

Abstract
BACKGROUND AND PURPOSE
Midazolam is a short-acting benzodiazepine that is widely used as an i.v. sedative and anticonvulsant. Besides interacting with the benzodiazepine site associated with GABAA receptors, some benzodiazepines act as agonists of translocator protein (18 kDa) (TSPO) to enhance the synthesis of steroids, including neurosteroids with positive modulatory actions on GABAA receptors. We sought to determine if neurosteroidogenesis induced by midazolam contributes to its anticonvulsant action.
EXPERIMENTAL APPROACH
Mice were pretreated with neurosteroid synthesis inhibitors and potentiators followed by midazolam or clonazepam, a weak TSPO ligand. Anticonvulsant activity was assessed with the i.v. pentylenetetrazol (PTZ) threshold test.
KEY RESULTS
Midazolam (500–5000 µg·kg−1, i.p.) caused a dose-dependent increase in seizure threshold. Pretreatment with the neurosteroid synthesis inhibitors finasteride, a 5α-reductase inhibitor, and a functional TSPO antagonist PK 11195, reduced the anticonvulsant action of midazolam. The anticonvulsant action of midazolam was enhanced by the neurosteroidogenic drug metyrapone, an 11β-hydroxylase inhibitor. In contrast, the anticonvulsant action of clonazepam (100 µg·kg−1) was reduced by finasteride but not by PK 11195, indicating a possible contribution of neurosteroids unrelated to TSPO.
CONCLUSION AND IMPLICATIONS
Enhanced endogenous neurosteroid synthesis, possibly mediated by an interaction with TSPO, contributed to the anticonvulsant action of midazolam. Enhanced neurosteroidogenesis may also be a factor in the actions of other benzodiazepines, even those that only weakly interact with TSPO.
Keywords: midazolam, clonazepam, peripheral benzodiazepine receptor, neurosteroid, finasteride, PK 11195, metyrapone, pentylenetetrazol
Introduction
Midazolam is a rapid onset, short-acting water-soluble benzodiazepine that is administered parenterally as a sedative, anxiolytic, hypnotic and amnestic agent. The drug is also administered i.m., i.v. or intranasally to terminate acute seizures and status epilepticus (Galvin and Jelinek, 1987). Midazolam exhibits anticonvulsant activity in a variety of chemoconvulsant seizure models, including the pentylenetetrazol (PTZ) model in various species (Pieri, 1983; Jaimovich et al., 1990; Raines et al., 1990; Orebaugh and Bradford, 1994). As is the case for other benzodiazepines, the pharmacological actions of midazolam are mediated predominantly through an interaction with a high affinity binding site in brain representing an allosteric modulatory site on GABAA receptors (Kucken et al., 2003; receptor nomenclature follows Alexander et al., 2011). Binding of midazolam and other classic benzodiazepines, such as clonazepam, to a recognition site at the interface between extracellular domains of the GABAA receptor γ subunit and one of the α subunits allosterically modulates gating of the GABAA receptor chloride channel complex leading to enhanced channel current (Rovira and Ben-Ari, 1993; Rüsch and Forman, 2005; Eom et al., 2011). This allosteric modulation of GABAA receptors is believed to account for the principal pharmacological actions of benzodiazepines, including their anticonvulsant activity (Rogawski, 2002).
Some benzodiazepines also bind to a pharmacologically distinct and unrelated binding site in brain and peripheral tissues that was formerly described as the peripheral-type benzodiazepine receptor (Gavish et al., 1999) but is now referred to as translocator protein (18 kDa) (TSPO) (Papadopoulos et al., 2006). TSPO is expressed predominantly in mitochondria, where it is localized to the outer mitochondrial membrane. TSPO binds cholesterol with high affinity and transports it from the outer mitochondrial membrane to the inner mitochondrial membrane, where it is converted to pregnenolone by the cytochrome P450 side-chain cleavage enzyme. This sequence represents the initial and rate-limiting step in the biosynthesis of all steroids. TSPO agonist ligands, including benzodiazepines with TSPO binding activity (Mukhin et al., 1989), stimulate steroidogenesis by facilitating cholesterol delivery to the cytochrome P450 side-chain cleavage enzyme in the inner mitochondrial membrane (Lacapère and Papadopoulos, 2003).
As is the case for hormonal steroids, a variety of evidence supports the concept that TSPO agonist ligands can also enhance the synthesis of endogenous neurosteroids, including allopregnanolone, that lack hormonal activity but serve as positive allosteric modulators of GABAA receptors (Romeo et al., 1993; Serra et al., 1999; Bitran et al., 2000; Verleye et al., 2005). In accordance with their effects on GABAA receptors, such neurosteroids exhibit anticonvulsant activity in various seizure models, including PTZ-induced seizures in mice (Kokate et al., 1994). The effect of TSPO agonist ligands on neurosteroidogenesis is believed to occur through enhanced mitochondrial synthesis of pregnenolone, which is converted to progesterone by microsomal 3β-hydroxysteroid dehydrogenase and then to neurosteroids by sequential A-ring reduction by 5α-reductase and 3α-hydroxysteroidoxidoreductase. Enhancement of endogenous neurosteroid synthesis, induced by TSPO agonist ligands, can produce pharmacological effects typical of neurosteroids, including anxiolytic actions (Kita and Furukawa, 2008; Rupprecht et al., 2009). In some experimental situations, the enhanced neurosteroid synthesis (Serra et al., 1999; Frye et al., 2009) and behavioural effects (Bitran et al., 2000; Kita et al., 2004; Rupprecht et al., 2009) of TSPO agonists can be reduced by the isoquinoline carboxamide TSPO antagonist, PK 11195.
Although there is some evidence that midazolam interacts with TSPO (Schoemaker et al., 1981; Bender and Hertz, 1987; Mak and Barnes, 1990; Matsumoto et al., 1994), until recently, it was not known if midazolam acts as a functional TSPO ligand to affect steroidogenesis. However, So et al. (2010) have now reported that midazolam enhances Leydig cell production of progesterone and testosterone. In addition, Tokuda et al. (2010) observed that midazolam enhances the synthesis of neurosteroids in hippocampal neurons in brain slices. These latter authors presented evidence that neurosteroidogenesis in addition to an interaction with the benzodiazepine binding site on GABAA receptors is required for the actions of midazolam on synaptic inhibition. We therefore hypothesized that the in vivo actions of midazolam are also due to a combination of direct effects on GABAA receptors (through the intrinsic benzodiazepine site) as well its indirect actions on neurosteroidogenesis mediated by TSPO. In the present study, we investigated this possibility with the use of various agents that modify endogenous neurosteroid synthesis.
Experimental procedures
Animals
All animal care and experimental protocols were approved by the Animal Care and Use Committee of the University of California, Davis in strict compliance with the Guide for the Care and Use of Laboratory Animals of the National Research Council (National Academy Press, Washington, D.C.; http://www.nap.edu/readingroom/books/labrats/). The animal facilities were fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. Male NIH Swiss mice (22–30 g) were kept in a vivarium under controlled environmental conditions (temperature, 22–26°C; humidity, 40–50%) with an artificial 12 h light/dark cycle. Wood chips were used in all cages. Experiments were performed during the light phase of the light/dark cycle after a minimum 30 min period of acclimation to the experimental room.
PTZ seizure threshold test
PTZ seizure threshold was determined according to a protocol used previously in our laboratory (Dhir et al., 2011). Given i.v., PTZ elicits a sequence of seizure signs beginning with twitch and progressing to clonus and tonic limb extension. In the present study, tonic extension was used as the endpoint. In preliminary experiments, tonic extension was found to be more sensitive to midazolam than the twitch or clonus phases. It is noteworthy that this is the endpoint originally used in studies characterizing the actions of midazolam against PTZ seizures in mice (Pieri, 1983). A 27-gauge, ¾ inch butterfly needle was inserted into the lateral tail vein, and the needle was secured with a narrow piece of adhesive tape. The animal was placed inside a 2 L glass beaker with free aeration from the top and allowed to move freely. The needle was connected by polyethylene tubing to a Beckton, Dickinson 1 mL syringe mounted on an infusion pump (Model ‘11’ plus syringe pump; Harvard Apparatus, Holliston, MA). PTZ solution (10 mg·mL−1) was infused at a constant rate of 0.5 mL·min−1. The infusion was stopped at 3 min or at the onset of tonic extension, whichever occurred first. The threshold value (mg·kg−1) for tonic extension was determined according to the following formula: [infusion duration (s) × infusion rate (mL·min−1) × PTZ concentration (mg·mL−1) × 1000] / [60 (s) × weight of mouse (g) ].
Treatment schedules
In an initial experiment, the dose–response relationship for midazolam elevation of seizure threshold was assessed at doses of 100 µg·kg−1 to 5 mg·kg−1 (15 min pretreatment time). The doses and time of administration of test substances in the remaining experiments with midazolam are shown in Figure 1. In the first protocol, the 5α-reductase inhibitor finasteride was used at a dose (100 mg·kg−1) that has previously been shown to partially inhibit neurosteroid synthesis (Kokate et al., 1999). Metyrapone was used at a dose (100 mg·kg−1) that has previously been shown to elevate seizure threshold through enhanced endogenous neurosteroid synthesis (Kaminski and Rogawski, 2011). PK 11195 was used at a dose (15 mg·kg−1) as in the study of Ugale et al. (2004). Clonazepam was administered at a dose of 100 µg·kg−1 (Akula et al., 2009). The volume of all i.p. injections was 10 mL·kg−1.
Figure 1.
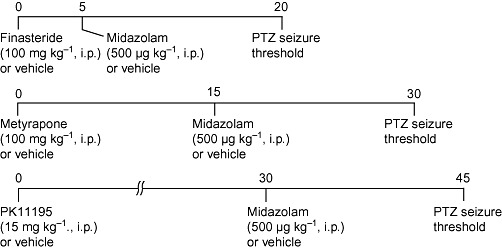
Drug treatment protocols. In some experiments, clonazepam (25 or100 µg·kg−1, i.p.) was used instead of midazolam.
Data analysis
Results are expressed as mean ± SEM; the significance of the difference in the responses of treatment groups with respect to control is based on one-way anova followed by specific post hoc comparisons using Tukey's test. Differences were considered statistically significant when the probability of type I error was less than 0.05.
Materials
Midazolam was administered as a commercially available injectable solution [midazolam hydrochloride, 10 mg·(10 mL)−1; APP Pharmaceuticals, Schaumburg, IL] containing sodium chloride (0.8 % w/v), disodium edetate (0.01% w/v), benzyl alcohol as preservative (1% v/v) with HCl to adjust the pH 3–3.6. Pentylenetetrazol (PTZ) (Sigma-Aldrich, St. Louis, MO) was dissolved in 0.9% w/v saline. The remaining compunds were all obtained from Tocris Bioscience (Ellisville, MO) except for clonazepam, which was from Sigma-Aldrich. Finasteride was dissolved in 25% hydroxypropyl-β-cyclodextrin (Trappsol; Cyclodextrin Technologies Development, High Springs, FL). Metyrapone was dissolved in double distilled water, and PK 11195 and clonazepam were suspended in 1% Tween 80 and adjusted to volume with saline.
Results
Midazolam causes a dose-dependent elevation in PTZ seizure threshold
I.v. infusion of PTZ (10 mg·mL−1) led to a sequence of seizure signs consisting of myoclonic jerk, clonus and tonic extension, followed by death. In the present study, cumulative dose to the onset of tonic extension was the measure of seizure threshold. Animals protected from tonic extension invariably survived, whereas those that experienced tonic extension expired immediately after the seizure. Figure 2 plots the fractional change in mean threshold for groups of animals that had been treated 15 min before the onset of the PTZ infusion with various doses of midazolam. There was a dose-dependent elevation in threshold with increasing midazolam dose that was significant for midazolam doses of 500 to 5000 µg·kg−1 but not 100 µg·kg−1.
Figure 2.
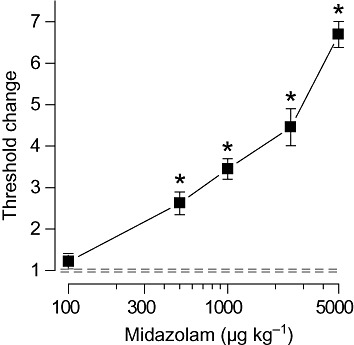
Dose–response relationship for midazolam protection against tonic extension in response to i.v. PTZ infusion in mice. Midazolam was administered i.p. 15 min before the beginning of the PTZ infusion. Data points indicate mean ± SEM of threshold values from six to eight mice normalized with respect to the mean threshold value in the vehicle-treated control group, which was 51.5 ± 4.0 mg·kg−1. Dashed lines indicate the limits of the SEM for the vehicle group. The mean threshold values for all groups other than the 100 µg·kg−1 group are significantly different from the value in the vehicle group. *P < 0.001; one-way anova followed by Tukey's test.
Finasteride pretreatment reduces the seizure threshold elevation induced by midazolam
To assess whether endogenous neurosteroid production contributes to the seizure threshold elevation induced by midazolam, mice were pretreated with the neurosteroid synthesis inhibitor finasteride (100 mg·kg−1, i.p.) before giving a dose of midazolam (500 µg·kg−1, i.p.) that in the experiment of Figure 2 caused a significant increase in threshold. As shown in Figure 3, finasteride pretreatment by itself did not alter the PTZ seizure threshold. However, finasteride did partially reduce the threshold elevation caused by midazolam.
Figure 3.
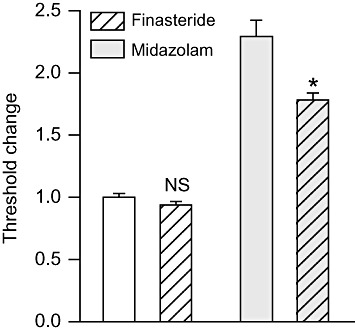
Finasteride pretreatment reduces the seizure threshold elevation induced by midazolam. Finasteride (100 mg·kg−1, i.p.) or vehicle was administered 5 min before the treatment with midazolam (500 µg·kg−1, i.p.) or vehicle; 15 min after the second pretreatment, all animals were infused with PTZ. Bars indicate mean ± SEM of fractional threshold change values for tonic extension from 6–14 mice normalized with respect to the mean threshold value in the vehicle only control group, which was 59.2 ± 1.8 mg·kg−1. In the absence of finasteride, midazolam caused a 2.3-fold increase in threshold (P < 0.001). Finasteride did not reduce the threshold significantly in the absence of midazolam (NS) but did reduce the threshold with midazolam pretreatment. *P < 0.001; one-way anova followed by Tukey's test.
Metyrapone enhances the seizure threshold elevation induced by midazolam
To provide further support for the involvement of neurosteroids in the anticonvulsant action of midazolam, the 11β-hydroxylase inhibitor metyrapone, which enhances endogenous neurosteroid synthesis (Rupprecht et al., 1998; Kaminski and Rogawski, 2011), was given before midazolam. By itself, metyrapone (100 mg·kg−1, i.p.) caused a significant elevation in the threshold (Figure 4), confirming our previous report (Kaminski and Rogawski, 2011). When metyrapone was administered before midazolam (500 µg·kg−1, i.p.), there was a further increment in threshold.
Figure 4.
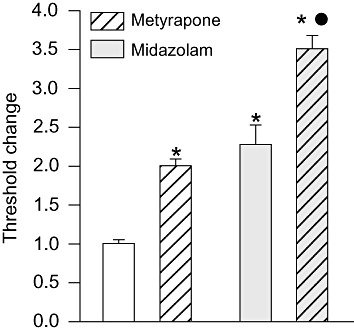
Metyrapone elevates seizure threshold in the absence and presence of midazolam. Metyrapone (100 mg·kg−1, i.p.) or vehicle was administered 15 min before treatment with midazolam or vehicle; 15 min after the second pretreatment, all animals were infused with PTZ. Bars indicate mean ± SEM of fractional change values in tonic extension threshold from six to nine mice normalized with respect to the mean threshold value in the vehicle only control group, which was 51.0 ± 4.4 mg·kg−1. *P < 0.001, significantly different from vehicle only control group; •P < 0.001, significantly different from midazolam only group; one-way anova followed by Tukey's test.
PK 11195 inhibits the seizure threshold elevation induced by midazolam
As an additional approach to assessing the role of neurosteroidogenesis in the action of midazolam we used PK 11195, a high-affinity ligand of TSPO that acts as an antagonist in some situations (Le Fur et al., 1983) and inhibits the behavioural effects of TSPO ligands that stimulate neurosteroidogenesis (Auta et al., 1993; Romeo et al., 1993; Tsuda et al., 1998; Frye et al., 2009). By itself, PK 11195 (15 mg·kg−1, i.p.) did not affect the seizure threshold. However, pretreatment with PK 11195 did significantly reduce the elevation in threshold produced by midazolam (500 µg·kg−1, i.p.) (Figure 5).
Figure 5.
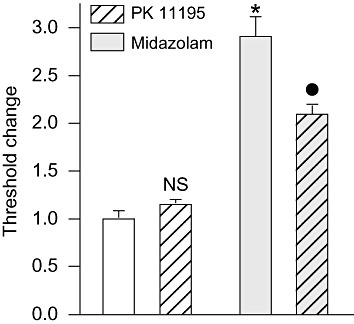
PK 11195 pretreatment reduces the seizure threshold elevation induced by midazolam. PK11195 (15 mg·kg−1, i.p.) was administered 30 min before the treatment with midazolam (500 µg·kg−1, i.p.) or vehicle; 15 min after the second pretreatment, all animals were infused with PTZ. Bars indicate mean ± SEM of fractional threshold change values for tonic extension from 6–12 mice normalized with respect to the mean threshold value in the vehicle only control group (same as Figure 4). *P < 0.001, significantly different from vehicle only control group; •P < 0.001, significantly different from midazolam only group; one-way anova followed by Tukey's test.
Effects of finasteride and PK 11195 on the seizure threshold elevation induced by clonazepam
To assess the specificity of the effects of finasteride and PK 11195, a series of experiments were conducted with clonazepam, a benzodiazepine that binds only weakly to TSPO (Schoemaker et al., 1981; Marangos et al., 1982; Bender and Hertz, 1987; Guarneri et al., 1995; McCauley et al., 1995) and does not enhance neurosteroid synthesis in some in vitro preparations (Mukhin et al., 1989; Papadopoulos et al., 1992; Tokuda et al., 2010). As shown in Figure 6 (upper panel), finasteride pretreatment did reduce the seizure threshold elevation produced by 100 µg·kg−1 clonazepam. In a second experiment with a lower does of clonazepam (25 µg·kg−1), there was a trend towards an effect of finasteride, although it did not reach statistical significance (data not shown). However, unlike the situation with midazolam, PK 11195 did not reduce the seizure threshold elevation produced by clonazepam (Figure 6, lower panel).
Figure 6.
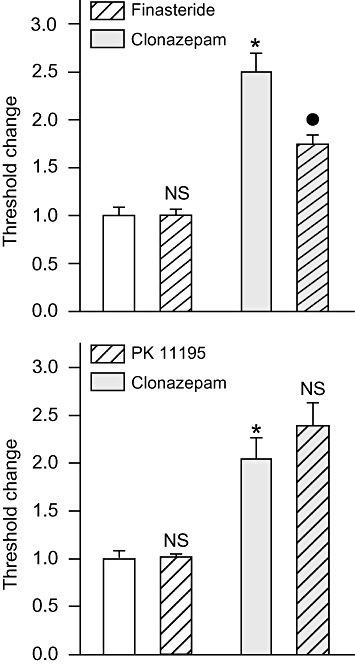
Finasteride (upper panel) but not PK 11195 (lower panel) pretreatment reduces the seizure threshold elevation induced by clonazepam. Finasteride (100 mg·kg−1, i.p.) or vehicle was administered 5 min before the treatment with clonazepam (100 µg·kg−1, i.p.) or vehicle; 15 min after the second pretreatment, all animals were infused with PTZ. In the absence of finasteride, clonazepam caused a 2.8-fold increase in threshold (P < 0.001). Finasteride did not reduce the threshold significantly in the absence of midazolam (NS) but did reduce the threshold with clonazepam pretreatment (P < 0.001). PK11195 (15 mg·kg−1, i.p.) was administered 30 min before the treatment with clonazepam or vehicle; 15 min after the second pretreatment, all animals were infused with PTZ. Bars indicate mean ± SEM of fractional threshold change values for tonic extension from 6–13 mice normalized with respect to the mean threshold value in the vehicle only control group, which was 46.9 ± 3.6 mg·kg−1 in the experiment with finasteride and 51.9 ± 2.7 mg·kg−1 in the experiment with PK 11195. *P < 0.001, significantly different from vehicle only control group; •P < 0.001, significantly different from clonazepam only group; one-way anova followed by Tukey's test.
Discussion
This study for the first time provides evidence for the involvement of neurosteroids in the anticonvulsant activity of midazolam. As expected, midazolam caused a dose-dependent anticonvulsant action in the i.v. PTZ threshold model. The anticonvulsant activity of midazolam was significantly reduced by finasteride, a 5α-reductase inhibitor that is well recognized to suppress neurosteroidogenesis in mice (Kokate et al., 1999; Finn et al., 2006). For example, at the dose used in the present study, finasteride eliminates the rise in plasma allopregnanolone induced by elevation of its precursor progesterone (Reddy et al., 2001) and also inhibits local neurosteroid synthesis in the brain (Tokuda et al., 2010). By itself, finasteride did not affect the seizure threshold, indicating that the effect on midazolam is not due to an enhancement of seizure susceptibility, unrelated to the action on neurosteroidogenesis. Moreover, our result is consistent with several other studies demonstrating that finasteride does not influence basal seizure susceptibility (Reddy and Rogawski, 2002; Lawrence et al., 2010), which lead to the conclusion that basal (unstimulated) neurosteroid levels do not have a tonic influence on seizure susceptibility.
In contrast to the effect of finasteride, metyrapone, an 11β-hydroxylase inhibitor, has been shown to increase neurosteroidogenesis by causing a buildup of neurosteroid precursors such as progesterone and 11-deoxycorticosterone that ordinarily flow to glucocorticoid synthesis (Kaminski and Rogawski, 2011). In the present study, metyrapone by itself elevated the seizure threshold consistent with our previous report (Kaminski and Rogawski, 2011). Midazolam caused a further and largely additive increment in threshold confirming that enhanced neurosteroidogenesis can augment the action of midazolam.
The TSPO ligand PK 11195 by itself did not affect PTZ seizure threshold. PK 11195 in some but not all situations acts as a TSPO antagonist (Le Fur et al., 1983; Mizoule et al., 1985; Matsumoto et al., 1994). As such, it inhibits TSPO agonist-induced steroidogenesis (Cavallaro et al., 1992). PK 11195 by itself has variable effects on basal steroidogenesis. In some situations it has no effect consistent with a role as a TSPO antagonist (Cavallaro et al., 1992), whereas in other cases, it may increase (Mukhin et al., 1989; McCauley et al., 1995) or decrease (Frye et al., 2009) steroidogenesis. These latter actions could be due to weak intrinsic (partial agonist) activity or to effects on endogenous TSPO ligands. Even though PK 11195 may reduce endogenous neurosteroid levels in some circumstances, the lack of effect of PK 11195 on basal seizure threshold is consistent with the results of several previous studies with finasteride discussed above that have concluded that basal neurosteroid levels do not influence seizure susceptibility. Here we took advantage of the ability of PK 11195 to antagonize neurosteroidogenesis activated by TSPO ligands. We observed that PK 11195 caused a significant inhibition of the seizure threshold increase produced by midazolam. This provides evidence that the anticonvulsant action of midazolam depends in part on its ability to interact with TSPO as an agonist.
To further explore the role of TSPO, we conducted experiments with clonazepam, a benzodiazepine that is a very weak TSPO ligand. Surprisingly, we observed that the seizure threshold increase produced by clonazepam was reduced by finasteride. This occurred at a higher dose of clonazepam but not at a lower dose. Given the weak affinity of clonazepam for TSPO, the concentrations achieved in brain with either dose are unlikely to produce substantial occupancy of TSPO. Accordingly, PK 11195 failed to reduce the effect of clonazepam on seizure threshold demonstrating that the activity of this benzodiazepine was not mediated by TSPO. At present, how finasteride inhibits the response to clonazepam is uncertain. There are no known pharmacokinetic interactions between finasteride and benzodiazepines, including clonazepam and midazolam. While clonazepam does not stimulate neurosteroid synthesis in mitochondria (Papadopoulos et al., 1992), isolated cell systems (Mukhin et al., 1989) or some brain regions (Tokuda et al., 2010), in at least one region of the CNS (retina), it can potently and rapidly (within minutes) enhance neurosteroid synthesis (Guarneri et al., 1995). This effect of clonazepam, which occurs in a PK 11195-independent fashion, appears to be mediated by a direct interaction with GABAA receptors. Whether a similar action occurs in brain regions relevant to the anticonvulsant activity of clonazepam remains to be determined.
In conclusion, our results demonstrate a role for neurosteroids in the anticonvulsant action of midazolam. We propose that in addition to directly activating GABAA receptors through an agonist interaction with the intrinsic benzodiazepine recognition site, midazolam enhanced neurosteroid synthesis through an agonist interaction with TSPO, although we cannot exclude the possibility that this occurred in part through a direct interaction with GABAA receptors, as is likely to be the case for clonazepam. Whether the enhanced neurosteroidogenesis occurred peripherally or directly in the brain was not defined in the present study. Although there is evidence that midazolam can influence neurosteroid synthesis locally in the brain (Tokuda et al., 2010), neurosteroids synthesized peripherally can readily enter the brain to influence seizure susceptibility. Therefore, enhanced peripheral neurosteroid synthesis could contribute to the neurosteroid-related component of the effect of midazolam on seizure threshold noted in the present study. Neurosteroids are known to bind to distinct sites on GABAA receptors through which they cause positive allosteric modulation of GABA responses (at low concentrations) and direct activation of the receptor (at higher concentrations) (Hosie et al., 2007). Unlike neurosteroids, agonists that act at the benzodiazepine recognition site do not directly activate GABAA receptors in the absence of GABA. Moreover, neurosteroids cause markedly greater maximal potentiation of GABA responses than agonists at the benzodiazepine recognition site (Kokate et al., 1994). The capacity of neurosteroids to cause large magnitude positive modulation of GABA responses and also to directly activate GABAA receptors confers potent anticonvulsant properties on neurosteroids (Rogawski and Reddy, 2004). The combination of the direct action of midazolam on synaptic GABAA receptors along with the indirect actions mediated by neurosteroids could account for the particularly effective anticonvulsant action of midazolam (Raines et al., 1990). Benzodiazepines only act on a restricted subset of GABAA receptor isoforms (Olsen and Sieghart, 2008). Neurosteroids, in contrast, act on all GABAA receptors subunit combinations and produce a particularly large augmentation in the activity of certain non-synaptic forms, such as those containing δ subunits, that mediate tonic inhibition (Stell et al., 2003; Farrant and Nusser, 2005). It is reasonable to speculate that an effect on non-synaptic, benzodiazepine-insensitive GABAA receptors mediated indirectly by neurosteroids also contributes to the powerful anticonvulsant action of midazolam. Neurosteroids may also contribute to the anticonvulsant actions of other benzodiazepines with TSPO binding activity, and there are benzodiazepines, most notably clonazepam, that may influence neurosteroids through mechanisms that do not involve TSPO.
Glossary
- PK11195
1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide
- PTZ
pentylenetetrazol
- TSPO
translocator protein (18 kDa)
Conflicts of interest
None.
References
- Akula KK, Dhir A, Kulkarni SK. Effect of various antiepileptic drugs in a pentylenetetrazol-induced seizure model in mice. Methods Find Exp Clin Pharmacol. 2009;31:423–432. doi: 10.1358/mf.2009.31.7.1393610. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auta J, Romeo E, Kozikowski A, Ma D, Costa E, Guidotti A. Participation of mitochondrial diazepam binding inhibitor receptors in the anticonflict, antineophobic and anticonvulsant action of 2-aryl-3-indoleacetamide and imidazopyridine derivatives. J Pharmacol Exp Ther. 1993;265:649–656. [PubMed] [Google Scholar]
- Bender AS, Hertz L. Pharmacological characteristics of diazepam receptors in neurons and astrocytes in primary cultures. J Neurosci Res. 1987;18:366–372. doi: 10.1002/jnr.490180215. [DOI] [PubMed] [Google Scholar]
- Bitran D, Foley M, Audette D, Leslie N, Frye CA. Activation of peripheral mitochondrial benzodiazepine receptors in the hippocampus stimulates allopregnanolone synthesis and produces anxiolytic-like effects in the rat. Psychopharmacology (Berl) 2000;151:64–71. doi: 10.1007/s002130000471. [DOI] [PubMed] [Google Scholar]
- Cavallaro S, Korneyev A, Guidotti A, Costa E. Diazepam-binding inhibitor (DBI)-processing products, acting at the mitochondrial DBI receptor, mediate adrenocorticotropic hormone-induced steroidogenesis in rat adrenal gland. Proc Natl Acad Sci U S A. 1992;89:10598–10602. doi: 10.1073/pnas.89.22.10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhir A, Zolkowska D, Murphy RB, Rogawski MA. Seizure protection by intrapulmonary delivery of propofol hemisuccinate. J Pharmacol Exp Ther. 2011;336:215–222. doi: 10.1124/jpet.110.173591. [DOI] [PubMed] [Google Scholar]
- Eom W, Lee JM, Park J, Choi K, Jung SJ, Kim HS. The effects of midazolam and sevoflurane on the GABAA receptors with alternatively spliced variants of the γ2 subunit. Korean J Anesthesiol. 2011;60:109–118. doi: 10.4097/kjae.2011.60.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Finn DA, Beadles-Bohling AS, Beckley EH, Ford MM, Gililland KR, Gorin-Meyer RE, et al. A new look at the 5α-reductase inhibitor finasteride. CNS Drug Rev. 2006;12:53–76. doi: 10.1111/j.1527-3458.2006.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye CA, Paris JJ, Rhodes ME. Increasing 3α,5α-THP following inhibition of neurosteroid biosynthesis in the ventral tegmental area reinstates anti-anxiety, social, and sexual behavior of naturally receptive rats. Reproduction. 2009;137:119–128. doi: 10.1530/REP-08-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin GM, Jelinek GA. Midazolam: an effective intravenous agent for seizure control. Arch Emerg Med. 1987;4:169–172. doi: 10.1136/emj.4.3.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavish M, Bachman I, Shoukrun R, Katz Y, Veenman L, Weisinger G, et al. Enigma of the peripheral benzodiazepine receptor. Pharmacol Rev. 1999;51:629–650. [PubMed] [Google Scholar]
- Guarneri P, Guarneri R, Cascio C, Piccoli F, Papadopoulos V. γ-Aminobutyric acid type A/benzodiazepine receptors regulate rat retina neurosteroidogenesis. Brain Res. 1995;683:65–72. doi: 10.1016/0006-8993(95)00343-o. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, Smart TG. Neurosteroid binding sites on GABAA receptors. Pharmacol Ther. 2007;116:7–19. doi: 10.1016/j.pharmthera.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Jaimovich DG, Shabino CL, Noorani PA, Bittle BK, Osborne JS. Intravenous midazolam suppression on pentylenetetrazol-induced epileptogenic activity in a porcine model. Crit Care Med. 1990;18:313–316. doi: 10.1097/00003246-199003000-00014. [DOI] [PubMed] [Google Scholar]
- Kaminski RM, Rogawski MA. 11β-Hydroxylase inhibitors protect against seizures in mice by increasing endogenous neurosteroid synthesis. Neuropharmacology. 2011;2011:133–137. doi: 10.1016/j.neuropharm.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita A, Furukawa K. Involvement of neurosteroids in the anxiolytic-like effects of AC-5216 in mice. Pharmacol Biochem Behav. 2008;89:171–178. doi: 10.1016/j.pbb.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Kita A, Kohayakawa H, Kinoshita T, Ochi Y, Nakamichi K, Kurumiya S, et al. Antianxiety and antidepressant-like effects of AC-5216, a novel mitochondrial benzodiazepine receptor ligand. Br J Pharmacol. 2004;142:1059–1072. doi: 10.1038/sj.bjp.0705681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokate TG, Svensson BE, Rogawski MA. Anticonvulsant activity of neurosteroids: correlation with γ-aminobutyric acid-evoked chloride current potentiation. J Pharmacol Exp Ther. 1994;270:1223–1229. [PubMed] [Google Scholar]
- Kokate TG, Banks MK, Magee T, Yamaguchi S, Rogawski MA. Finasteride, a 5α-reductase inhibitor, blocks the anticonvulsant activity of progesterone in mice. J Pharmacol Exp Ther. 1999;288:679–684. [PubMed] [Google Scholar]
- Kucken AM, Teissére JA, Seffinga-Clark J, Wagner DA, Czajkowski C. Structural requirements for imidazobenzodiazepine binding to GABA(A) receptors. Mol Pharmacol. 2003;63:289–296. doi: 10.1124/mol.63.2.289. [DOI] [PubMed] [Google Scholar]
- Lacapère JJ, Papadopoulos V. Peripheral-type benzodiazepine receptor: structure and function of a cholesterol-binding protein in steroid and bile acid biosynthesis. Steroids. 2003;68:569–585. doi: 10.1016/s0039-128x(03)00101-6. [DOI] [PubMed] [Google Scholar]
- Lawrence C, Martin BS, Sun C, Williamson J, Kapur J. Endogenous neurosteroid synthesis modulates seizure frequency. Ann Neurol. 2010;67:689–693. doi: 10.1002/ana.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Fur G, Vaucher N, Perrier ML, Flamier A, Benavides J, Renault C, et al. Differentiation between two ligands for peripheral benzodiazepine binding sites, [3H]RO5-4864 and [3H]PK 11195, by thermodynamic studies. Life Sci. 1983;33:449–457. doi: 10.1016/0024-3205(83)90794-4. [DOI] [PubMed] [Google Scholar]
- Mak JC, Barnes PJ. Peripheral type benzodiazepine receptors in human and guinea pig lung: characterization and autoradiographic mapping. J Pharmacol Exp Ther. 1990;252:880–885. [PubMed] [Google Scholar]
- Marangos PJ, Patel J, Boulenger JP, Clark-Rosenberg R. Characterization of peripheral-type benzodiazepine binding sites in brain using [3H]Ro 5-4864. Mol Pharmacol. 1982;22:26–32. [PubMed] [Google Scholar]
- Matsumoto T, Ogata M, Koga K, Shigematsu A. Effect of peripheral benzodiazepine receptor ligands on lipopolysaccharide-induced tumor necrosis factor activity in thioglycolate-treated mice. Antimicrob Agents Chemother. 1994;38:812–816. doi: 10.1128/aac.38.4.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley LD, Park CH, Lan NC, Tomich JM, Shively JE, Gee KW. Benzodiazepines and peptides stimulate pregnenolone synthesis in brain mitochondria. Eur J Pharmacol. 1995;276:145–153. doi: 10.1016/0014-2999(95)00036-k. [DOI] [PubMed] [Google Scholar]
- Mizoule J, Gauthier A, Uzan A, Renault C, Dubroeucq MC, Guérémy C, et al. Opposite effects of two ligands for peripheral type benzodiazepine binding sites, PK 11195 and RO5-4864, in a conflict situation in the rat. Life Sci. 1985;36:1059–1068. doi: 10.1016/0024-3205(85)90491-6. [DOI] [PubMed] [Google Scholar]
- Mukhin AG, Papadopoulos V, Costa E, Krueger KE. Mitochondrial benzodiazepine receptors regulate steroid biosynthesis. Proc Natl Acad Sci U S A. 1989;86:9813–9816. doi: 10.1073/pnas.86.24.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W. International Union of Pharmacology. LXX. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev. 2008;60:243–260. doi: 10.1124/pr.108.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orebaugh SL, Bradford SM. Intravenous versus intramuscular midazolam in treatment of chemically induced generalized seizures in swine. Am J Emerg Med. 1994;12:284–287. doi: 10.1016/0735-6757(94)90139-2. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Guarneri P, Kreuger KE, Guidotti A, Costa E. Pregnenolone biosynthesis in C6-2B glioma cell mitochondria: regulation by a mitochondrial diazepam binding inhibitor receptor. Proc Natl Acad Sci U S A. 1992;89:5113–5117. doi: 10.1073/pnas.89.11.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapère JJ, Lindemann P, et al. Translocator protein (18 kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27:402–409. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Pieri L. Preclinical pharmacology of midazolam. Br J Clin Pharmacol. 1983;16(Suppl. 1):17S–27S. doi: 10.1111/j.1365-2125.1983.tb02267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines A, Henderson TR, Swinyard EA, Dretchen KL. Comparison of midazolam and diazepam by the intramuscular route for the control of seizures in a mouse model of status epilepticus. Epilepsia. 1990;31:313–317. doi: 10.1111/j.1528-1157.1990.tb05381.x. [DOI] [PubMed] [Google Scholar]
- Reddy DS, Rogawski MA. Stress-induced deoxycorticosterone-derived neurosteroids modulate GABAA receptor function and seizure susceptibility. J Neurosci. 2002;22:3795–3805. doi: 10.1523/JNEUROSCI.22-09-03795.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Kim HY, Rogawski MA. Neurosteroid withdrawal model of perimenstrual catamenial epilepsy. Epilepsia. 2001;42:328–336. doi: 10.1046/j.1528-1157.2001.10100.x. [DOI] [PubMed] [Google Scholar]
- Rogawski MA. Principles of antiepileptic drug action. In: Levy RH, Mattson RH, Meldrum BS, Perucca E, editors. Antiepileptic Drugs. 5th edn. Philadelphia, PA: Lippincott Williams & Wilkins; 2002. pp. 3–22. [Google Scholar]
- Rogawski MA, Reddy DS. Neurosteroids: endogenous modulators of seizure susceptibility. In: Rho JM, Sankar R, Cavazos J, editors. Epilepsy: Scientific Foundations of Clinical Practice. New York: Marcel Dekker; 2004. pp. 319–355. [Google Scholar]
- Romeo E, Cavallaro S, Korneyev A, Kozikowski AP, Ma D, Polo A, et al. Stimulation of brain steroidogenesis by 2-aryl-indole-3-acetamide derivatives acting at the mitochondrial diazepam-binding inhibitor receptor complex. J Pharmacol Exp Ther. 1993;267:462–471. [PubMed] [Google Scholar]
- Rovira C, Ben-Ari Y. Developmental study of benzodiazepine effects on monosynaptic GABAA-mediated IPSPs of rat hippocampal neurons. J Neurophysiol. 1993;70:1076–1085. doi: 10.1152/jn.1993.70.3.1076. [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Ströhle A, Hermann B, di Michele F, Spalletta G, Pasini A, et al. Neuroactive steroid concentrations following metyrapone administration in depressed patients and healthy volunteers. Biol Psychiatry. 1998;44:912–914. doi: 10.1016/s0006-3223(97)00521-0. [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Rammes G, Eser D, Baghai TC, Schüle C, Nothdurfter C, et al. Translocator protein (18 kD) as target for anxiolytics without benzodiazepine-like side effects. Science. 2009;325:490–493. doi: 10.1126/science.1175055. [DOI] [PubMed] [Google Scholar]
- Rüsch D, Forman SA. Classic benzodiazepines modulate the open-close equilibrium in α1β2γ2Lγ-aminobutyric acid type A receptors. Anesthesiology. 2005;102:783–792. doi: 10.1097/00000542-200504000-00014. [DOI] [PubMed] [Google Scholar]
- Schoemaker H, Bliss M, Yamamura HI. Specific high-affinity saturable binding of [3H] Ro5-4864 to benzodiazepine binding sites in the rat cerebral cortex. Eur J Pharmacol. 1981;71:173–175. doi: 10.1016/0014-2999(81)90405-2. [DOI] [PubMed] [Google Scholar]
- Serra M, Madau P, Chessa MF, Caddeo M, Sanna E, Trapani G, et al. 2-Phenyl-imidazo[1,2-a]pyridine derivatives as ligands for peripheral benzodiazepine receptors: stimulation of neurosteroid synthesis and anticonflict action in rats. Br J Pharmacol. 1999;127:177–187. doi: 10.1038/sj.bjp.0702530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So EC, Chang YT, Hsing CH, Poon PW, Leu SF, Huang BM. The effect of midazolam on mouse Leydig cell steroidogenesis and apoptosis. Toxicol Lett. 2010;192:169–178. doi: 10.1016/j.toxlet.2009.10.017. [DOI] [PubMed] [Google Scholar]
- Stell BM, Brickley SG, Tang CY, Farrant M, Mody I. Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by δ subunit-containing GABAA receptors. Proc Natl Acad Sci U S A. 2003;100:14439–14444. doi: 10.1073/pnas.2435457100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuda K, O'Dell KA, Izumi Y, Zorumski CF. Midazolam inhibits hippocampal long-term potentiation and learning through dual central and peripheral benzodiazepine receptor activation and neurosteroidogenesis. J Neurosci. 2010;30:16788–16795. doi: 10.1523/JNEUROSCI.4101-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Suzuki T, Misawa M. Subsensitivity to mitochondrial diazepam binding inhibitor receptor agonist FGIN-1-27-induced antiseizure effect in diazepam-withdrawn mice. Life Sci. 1998;62:PL213–PL217. doi: 10.1016/s0024-3205(98)00060-5. [DOI] [PubMed] [Google Scholar]
- Ugale RR, Mittal N, Hirani K, Chopde CT. Essentiality of central GABAergic neuroactive steroid allopregnanolone for anticonvulsant action of fluoxetine against pentylenetetrazole-induced seizures in mice. Brain Res. 2004;1023:102–111. doi: 10.1016/j.brainres.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Verleye M, Akwa Y, Liere P, Ladurelle N, Pianos A, Eychenne B, et al. The anxiolytic etifoxine activates the peripheral benzodiazepine receptor and increases the neurosteroid levels in rat brain. Pharmacol Biochem Behav. 2005;82:712–720. doi: 10.1016/j.pbb.2005.11.013. [DOI] [PubMed] [Google Scholar]