA6 is cardioprotective
To examine the function of PDIA6, AdV-PDIA6 and AdV-miPDIA6 were used in cultured cardiac myocytes to assess the effects of PDIA6 gain- and loss-of-function, respectively. Compared to AdV-Con, cultures infected with AdV-PDIA6 (Fig. 7A, top) exhibited significantly less cell death when subjected to sI/R (Fig. 7A, bars 4 and 5). Furthermore, infection with an AdV that encodes a form of PDIA6 that is catalytically inactive (AdV-PDIA6-CD) (Fig. 7A, top) did not protect the cells from sI/R-induced cell death (Fig. 7A, bar 6). Conversely, knocking down PDIA6 using AdV-miPDIA6 (Fig. 1D) resulted in increased cell death under control conditions and upon sI/R (Fig. 7B). These results demonstrate that PDIA6 can protect cardiac myocytes from sI/R induced death and this protection is dependent on the oxidoreductase activity of PDIA6.
Fig 7. Effect of PDIA6 on sI/R-mediated cell death.
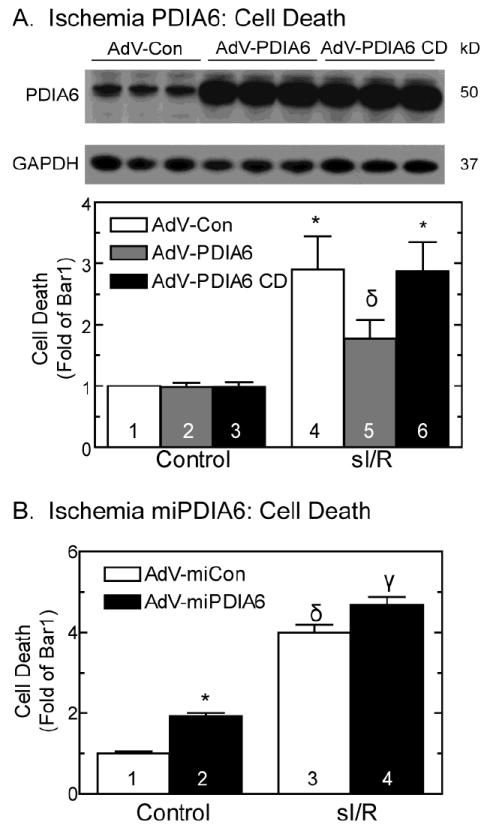
Panel A- Effect of PDIA6 or PDIA6 catalytically dead (CD) on sI/R-mediated cell death: NRVMCs were infected with a control adenovirus (AdV-Con), an adenovirus encoding PDIA6 (AdV-PDIA6), or a catalytically dead mutant of PDIA6 (AdV-PDIA6 CD) and then analyzed for PDIA6 and GAPDH by immunoblotting (top). NRVMCs infected with AdV-Con, AdV-PDIA6 or AdV-PDIA6 CD were subjected to 20hrs of sI followed by 24hrs of reperfusion (sI/R), and then examined for cell death (bottom). Shown are the results of a cell death assay, mean ± SE, n=3 cultures per treatment.
Panel B- Effect of PDIA6 knockdown on sI/R-mediated cells death: NRVMCs were infected with AdV-Con or AdV-miPDIA6, subjected to 20hrs of sI followed by 24hrs of reperfusion (sI/R), then examined for cell death. Shown are the results of a cell death assay, mean ± SE, n=3 cultures per treatment.
4. Discussion
Secretory proteins, including growth factors, hormones, and stem cell homing factors, as well as membrane proteins, such as receptors and ion channels, are made in the ER, and sarcoplasmic reticulum (SR) of cardiac myocytes [41]. In most cells, secretory and membrane proteins account for at least one-third of all proteins produced. Most secretory and membrane proteins contain disulfide bonds that are required for their functions [42]. Therefore, protein disulfide bond formation in the ER/SR is critical for a significant portion of protein synthesis and, thus, is required for proper function of all organs, including the heart. This emphasizes the essential role played by the proteins responsible for disulfide bond formation, the protein disulfide isomerases (PDIs), which have not been studied extensively in the heart.
In this study, we characterized the PDI family member, PDIA6, also called ERp5, PDI P5, TXNDC7, in cultured cardiac myocytes, and in the mouse heart. We showed that PDIA6 expression was increased by chemical ER stressors, as well as by simulated ischemia-mediated ER stress. The nodal ER stress-activated transcription factor, ATF6, which is cardioprotective [9] and activated by ischemia [15], was shown to induce PDIA6 in cultured cardiac myocytes, and in the myocardium, in vivo. Moreover, wild type PDIA6 protected cardiac myocytes from ischemia-mediated cell death, while a mutant form of PDIA6 that lacks disulfide isomerase activity was not protective. These results suggest that by serving a central role in secretory and membrane protein folding, PDIA6 contributes to the mechanisms by which ATF6 protects the heart from damage during ischemia.
Although little is known about PDIA6, and it has not been studied in the heart, the PDI family has been extensively studied [43, 44]. The PDI family comprises 20 diverse members, ranging in molecular weight from about 18 to 80 kD. The PDIs have been shown to participate in oxidative disulfide bond formation of nascent proteins in the ER, but also many other functions, such as chaperone and, perhaps, regulation of calcium levels in the ER. PDI family members vary in domain arrangement, but have at least one domain that resembles thioredoxin, as well as -Cys-X-X-Cys- motifs that participate in the catalysis of disulfide bond formation in newly synthesized proteins. Like other PDI family members, PDIA6 is ubiquitously expressed in human tissue (BioGPS, 207668_x_at), albeit at different levels, depending upon the tissue [45], and, although each member can facilitate disulfide bond formation in numerous proteins, they appear to exhibit a certain degree of client selectivity [46]. Moreover, the various PDI family members exhibit cell- and stimulus-specific induction responses. For example, hypoxia was shown to induce PDIA6 in head and neck carcinoma cells, but not in cervical carcinoma cells [43].
Although the precise function of PDIA6 in cardiac myocytes is yet to be determined, it is safe to speculate that it most likely contributes to oxidative protein disulfide bond formation in nascent secretory and cell-membrane proteins made in the ER/SR of cardiac myocytes. Since PDI-mediated disulfide bond formation requires oxygen, the induction of PDIA6 by ischemia, which we showed to be ATF6-dependent, likely represents a compensatory response to the ER stress that is generated upon the accumulation of misfolded proteins in the ER/SR of ischemic cardiac myocytes. It is of further interest to note that in the sole previous paper in which hypoxia was shown to induce PDIA6, compared to atmospheric oxygen levels of 21%, 1% oxygen resulted in minimal PDIA6 induction, while 0% oxygen maximally induced PDIA6 [47]. This further supports the hypothesis that PDIA6 is induced by hypoxia-mediated ER stress, because ER stress is not activated until oxygen levels decrease to about 0.1% [15]. Indeed, ER stress is activated during myocardial infarction and stroke [9, 15, 48] and some studies support the notion that under these conditions, ER stress promotes survival [9, 12, 19]. Moreover, we previously showed that the 50 kD anti-KDEL cross-reactive protein identified in this study as PDIA6, was strongly upregulated in cultured cardiac myocytes exposed to simulated ischemia [12].
In addition to its role in oxidative protein disulfide bond formation in the ER, PDIA6 has been shown to have other functions. For example, in agonist-treated platelets, PDIA6 translocates from the ER to the cell surface, where it participates in the final aspects of platelet activation, such as α-granule secretion and the binding of platelets to fibrinogen; these activities were shown to depend upon PDIA6 enzyme activity [49]. Interestingly, in the same study, cell-surface PDIA6 was found to associate with integrins, suggesting that PDIA6 may contribute to platelet activation by affecting the configuration of disulfide bonds of several integrins. These intriguing effects were extended further when it was shown that PDIA6 was one of several other ER proteins that relocated to the cell surface upon plasma membrane wounding [50]. Cell-surface PDIA6 has also been shown to promote tumor immune evasion through the shedding of tumor associated ligands [51]. Additionally, PDIA6 has been found on the inner side of the inner mitochondrial membrane, but currently the function of PDIA6 in mitochondria is not known [52]. Thus, in the heart, PDIA6 may play diverse roles beyond ER/SR protein disulfide bond formation, including functioning on the cell surface and in mitochondria in ways that regulate cardiac myocyte signaling from both the inside and outside of the cell.
5. Conclusions
In this study we identified PDIA6 as a previously uncharacterized 50 kD anti-KDEL cross-reactive protein that is induced by hypoxic and ischemic stress in various cells and tissues, including the heart. Moreover, this study showed that, upon simulated ischemia, PDIA6 was induced in cardiac myocytes in an ATF6-dependent manner. These findings are consistent with another recent study which showed that another member of the ER protein disulfide isomerization coupling reaction, ERO1, is also strongly induced in cultured cardiac myocytes by hypoxia [53]. Thus, the ER/SR thiol oxidoreductase enzyme system is likely to serve important roles in the biosynthesis of numerous secreted and membrane proteins that are required for proper cardiac myocyte and heart function, supporting the hypothesis that the induction of enzymes, such as PDIA6, may help protect the myocardium in response to pathological ER stresses, such as ischemia.
Supplementary Material
Highlights.
The protein disulfide isomerase, PDIA6, was identified in cultured cardiac myocytes.
PDIA6 is involved in oxidative disulfide bond formation and ER protein folding.
In cardiac myocytes, PDIA6 was upregulated by ischemia and ATF6.
PDIA6 protected cardiac myocytes from death upon ischemia reperfusion.
PDIA6-mediated protection may be due to its ability to enhance protein folding.
Acknowledgments
This work was supported by grants from the National Institutes of Health, HL-075573, HL-085577, HL104535 and RO3 EB011698. The authors wish to thank Shirin Doroudgar and Archana Tadimalla for their scientific discussions, and for their insightful and critical suggestions.
Footnotes
Disclosures
None declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001;26:504–10. doi: 10.1016/s0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- 2.Wu J, Kaufman RJ. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006;13:374–84. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 3.Glembotski CC. Endoplasmic reticulum stress in the heart. Circ Res. 2007;101:975–84. doi: 10.1161/CIRCRESAHA.107.161273. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273:33741–9. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- 5.Zhu C, Johansen FE, Prywes R. Interaction of ATF6 and serum response factor. Mol Cell Biol. 1997;17:4957–66. doi: 10.1128/mcb.17.9.4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13:365–76. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 7.Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J. 2002;366:585–94. doi: 10.1042/BJ20020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belmont PJ, Tadimalla A, Chen WJ, Martindale JJ, Thuerauf DJ, Marcinko M, et al. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (RCAN1), a novel ATF6-inducible gene. J Biol Chem. 2008;283:14012–21. doi: 10.1074/jbc.M709776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98:1186–93. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
- 10.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–30. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 11.Belmont PJ, Chen WJ, San Pedro MN, Thuerauf DJ, Gellings Lowe N, Gude N, et al. Roles for endoplasmic reticulum-associated degradation and the novel endoplasmic reticulum stress response gene Derlin-3 in the ischemic heart. Circ Res. 2009;106:307–16. doi: 10.1161/CIRCRESAHA.109.203901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99:275–82. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- 13.Ferrari DM, Soling HD. The protein disulphide-isomerase family: unravelling a string of folds. Biochem J. 1999;339(Pt 1):1–10. [PMC free article] [PubMed] [Google Scholar]
- 14.Tu BP, Weissman JS. The FAD- and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol Cell. 2002;10:983–94. doi: 10.1016/s1097-2765(02)00696-2. [DOI] [PubMed] [Google Scholar]
- 15.Doroudgar S, Thuerauf DJ, Marcinko MC, Belmont PJ, Glembotski CC. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J Biol Chem. 2009;284:29735–45. doi: 10.1074/jbc.M109.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azfer A, Niu J, Rogers LM, Adamski FM, Kolattukudy PE. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am J Physiol Heart Circ Physiol. 2006;291:H1411–20. doi: 10.1152/ajpheart.01378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang M, Ye R, Barron E, Baumeister P, Mao C, Luo S, et al. Essential role of the unfolded protein response regulator GRP78/BiP in protection from neuronal apoptosis. Cell Death Differ. 2010;17:488–98. doi: 10.1038/cdd.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toldo S, Severino A, Abbate A, Baldi A. The role of PDI as a survival factor in cardiomyocyte ischemia. Methods Enzymol. 2011;489:47–65. doi: 10.1016/B978-0-12-385116-1.00003-0. [DOI] [PubMed] [Google Scholar]
- 19.Severino A, Campioni M, Straino S, Salloum FN, Schmidt N, Herbrand U, et al. Identification of protein disulfide isomerase as a cardiomyocyte survival factor in ischemic cardiomyopathy. J Am Coll Cardiol. 2007;50:1029–37. doi: 10.1016/j.jacc.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 20.Toldo S, Boccellino M, Rinaldi B, Seropian IM, Mezzaroma E, Severino A, et al. Altered Oxido-Reductive State in the Diabetic Heart: Loss of Cardioprotection due to Protein Disulfide Isomerase. Mol Med. 2011;17:1012–21. doi: 10.2119/molmed.2011.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyamoto S, Purcell NH, Smith JM, Gao T, Whittaker R, Huang K, et al. PHLPP-1 negatively regulates Akt activity and survival in the heart. Circ Res. 2010;107:476–84. doi: 10.1161/CIRCRESAHA.109.215020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim NN, Villarreal FJ, Printz MP, Lee AA, Dillmann WH. Trophic effects of angiotensin II on neonatal rat cardiac myocytes are mediated by cardiac fibroblasts. Am J Physiol. 1995;269:E426–37. doi: 10.1152/ajpendo.1995.269.3.E426. [DOI] [PubMed] [Google Scholar]
- 23.Thuerauf DJ, Morrison LE, Hoover H, Glembotski CC. Coordination of ATF6-mediated transcription and ATF6 degradation by a domain that is shared with the viral transcription factor, VP16. J Biol Chem. 2002;277:20734–9. doi: 10.1074/jbc.M201749200. [DOI] [PubMed] [Google Scholar]
- 24.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–8. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 25.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–14. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Craig R, Larkin A, Mingo AM, Thuerauf DJ, Andrews C, McDonough PM, et al. p38 MAPK and NF-kappa B collaborate to induce interleukin-6 gene expression and release. Evidence for a cytoprotective autocrine signaling pathway in a cardiac myocyte model system. J Biol Chem. 2000;275:23814–24. doi: 10.1074/jbc.M909695199. [DOI] [PubMed] [Google Scholar]
- 27.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. 2000;20:6755–67. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee AS, Bell J, Ting J. Biochemical characterization of the 94- and 78-kilodalton glucose-regulated proteins in hamster fibroblasts. J Biol Chem. 1984;259:4616–21. [PubMed] [Google Scholar]
- 30.Munro S, Pelham HR. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- 31.Kang HS, Welch WJ. Characterization and purification of the 94-kDa glucose-regulated protein. J Biol Chem. 1991;266:5643–9. [PubMed] [Google Scholar]
- 32.Vaux D, Tooze J, Fuller S. Identification by anti-idiotype antibodies of an intracellular membrane protein that recognizes a mammalian endoplasmic reticulum retention signal. Nature. 1990;345:495–502. doi: 10.1038/345495a0. [DOI] [PubMed] [Google Scholar]
- 33.Tadimalla A, Belmont PJ, Thuerauf DJ, Glassy MS, Martindale JJ, Gude N, et al. Mesencephalic astrocyte-derived neurotrophic factor is an ischemia-inducible secreted endoplasmic reticulum stress response protein in the heart. Circ Res. 2008;103:1249–58. doi: 10.1161/CIRCRESAHA.108.180679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wooden SK, Li LJ, Navarro D, Qadri I, Pereira L, Lee AS. Transactivation of the grp78 promoter by malfolded proteins, glycosylation block, and calcium ionophore is mediated through a proximal region containing a CCAAT motif which interacts with CTF/NF-I. Mol Cell Biol. 1991;11:5612–23. doi: 10.1128/mcb.11.11.5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Chen B, Li Y, Chen J, Lou G, Chen M, et al. Transcriptional regulation of the human PNRC promoter by NFY in HepG2 cells. J Biochem. 2008;143:675–83. doi: 10.1093/jb/mvn019. [DOI] [PubMed] [Google Scholar]
- 36.Thuerauf DJ, Morrison L, Glembotski CC. Opposing roles for ATF6alpha and ATF6beta in endoplasmic reticulum stress response gene induction. J Biol Chem. 2004;279:21078–84. doi: 10.1074/jbc.M400713200. [DOI] [PubMed] [Google Scholar]
- 37.Haze K, Okada T, Yoshida H, Yanagi H, Yura T, Negishi M, et al. Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem J. 2001;355:19–28. doi: 10.1042/0264-6021:3550019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li M, Baumeister P, Roy B, Phan T, Foti D, Luo S, et al. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol Cell Biol. 2000;20:5096–106. doi: 10.1128/mcb.20.14.5096-5106.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thuerauf DJ, Marcinko M, Belmont PJ, Glembotski CC. Effects of the isoform-specific characteristics of ATF6 alpha and ATF6 beta on endoplasmic reticulum stress response gene expression and cell viability. J Biol Chem. 2007;282:22865–78. doi: 10.1074/jbc.M701213200. [DOI] [PubMed] [Google Scholar]
- 40.Liu XM, Peyton KJ, Ensenat D, Wang H, Schafer AI, Alam J, et al. Endoplasmic reticulum stress stimulates heme oxygenase-1 gene expression in vascular smooth muscle. Role in cell survival. J Biol Chem. 2005;280:872–7. doi: 10.1074/jbc.M410413200. [DOI] [PubMed] [Google Scholar]
- 41.Doroudgar S, Glembotski CC. The cardiokine story unfolds: ischemic stress-induced protein secretion in the heart. Trends Mol Med. 2011;17:207–14. doi: 10.1016/j.molmed.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bulleid NJ, Ellgaard L. Multiple ways to make disulfides. Trends Biochem Sci. 2011;36:485–92. doi: 10.1016/j.tibs.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Kozlov G, Maattanen P, Thomas DY, Gehring K. A structural overview of the PDI family of proteins. Febs J. 2010;277:3924–36. doi: 10.1111/j.1742-4658.2010.07793.x. [DOI] [PubMed] [Google Scholar]
- 44.Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–50. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 45.Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U S A. 2002;99:4465–70. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hatahet F, Ruddock LW. Substrate recognition by the protein disulfide isomerases. Febs J. 2007;274:5223–34. doi: 10.1111/j.1742-4658.2007.06058.x. [DOI] [PubMed] [Google Scholar]
- 47.Sorensen BS, Horsman MR, Vorum H, Honore B, Overgaard J, Alsner J. Proteins upregulated by mild and severe hypoxia in squamous cell carcinomas in vitro identified by proteomics. Radiother Oncol. 2009;92:443–9. doi: 10.1016/j.radonc.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 48.Krajewska M, Xu L, Xu W, Krajewski S, Kress CL, Cui J, et al. Endoplasmic reticulum protein BI-1 modulates unfolded protein response signaling and protects against stroke and traumatic brain injury. Brain Res. 2011;1370:227–37. doi: 10.1016/j.brainres.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jordan PA, Stevens JM, Hubbard GP, Barrett NE, Sage T, Authi KS, et al. A role for the thiol isomerase protein ERP5 in platelet function. Blood. 2005;105:1500–7. doi: 10.1182/blood-2004-02-0608. [DOI] [PubMed] [Google Scholar]
- 50.Mellgren RL. A plasma membrane wound proteome: reversible externalization of intracellular proteins following reparable mechanical damage. J Biol Chem. 2010;285:36597–607. doi: 10.1074/jbc.M110.110015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH, et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature. 2007;447:482–6. doi: 10.1038/nature05768. [DOI] [PubMed] [Google Scholar]
- 52.Kimura T, Horibe T, Sakamoto C, Shitara Y, Fujiwara F, Komiya T, et al. Evidence for mitochondrial localization of P5, a member of the protein disulphide isomerase family. J Biochem. 2008;144:187–96. doi: 10.1093/jb/mvn057. [DOI] [PubMed] [Google Scholar]
- 53.Chin KT, Kang G, Qu J, Gardner LB, Coetzee WA, Zito E, et al. The sarcoplasmic reticulum luminal thiol oxidase ERO1 regulates cardiomyocyte excitation-coupled calcium release and response to hemodynamic load. Faseb J. 2011;25:2583–91. doi: 10.1096/fj.11-184622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.