

Abstract
BACKGROUND AND PURPOSE
The cannabinoid CB1 receptor is the chief mediator of the CNS effects of cannabinoids. In cell culture model systems, CB1 receptors both desensitize and internalize on activation. Previous work suggests that the extreme carboxy-terminus of this receptor regulates internalization via phosphorylation of residues clustered within this region. Mutational analysis of the carboxy-terminus of CB1 receptors has demonstrated that the last six serine/threonine residues are necessary for agonist-induced internalization. However, the structural determinants of CB1 receptor internalization are also dependent on the local cellular environment. The importance of cell context on CB1 receptor function calls for an investigation of the functional roles of these residues in neurones.
EXPERIMENTAL APPROACH
To determine the structural requirements of CB1 internalization in neurones, we evaluated the signalling properties of carboxy-terminal mutated CB1 receptors expressed in cultured autaptic hippocampal neurones, using electrophysiological methods.
KEY RESULTS
CB1 receptors transfected into CB1 knockout neurones signalled and desensitized as did wild-type neurones, allowing us to test specific CB1 receptor mutations. Deletion of the last 13 residues yielded a CB1 receptor that inhibited excitatory postsynaptic currents but did not desensitize. Furthermore, mutation of the final six serine and threonine residues to alanines resulted in a non-desensitizing receptor. In contrast, CB1 receptors lacking residues 419–460, leaving the last 14 residues intact, did desensitize.
CONCLUSIONS AND IMPLICATIONS
The distal thirteen residues of CB1 receptors are crucial for their desensitization in cultured neurones. Furthermore, this desensitization is likely to follow phosphorylation of serines and threonines within this region.
LINKED ARTICLES
This article is part of a themed section on Cannabinoids in Biology and Medicine. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2012.165.issue-8. To view Part I of Cannabinoids in Biology and Medicine visit http://dx.doi.org/10.1111/bph.2011.163.issue-7
Keywords: endocannabinoid, DSE, 2-AG, retrograde transmission
Introduction
The cannabinoid CB1 receptor is the chief mediator of the CNS effects of cannabinoids and it is these receptors that are activated by exogenous cannabinoids such as THC, the chief psychoactive component of marijuana and hashish (Gaoni and Mechoulam, 1964; receptor nomenclature follows Alexander et al., 2011). As such, an understanding of the function and regulation of these receptors is critical to understanding the nature of this psychoactivity as well as potential therapeutic consequences of CB1 receptor activation. CB1 receptors are members of the GPCR superfamily. More specifically, they are members of the class A subfamily, consisting of rhodopsin-like GPCRs. Activation of CB1 receptors has been shown to act chiefly via Gi/o-type G-proteins, resulting in inhibition of calcium channels and activation of inwardly rectifying potassium channels; CB1 receptors have also been shown to activate MAPKs and inhibit adenylyl cyclase (Howlett and Fleming, 1984; Mackie and Hille, 1992; Bouaboula et al., 1995; Mackie et al., 1995). CB1 receptors are widely distributed throughout the CNS (Herkenham et al., 1991), where they mediate a diverse array of behavioural effects in animal models, including hypolocomotion, hypothermia, anti-nociception and catalepsy (Howlett et al., 2002).
Behavioural and physiological tolerance to cannabinoids develops over the course of minutes to hours and is chiefly mediated by changes in the CB1 receptor (Bass and Martin, 2000). Chronic cannabinoid exposure has generally been found to decrease CB1 receptor function and/or binding sites (Breivogel et al., 1999; 2003), most likely via uncoupling of the receptor from its G proteins and/or internalization (Rinaldi-Carmona et al., 1998; Hsieh et al., 1999; Jin et al., 1999; Coutts et al., 2001; Kouznetsova et al., 2002; Martin et al., 2004). For this reason, it is important to identify the molecular events involved in CB1 receptor desensitization to grasp the implications of chronic cannabis use and to fully exploit the therapeutic potential of the cannabinoid signalling system.
Activation of GPCRs induces the dissociation of G-proteins into α and βγ subunits. The βγ subunits then typically recruit members of the G-protein receptor kinase (GRK) family, which in turn phosphorylate specific serine and threonine residues on the GPCR. This phosphorylated region, generally in the carboxy-terminus of the receptor, binds β-arrestin, thereby uncoupling the receptor from further G-protein signalling. β-Arrestin additionally promotes clathrin-dependent internalization of GPCRs (see Claing et al., 2002; Gainetdinov et al., 2004), although internalization can also occur via non-clathrin-mediated pathways (Wu et al., 2008). Because of the considerable variations between GPCRs (∼500), G-proteins (∼25) and effector/modulator proteins, the mechanisms of desensitization vary considerably among GPCRs. As a consequence, it is necessary to study the desensitization of each receptor individually.
The essential structural requirements of the CB1 receptor for agonist-induced desensitization remain to be elucidated in neurones. In cell lines, the last 14 residues of the CB1 receptor are involved in internalization-dependent desensitization. In AtT20 cells, a murine cell line with pituitary origins, the deletion of these last residues completely prevents internalization (Hsieh et al., 1999; Daigle et al., 2008). This analysis is complicated by the observation that, in HEK293 cells, deletion of these residues had no impact on internalization (Daigle et al., 2008). A cluster of six serines and threonines within this tail region represent candidate GRK phosphorylation sites and, in HEK293 cells, mutation of these six residues to alanine prevents internalization, suggesting that phosphorylation of these residues might be involved in CB1 receptor internalization.
It is not surprising that different cell lines, expressing distinct complements of signalling machinery (Atwood et al., 2011), should differentially regulate GPCR desensitization. Certainly heterologous expression systems serve as excellent models for a first-pass study of GPCR signalling. However, recent advances in neuronal transfection and the discovery of an intact retrograde cannabinoid signalling system in cultured neurones, offer an opportunity for detailed investigation of the mechanisms of CB1 receptor-mediated signalling in a neuronal context (Ohno-Shosaku et al., 2001; 2002; Straiker and Mackie, 2005; 2007; 2009). Consequently, we transfected wild-type rat CB1 (WT rCB1) and various mutant CB1 receptors into neurones cultured from CB1 receptor knockout (CB1−/−) mice. WT CB1 receptors transfected into CB1−/− neurones signalled and desensitized as effectively as endogenously expressed WT CB1 receptors, while transfection of specific CB1 receptor mutants resulted in pronounced, mutation-specific effects on both baseline signalling and desensitization.
Methods
Culture preparation
All animal care and experimental procedures used in this study were approved by the Animal Care Committee of the Indiana University and conform to the Guidelines of the National Institutes of Health on the Care and Use of Animals. Mouse (CD1 strain) hippocampal neurones isolated from the CA1–CA3 region were cultured on microislands as described previously (Furshpan et al., 1976; Bekkers and Stevens, 1991). Neurones were obtained from animals (age postnatal day 0–2) and plated onto a feeder layer of hippocampal astrocytes that had been laid down previously (Levison and McCarthy, 1991). Cultures were grown in high-glucose (20 mM) medium containing 10% horse serum, without mitotic inhibitors and used for recordings after 8 days in culture and for no more than 3 h after removal from culture medium.
Electrophysiology
When a single neuron is grown on a small island of permissive substrate, it forms synapses – or ‘autapses’– onto itself. All experiments were performed on isolated autaptic neurones. Whole cell voltage-clamp recordings from autaptic neurones were carried out at room temperature using an Axopatch 200A amplifier (Axon Instruments, Burlingame, CA). The extracellular solution contained (in mM) 119 NaCl, 5 KCl, 2.5 CaCl2, 1.5 MgCl2, 30 glucose and 20 HEPES. Continuous flow of solution through the bath chamber (∼2 mL·min−1) ensured rapid drug application and clearance. Drugs were typically prepared as stocks then diluted into extracellular solution at their final concentration and used on the same day.
Recording pipettes of 1.8–3 MΩ were filled with (in mM) 121.5 K gluconate, 17.5 KCl, 9 NaCl, 1 MgCl2, 10 HEPES, 0.2 EGTA, 2 MgATP and 0.5 LiGTP. Access resistance and holding current were monitored, and only cells with both stable access resistance and holding current were included for data analysis. Conventional stimulus protocol: The membrane potential was held at −70 mV and excitatory postsynaptic currents (epscs) were evoked every 20 s by triggering an unclamped action current with a 1.0 ms depolarizing step. The resultant evoked waveform consisted of a brief stimulus artifact and a large downward spike representing inward sodium currents, followed by the slower epsc. The size of the recorded epscs was calculated by integrating the evoked current to yield a charge value (in pC). Calculating the charge value in this manner yields an indirect measure of the amount of neurotransmitter released while minimizing the effects of cable distortion on currents generated far from the site of the recording electrode (the soma). Data were acquired at a sampling rate of 5 kHz.
Depolarization-induced suppression of excitation (DSE) stimuli: After establishing a 10–20 s 0.5 Hz baseline, DSE was evoked by depolarizing to 0 mV for 1–10 s, followed by resumption of a 0.5 Hz stimulus protocol for 10–80+ s, until epscs recovered to baseline values.
Neuronal transfection
We transfected neurones using a calcium phosphate-based method adapted from Jiang et al. (2004). Briefly, plasmids for the protein of interest and enhanced yellow fluorescent protein (EYFP; 2 µg per well) were combined with 2 M CaCl2 in water and gradually added to HEPES buffered saline (HBS); the mixture was added to the serum-free neuronal media. Coverslips were incubated with this mixture for 2.5 h, while additional serum-free media was placed in a 10% CO2 incubator to induce acidification. At the end of 2.5 h, the reaction mixture was replaced with acidified serum-free media for 20 min. After this cells were returned to their home wells containing the original media. Each data set was taken from at least two different neuronal platings.
The rCB1 receptor as well as the V460Z and six-point mutant plasmids were described previously (Hsieh et al., 1999; Jin et al., 1999; Daigle et al., 2008). The 419–460 mutant was made by overlap extension (Ho et al., 1989), and BamHI and NotI restriction sites were introduced. Amplicons were cut, gel purified and ligated into a pcDNA3.0 expression vector previously altered to contain a Kozak start site followed by an HA11 epitope tag between BamHI and EcoRI restriction sites on the MCS. All inserts were confirmed via ABI sequencing.
Experimental design and statistical analyses
As a rule, experiments were accompanied by same-day controls (e.g. rCB1 receptor transfected responses were accompanied by non-transfected negative controls). For desensitization experiments, neurones were treated overnight with WIN 55212-2 (WIN) in vehicle (0.001% DMSO). We conducted a vehicle control for rCB1 (see Results); the remaining controls consisted of untreated neurones. We have reported that WIN washes out with a half-life of 5.5 min (Straiker and Mackie, 2005). As a consequence, after overnight treatment with WIN, we allowed a washout period of at least 20 min before testing for DSE.
As noted previously (Straiker and Mackie, 2005), due to inherent variability of baseline epscs, we have normalized responses to unity (the epsc charge at the beginning of the recording session). However, we did not observe a statistically significant difference in baseline epsc size after transfection with rCB1 or any of the mutant receptors [epsc sizes (in nA): rCB1: 1.3 ± 0.2; 6Pt mutant: 1.6 ± 0.3; V460Z: 2.2 ± 0.4; G→K: 1.8 ± 0.4, P > 0.05, one-way anova]. Nor did overnight WIN treatment result in a reduction in epsc size (epsc sizes for rCB1: 3.0 ± 1.3 nA ).
Values are presented as means with SEM or 95% confidence intervals (CI). Statistical comparisons of ‘dose–response’ curves used two-way anova, with a subsequent Bonferroni post hoc test, to assess pairs of responses along the curve. In Figure 5, we instead used a one-way anova with a Dunnett's post hoc test, because this allowed multiple comparisons against a single control group (rCB1). The remaining statistical analyses were with unpaired t-tests.
Figure 5.
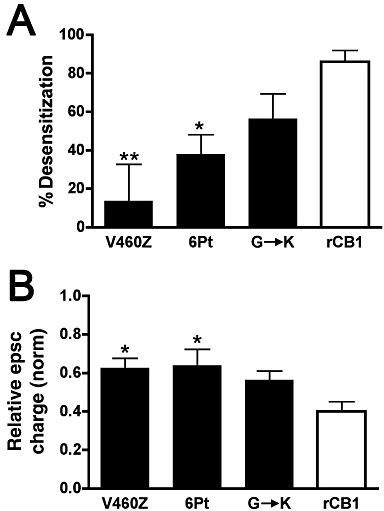
Carboxy-terminal mutations of CB1 receptors have the most effect on desensitization. (A) Bar graph shows percent desensitization for each mutant and rCB1 receptor relative to their respective baseline epsc inhibition after overnight treatment with 100 nM WIN 55212-2. (B) Bar graph shows maximal inhibition in response to a 10 s depolarization for each mutant and rCB1 receptor. 1.0 = no inhibition. *P < 0.05, **P < 0.01, significantly different from values for rCB1 receptors; one-way anova with Dunnett's post hoc test.
Because in these neurones an increasing duration of depolarizing stimulus results in progressively stronger inhibition via DSE, we have found it is convenient to use stimulus time (in seconds) as a ‘dose’, plotted on a log scale to obtain a log ‘stimulus duration–response curve’ with properties similar to a classical dose–response curve. Taking the largest maximal slope of the curve in combination with observed baseline and maximal responses, allows us to derive an ‘ED50’. This ED50 represent the duration of depolarization required to induce a response halfway between the baseline and the maximum response.
In some cases, the maximal response was difficult to assess with confidence (especially where there was little difference between the maximal response and baseline), making interpretation of some curve parameters challenging. In this situation, we made use of anova tests rather than relying on curve parameters.
Materials
CB1+/− mice to found a CB1−/− colony were generously provided by Catherine Ledent (Ledent et al., 1999). WIN 55212-2 was purchased from Sigma-Aldrich (St. Louis, MO), dissolved in DMSO as a 10 mM stock, stored at −20°C and diluted shortly before use. We have previously reported that acute DMSO (0.1%) has no effect on epscs or DSE (Straiker and Mackie, 2005). 2-Arachidonoyl glycerol (2-AG) was obtained from Cayman Chemical (Ann Arbor, MI) as a 26 mM stock in acetonitrile (stored at −80°C) and diluted shortly before use. In our hands, 0.02% acetonitrile had no effect on epscs or DSE (data not shown).
Results
CB1 receptor transfection into CB1−/− neurones rescues DSE and desensitization
We have previously reported that depolarization of autaptic hippocampal neurones results in depolarization-induced suppression of excitation (DSE) (Straiker and Mackie, 2005), a form of retrograde inhibition involving release of 2-AG onto presynaptic CB1 receptors. A depolarization for as little as 100 ms is sufficient to induce measurable DSE in these cultures, and a series of depolarizations over a range of 50 ms to 10 s yields a sigmoidal depolarization-response curve that shares many useful properties with a classical dose–response curve. In neurones cultured from wild-type mice, the depolarization required to reach 50% of maximal inhibition (ED50) is ∼1.3 s (Figure 1A, adapted from Straiker and Mackie, 2009).
Figure 1.
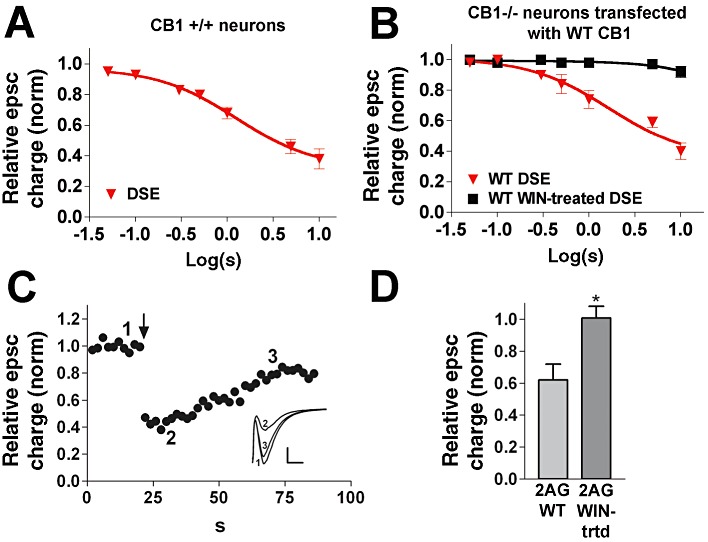
Transfection of WT CB1 receptor rescues DSE in CB1−/− autaptic hippocampal neurones. (A) WT untransfected DSE depolarization–response curve, representing inhibition in response to increasing duration of depolarization (50 ms, 100 ms, 300 ms, 500 ms, 1 s, 3 s, 10 s). (B) DSE depolarization–response curve for CB1−/− neurones transfected with WT CB1 receptors under control or WIN-treated (overnight, 100 nM) conditions. (C) Typical DSE time course from panel B (3 s depolarization at time point designated by arrow). Inset: sample epscs at time points indicated on DSE time course. (D) Responses to 2-AG (5 µM) before and after overnight treatment with 100 nM WIN 55212-2. *P < 0.05, unpaired t-test.
Importantly, we found that transfection of wild-type (WT) rCB1 receptors into autaptic hippocampal neurones derived from CB1 receptor knockout (CB1−/−) mice fully rescued DSE. The resultant depolarization–response curve closely mimicked that of DSE in WT neurones (Figure 1B,C; ED50: 1.7 s depolarization for neurones transfected with rCB1, 95% CI: 1.4–2.3; n= 6). In autaptic neurones, this CB1 response almost completely desensitizes with prolonged (>18 h) treatment with the CB1 receptor agonist WIN (100 nM) (Straiker and Mackie, 2005). WT (rCB1) receptors transfected into neurones cultured from CB1−/− mice desensitized in a similar manner (Figure 1B and 95% confidence intervals are non-overlapping, n= 6). The dose–response curve for vehicle-treated (0.001% DMSO, overnight), rCB1 receptor transfected neurones closely mimicked that of untreated rCB1-transfected neurones (ED50: 1.6 s, CI: 1.3–2.4; n= 5).
Direct application of 2-AG (5 µM) also inhibited epscs in autaptic hippocampal neurones transfected with wild-type receptors, an inhibition that was eliminated in WIN-treated neurones (Figure 1D; P < 0.05, unpaired t-test).
We conclude that transfection of wild-type CB1 receptors into CB1−/− neurones fully rescued the endogenous cannabinoid signalling system and its desensitization by prolonged CB1 receptor activation.
Deletion of the carboxy terminal of CB1 receptors alters both baseline signalling and desensitization
We next examined a truncation mutant where the last 13 residues (461–473, ‘V460Z’; shown in Figure 2A, adapted from Bramblett et al., 1995) were removed. In earlier studies, this mutation prevented internalization in AtT20 cells, but not in HEK293 cells (Hsieh et al., 1999; Daigle et al., 2008). Here, the V460Z truncation mutant, expressed in CB1−/− neurones, clearly rescued DSE, but the magnitude of the DSE and its sensitivity to depolarization were about half of that seen with the WT receptor (Figures 2B and 5B; ED50: 3.1 s, 95% CI: 0.8 to 12.9; n= 5).
Figure 2.

A carboxy terminus truncation mutant receptor signals less well than WT CB1 receptors but does not desensitize. (A) A helix-net diagram of the V460Z mutant CB1 receptor showing the deleted portion (last 14 residues). (B) V460Z DSE depolarization–response curve under control and WIN-treated (100 nM overnight) conditions. P > 0.05, two-way anova. rCB1 (untreated) and rCB1 (WIN-treated) responses (grey lines) are included for reference. (C) Sample DSE time course from panel B. (D) Inhibition of epscs in response to 5 µM 2-AG application in V460Z mutant-expressing neurones under control and WIN-treated conditions. *P < 0.05, unpaired t-test.
In WIN-treated cultures, DSE mediated by the truncated receptor did not significantly desensitize relative to untreated neurones (Figures 2B and 5A, P > 0.05, two-way anova). The extent of CB1 receptor desensitization for this and the other mutants is summarized in Figure 5. Direct 2-AG (5 µM) application also inhibited epscs under control conditions (Figure 2D), as did WIN (100 nM, relative epsc charge: 0.59 ± 0.06, n= 5). The preserved response to WIN indicates that the lack of desensitization was not due to a ligand-specific signalling deficit. However, in contrast to our DSE data, inhibition of epscs by 2-AG, after overnight WIN treatment, was reduced, indicating an intermediate degree of desensitization of V460Z CB1 receptor signalling by this agonist (Figure 2D; P < 0.05). However, because responses to 2-AG of the rCB1 (WIN-treated) receptors and V460Z (WIN-treated) receptors were different and statistically significant (P < 0.05, unpaired t-test), it may be said that the 2-AG response desensitized to a lesser extent in the V460Z mutant than in the WT receptor.
Mutation of six serines and threonines to alanines in the distal carboxy tail of CB1 receptors attenuates signalling and prevents desensitization
The distal carboxy terminus of the CB1 receptor is the site of a cluster of six serine and threonine residues (Figure 3A). Phosphorylation of these residues may play a role in desensitization of CB1 receptors, as mutation of these six serines and threonines to alanines prevented internalization and β-arrestin2 recruitment in HEK293 cells (Daigle et al., 2008). We therefore tested the effects of these mutations (‘six-point mutant’) on cannabinoid signalling in autaptic neurones.
Figure 3.
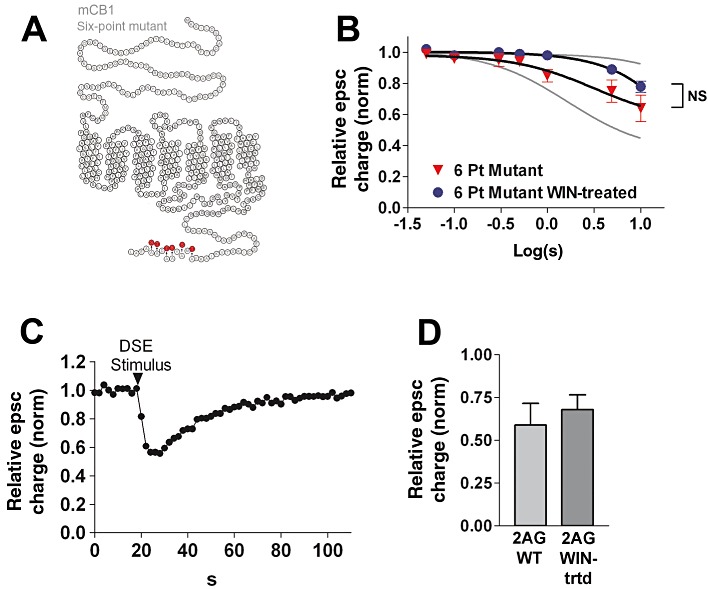
Mutation of the terminal six serines and threonines to alanines suppresses desensitization. (A) A helix-net diagram of the 6 point mutant CB1 receptor showing the alanine substitutions at the carboxy-terminus. (B) Six-point mutant DSE depolarization–response curve under control and WIN-treated (100 nM overnight) conditions, P > 0.05, two-way anova. rCB1 (untreated) and rCB1 (WIN-treated) responses (grey lines) are included for reference. (C) Typical DSE time course from panel B. (D) Inhibition of epscs in response to 5 µM 2-AG application in neurones expressing 6 point mutant CB1 receptors, under control and WIN-treated conditions. P > 0.05, unpaired t-test.
This six-point mutation of the CB1 carboxy terminus also rescues cannabinoid signalling, with a response profile similar to that of the V460Z mutant receptor (Figures 3B,C and 5B; ED50: 3.2 s; 95% CI: 2.2–4.6, n= 5). 2-AG responses were also intact in neurones transfected with the six-point mutant receptor (Figure 3D), as were the effects of WIN (100 nM; relative epsc charge after WIN: 0.54 ± 0.15, n= 5).
The six-point mutant receptors did not significantly desensitize relative to untreated neurones [Figures 3B and 5A, P > 0.05, two-way anova six-point mutant (untreated) vs. six-point mutant (WIN-treated) ]. Similarly, 2-AG inhibition was not altered by overnight WIN treatment (Figure 3D; P > 0.05). We conclude that this group of serines and threonines may be responsible for the lack of DSE desensitization seen in the V460Z truncation.
Lastly, we studied the impact of a deletion that leaves the last 14 residues intact but removes ∼40 residues upstream of them (Figure 4A). This 419–460 deletion (‘G→K’ mutant) allowed us to test the following: (1) whether baseline signalling was rescued by re-attachment of an intact C-terminus and (2) whether desensitization was rescued by reattachment of a section including the 6 terminal serine and threonine residues described above. It also tested the hypothesis that the domain involving the probable β-arrestin-interacting site for uncoupling from G-proteins, is important for desensitization in neurones (Jin et al., 1999). In our experiments, the G→K mutant did rescue DSE and EPSC inhibition by 2-AG (Figure 4B, D). In contrast to the other mutants studied, the G→K mutant did desensitize (Figures 4B and 5A: P < 0.01 and P < 0.001 at 3 and 10 s depolarizations, respectively). Similarly, 2-AG responses were diminished after WIN treatment, relative to control (Figure 4D; P < 0.05).
Figure 4.
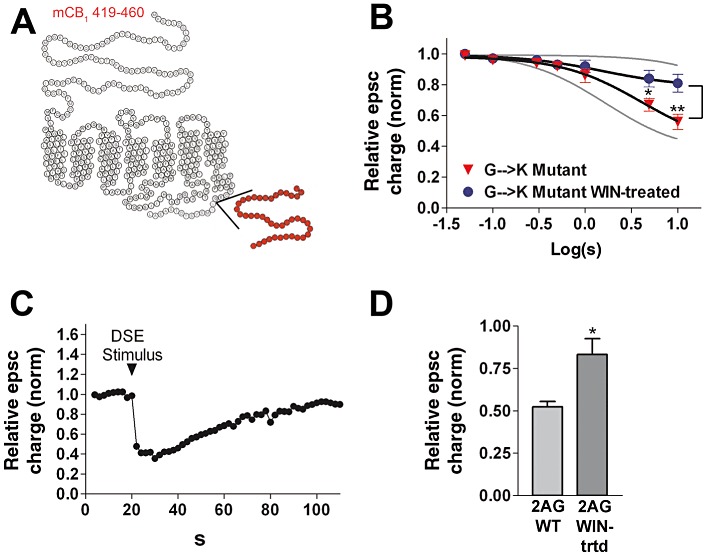
The 419–460 deletion ‘G→K’ in the CB1 receptor has little effect on desensitization. (A) A helix-net diagram of the 419-460 CB1 deletion mutant showing the deletion near the cytosolic side of the seventh transmembrane region. (B) 419–460 DSE depolarization–response curve under control and WIN-treated (100 nM overnight) conditions. *P < 0.05, **P < 0.001, two-way anova with Bonferroni post hoc test. rCB1 (untreated) and rCB1 (WIN-treated) responses (grey lines) are included for reference. (C) Typical DSE time course from panel B. (D) Inhibition of epscs in response to 5 µM 2-AG application in neurones expressing 419–460 mutant CB1 receptors, under control and WIN-treated conditions. *P < 0.05, unpaired t-test.
The extent of desensitization for each mutant is summarized graphically in Figure 5A as percent of their respective baseline inhibitions in response to a 10 s depolarization (P < 0.01 for V460Z vs. rCB1; P < 0.05 for six-point vs. rCB1). Because the maximal inhibition differs among the mutations, Figure 5B additionally shows these maximal responses to a 10 s depolarization (no WIN pretreatment; P < 0.05 for V460Z and six-point vs. rCB1) The latter result shows that among the three mutations, baseline activation is best rescued by the G→K mutation.
Discussion
Since the 1970s, cell lines such as HEK293s have been the most popular cellular model used to study the function and regulation of GPCRs. Cell lines have made important contributions to the field, as attested by the sheer number of studies (>10 000) making use of HEK cells, as just one example. However, different cell lines have different repertoires of signalling proteins, and thus regulation of GPCR signalling is expected to vary between cell lines and may not represent the signalling environment within a neuron. With the advent of improved neuronal culturing and transfection techniques, it has become feasible to revisit and extend mutational studies of GPCR signal transduction to a neuronal setting. This is particularly important when – as is the case here – results differ dramatically depending on the cell line used (i.e. AtT20 vs. HEK293 cells).
In this regard, the autaptic hippocampal preparation is well suited to study the full extent of cannabinoid signalling, expressing not only CB1 receptors but also the machinery to produce endocannabinoids (probably 2-AG) in response to depolarization (Straiker and Mackie, 2005), and metabotropic GPCR activation (Straiker and Mackie, 2007). They also express a form of CB1-dependent long-term depression (Kellogg et al., 2009). The current work represents the first use of neurones for the study of the effects of specific CB1 receptor domains on endogenous cannabinoid signalling. Importantly, transfection of non-mutated CB1 receptors results in DSE that is nearly identical to that of native neurones, including sensitivity to depolarization, suggesting the transfected receptors are properly trafficked and regulated. Desensitization of CB1 receptors by overnight treatment with WIN 55212 is also closely mimicked in transfected neurones, making this an appropriate model system to study desensitization of CB1 receptor signalling.
Our results strongly support the notion that the distal carboxy terminus of CB1 receptors constitutes an important regulatory domain for agonist-induced desensitization. Previous results using both HEK and AtT20 cells showed that the six serine and threonine residues within the extreme terminal region played an essential role in regulating desensitization (Hsieh et al., 1999; Daigle et al., 2008). Our results are in full agreement with this set of findings. However, the same studies also reported differential effects of simple truncation of the final 13 residues, with prevention of desensitization in AtT20 but not HEK cells. This likely reflects the different expression of structural and signalling pathway components in these cell lines and emphasizes the importance of assessing signalling pathways in a neuronal setting. Interestingly, AtT20 cells have a strongly neuronal phenotype (Tooze et al., 1989). As shown in Figure 5, the constructs with the least desensitization are also those with the least baseline inhibition. Nonetheless, this lesser maximal inhibition still represents ∼40% inhibition of neurotransmission, relative to ∼60% inhibition seen in wild-type rCB1-transfected neurones. However, it is possible that the diminished baseline activation via V460Z and the six-point mutant contributes to their relatively poor desensitization. The relationship between baseline activation and net desensitization may be a fruitful line of future inquiry.
In summary, we conclude that the distal carboxy terminal is required for agonist-induced desensitization of CB1 receptor signalling in cultured neurones and that phosphorylation of six serine and threonine residues in this domain is likely to regulate this desensitization.
Acknowledgments
Excellent technical assistance was provided by Ben Cornett. This work was supported by National Institutes of Health (grants DA11322, DA021696, DA024122); the Indiana METACyt Initiative of Indiana University, funded in part through a major grant from the Lilly Endowment, Inc.
Glossary
- DSE
depolarization-induced suppression of excitation
- DSI
depolarization-induced suppression of inhibition
- 2-AG
2-arachidonyl glycerol
Conflict of interest
There are no conflicts of interest regarding this manuscript.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Lopez J, Wager-Miller J, Mackie K, Straiker A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2 and N18 cell lines as revealed by microarray analysis. BMC Genomics. 2011;12:14. doi: 10.1186/1471-2164-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass CE, Martin BR. Time course for the induction and maintenance of tolerance to Delta(9)-tetrahydrocannabinol in mice. Drug Alcohol Depend. 2000;60:113–119. doi: 10.1016/s0376-8716(99)00150-7. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci U S A. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaboula M, Poinot-Chazel C, Bourrie B, Canat X, Calandra B, Rinaldi-Carmona M, et al. Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J. 1995;312(Pt 2):637–641. doi: 10.1042/bj3120637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramblett RD, Panu AM, Ballesteros JA, Reggio PH. Construction of a 3D model of the cannabinoid CB1 receptor: determination of helix ends and helix orientation. Life Sci. 1995;56:1971–1982. doi: 10.1016/0024-3205(95)00178-9. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Vogt LJ, Sim-Selley LJ. Chronic delta9-tetrahydrocannabinol treatment produces a time-dependent loss of cannabinoid receptors and cannabinoid receptor-activated G proteins in rat brain. J Neurochem. 1999;73:2447–2459. doi: 10.1046/j.1471-4159.1999.0732447.x. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Scates SM, Beletskaya IO, Lowery OB, Aceto MD, Martin BR. The effects of delta9-tetrahydrocannabinol physical dependence on brain cannabinoid receptors. Eur J Pharmacol. 2003;459:139–150. doi: 10.1016/s0014-2999(02)02854-6. [DOI] [PubMed] [Google Scholar]
- Claing A, Laporte SA, Caron MG, Lefkowitz RJ. Endocytosis of G protein-coupled receptors: roles of G protein-coupled receptor kinases and beta-arrestin proteins. Prog Neurobiol. 2002;66:61–79. doi: 10.1016/s0301-0082(01)00023-5. [DOI] [PubMed] [Google Scholar]
- Coutts AA, Anavi-Goffer S, Ross RA, MacEwan DJ, Mackie K, Pertwee RG, et al. Agonist-induced internalization and trafficking of cannabinoid CB1 receptors in hippocampal neurons. J Neurosci. 2001;21:2425–2433. doi: 10.1523/JNEUROSCI.21-07-02425.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle TL, Kwok ML, Mackie K. Regulation of CB(1) cannabinoid receptor internalization by a promiscuous phosphorylation-dependent mechanism. J Neurochem. 2008;106:70–82. doi: 10.1111/j.1471-4159.2008.05336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furshpan EJ, MacLeish PR, O'Lague PH, Potter DD. Chemical transmission between rat sympathetic neurons and cardiac myocytes developing in microcultures: evidence for cholinergic, adrenergic, and dual-function neurons. Proc Natl Acad Sci U S A. 1976;73:4225–4229. doi: 10.1073/pnas.73.11.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Gaoni Y, Mechoulam R. Isolation, structure and partial synthesis of an active constituent of hashish. J Am Chem Soc. 1964;86:1646–1647. [Google Scholar]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Fleming RM. Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Mol Pharmacol. 1984;26:532–538. [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Hsieh C, Brown S, Derleth C, Mackie K. Internalization and recycling of the CB1 cannabinoid receptor. J Neurochem. 1999;73:493–501. doi: 10.1046/j.1471-4159.1999.0730493.x. [DOI] [PubMed] [Google Scholar]
- Jiang M, Deng L, Chen G. High Ca(2+)-phosphate transfection efficiency enables single neuron gene analysis. Gene Ther. 2004;11:1303–1311. doi: 10.1038/sj.gt.3302305. [DOI] [PubMed] [Google Scholar]
- Jin W, Brown S, Roche JP, Hsieh C, Celver JP, Kovoor A, et al. Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J Neurosci. 1999;19:3773–3780. doi: 10.1523/JNEUROSCI.19-10-03773.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg R, Mackie K, Straiker A. Cannabinoid CB1 receptor-dependent long-term depression in autaptic excitatory neurons. J Neurophysiol. 2009;102:1160–1171. doi: 10.1152/jn.00266.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouznetsova M, Kelley B, Shen M, Thayer SA. Desensitization of cannabinoid-mediated presynaptic inhibition of neurotransmission between rat hippocampal neurons in culture. Mol Pharmacol. 2002;61:477–485. doi: 10.1124/mol.61.3.477. [DOI] [PubMed] [Google Scholar]
- Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, et al. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science. 1999;283:401–404. doi: 10.1126/science.283.5400.401. [DOI] [PubMed] [Google Scholar]
- Levison SW, McCarthy KD. Characterization and partial purification of AIM: a plasma protein that induces rat cerebral type 2 astroglia from bipotential glial progenitors. J Neurochem. 1991;57:782–794. doi: 10.1111/j.1471-4159.1991.tb08220.x. [DOI] [PubMed] [Google Scholar]
- Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci U S A. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K, Lai Y, Westenbroek R, Mitchell R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci. 1995;15:6552–6561. doi: 10.1523/JNEUROSCI.15-10-06552.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Sim-Selley LJ, Selley DE. Signaling pathways involved in the development of cannabinoid tolerance. Trends Pharmacol Sci. 2004;25:325–330. doi: 10.1016/j.tips.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Tsubokawa H, Mizushima I, Yoneda N, Zimmer A, Kano M. Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J Neurosci. 2002;22:3864–3872. doi: 10.1523/JNEUROSCI.22-10-03864.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Le Duigou A, Oustric D, Barth F, Bouaboula M, Carayon P, et al. Modulation of CB1 cannabinoid receptor functions after a long-term exposure to agonist or inverse agonist in the Chinese hamster ovary cell expression system. J Pharmacol Exp Ther. 1998;287:1038–1047. [PubMed] [Google Scholar]
- Straiker A, Mackie K. Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol. 2005;569(Pt 2):501–517. doi: 10.1113/jphysiol.2005.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Mackie K. Metabotropic suppression of excitation in murine autaptic hippocampal neurons. J Physiol. 2007;578(Pt 3):773–785. doi: 10.1113/jphysiol.2006.117499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Mackie K. Cannabinoid signaling in inhibitory autaptic hippocampal neurons. Neuroscience. 2009;163:190–201. doi: 10.1016/j.neuroscience.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze J, Hollinshead M, Fuller SD, Tooze SA, Huttner WB. Morphological and biochemical evidence showing neuronal properties in AtT-20 cells and their growth cones. Eur J Cell Biol. 1989;49:259–273. [PubMed] [Google Scholar]
- Wu DF, Yang LQ, Goschke A, Stumm R, Brandenburg LO, Liang YJ, et al. Role of receptor internalization in the agonist-induced desensitization of cannabinoid type 1 receptors. J Neurochem. 2008;104:1132–1143. doi: 10.1111/j.1471-4159.2007.05063.x. [DOI] [PubMed] [Google Scholar]