

Abstract
Alpha-methylacyl coenzyme A racemase (AMACR) is a metabolic enzyme whose over-expression has been shown to be a diagnostic indicator of prostatic adenocarcinoma as well as other solid tumors. Here we confirm that attenuation of AMACR expression diminishes the growth of prostate cancer cell lines using stably expressed shRNA constructs. This observation strongly suggests that the AMACR enzyme may be a target for therapeutic inhibition in prostate cancer. To this end, we report here a novel assay capable of screening libraries of diverse small molecules for inhibitors of AMACR activity. This assay facilitated the screening of approximately 5,000 unique compounds and the discovery of seven distinct chemical entities capable of inhibiting AMACR at low micromolar concentrations. The most potent inhibitor discovered is the seleno-organic compound ebselen oxide (IC50:0.80 μM). The parent compound, ebselen (IC50:2.79 μM), is a covalent inactivator of AMACR (KI(inact):24 μM). Two of the AMACR inhibitors appear selectively toxic to prostate cancer cell lines (LAPC4/LNCaP/PC3) that express AMACR compared to a normal prostate fibroblast cell line (WPMY1) that does not express the protein. This report demonstrates the first high-throughput screen for the discovery of novel AMACR inhibitors, characterizes the first non-substrate based inhibitors, and validates that AMACR is a viable chemotherapeutic target in-vitro.
Keywords: High-Throughput, Inhibitor, AMACR, Prostate Cancer, Racemase, Screen, Assay, Imaging
Introduction
α-Methylacyl-Coenzyme A Racemase (AMACR, EC 5.1.99.4) is a cofactor independent metabolic enzyme important for the catabolism of branch chained fatty acids as well as the maturation of bile acids from cholesterol precursors (1). The natural substrates for AMACR include (2R)/(2S) pristanoyl coenzyme A (Fig. 1A) and the bile acid precursor molecule (25R)/(25S)-trihydroxycholestanoyl coenzyme A (Fig. 1B) (1–5). Acting upon its substrate, the enzyme has the ability to catalyze the bi-directional stereoconversion (from S to R and the reverse) of the α-methyl proton via a 1,1-proton transfer thought to proceed through an enolate intermediate (3,6).
Figure 1. Well characterized AMACR substrates.
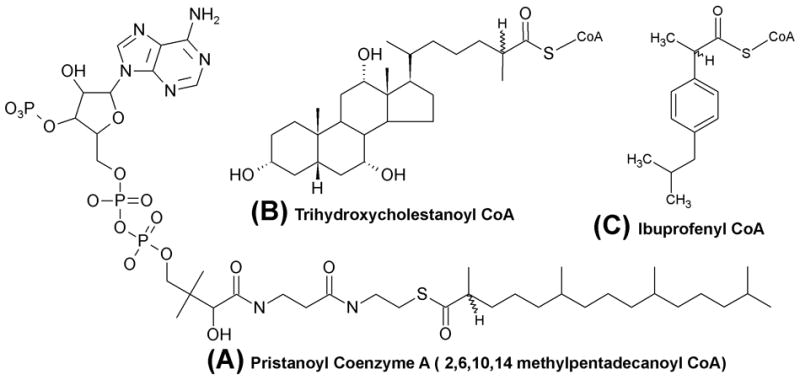
(A) Pristanoyl-Coenzyme A (Pri-CoA) (with complete chemical structure of coenzyme A (CoA) moiety). (B) 3,7,12-Trihydroxycholestanoyl-CoA (THCA-CoA) (C) Ibuprofenyl-CoA
In mammals, branch-chain lipids are acquired either directly through the degradation of chlorophyll into phytanic acid (ruminants) or by the intake of ruminant byproducts (milk, beef, etc.) (1, 2). These lipids (and cholesterol precursors of bile acids) naturally occur as a racemic mixture, and their complete oxidation requires that they be in the S-conformation (1,2). AMACR carries out that conversion prior to further oxidation. In addition to natural substrates, AMACR is believed to catalyze the stereoconversion of ibuprofenyl-CoA (Fig 1C) from its inactive R-conformer to its biologically active S-enantiomer (3).
Two crystal structures of the homologous α-methylacyl coenzyme A racemase from Mycobacterium tuberculosis, called MCR, have been published (7,8). Structurally MCR (43% homologous to AMACR by protein sequence) belongs to the type III CoA transferase superfamily of enzymes. The crystal structure indicates that the enzyme forms a dimer of interlocking dimers with the active site at the interface of the large domain of one monomer and the small domain of the other monomer. The identified catalytic residues in MCR are H126 and D156 (homologous residues H122 and D152 in AMACR). The size and lack of prominent topography in the hydrophobic substrate binding pocket accommodates the binding of either enantiomer from a diverse array of substrates (Fig. 1) (7, 8). A recent kinetic study using recombinant human AMACR and a deuterium-labeled substrate assay observed that the rate of solvent exchange (without stereoconversion) is twice the rate of stereoconversion, implying that the mechanism of racemization is inefficient (3). This is consistent with data showing little to no preference for one chiral center (R or S) over the other.
Beyond its metabolic significance, AMACR is important in several human diseases. Patients with peroxisomal deficiency (Zellweger’s syndrome) are deficient in AMACR activity (1), and patients with inactivating AMACR mutations (S52P and L107P) accumulate toxic levels of the R-conformer of branch chain fatty acids in their blood, resulting in neuropathy similar to Refsum disease (9). Additionally, the specific up-regulation of AMACR at both transcript and protein levels in prostatic adenocarcinoma and its precursor lesions, including putative prostate cancer progenitor populations, has been reported (10, 11, 12). Immunohistochemical detection of AMACR has become a valuable tool for the positive diagnosis of prostate cancer in tissue samples (11, 13). AMACR over-expression correlates with increased AMACR activity, indicating that the protein being expressed is enzymatically active and may be contributing to cancer growth (14, 15).
Decreasing the expression of AMACR through the use of siRNA constructs has been shown to slow the growth of prostate cancer cell lines, indicating that not only may AMACR expression directly be supporting cancer growth, but also that AMACR may be a new target for chemotherapeutic inhibition (14, 16). For targeted therapy, AMACR offers several advantages. First, an AMACR knockout mouse model has been generated and beyond the expected problem of branch chain lipid accumulation (safely regulated by diet alone) the mice have been reported to be healthy and fertile (17). This finding is in agreement with the observation that individuals with AMACR deficiency may remain asymptomatic for extended periods (9). Those data suggest that targeted inhibition of AMACR may offer promise as chemotherapy against prostate cancer without major inhibition related side effects (14, 16). In addition to prostate cancer, AMACR has been shown to be over-expressed in a variety of solid tumors, suggesting that targeted inhibition may be translatable to other cancers (18).
In addition to exploiting the over-expression of AMACR for cancer chemotherapy, it is becoming increasingly clear that AMACR represents an excellent target for imaging of prostate cancer (19). While several imaging agents exist for detecting prostate cancer after it has disseminated, there is still a need for imaging agents to detect intraprostatic lesions (20, 21). Given that prostate cancer is the most commonly diagnosed cancer in men, as well as the second leading cause of cancer-related death in men (22) and that an increasing number of cases are being followed by active surveillance, finding low molecular weight agents that target AMACR could represent an important advance for managing this disease (23, 24). An important distinction between AMACR and other candidate prostate cancer imaging targets is that AMACR expression is largely cancer specific, whereas PSA and PSMA are prostate specific, being expressed by both normal and cancerous prostate epithelial cells (20, 21).
There are few reported AMACR inhibitors. The scarcity of inhibitors relates in large part to the unwieldy substrate requirements of the enzyme, namely: the presence of a coenzyme A thioester (rendering the molecule impermeable to cells due to the presence of three phosphate molecules) and a minimum carbon chain length of 8 carbons (with the exception of ibuprofenyl coenzyme A) for the acyl portion of the substrate (1, 2, 8). Multiple assays exist to quantify AMACR activity (1–8, 15, 25, 26). The most common assay relies on the production of radiolabeled water after incubation of AMACR with substrates containing tritium or deuterium at the α-position (1–3). Also, it is possible to monitor the stereoconversion of one enantiomer to another by incubating the enzyme with a stereochemically pure pool of substrate and then measuring the production of the opposite stereoisomer after diastereomeric separation using either gas chromatography (GC) or high-performance liquid chromatography (HPLC) (1–2, 6). Using these assays, inhibitors of AMACR activity have been identified (including mercury, copper(II), diethylpyrocarbonate, Ellman’s reagent, and N-ethylmaleimide) (1, 2). Recently it has been shown that fluorine for hydrogen substitutions of known substrates near the α-position can result in the generation of competitive inhibitors. However, such molecules still require the presence of the coenzyme A (SCoA) moiety, limiting their therapeutic potential (26).
While the aforementioned assays have single time point dependence, two continuous assays have been published (4, 25). One assay is an indirect coupled assay that monitors hydrogen peroxide production by the stereospecific oxidase immediately downstream of AMACR (25). That assay is limited in that it requires two different enzymes be present, and a stereochemically pure substrate (25). Recently a circular dichroism (CD) assay for MCR activity has been published (4). In this assay, recombinant MCR is incubated with stereochemically pure R- or S-ibuprofenyl-coenzyme A and circular dichroism measurements are made as the enzyme converts one diastereomer to the other (4). As with all prior assays for measuring AMACR activity or inhibition, the CD assay cannot be used to screen more than one reaction condition at a time. The arduous substrate (single optical isomers) or product preparations (diastereomer derivitization for analytic GC/HPLC separation), precludes the use of all existing assays in screening large libraries of diverse compounds.
We have designed a 96-well based assay for the detection and testing of AMACR inhibitors. Using this assay we discovered and subsequently characterized AMACR inhibitors that are not substrate-based. None of the inhibitors identified require the presence of the coenzyme A moiety for activity, rendering them superior to previously identified compounds with respect to expected pharmacokinetic properties, thereby enabling further optimization and implementation in vitro and in vivo. Unlike previous inhibitors these compounds do not behave in a competitive fashion, offering unique opportunities to gain insight into the structural requirements for AMACR inhibition. Additionally, we report the development of the first stable AMACR knockdown cell line of prostate cancer cells (LAPC4-AMACRKO). These cells exhibit a statistically significant decreased growth rate and will offer an ideal syngeneic control for further AMACR related in vitro and in vivo studies.
Experimental Procedures
Reagents
Ebselen and ebselen oxide were purchased from Cayman Chemical Co (Ann Arbor, MI), 3,7,12-trihydroxycholestanoic acid and pristanic acid were purchased from Larodan Fine Chemicals (Malmo, Sweden), coenzyme A trilithium salt was purchased from MP Biomedicals (Solon, OH), solid phase extraction plates (Strata C18C) were purchased from Phenomenex Inc. (Torrence, CA), Acetonitrile was purchased from ThermoFisher Scientific (Newark, DE). [2, 3-3H]-pristanoyl coenzyme A was synthesized by Moravek Biochemicals (Brea, CA). Unless otherwise indicated, all other reagents were purchased from Sigma-Aldrich Corp (St. Louis, MO). All graphs and data analysis was accomplished using GraphPad Prism software (GraphPad Software, San Diego, CA). All cell lines used have been authenticated as of November 2010 using short tandem repeat (STR) DNA analysis according to the manufacturer’s protocol for the PowerPlex 1.2 System (Promega Corp., Madison, WI).
shRNA Mediated AMACR Knockdown
The human prostate cancer cell line LAPC4 was acquired from the laboratory of Dr. John T. Isaacs (14). 1×106 cells were plated in 100 mm dishes in normal culture media (Iscove’s Modified Dulbecco’s Media (IMDM), Invitrogen Corp, Carlsbad, CA) supplemented with 10% Fetal Bovine Serum (Invitrogen) and 10 nm R1881 (PerkinElmer, Waltham, MA). The cells were allowed to establish and reach 80% confluency. The media was then replaced with fresh media containing 8 μg/mL hexadimethrine. Lentiviral transduction particles encoding shRNA either targeting the AMACR transcript (AMACRKO, Sigma-Aldrich, clone ID TRCN0000084114) or not targeting any known human gene (Vector control, Sigma-Aldrich, SHC002V) were added to this media. Cells were incubated with particles overnight before replacement with fresh media without hexadimethrine or particles. After a 24 hour recovery period, the media was exchanged for selection media containing 5 μg/mL puromycin and were allowed to undergo selection for ten days before harvesting the entire population of viable cells and expanding them into a 300 mL tissue culture flask. Selection media was maintained henceforth.
After both the AMACRKO and the Vector cell lines adequately expanded, Western blot analysis was performed. Parental, AMACRKO, and Vector cells were trypsinized, collected, washed with phosphate buffered saline (PBS) pH 7.4, and lysed for 30 minutes in Cell Extraction Buffer (Invitrogen). Insoluble material was spun down, and the lysate retained and quantified using the BCA assay kit (ThermoFisher). 50μg of each lysate was then loaded into adjacent wells in duplicate SDS-PAGE gels, 4–20% gradient (ThermoFisher). The gels were electrophoresed according to manufacturer’s protocols and were then transferred to nitrocellulose membranes for Western blotting. After electrophoretic transfer, 1.5 hours at 100V, the blots were blocked for 30 minutes with blocking buffer (Li-Cor Biosciences, Lincoln, NE). The membranes were then incubated with mouse anti-AMACR antibodies (1:2000, Invitrogen) and rabbit anti-tubulin (1:10,000, Millipore Corp, Billerica, MA) or mouse anti-actin (1:25000 Sigma-Aldrich) overnight at 4°C. The membranes were then washed with PBS (+0.1% Tween-20, USB Corp., Cleveland, OH) and incubated for 4 hours with anti-mouse and anti-rabbit or anti-mouse only secondary antibodies (1:25,000 Li-Cor). The membrane were then developed using the Li-Cor Odyssey infrared imaging system (Li-Cor). Cropped results are shown inset within figure 2 and the complete blots are included in the supplementary figure S5.
Figure 2. shRNA mediated AMACR knockout in LAPC4 cells diminishes cell growth.
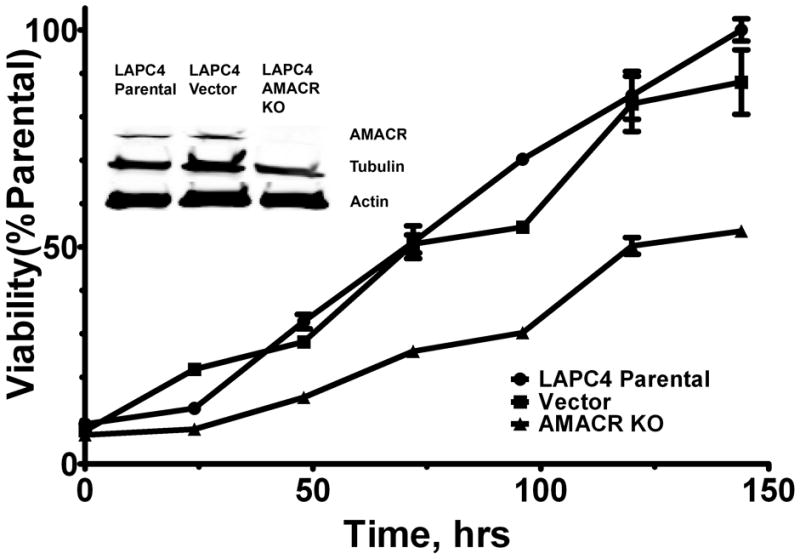
Diminished AMACR expression affects cell growth as tested using the alamarBlue assay (p-value<0.005, vector vs. AMACR KO, two-tailed t-test, r=0.9875, P-value =0.2505 vector vs. parental, r=0.9810), all data points are normalized as a percentage of the maximum parental control. Inset, Western blot analysis showing AMACR expression in parental, vector control, and LAPC4 cells containing shRNA targeting AMACR protein. Tubulin and actin used as loading controls. AMACR is undetectable in AMACR KO cell line. Full length blots in Supplementary Fig. S5.
To determine the effect that shRNA mediated AMACR knockout has on cell growth and viability, LAPC4 parental, AMACRKO, and vector cells were assayed for their growth rate using the alamarBlue assay (AbD serotec, Oxford, UK). 5,000 cells of each type were plated in triplicate in seven different tissue culture treated zero fluorescence 96-well plates (BD Biosciences, San Jose, CA.) in 100 μL of media. Beginning with the initial plating and at subsequent 24 hour intervals 10 μL alamarBlue reagent was added to each well (including media only controls) and incubated for 4 hours. The plate was then read on a Fluostar Omega plate reader (544 excitation/590 emission, BMG Labtech Inc., Cary, NC), background fluorescence removed and the change in fluorescence was measured for each cell type overtime. The fluorescence measured in the parental line at 144 hours post-plating was taken as the maximum reading (100%) and all prior readings were calculated as a percentage of this time-point (normalized as a percentage of the parental line). The results are depicted in figure 2.
Recombinant Human AMACR-MBP fusion purification
The plasmid vector (pMal-C2x, New England Biolabs (NEB), Ipswich, MA) with human AMACR cloned after an N-terminal Maltose Binding Protein (MBP) was a generous gift of Dr. Sacha Ferdinandusse (9). The plasmid was transformed into chemically competent E. coli (TB1 strain) and recombinant protein was purified according to the manufacturer’s protocol (pMal Purification Kit, NEB). After elution from the maltose column (MBPHiTrap HP maltose column, GE Healthcare Biosciences Corp, Piscataway, NJ), the single eluted peak of recombinant protein was dialyzed for 1–4 h against 1 L of 100mM Na/K/Pi buffer pH 7.25 (referred to hereafter as “reaction buffer”) using slide-a-lyzer dialysis cassettes (10 kDa cutoff, ThermoFisher). The dialysis cassette was then transferred to 3 L of reaction buffer and dialyzed overnight. The following day the dialyzed lysate was quantified using the BCA protein assay kit. Additionally, the recombinant AMACR-MBP protein was quantified by densitometry analysis.
High Throughput Screen
(HTS) [2,3-3H]-pristanoyl coenzyme A (Pri-CoA) was purchased from Moravek Biochemicals (Brea, CA) with a specific radioactivity of 4.7 Ci/mmol. For the purposes of this assay the specific radioactivity was reduced to 60 Ci/mol by dilution with unlabeled Pri-CoA. Pristanic acid was purchased from Larodan and was ligated with coenzyme A and purified according to published procedures (1, 2). This substrate exhibited non-specific binding to polypropylene. To improve recoveries, 0.125 mg/mL agarose purified BSA (Sigma-Aldrich) was added to the reaction buffer for both experimental and control wells (referred to as “complete reaction buffer”). Library Preparation-The Johns Hopkins Drug Library (JHDL), was diluted from the master plates (100% DMSO) into 1X phosphate buffered saline (PBS) at a final concentration of 500 μM. The requested compounds (Diversity Set 2 and Natural Products Set) from the NCI/DTP Open Chemical Repository (http://dtp.cancer.gov) arrived as 10 mM solutions in 100% DMSO and were diluted to 500 μM in reaction buffer (without BSA, final DMSO concentration of 5%). All library plates were stored at −20°C until use, at which time they were thawed and vigorously shaken to redissolve any precipitates. The complete assay setup is described in detail in the Supplementary Material but is summarized here and in figure 3. The enzymatic reaction is setup in a 96 well PCR plate with a total reaction volume of 25μL. 0.3μM AMACR-MBP is incubated with 100μM of library compound (1% DMSO) in the presence of 150μM [2, 3 3H] Pri-CoA for 30 minutes at 37°C on a 96-well PCR machine. Buffer only (zero AMACR) controls are included in each plate setup as background controls. Additionally, column A for each plate is used as an AMACR only (100% activity) control and column H is used as a positive AMACR inhibitor (DEPC, (2)) control. During the incubation, a 96-well solid phase extraction plate (SPE) (Strata C18C Plate, Phenomenex) is conditioned with acetonitrile and equilibrated with reaction buffer on a vacuum manifold. After 30 minutes, the reaction is acid quenched, neutralized, diluted to 100 μL, and then transferred to the SPE plate. Each well of the reaction plate is then washed twice and the washes are transferred to their respective wells. The total post reaction volume is 300 μL per SPE well. Vacuum is applied and the reaction is pulled through the SPE, retaining unreacted substrate and allowing [3H] H2O product to pass into the collection plate. 60 μL of the eluate from each well is then transferred to a reading plate containing liquid scintillation fluid. The read plate is then sealed, vortexed, and read in a 96-well scintillation detector (MicroBeta Jet, Perkin-Elmer) using a dwell time of 2 min per well and the same set of efficiency (CPM to DPM) standards for every plate. After the entire plate is read, the background is corrected by removing the average counts from background wells. The fractional activity (AI/A0) was then calculated for each library position by dividing the library DPM by the 100% activity control for that row (A column). Any well with fractional activity less than 0.8 indicates a compound that is least 20% inhibitory. Such compounds were considered worthy of further validation (Supplementary Tables S1 and S2, Table S1 and S2).
Figure 3.

Schematic Flow Chart of Novel High Throughput Screen (HTS) for AMACR Inhibitors
HTS Validation
The Km and the Vmax for [2, 3 3H] Pri-CoA (Fig. 4) was determined by setting up 185 μL reactions at various concentrations of substrate (1, 5, 10, 15, 25, 50, 75, 100, 150, 200, 300, and 400 μM) from which 20 μL aliquots were taken at time points of 0, 15, 30, 60, 120, 300, 420, 600, and 900 sec. All of the concentrations tested had the same specific radioactivity (60 Ci/mol). The reactions were carried out using 0.3 μM AMACR-MBP in microcentrifuge tubes at 37°C using complete reaction buffer (+BSA) and were initiated by the addition of enzyme to the reaction tube. After the withdrawal of each 20 μL aliquot at its designated time, the aliquot was immediately dispensed into one well of a 96 well plate containing 10 μL of 2 M HCl to quench the reaction. Also, background controls without AMACR-MBP were set up for each time point and processed along with each experimental time point. Once the aliquots were added to the quench plate, they were processed as described for the wells in the library screen (e.g. neutralization, dilution to 100 μL, transfer to SPE plate, etc.). Using specific CPM to DPM standards, the number of DPM produced per unit of time was calculated and the rate of reaction determined for all concentrations tested. Those concentration dependent rates were then directly plotted and were fit to the Michaelis-Menten equation to calculate the kinetic parameters shown in Fig. 4 (GraphPad).
Figure 4. Kinetic Analysis of AMACR-MBP Purification.
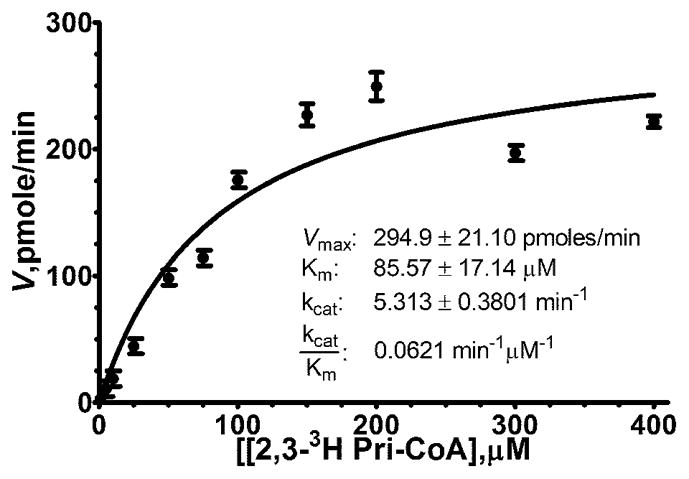
Direct Plot with non-linear regression of substrate dependent velocity curve of AMACR-MBP incubated with [2, 3-3H] Pristanoyl-Coenzyme A. Error bars indicate standard error of the mean.
HTS Candidate Inhibitor Validation
All library compounds that demonstrated fractional activity of less than 0.8 were verified first by reproducing the initial HTS screen. The initial candidate inhibitors were verified at least twice before further validation (data not shown). In the case of the JHDL, candidate inhibitors that were distinct single chemical entities and were available for purchase were obtained and their inhibitory capacities tested in a small scale IC50 experiment using conditions identical to the HTS assay except that the compound concentration included 1, 10, 50, 100, 200, 500, and 1000 μM concentrations (data not shown). If a dose dependent inhibition could be observed in that assay, then more rigorous follow up was pursued. Based on this analysis and on considerations of chemical structures, only ebselen, congo red, and rose bengal (Fig. 5) warranted further study. For compounds identified from the NCI/DTP Open Chemical Repository, validation was carried out in the exactly as for the JHDL. DMPMB, MSDTP, DPZBD, and DPTD (Fig. 5) were chosen for rigorous follow up and 1–10 mg requests were made from the NCI/DTP Open Chemical Repository (http://dtp.cancer.gov). All of the received compounds were dissolved in 100% DMSO at 25 mM concentrations kept at −20°C until use. Any subsequent dilutions were also made into 100% DMSO to maintain solubility.
Figure 5. Novel Inhibitors of AMACR discovered by HTS.

Structures, IC50 values, and LD50 values of best inhibitors (1–8) discovered. Capitalized letters represent the acronym of the compound as used in the paper. Compound 9 included for reference as a known substrate
AMACR Inhibitor IC50 Determination
Library compounds (Fig. 5) were tested over a broad range of concentrations (1–1000 μM, and additionally 1–750 nM for ebselen/ebselen oxide) in triplicate. While the inhibitor concentration for those experiments was varied, the substrate concentration was held constant at 100 μM, the AMACR-MBP at 0.3 μM, and the DMSO concentration varied from 2 to 6%. Each set of reactions was set up on the same reaction plate to keep variability to a minimum. The reactions were setup by adding the necessary amount of complete reaction buffer (13.5 to 14.25 μL) to the wells of the reaction plate, then the inhibitor was added at the desired concentration (0.5 to 1.5 μL) giving a total volume of 15 μL. Next 5 μL of 500 μM [2, 3 3H] Pri-CoA was added to the wells, and finally the reaction was initiated by the addition of 5 μL of 1.5μM AMACR-MBP to each well. The reactions were mixed via pipetting. All downstream processing procedures were carried out as detailed for the HTS assay. After the reading step, the fractional activity was calculated as described above and the data were analyzed using 3 parameter log(inhibitor) vs. Response analysis (GraphPad). The results of those analyses are shown Fig. 5 (All IC50 curves are given in Supplementary Figure S3).
Dialysis Resistant Inhibition by Candidate Inhibitors
400 μL pre-incubations were set up with 0.375 μM AMACR-MBP and the concentration of each inhibitor was chosen so that inhibition would be measurable prior to 1 L dialysis but would be below an inhibitory concentration after dialysis. The concentrations employed were as follows: 25 μM ebselen, 200 μM DMPMB, 400 μM MSDTP, 50 μM DPZBD, 50 μM DPTD, and a 1.6% DMSO control. Pre-incubations were allowed to continue for 30 min on ice and then 200 μL was removed from each pre-incubation and injected into a slide-a-lyzer dialysis cassette (10 kDa cutoff, ThermoFisher). The cassettes were then separately dialyzed against 1 L of reaction buffer overnight. The remaining pre-incubation reaction was placed at 4°C overnight Assuming complete dialysis, the post dialysis concentration of each compound is: 5 nM ebselen, 40 nM DMPMB, 80 nM MSDTP, 10 nM DPZBD, and 10 nM DPTD. The following day the ultraviolet-visible (UV-Vis) spectra for each compound, both before and after dialysis, were scanned to determine if there was any absorbance attributable to the inhibitor remaining in the dialyzed samples. The presence of remaining inhibitor after dialysis could not be detected for any compounds (DPZBD had no detectable absorbance peak before or after dialysis) (data not shown). Next, 20 μL of both the dialyzed and undialyzed pre-incubations was added to 5 μL of 500 μM [2, 3 3H] Pri-CoA in a 96-well plate (final concentration of AMACR-MBP 0.3 μM and 100 μM [2, 3 3H] Pri-CoA), and the reactions with AMACR-MBP were performed as described above for the HTS assay. These dialysis experiments were undertaken on three different occasions. Dialysis against a greater volume of reaction buffer was attempted and did not change the results (data not shown). The fractional activity for both the undialyzed and dialyzed samples were calculated as described above. Dialysis could not recover AMACR activity after pre-incubation (Fig. 6A).
Figure 6. Time Dependent Inactivation of AMACR.

(A) AMACR-MBP was incubated with each inhibitor at the following concentrations: 25 μM ebselen, 200 μM DMPMB, 400 μM MSDTP, 50 μM DPZBD, 50 μM DPTD, or 1.6% DMSO control and then dialyzed overnight against 1L reaction buffer. This dialysis was not able to recover enzymatic activity indicating inactivation of the enzyme by the inhibitor. (B) Inhibition progress curves for DMPMB. DMPMB is pre-incubated with AMACR-MBP prior reaction initiation. Fractional activity (enzymatic activity with inhibitor/activity without) decreases with increasing pre-incubation time. Kitz-Wilson transformation of inhibition progress curves allows calculation of kinact and KI(inact).
Kitz-Wilson Analysis
In a 37°C 96-well plate, inhibition reactions were setup containing DMPMB concentrations from 0 to 400 μM and 5 μM AMACR-MBP. Inhibition was initiated by the addition of AMACR-MBP; immediately after which (time-point zero), a 2 μL aliquot was removed and diluted into 98 μL of reaction buffer containing 150 μM [2, 3 3H] Pri-CoA (a 50 fold dilution reducing the final AMACR-MBP concentration to 100 nM). These enzymatic reactions were allowed to proceed for thirty minutes before quenching and processing as described above for the HTS. Additional aliquots were withdrawn from the inhibition reaction at subsequent time points and diluted. Data analysis was performed as described by Kitz and Wilson (27, 28). The extent of inactivation occurring during pre-incubation in the inhibition reaction is measured relative to time matched control wells without inhibitor by simply dividing the background corrected DPM from the experimental (%AI) well by their control well (%AC) (expressed as a percentage), giving a percent activity remaining. Enzymatic reaction progress curves were generated by graphing the log of the percent of activity remaining versus pre-incubation time prior to dilution, Fig. 6B. Linear regression analysis of the reaction progress curves allows for the calculation of the inactivation half-life (T1/2, time to a 50 percent reduction in experimental activity). Linear regression of the T1/2 values versus [I]−1 yields an equation allowing for the calculation of the inactivation rate (kinact = ln2/y-intercept) as well as the KI(inact) (−1/x-intercept), figure 6B inset. Identical experiments were carried out for ebselen, Supplementary. Fig. S4.
In-Vitro LD50 Measurements for DMPMB and Ebselen
PC3 and LNCaP cell lines were purchased from ATCC (Manassas, Va.), WPMY1 cells were acquired from the laboratory of Dr. Samuel R. Denmeade (29). LAPC4 are described above. Western blotting for AMACR expression and loading controls for these cell lines was done identically as for LAPC4 above and the results are depicted in the inset of figure 7A inset (complete blots are in S. Fig. S5).
Figure 7. In-Vitro Cytotoxicity Studies of Ebselen and DMPMB.

(A) LD50 curves for ebselen in 3 prostate cancer cell lines (PC3, LNCaP, LAPC4) and one normal prostate fibroblast cell line (WPMY1), all data points are normalized as a percentage of their respective vehicle controls. Inset Western blot analysis of AMACR expression in 4 cell lines demonstrating increasing ebselen sensitivity with increasing AMACR expression (tubulin and actin blotted as loading controls). (B) LD50 curves for DMPMB as in (A).
To determine the concentration of ebselen or DMPMB that is lethal to 50% of cells (LD50), 10,000 cells of each type were plated in triplicate in zero fluorescence tissue culture treated 96-well plates (BD Biosciences). The cells established overnight and the following day their media was replaced with media containing either ebselen or DMPMB in a concentration range of 0–500 μM or 0–400 μM respectively. After 48 hours the alamarBlue assay for viability was performed as described for LAPC4 above. The viability was calculated as a percent of the no inhibitor control for each cell type, after correcting for media only background fluorescence. The percent viability as a function of increasing log of inhibitor concentration was then analyzed using a least squares fit for non-linear regression with a variable slope (4 parameter) and constraining the bottom to zero (GraphPad). The results of these analyses can be seen in figures 7A and 7B.
Results
Stable shRNA mediated AMACR knockdown validates AMACR as a therapeutic target
Previously, we have studied the effect of substrate-based AMACR inhibitors on different prostate cancer cell lines with variable levels of AMACR expression (26). In addition we have transiently attenuated AMACR expression through the use of siRNA (14). To extend these analyses, here we report the use of commercial lentiviral particles to stably reduce the expression of AMACR.
LAPC4 cells were transduced with lentivirus containing an shRNA construct targeting AMACR (LAPC4 AMACRKO). As a parallel control, cells were also transduced with lentivirus containing an shRNA construct targeting no known gene in the human genome (vector). To assess the ability of the shRNA to reduce AMACR expression, a Western blot was prepared using lysates from the parental, vector, and AMACRKO cell lines (Fig. 2 inset). As seen in Figure 2, the shRNA construct rendered AMACR protein expression undetectable by immunoblotting. In order to test the growth effect of biochemical AMACR depletion, parental, vector, and AMACRKO cells were plated at equal density in 96 well plates and their growth was measured for 6 days using the Alamar blue assay. There is a statistically significant difference between the vector and AMACRKO cell lines (P-value < 0.05, two-tailed, t-test); while no significant difference exists between vector and LAPC4 parental cell lines (Fig. 2). Identical studies were carried out using the prostate cancer cell line LNCaP (Supplementary Figure S1). Our observations confirm that reducing AMACR expression slows prostate cancer cell growth, providing further impetus to develop small molecules that can inhibit AMACR activity.
Purification of Enzymatically Active AMACR from E. coli
A recombinant expression vector containing the human AMACR cDNA with an N-terminal maltose-binding protein (MBP) tag was obtained (AMACR-MBP) (9). Using that vector, milligram quantities of AMACR-MBP were routinely purified. To guard against the possibility that non-specific carryover from the E coli host strain may be providing enzymatic activity similar to that of AMACR, site-directed mutagenesis was employed to create a known inactivating mutation within the AMACR construct, changing serine 52 to proline (9). That mutation eliminated all measurable AMACR activity from our preparations indicating that all measured AMACR activity was from AMACR-MBP and not from a contaminant (data not shown).
To ensure that we were purifying enzymatically active AMACR-MBP, an HPLC-based assay was employed (6). It has been shown that diastereomers of the AMACR substrate (25 R,S)-THCA-CoA can be separated and collected independently of each other via reverse phase C18 HPLC. The separated diastereomers (either 25R or 25S) can then be incubated in the presence of AMACR and the conversion of one enantiomer to the other can be monitored over time via HPLC. Using that assay AMACR-MBP was confirmed as enzymatically active (or inactive in the case of the S52P mutant) (S. Fig. S2). Although this assay is adequate for validation and comparison of enzymatic preparations, the retention times for the separation of diastereomers, as well as the amount of processing involved make this assay impractical for use in a high-throughput screen.
Development of a High-Throughput Screen (HTS) for AMACR Inhibitors
None of the current assays for AMACR activity have been adapted to a multi-well screening format (1–6, 9, 15, 25, 26). We established a 96-well plate assay for AMACR activity based on a previously published assay for measuring cytochrome p450 activity (30, 31). In the plate-based assay, the enzyme (0.3 μM) is incubated with 150 μM labeled substrate [2, 3-3H] pristanoyl coenzyme A (Pri-CoA) (Fig. 1), in the presence or absence of 100 μM of a library compound producing 3H2O as a measure of enzymatic activity (Fig. 3). The reaction mixture is transferred to a C18 solid phase extraction (SPE) plate which allows the liberated tritiated water to be separated from any unmetabolized substrate. The level of enzymatic activity can be measured through the use of 96-well microplate scintillation counting. AMACR inhibition is quantified by comparing the number of disintegrations per min (DPM) from enzymatic product to plate controls containing no inhibitor or 100 μM of a known inhibitor (diethylpyrocarbonate (DEPC)) (2).
Kinetic Parameters of the High-Throughput Screen
The kinetic parameters for labeled substrate in the assay were determined (Fig. 4). Initial reaction rates were determined by incubating 0.3 μM AMACR-MBP with varying concentrations of [2, 3-3H] Pri-CoA (specific radioactivity was maintained at 60 Ci/mole) from which aliquots were taken at time points from 0 to 900 sec. Those samples were then treated as individual wells in our assay and processed as described above. The concentration-dependent time points were used to calculate reaction rates. Those rates were then directly plotted against concentrations and kinetic parameters calculated by non-linear regression (Fig. 4). The calculated kinetic values (Vmax: 294.9 ± 21.10 pmoles/min (standard error of the mean, SEM); Km: 85.57 ± 17.14 μM (SEM)) are similar to other published reports using human AMACR (1, 3).
High-Throughput Screen of 4896 Unique Small Molecules
The Johns Hopkins Drug Library (JHDL) contains 3280 distinct molecules the majority of which are drugs currently approved by the U.S. Food and Drug Administration, or its foreign counterparts, in addition to numerous other bioactive compounds (32). Those compounds were screened at a final concentration of 100 μM. Any compound that diminished the fractional activity by ≥ 20% was considered a candidate inhibitor (Supplementary Table S1). Of the 3,280 compounds screened in this library, 167 met the criteria of a minimum of 20% inhibition (5.1% positive hit rate), and of these, three were chosen for further analysis (Rose Bengal, Congo Red, and Ebselen) (Fig. 5). In screening the JHDL, the detection of mercury and copper containing compounds was an internal validation of the reliability of the assay (along with N-ethylmaleimide), as these compounds have been reported to be inhibitory elsewhere (S. Table 1) (2). AMACR has been shown to have an active site histidine (His122) (8). Therefore, the discovery of rose bengal, a compound known to selectively inhibit enzymes with active site histidine residues, indicated that the discovered compounds were relevant to AMACR biochemistry (33).
After screening the JHDL, two other small libraries of compounds from the NCI/DTP Open Chemical Repository (http://dtp.cancer.gov/) were obtained. The ‘natural products’ set is composed of 235 structurally diverse compounds. The ‘diversity set 2’ library contains 1,324 compounds that were chosen by the NCI/DTP based on several criteria as potential pharmacophores for biological activity. The screen of both of those libraries was performed identically to that of the JHDL, and the candidate inhibitors meeting the 20% inhibitory cutoff are listed in S. Table 2. Of the 1,559 compounds from the NCI/DTP Open Chemical Repository, 37 met the cutoff of a minimum of 20% inhibition (2.4% positive hit rate) and upon subsequent validation only four were chosen for further follow-up (Fig. 5). The detection of the inhibition of AMACR-MBP by mitoxantrone in both the JHDL and the NCI/DTP libraries indicates good reproducibility across library platforms (S. Tables 1 and 2).
Candidate Inhibitor Validation
After reproducing the results from the initial screen, several of the most potent inhibitors were subjected to subsequent analysis (Fig. 5). The potency of the candidate inhibitors was validated by determining their IC50 values. The IC50 values for characterized candidate inhibitors are listed in Fig. 5 and the IC50 curves themselves are in S. Fig. S3. Trihydroxycholestanoyl-Coenzyme A (compound 9), is a well characterized substrate and was included as a positive control for inhibition and as external validation of the SPE based assay (1, 2, 6). Ebselen oxide was tested as a commercially available derivative of the most potent inhibitor, ebselen, and showed an equivalent ability to inhibit AMACR (Fig. 5). As the first reported inhibitors of AMACR that are not substrates containing SCoA, the IC50 values for these compounds (1–8) ranged from 0.80 μM to 84.69 μM, which compares favorably to recently reported competitive inhibitors of the enzyme (Ki:0.9 – 137 μM), all of which are based on known AMACR substrates and contain the cell impermeable coenzyme A moiety (10).
Time-Dependent, Non-Dialyzable Inhibition by Candidate Inhibitors
During the validation experiments for ebselen, it was observed that the degree of inhibition increased depending on whether the reaction was initiated with substrate or enzyme. The only difference between those two conditions is the amount of time that the enzyme is pre-incubated with inhibitor prior to the addition of substrate (reaction initiation). Enzyme initiated reactions had no pre-incubation period and substrate initiated reactions had pre-incubation times of five minutes or more. In the presence of ebselen, a pre-incubation time of ≥ 5 minutes reduces the fractional activity of AMACR-MBP from ~50% to just above background, demonstrating a time-dependent component to ebselen inhibition (S. Fig. S3, inset).
Published reports demonstrating that ebselen is capable of covalent modification of proteins suggest that ebselen may cause an irreversible inhibition of AMACR that is resistant to dialysis (34, 35). In order to assess this possibility, experiments were setup whereby the AMACR-MBP was pre-incubated with inhibitor for 30 minutes at 4°C, dialyzed overnight, and then exposed to the substrate. The amount of activity remaining after dialysis was then compared to controls to determine to what extent the dialysis was able to recover AMACR-MBP activity. As shown in Fig. 6A, for none of the inhibitors tested, could activity be recovered. The only inhibitor for which there seemed to be any statistically significant recovery of activity was DPZBD, and even this was only marginal (Fig. 6A). To ensure that the compounds themselves were capable of being dialyzed, the UV-Vis spectrum of each reaction was checked before and after dialysis and in all cases the absorbance peaks of the inhibitors seen prior to dialysis were not seen after dialysis (data not shown). Neither congo red nor rose bengal could be included in that analysis as both have been shown to form non-dialyzable aggregates (36, 37).
The dialysis data (Fig 6A) suggests that the AMACR inhibitors may be acting as covalent inactivators. To further investigate this possibility we chose to carry out Kitz-Wilson analyses for our top two inhibitors (compounds 1–2, Fig. 5) (27, 28). Inhibition progress curves for compound 2 (DMPMB) were linear with increasing time and concentration (Fig. 6B, inset), and upon Kitz-Wilson transformation yielded an inactivation rate (ki(inact)) of 0.07521(±0.01067) min−1 and a KI(inact) of 24.32(±3.07) μM (Fig 6B). Unfortunately, the inhibition progress curves for ebselen are curvilinear and do not fit the standard Kitz-Wilson model; and transformation of the linear portion of these curves results in a negative ki(inact) (S. Fig. S5). It has been suggested in the literature that inhibitors with this pattern of Kitz-Wilson analysis may be indicative of multiple binding events (either multiple inactivator binding sites or multiple inactivator molecules binding to a single site) (38).
Non-Cysteine Dependent Inactivation by Ebselen
Ebselen has been shown to covalently modify cysteine residues in several proteins (34, 35). However, it has not been shown that any cysteine residues play a role in AMACR catalysis (7, 8). To gain further insight into the mechanism of inhibition by ebselen, we sought determine whether or not ebselen (100 μM) mediated inhibition could be prevented by pre-incubation of AMACR with cysteine specific alkylating agents (5 mM tributylphosphine (TBP), 15 mM iodoacetamide (IAA), TBP + IAA, 20 mM dithithreitol (DTT)). The enzyme was either first exposed to the alkylating agents and then directly incubated with ebselen prior to enzymatic measurement; or, the AMACR was dialyzed after exposure to alkylators (removing excess alkylator) and then ebselen was added. Both the enzymatic activity and the sensitivity to inhibition with alkylator/ebselen were tested (S. Fig. S6). The data for TBP indicate that TBP co-incubated with ebselen can protect AMACR from inhibition, presumably by alkylation of ebselen by excess TBP rather than direct protection of AMACR. When the excess TBP is dialyzed away, the enzyme is still sensitive to inhibition by ebselen. While IAA itself did slightly inhibit AMACR activity (~83% activity remaining after dialysis), it was not able to protect AMACR from ebselen mediated inhibition. The combination of TBP and IAA was slightly inhibitory after dialysis (~81% activity, similar to IAA alone) and did offer some degree of protection from ebselen prior to dialysis (~50% activity remaining, most likely attributable to the reaction of TBP with ebselen) but this protection was not present after dialysis. DTT did not protect AMACR from inhibition during co-incubation of both ebselen and DTT. The fact that none of the cysteine-specific reactive compounds affected AMACR activity confirms that there is not likely a cysteine mediated contribution to catalysis. Their failure to protect AMACR from ebselen inhibition indicates that cysteine modification by ebselen may not be the cause of AMACR inhibition
In Vitro Toxicity for Ebselen and DMPMB
AMACR inhibitors may have differential effects on cells based on the amount of AMACR protein being expressed. As a baseline for comparison, 4 immortalized human prostate cell lines with differing levels of AMACR expression were tested for toxicity in the presence of ebselen and DMPMB (Fig. 7A and 7B). WPMY1 is a non-tumorgenic human prostate stromal myofibroblast cell line that has undetectable AMACR protein by Western blot analysis (Fig. 7A, inset) (29). PC3, LAPC4, and LNCaP are three commonly used prostate cancer cell lines listed in order of increasing AMACR expression (Fig 7A, inset). For all four cell lines LD50 experiments were carried out by plating each line in triplicate at equal density in 96 well plates. The cells were then allowed to establish overnight before exposure to increasing quantities of either ebselen or DMPMB. For ebselen, the measured LD50 decreases with increasing AMACR expression (LD50’s, WPMY1:467.1±1.017μM, PC3:332.3±1.015μM, LAPC4:98.43±1.038μM, LNCaP: 71.96±1.106μM, Fig. 7A). This trend did not appear when measuring the LD50 for DMPMB in these cell lines: WPMY1:117.6±1.011μM, PC3:101.1±1.034μM, LAPC4: 58.78±1.058μM, 116.9±1.033μM (Fig. 7B).
Discussion
Recent controversy about prostate cancer screening has highlighted the need for non-invasive intraprostatic detection and monitoring of cancer (23, 24, 39). A tissue biomarker of prostate cancer like AMACR provides an ideal candidate for targeted molecular imaging and chemotherapy (19, 21). AMACR has been shown to be over-expressed in prostate adenocarcinoma by at least 10-fold over normal tissue and is used as an immunohistochemical marker of prostate cancer (11). To further exploit AMACR over-expression for both therapeutic and imaging purposes, we have created the first published 96-well based assay to screen for inhibitors, and report the first inhibitors for AMACR that are not SCoA-containing substrate analogs (Figs. 3 and 5).
Unlike its predecessors, our assay is not a single condition assay and can therefore be used to screen large numbers of compounds, concentrations, and conditions at the same time. Until now a high-throughput assay for AMACR activity has remained elusive for a variety of reasons, the most obvious being the fact that AMACR is a racemase that requires an SCoA moiety in its substrates (Fig. 1). As a racemase, AMACR is responsible for the stereoinversion of a single chiral center, and as such does not produce a product that is easily distinguishable from its substrate. There have been only two other published multi-well screens for inhibitors of any known racemase (40, 41). We demonstrate here a 96-well plate based AMACR assay capable of screening thousands of compounds and finding unique chemical entities that do not face the limitations of substrate based competitive inhibitors (26).
While validating the most potent inhibitors (ebselen and DMPMB, Fig. 5) we observed that this inhibition is both time-dependent and irreversible by dialysis (Fig. 6, S. Figs. S3 and S4). Furthermore, we demonstrate that both DMPMB and ebselen are irreversible inactivators of AMACR, the first uncompetitive AMACR inhibitors reported.
Ebselen has been previously reported to covalently modify several other proteins, act as a peroxidase, have anti-inflammatory properties, and be neuroprotective (34, 35, 42–44). It is important to consider that all of the above characteristics are not only beneficial but considered chemopreventative for cancer (45). In addition, ebselen has been used in two different clinical trials as a neuroprotective agent (44, 46). Neuroprotective doses of ebselen (300 mg/day) were well tolerated and could reach therapeutic levels through oral dosing (44, 46). Those pharmacologic properties make ebselen amenable to use in the setting of prostate cancer. To this end we have determined the LD50 for ebselen and DMPMB in a variety of prostate cell lines and have observed some interesting trends. For ebselen, cells that express more AMACR seem to be more sensitive to ebselen mediated cell death (Fig. 7). There is a 5-fold difference in LD50 between the highest AMACR expressing cell line (LNCaP) and the lowest AMACR expressor (WPMY1). Additionally, we demonstrate in Supplementary Figure S7 that both ebselen and DMPMB induced necrotic cell death. One possible explanation for this observation is that cell lines with high AMACR expression may become dependent on AMACR activity (for example branch chain lipid oxidation may provide additional reducing equivalents or intermediary metabolites like acetyl-coenzyme A) and are then sensitized to ebselen mediated AMACR inhibition. The observation that the AMACR inhibitors ebselen and DMPMB cause necrotic cell death (S. Fig. S7) is consistent with another published report of metabolic inhibitors causing necrosis rather than apoptosis, and supports the possibility that AMACR over-expression may make cells dependent on metabolites derived from AMACR reliant pathways (47). This ‘AMACR addiction’ is consistent with the observation that LAPC4 cells with decreased AMACR expression grow much more slowly than do the vector controls (Fig. 2). That there is a difference in sensitivity to ebselen between cells expressing AMACR and those that do not suggests that there is a therapeutic window for specifically targeting AMACR expressing cells (cancer) while sparing the non-AMACR expressing normal tissue.
Ebselen is known to inhibit other enzymes through covalent modification of cysteine residues (34, 35). We demonstrate that pre-incubation of AMACR with cysteine specific reagents (20 mM dithithreitol, 5 mM tributylphosphine, or 15 mM iodoacetamide) does not abrogate AMACR-MBP activity, nor does it prevent ebselen- mediated inhibition (S. Fig. S6). That suggests that ebselen is not simply alkylating an exposed cysteine (thought to be the mechanism for ebselen-mediated inhibition of other enzymes), but we hypothesize rather that it may alkylate the active site histidine (His122) specifically. Rose bengal has been reported to inhibit enzymes with an active site histidine reside, so it is perhaps not surprising that our assay discovered it to inhibit AMACR (33).
Along with its role in prostate cancer, the AMACR homolog in Mycobacterium tuberculosis (MCR) has been intimately tied to the field of AMACR research through use of both the MCR crystal structure and the enzyme itself as a stand-in for AMACR (7,8). It has not been established whether MCR is required for M. tuberculosis survival, but it will be interesting to see if our assay can be used to screen for MCR directed antibiotics and whether any of the compounds that we have outlined here may show antibiotic activity.
Intraprostatic tumor imaging remains elusive as the conventional agent, [18F] fluorodeoxyglucose with positron emission tomography (PET), has not proved useful in prostate cancer and all other agents remain at the experimental stage (20). As covalent inactivators of AMACR, ebselen, DMPMB, and MSDTP (unpublished observation), may accumulate in areas where AMACR is highly expressed (like prostate cancer) compared to areas with little to no AMACR expression (normal tissue), suggesting that PET/computed tomography imagible analogs of ebselen could represent an advance over non-covalent inhibitors. While we have only studied the properties of a single ebselen analog (ebselen oxide), there are numerous other synthetically available analogs (42, 48). We believe that our discovery of these potent inhibitors of AMACR activity warrant further preclinical investigation as imaging and chemotherapeutic agents for prostate cancer. Furthermore, the high-throughput assay that we have developed lends itself to the rapid screening of both new libraries as well as derivatives of the inhibitors described above.
Supplementary Material
Acknowledgments
Financial Support: This work is supported by funding from the U.S. Department of Defense (W81XWH-08-1-0073, B.A.P.W.), the Patrick C. Walsh Prostate Cancer Research Foundation (W.B.I, B.A.P.W.), and the National Institutes of Health (CA131702, CA92871, H.W., R.C.M., M.G.P.). The maintenance of JHDL (B.A.N., J.O.L.) is supported by NCI (CA122814), FAMRI, CTSA, and the Patrick C. Walsh Prostate Cancer Research Foundation.
We would like to thank Marc Rosen and Dr. Samuel R. Denmeade for training and use of HPLC equipment and reagents, and Kellen H. Burgwin for cytotoxicity discussions.
Abbreviations
- AMACR
α-Methylacyl Coenzyme A Racemase
- JHDL
Johns Hopkins Drug Library
- SCoA
Coenzyme A
- THCA-CoA
Trihydroxycoprostanoyl-Coenzyme A
- Pri-CoA
Pristanoyl-Coenzyme A
- MCR
Mycobacterium homolog of α-Methylacyl Coenzyme A Racemase
- HTS
High-Throughput Screen
- PET
Positron Emission Tomography
- DMPMB
2-(2,5-dihydroxy-4-methylphenyl)-5-methyl benzene-1,4-diol
- MSDTP
2-methylsulfanyl-7,9-dihydro-3H-purine-6,8-dithione
- DPZBD
2,5-di(pyrazol-1-yl)benzene-1,4-diol
- DPTD
3,5-di(pyridin-4-yl)-1,2,4-thiadiazole
- MBP
Maltose Binding Protein
- shRNA
Short-Hairpin Ribonucleic Acid
- HPLC
High Performance Liquid Chromatography
Footnotes
Potential Conflicts of Interest:
None
References
- 1.Schmitz W, Albers C, Fingerhut R, Conzelmann E. Purification and characterization of an α-methylacyl-CoA racemase from human liver. European Journal of Biochemistry. 1995;231(3):815–822. doi: 10.1111/j.1432-1033.1995.tb20766.x. [DOI] [PubMed] [Google Scholar]
- 2.Schmitz W, Fingerhut R, Conzelmann E. Purification and properties of an alpha-methylacyl-CoA racemase from rat liver. Eur J Biochem. 1994;222:313–323. doi: 10.1111/j.1432-1033.1994.tb18870.x. [DOI] [PubMed] [Google Scholar]
- 3.Darley DJ, Butler DS, Prideaux SJ, et al. Synthesis and use of isotope-labelled substrates for a mechanistic study on human α-methylacyl-CoA racemase 1A (AMACR; P504S) Org Biomol Chem. 2009;7:543–552. doi: 10.1039/b815396e. [DOI] [PubMed] [Google Scholar]
- 4.Ouazia D, Bearne SL. A continuous assay for α-methylacyl-coenzyme A racemase using circular dichroism. Analytical Biochemistry. 2010;398:45–51. doi: 10.1016/j.ab.2009.10.039. [DOI] [PubMed] [Google Scholar]
- 5.Sattar FA, Darley DJ, Politano F, Woodman TJ, Threadgill MD, Lloyd MD. Unexpected stereoselective exchange of straight-chain fatty acyl-CoA α-protons by human α-methylacyl-CoA racemase 1A (P504S) Chem Commun. 2010;46:3348–3350. doi: 10.1039/c002509g. [DOI] [PubMed] [Google Scholar]
- 6.Cuebas DA, Phillips C, Schmitz W, Conzelmann E, Novikov DK. The role of alpha-methylacyl-CoA racemase in bile acid synthesis. Biochemical Journal. 2002;363(Pt 3):801–807. doi: 10.1042/0264-6021:3630801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salvolainen K, Bhaumik P, Schmitz W, et al. α-Methylacyl-CoA Racemase from Mycobacterium tuberculosis: Mutational and structural characterization of the active site and the fold. Journal of Biological Chemistry. 2004;280:12611–12620. doi: 10.1074/jbc.M409704200. [DOI] [PubMed] [Google Scholar]
- 8.Bhaumik P, Schmitz W, Hassinen A, Hiltunen JK, Conzelmann E, Wierenga RK. The Catalysis of the 1, 1-Proton Transfer by [alpha]-Methyl-acyl-CoA Racemase Is Coupled to a Movement of the Fatty Acyl Moiety Over a Hydrophobic, Methionine-rich Surface. Journal of molecular biology. 2007;367:1145–1161. doi: 10.1016/j.jmb.2007.01.062. [DOI] [PubMed] [Google Scholar]
- 9.Ferdinandusse S, Denis S, Clayton PT, et al. Mutations in the gene encoding peroxisomal α-methylacyl-CoA racemase cause adult-onset sensory motor neuropathy. Nature genetics. 2000;24:188–191. doi: 10.1038/72861. [DOI] [PubMed] [Google Scholar]
- 10.Xu J, Stolk JA, Zhang X, et al. Identification of differentially expressed genes in human prostate cancer using subtraction and microarray. Cancer research. 2000;60:1677–1682. [PubMed] [Google Scholar]
- 11.Luo J, Zha S, Gage WR, et al. α-Methylacyl-CoA racemase: a new molecular marker for prostate cancer. Cancer research. 2002;62:2220–2226. [PubMed] [Google Scholar]
- 12.Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a Cell of Origin for Human Prostate Cancer. Science. 2010;329:568–571. doi: 10.1126/science.1189992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rubin MA, Zhou M, Dhanasekaran SM, et al. α-Methylacyl Coenzyme A Racemase as a Tissue Biomarker for Prostate Cancer. JAMA. 2002;287:1662–1670. doi: 10.1001/jama.287.13.1662. [DOI] [PubMed] [Google Scholar]
- 14.Zha S, Ferdinandusse S, Denis S, et al. α-Methylacyl-CoA Racemase as an Androgen-Independent Growth Modifier in Prostate Cancer. Cancer Research. 2003;63:7365–7376. [PubMed] [Google Scholar]
- 15.Kumar-Sinha C, Shah RB, Laxman B, et al. Elevated α-methylacyl-CoA racemase enzymatic activity in prostate cancer. American Journal of Pathology. 2004;164:787–793. doi: 10.1016/s0002-9440(10)63167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahara K, Azuma H, Sakamoto T, et al. Conversion of Prostate Cancer from Hormone Independency to Dependency Due to AMACR Inhibition: Involvement of Increased AR Expression and Decreased IGF1 Expression. Anticancer Research. 2009;29:2497–2505. [PubMed] [Google Scholar]
- 17.Savolainen K, Kotti TJ, Schmitz W, et al. A mouse model for α-methylacyl-CoA racemase deficiency: adjustment of bile acid synthesis and intolerance to dietary methyl-branched lipids. Human Molecular Genetics. 2004;13:955–965. doi: 10.1093/hmg/ddh107. [DOI] [PubMed] [Google Scholar]
- 18.Zhou M, Chinnaiyan AM, Kleer CG, Lucas PC, Rubin MA. Alpha-methylacyl-CoA racemase: a novel tumor marker over-expressed in several human cancers and their precursor lesions. The American journal of surgical pathology. 2002;26(7):926–931. doi: 10.1097/00000478-200207000-00012. [DOI] [PubMed] [Google Scholar]
- 19.Zlatopolskiy B, Morgenroth A, Urusova E, et al. IVC-11: A tracer for targeting of α-methylacyl-CoA racemase (AMACR) J Nucl Med Meeting Abstracts. 2009;50(2):1913. [Google Scholar]
- 20.Zaheer A, Cho SY, Pomper MG. New Agents and Techniques for Imaging Prostate Cancer. Journal of Nuclear Medicine. 2009;50:1387–1390. doi: 10.2967/jnumed.109.061838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turkbey B, Albert PS, Kurdziel K, Choyke PL. Imaging Localized Prostate Cancer: Current Approaches and New Developments. Am J Roentgenol. 2009;192:1471–1480. doi: 10.2214/AJR.09.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116:544–573. doi: 10.1002/cncr.24760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welch HG, Albertsen PC. Prostate Cancer Diagnosis and Treatment After the Introduction of Prostate-Specific Antigen Screening: 1986–2005. Journal of the National Cancer Institute. 2009;101:1325–1329. doi: 10.1093/jnci/djp278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brawley OW. Prostate Cancer Screening; Is This a Teachable Moment? Journal of the National Cancer Institute. 2009;101:1295–1297. doi: 10.1093/jnci/djp310. [DOI] [PubMed] [Google Scholar]
- 25.Van Veldhoven P, Croes K, Casteels M, Mannaerts G. 2-methylacyl racemase: a coupled assay based on the use of pristanoyl-CoA oxidase/peroxidase and reinvestigation of its subcellular distribution in rat and human liver. Biochim Biophys Acta. 1997;1347:62–8. doi: 10.1016/s0005-2760(97)00053-2. [DOI] [PubMed] [Google Scholar]
- 26.Carnell AJ, Hale I, Denis S, et al. Design, Synthesis, and In Vitro Testing of α-Methylacyl-CoA Racemase Inhibitors. J Med Chem. 2007;50:2700–2707. doi: 10.1021/jm0702377. [DOI] [PubMed] [Google Scholar]
- 27.Silverman RB. Enzyme Kinetics and Mechanism Part D: Developments in Enzyme Dynamics. Academic Press; 1995. Mechanism-based enzyme inactivators; pp. 240–283. [DOI] [PubMed] [Google Scholar]
- 28.Kitz R, Wilson IB. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. Journal of Biological Chemistry. 1962;237:3245–3250. [PubMed] [Google Scholar]
- 29.Webber MM, Trakul N, Thraves PS, et al. A human prostatic stromal myofibroblast cell line WPMY-1: a model for stromal–epithelial interactions in prostatic neoplasia. Carcinogenesis. 1999;20:1185–1192. doi: 10.1093/carcin/20.7.1185. [DOI] [PubMed] [Google Scholar]
- 30.Marco AD, Marcucci I, Verdirame M, Pérez J, et al. Development and Validation of a High-Throughput Radiometric CYP3A4/5 Inhibition Assay Using Tritiated Testosterone. Drug Metabolism and Disposition. 2005;33:349–358. doi: 10.1124/dmd.104.002873. [DOI] [PubMed] [Google Scholar]
- 31.Di Marco A, Cellucci A, Chaudhary A, Fonsi M, Laufer R. High-Throughput Radiometric CYP2C19 Inhibition Assay Using Tritiated (S)-Mephenytoin. Drug Metabolism and Disposition. 2007;35:1737–1743. doi: 10.1124/dmd.107.016345. [DOI] [PubMed] [Google Scholar]
- 32.Chong CR, Chen X, Shi L, Liu JO, Sullivan DJ. A clinical drug library screen identifies astemizole as an antimalarial agent. Nat Chem Biol. 2006;2:415–416. doi: 10.1038/nchembio806. [DOI] [PubMed] [Google Scholar]
- 33.Grillo FG, Aronson PS. Inactivation of the renal microvillus membrane Na+-H+ exchanger by histidine-specific reagents. Journal of Biological Chemistry. 1986;261:1120. [PubMed] [Google Scholar]
- 34.Walther M, Holzhutter HG, Kuban RJ, Wiesner R, Rathmann J, Kuhn H. The inhibition of mammalian 15-lipoxygenases by the anti-inflammatory drug ebselen: dual-type mechanism involving covalent linkage and alteration of the iron ligand sphere. Molecular Pharmacology. 1999;56:196. doi: 10.1124/mol.56.1.196. [DOI] [PubMed] [Google Scholar]
- 35.Sakurai T, Kanayama M, Shibata T, Itoh K, Kobayashi A, Yamamoto M, Uchida K. Ebselen, a Seleno-organic Antioxidant, as an Electrophile. Chem Res Toxicol. 2006;19:1196–1204. doi: 10.1021/tx0601105. [DOI] [PubMed] [Google Scholar]
- 36.Weiser HB, Radcliffe RS. The Physical Chemistry of Color Lake Formation. IV. Red Congo Acids and Red Congo Lakes. The Journal of Physical Chemistry. 1928;32:1875–1885. [Google Scholar]
- 37.Valdes-Aguilera O, Neckers DC. Aggregation of rose bengal ethyl ester induced by alkali metal cations in aqueous solution. Journal of Photochemistry and Photobiology A: Chemistry. 1989;47:213–222. [Google Scholar]
- 38.Kuo DJ, Jordan F. Active site directed irreversible inactivation of brewers’ yeast pyruvate decarboxylase by the conjugated substrate analog (E)-4-(4-chlorophenyl)-2-oxo-3-butenoic acid: development of a suicide substrate. Biochemistry. 1983;22:3735–3740. doi: 10.1021/bi00285a003. [DOI] [PubMed] [Google Scholar]
- 39.Schroder FH, Hugosson J, Roobol MJ, et al. Screening and prostate-cancer mortality in a randomized European study. The New England journal of medicine. 2009;360:1320–1328. doi: 10.1056/NEJMoa0810084. [DOI] [PubMed] [Google Scholar]
- 40.Henderickx HJW, Duchateau ALL, Raemakers-Franken PC. Chiral liquid chromatography-mass spectrometry for high-throughput screening of enzymatic racemase activity. Journal of Chromatography A. 2003;1020:69–74. doi: 10.1016/s0021-9673(03)00427-8. [DOI] [PubMed] [Google Scholar]
- 41.Dixon SM, Li P, Liu R, et al. Slow-Binding Human Serine Racemase Inhibitors from High-Throughput Screening of Combinatorial Libraries. J Med Chem. 2006;49:2388–2397. doi: 10.1021/jm050701c. [DOI] [PubMed] [Google Scholar]
- 42.Bhabak K, Mugesh G. Synthesis, Characterization, and Antioxidant Activity of Some Ebselen Analogues. Chem Eur J. 2007;13:4594–4601. doi: 10.1002/chem.200601584. [DOI] [PubMed] [Google Scholar]
- 43.Zhao R, Masayasu H, Holmgren A. Ebselen: a substrate for human thioredoxin reductase strongly stimulating its hydroperoxide reductase activity and a superfast thioredoxin oxidant. Proceedings of the National Academy of Sciences. 2002;99:8579–8584. doi: 10.1073/pnas.122061399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saito I, Asano T, Sano K, et al. Neuroprotective Effect of an Antioxidant, Ebselen, in Patients with Delayed Neurological Deficits after Aneurysmal Subarachnoid Hemorrhage. Neurosurgery. 1998;42:269–277. doi: 10.1097/00006123-199802000-00038. [DOI] [PubMed] [Google Scholar]
- 45.Bardia A, Platz EA, Yegnasubramanian S, De Marzo AM, Nelson WG. Anti-inflammatory drugs, antioxidants, and prostate cancer prevention. Current Opinion in Pharmacology. 2009;9:419–426. doi: 10.1016/j.coph.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamaguchi T, Sano K, Takakura K, Saito I, Shinohara Y, Asano T, Yasuhara H. Ebselen in Acute Ischemic Stroke: A Placebo-Controlled, Double-blind Clinical Trial. Stroke. 1998;29:12–17. doi: 10.1161/01.str.29.1.12. [DOI] [PubMed] [Google Scholar]
- 47.Zhang D, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 48.Parnham M, Biedermann J, Bittner C, Dereu N, Leyck S, Wetzig H. Structure-activity relationships of a series of anti-inflammatory benzisoselenazolones (BISAs) Agents Actions. 1989;27(3–4):306–8. doi: 10.1007/BF01972806. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.