

Abstract
In this Letter, we describe a novel three-step, one-pot procedure for the enantioselective synthesis of N-benzyl protected morpholines and orthogonally N,N′-protected piperazines with chiral alkyl groups installed at the C2 position of each heterocyclic core via organocatalysis. This methodology allows for the rapid preparation of functionalized morpholines and piperazines that are not readily accessible through any other chemistry in good to excellent % ee (55–98% ee).
Keywords: Morpholine, Piperazine, Enantioselective, Organocatalysis, Cyclization
Both morpholines (1) and piperazines (2) are widely used azaheterocyclic bases in organic synthesis;1 furthermore, they are a frequently found component in both natural products and pharmaceutical compositions,1–3 finding applications as key pharmacophores in antidepressants (3),4 antibiotics (4),5 antipsychotics (5),6 anticancer agents (6)7 and antihypertensive agents (7)8 (Fig. 1). Many of the pharmacologically relevant congeners possess chiral C-functionalization, frequently at C2.1 Importantly, synthetic chemistry to access enantiomerically pure C2-functionalized morpholines and piperazines is limited.1 The majority of synthetic efforts rely either on the resolution of racemic mixtures, or employ enantiopure amino alcohols and amino acids accessible from the chiral pool.1,9 Alas, reliance on the chiral pool limits the structural diversity of C2-functionalized congeners, decreasing the synthetic versatility of these methods.1 Recently, alternative approaches to these scaffolds are beginning to appear.10–13 With our NIH sponsored Molecular Probe Center Network (MLPCN) efforts,14 many screening hits are based on these motifs, and we required new synthetic methods to enable rapid lead optimization campaigns. In this Letter, we describe a novel three-step, one-pot procedure for the enantioselective synthesis of N-benzyl protected morpholines and orthogonally N,N′-protected piperazines with chiral alkyl groups installed at the C2 position of each heterocyclic core via organocatalysis.
Figure 1.

Structures of morpholine (1), piperazine (2) and pharmaceutical compositions 3–7, possessing C2-functionalization of these aza-heterocycles.
Our approach for the enantioselective synthesis of C2-functionalized morpholines and piperazines is based upon our recent application of organocatalysis to access chiral β-fluoroamines,15,16 which in turn, led us to develop a one-pot protocol for the enantioselective synthesis of N-alkyl terminal aziridines (Scheme 1).17 Here, commercial aldehydes 8 are subjected to an organocatalytic, enantioselective α-chlorination to produce 9,18 which then undergoes a reductive amination reaction with a primary amine, followed by base induced cyclization to deliver N-alkyl terminal aziridines 10 in good yields (40–65% for the one-pot, three step sequence) and good to excellent enantioselectivity (55–96% ee).17
Scheme 1.

Organocatalytic approach to chiral N-alkyl terminal aziridines 10.
Based on the precedent, we hypothesized that we should be able to employ an amine in the reductive amination step containing an embedded nucleophile (11 or 12), such that after based-induced cyclization of either 13 or 14, N-benzyl protected morpholines 15 and orthogonally N,N′-protected piperazines 16, respectively, with chiral alkyl groups (with inversion of stereochemistry) at the C2 position could be obtained (Scheme 2). Here, we recount our efforts toward the realization of this hypothesis.
Scheme 2.

Proposed organocatalytic approach to C-functionalized, N-protected morpholines 15 and orthogonally N,N′-protected piperazine 16.
We initially proceeded with a racemic chlorination employing 10 mol % d,l-proline as catalyst to validate the approach, and to provide access to racemic analogs to optimize separation of the enantiomers via chiral supercritical fluid chromatography (SFC) to determine % ee.15–17 Dodecanal 17 smoothly underwent the desired racemic α-chlorination to provide 18 after a quick pentane work-up. Reductive amination with 11 and NaB(OAc)3H at −78 °C, followed by KOtBu-induced cyclization in CH3CN at −10 °C provided racemic C2-functionalized, benzyl-protected morpholine 19 (Scheme 3). While we were thrilled to see the approach successful, the overall yield for the three step, one-pot process was low (11% overall, or ~48% yield per step). Of note, the reductive amination step required low temperature, −78 °C. Above −78 °C, we observed severe racemization of the α-stereocenter, elimination, alkylation and other by-products, while at −78 °C, reductive aminations proceeded smoothly with amines such as 11 and 12.
Scheme 3.

Three step, one-pot synthesis of C2-functionalized, N-benzyl protected morpholine 19.
Efforts now focused on optimization of the reaction sequence. The racemic α-chlorination was a robust step, affording 18 and related congeners in yields of >90% routinely; thus, the low yields had to be the result of either the reductive amination and/or the base-induced cyclization step. In addition to 19, the major isolated by-product from the one-pot process was the α-chloroaldehyde 18; however, by TLC, LCMS, and NMR analysis of the crude reactions, 18 was consumed. This led us to speculate that the incipient iminium ion 20 could either be reduced by the hydride to provide the desired 13, or the free hydroxyl could attack 20 producing an oxazolidine 21, which could be hydrolyzed back to 18 upon aqueous work-up and/or chromatography (Scheme 4). To explore this possibility, an NMR study was conducted and confirmed that a stereoisomeric mixture of oxazolidines 21 was being formed, as shown by the shift and coupling of the two characteristic downfield diastereotopic protons indicated below. To improve the yield of the three step, one-pot process, we would need to prevent this undesired oxazolidine pathway.
Scheme 4.

Competing pathways: undesired oxazolidine 21 formation.
Two approaches were pursued to prevent the formation of 21. First, we protected the hydroxyl moiety as a TBDMS ether such that the silyl analog of 13 was generated without the ability to produce 21. A subsequent TBAF deprotection and cyclization afforded race-mic morpholine 19 in 35% overall yield, a notable improvement; however, this route added an additional protection/deprotection sequence. Alternatively, we focused on the reducing agent. Simply replacing NaB(OAc)3H/DCM with NaCNBH3/5% HOAc/THF provided morpholine 19 once again in 35% overall yield for the three steps (~70%/step); therefore, these latter conditions were employed to generate the racemic examples for SFC separation and % ee determination. The enantioselective congeners utilized the same protocol, except the d,l-proline catalyst was replaced with 10 mol % (2R,5R)-diphenylpyrrolidine.18,19 This proved to be the optimal catalyst/chlorination system, as it proceeds in DCM; the majority of other organocatalysts/chlorination systems require acetone as solvent, which is not compatible with our one-pot reductive amination sequence, as described in our earlier aziridine work.17 Therefore, asymmetric α-chlorination, reductive amination and cyclization afforded enantioenriched C2-functionalized morpholines 19, 22 and 23 in 76–98% ee (Fig. 2).20 Interestingly, the one-pot, three step yields for the racemic analogs were higher (35–40% overall or 70–75% per step) than for the enantioenriched congeners (13–19% overall or 50–58% per step).
Figure 2.

Structures and yields for the one-pot, three step synthesis of racemic and enantioenriched C2-functionalized, N-benzyl protected morpholines.
We next explored the synthesis of orthogonally N,N′-protected piperazines 16. Here, we did not need to worry about the undesired oxazolidine pathway, so we piloted our initial morpholine conditions. To our surprise, the one-pot protocol afforded 24 in good yield, with very little of the desired 25 (Scheme 5), suggesting the cyclization conditions required optimization. After surveying a number of solvents (THF, CH3CN and DMF), bases (NaH, KOtBu, KHMDS) and temperatures, we found that simply replacing the CH3CN with DMF and employing KOtBu at −20 °C enabled the formation of 25 from 24 in 98% conversion. Application of this modification to the one-pot, three step protocol (Scheme 6) delivered racemic 25 in 47% overall yield (78% per step).
Scheme 5.

First attempt at a three step, one-pot synthesis of a C2 functionalized, orthogonally N,N′-protected piperazine 25.
Scheme 6.

Three step, one-pot synthesis of C2-functionalized, orthogonally N,N′-protected piperazine 25.
As shown in Table 1, the three step, one-pot yields for orthogonally N,N′-protected piperazines were uniformly better (15–50% overall or 53–79% per step) than for the analogous morpholines, and % ee, obtained by chiral SFC analysis, remained good to excellent (55–96% ee).21
Table 1.
Structures and yields for enantioenriched C2-functionalized, orthogonally N,N′-protected piperazines
| Compd | Piperazines | 3-Step, one-pot yield % (racemic) |
% eea |
|---|---|---|---|
| 25 | 50 (47) | 96 | |
| 26 | 45 (55) | 92 | |
| 27 | 21 (31) | 55 | |
| 28 | 15 (21) | 70 |
% ee determined by chiral SFC.
Finally, since the piperazines worked well, we elected to see if this approach would allow entry into enantioenriched homopiperazines, the seven-membered ring congeners of 16. Beginning with 17, racemic organocatalytic α-chlorination, followed by reductive amination with the N-benzyl-N-Boc protected diaminopropane delivered 29 in 55% overall yield for the two steps. Our standard base-induced cyclization conditions for piperazines (KOtBu, DMF, −20 °C) did deliver the desired C2-functionalized, orthogonally N,N′-protected homopiperazine 30, but as a 4:1 mixture with the elimination product 31 in 65% yield (Scheme 7). This was not totally unexpected, as the cyclization to produce the seven-membered ring was anticipated to be slow, allowing the competing elimination pathway to the alkene to compete. All attempts to modify the reaction conditions to increase the rate of cyclization leading to 30 and diminish the production of 31 were unsuccessful; therefore we did not produce an enantioselective variant of 30.
Scheme 7.

Three step, one-pot synthesis of C2-functionalized, orthogonally N,N′-protected homopiperazine 30.
In summary, we have developed a novel three-step, one-pot procedure for the enantioselective synthesis of N-benzyl protected morpholines and orthogonally N,N′-protected piperazines with chiral alkly groups installed at the C2 position of each heterocyclic core via organocatalysis. Notably, this methodology does not rely on the chiral pool; instead we can employ simple aldehydes and commercial organocatalysts. Thus, either enantiomer of the corresponding morpholines and piperazines can be arrived at by employing either the (R)- or (S)-organocatalyst. This methodology allows for the rapid preparation of functionalized, pharmaceutically relevant morpholines and piperazines in 13–50% overall yield (50–79% per step) that are not readily accessible through any other chemistry in good to excellent % ee (55–98% ee). Further refinements and improvements are under development and will be reported in due course.
Acknowledgments
The authors gratefully acknowledge funding from the Department of Pharmacology, Vanderbilt University Medical Center and the NIH/Molecular Libraries Production Center Network (MLPCN) (U54MH084659). M.C.O. acknowledges a predoctoral fellowship (2009–2010) from the Vanderbilt Institute of Chemical Biology (VICB) and an ACS Division of Medicinal Chemistry Predoctoral Fellowship (2011–2012) for support.
References and notes
- 1.Wijtmans R, Vink MKS, Schoemaker HE, van Delft FL, Blaauw RH. Synthesis. 2004;5:641–662. and references therein. [Google Scholar]
- 2.Elliott S. Drug Test. Anal. 2011;3:430–438. doi: 10.1002/dta.307. [DOI] [PubMed] [Google Scholar]
- 3.Loeber S, Heubner H, Tschammer N, Gmeiner P. Trends Pharmacol. Sci. 2011;32:148–157. doi: 10.1016/j.tips.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Hajos M, Fleishaker JC, Filipak-Reisner JK, Brown MT, Wong EHF. CNS Drug Rev. 2004;10:23–44. doi: 10.1111/j.1527-3458.2004.tb00002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakurai N, Sano M, Hirayama F, Kuroda T, Uemori S, Moriguchi A, Yamamoto K, Ikeda Y, Kawakita T. Bioorg. Med. Chem. Lett. 1998;8:2185–2190. doi: 10.1016/s0960-894x(98)00390-4. [DOI] [PubMed] [Google Scholar]
- 6.Kulagowski JJ, Broughton HB, Curtis NR, Mawer IM, Ridgill MP, Baker R, Emms F, Freedman SB, Marwood R, Patel S, Ragan CI, Leeson PD. J. Med. Chem. 1996;39:1941–1942. doi: 10.1021/jm9600712. [DOI] [PubMed] [Google Scholar]
- 7.Lawrence NJ, Pireddu RS, Sebti SM. 2011 WO 130740. [Google Scholar]
- 8.Scheiper B, Matter H, Steinhagen H, Bocskei Z, Fleury V, McCort G. Bioorg. Med. Chem. Lett. 2011;21:5480–5486. doi: 10.1016/j.bmcl.2011.06.114. [DOI] [PubMed] [Google Scholar]
- 9.Otto WG. Angew. Chem. 1956;68:181–183. [Google Scholar]
- 10.Yar M, McGarrigle EM, Aggarwal VK. Angew. Chem. Int. Ed. 2008;47:3784–3786. doi: 10.1002/anie.200800373. [DOI] [PubMed] [Google Scholar]
- 11.Yar M, McGarrigle EM, Aggarwal VK. Org. Lett. 2009;11:257–260. doi: 10.1021/ol8023727. [DOI] [PubMed] [Google Scholar]
- 12.Lanman BA, Myers AG. Org. Lett. 2004;6:1045–1047. doi: 10.1021/ol049861t. [DOI] [PubMed] [Google Scholar]
- 13.Bornholdt J, Felding J, Kristensen JL. J. Org. Chem. 2010;75:7454–7457. doi: 10.1021/jo101339g. [DOI] [PubMed] [Google Scholar]
- 14.For information on the MLPCN see: http://mli.nih.gov/mli/mlpcn.
- 15.Fadeyi OO, Lindsley CW. Org. Lett. 2009;11:943–946. doi: 10.1021/ol802930q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulte ML, Lindsley CW. Org. Lett. 2011;13:5684–5687. doi: 10.1021/ol202415j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fadeyi OO, Schulte ML, Lindsley CW. Org. Lett. 2010;12:3276–3278. doi: 10.1021/ol101276x. [DOI] [PubMed] [Google Scholar]
- 18.Halland N, Braunton A, Bachmann S, Marigo M, Jørgensen KA. J. Am. Chem. Soc. 2004;126:4790–4791. doi: 10.1021/ja049231m. [DOI] [PubMed] [Google Scholar]
- 19.Marigo M, Bachmann S, Halland N, Braunton A, Jørgensen KA. Angew Chem. Int. Ed. 2004;43:5507–5510. doi: 10.1002/anie.200460462. [DOI] [PubMed] [Google Scholar]
-
20.Representative experimental for the synthesis of enantioenriched C2-functionalized morpholines: To a solution of aldehyde (1 mmol, 1 equiv) and (2R,5R)-2,5-diphenylpyrrolidine (22.3 mg, 0.1 mmol, 0.1 equiv) in DCM (2.0 mL) was added N-chlorosuccinimide (173 mg, 1.3 mmol, 1.3 equiv) at 0 °C. The reaction was kept at 0 °C for 1 h at which point it was allowed to warm to ambient temperature. It was then stirred until the aldehyde was completely consumed as determined by 1H NMR spectroscopy of the reaction mixture. Pentane was added to the reaction mixture at −78 °C and the precipitated NCS, succinimide, and catalyst were filtered off. After removal of the solvent at 0 °C, the filtration process was repeated one time. The crude oil was redissolved in THF (3 mL) and 4 Å molecular sieves (500 mg) were added. The reaction was cooled to −78 °C and a solution of amine (1.5 mmol, 1.5 equiv) in THF (2 mL) was added followed by the addition of NaBH3CN (100.5 mg, 1.6 mmol, 1.6 equiv) and acetic acid (120.1 mg, 2 mmol, 2 equiv). The reaction vessel was purged and left to stir at −78 °C for greater than 16 h at which time it was filtered through a pad of celite eluting with ethyl acetate. It was extracted with ethyl acetate and washed with saturated NaHCO3 and brine, and the organic layer was dried over MgSO4 followed by concentration in vacuo resulting in a crude oil, which was then redissolved in acetonitrile (50 mL). The solution was cooled to −20 °C followed by the addition of KOtBu (560 mg, 5 mmol, 5 equiv). It was then stirred until the β-chloroaminoalcohol was completely consumed as determined by thin layer chromatography. The reaction was extracted with diethyl ether (3×) and washed with brine. The crude oil was concentrated and purified by flash column chromatography (Hexanes/Ethyl Acetate) to afford the product. Enantiomeric Excess was determined via chiral SFC analysis.
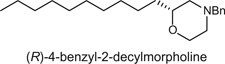 The product was prepared according to the above description and was purified by silica chromatography (9:1 Hexanes/EtOAc) to afford the product as a clear oil (57 mg, 18%). 1H NMR (400.1 MHz, CDCl3) δ (ppm): 1HNMR (400.1 MHz, CDCl3) δ (ppm): 7.32-7.24 (m, 5H); 3.84 (dq, J1 = 11.5 Hz, J2 = 1.8, 1H); 3.65 (td, J1 = 11.5, J2 = 2.5, 1H); 3.52-3.45 (m, 3H), 2.74-2.71, (m, 1H), 2.65 (dq, J1 = 11.4, J2 = 1.8, 1H), 2.14 (td, J1 = 11.4, J2 = 3.3, 1H), 1.87-1.82 (m, 1H), 1.5-1.25 (m, 18H), 0.87 (t, J = 7.1, 3H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 137.76, 129.11, 128.17, 127.03, 75.76, 66.73, 63.29, 58.80, 53.12, 33.67, 31.83, 29.60, 29.52, 29.51, 29.45, 29.24, 25.33, 22.60, 14.04. Specific rotation (c 100, MeOH).
The product was prepared according to the above description and was purified by silica chromatography (9:1 Hexanes/EtOAc) to afford the product as a clear oil (57 mg, 18%). 1H NMR (400.1 MHz, CDCl3) δ (ppm): 1HNMR (400.1 MHz, CDCl3) δ (ppm): 7.32-7.24 (m, 5H); 3.84 (dq, J1 = 11.5 Hz, J2 = 1.8, 1H); 3.65 (td, J1 = 11.5, J2 = 2.5, 1H); 3.52-3.45 (m, 3H), 2.74-2.71, (m, 1H), 2.65 (dq, J1 = 11.4, J2 = 1.8, 1H), 2.14 (td, J1 = 11.4, J2 = 3.3, 1H), 1.87-1.82 (m, 1H), 1.5-1.25 (m, 18H), 0.87 (t, J = 7.1, 3H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 137.76, 129.11, 128.17, 127.03, 75.76, 66.73, 63.29, 58.80, 53.12, 33.67, 31.83, 29.60, 29.52, 29.51, 29.45, 29.24, 25.33, 22.60, 14.04. Specific rotation (c 100, MeOH).
-
21.Representative experimental for the synthesis of enantioenriched C2-functionalized orthogonally N,N′-protected piperazines: To a solution of aldehyde (1 mmol, 1 equiv) and (2R,5R)-2,5-diphenylpyrrolidine (22.3 mg, 0.1 mmol, 0.1 equiv) in DCM (2.0 mL) was added N-chlorosuccinimide (173 mg, 1.3 mmol, 1.3 equiv) at 0 °C. The reaction was kept at 0 °C for 1 h at which point it was allowed to warm to ambient temperature. It was then stirred until the aldehyde was completely consumed as determined by 1H NMR spectroscopy of the reaction mixture. Pentane was added to the reaction mixture at −78 °C and the precipitated NCS, succinimide, and catalyst were filtered off. After removal of the solvent at 0 °C, the filtration process was repeated one time. The crude oil was redissolved in DCM (3 mL) and 4 Å molecular sieves (500 mg) were added. The reaction was cooled to −78 °C and a solution of amine (1.5 mmol, 1.5 equiv) in DCM (2 mL) was added followed by the addition of NaBH(OAc)3 (339 mg, 1.6 mmol, 1.6 equiv). The reaction vessel was purged and left to stir at −78 °C for greater than 16 h at which time it was filtered through a pad of celite eluting with ethyl acetate. It was extracted with ethyl acetate and washed with saturated NaHCO3 and brine, and the organic layer was dried over MgSO4 followed by concentration in vacuo resulting in a crude oil, which was then redissolved in DMF (50 mL). The solution was cooled to −20 °C followed by the addition of KOtBu (560 mg, 5 mmol, 5 equiv). It was then stirred until the β-chlorodiamine was completely consumed as determined by thin layer chromatography. The reaction was extracted with diethyl ether (3 ×) and washed with brine. The crude oil was concentrated and purified by flash column chromatography (9:1 Hexanes/Ethyl Acetate) to afford the product. Enantiomeric Excess was determined via chiral SFC analysis.
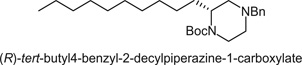 The product was prepared according to the above description and was purified by silica chromatography (9:1 Hexanes/EtOAc) to afford the product as a clear oil (208 mg, 50%). 1H NMR (400.1 MHz, CDCl3) δ (ppm): 1HNMR (400.1 MHz, CDCl3) δ (ppm): 7.32-7.21 (m, 5H); 3.99 (s, br r, 1H); 3.90-3.82 (m, 1H); 3.53 (d, J = 13.2, 1H); 3.37 (d, J = 13.3, 1H); 3.05 (t, J = 12.5, 1H); 2.75-2.65 (m, 2H); 2.06-2.00 (m, 2H); 1.78-1.73 (m, 1H); 1.66-1.61 (m, 1H); 1.45 (s, 9H); 1.30-1.15 (m, 16H); 0.88 (t, J = 7.5, 3H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 154.81, 138.39, 128.68, 128.12, 126.93, 79.18, 62.80, 55.39, 53.29, 51.37, 39.24, 31.84, 29.74, 29.59, 29.56, 29.53, 29.50, 29.27, 28.38, 26.20, 22.52, 14.04. Specific rotation (c 100, MeOH).
The product was prepared according to the above description and was purified by silica chromatography (9:1 Hexanes/EtOAc) to afford the product as a clear oil (208 mg, 50%). 1H NMR (400.1 MHz, CDCl3) δ (ppm): 1HNMR (400.1 MHz, CDCl3) δ (ppm): 7.32-7.21 (m, 5H); 3.99 (s, br r, 1H); 3.90-3.82 (m, 1H); 3.53 (d, J = 13.2, 1H); 3.37 (d, J = 13.3, 1H); 3.05 (t, J = 12.5, 1H); 2.75-2.65 (m, 2H); 2.06-2.00 (m, 2H); 1.78-1.73 (m, 1H); 1.66-1.61 (m, 1H); 1.45 (s, 9H); 1.30-1.15 (m, 16H); 0.88 (t, J = 7.5, 3H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 154.81, 138.39, 128.68, 128.12, 126.93, 79.18, 62.80, 55.39, 53.29, 51.37, 39.24, 31.84, 29.74, 29.59, 29.56, 29.53, 29.50, 29.27, 28.38, 26.20, 22.52, 14.04. Specific rotation (c 100, MeOH).

