

Abstract
BACKGROUND AND PURPOSE
Wherever they are located, dopamine transporters (DATs) clear dopamine (DA) from the extracellular milieu to help regulate dopaminergic signalling. Exposure to amphetamine (AMPH) increases extracellular DA in the synaptic cleft, which has been ascribed to DAT reverse transport. Increased extracellular DA prolongs postsynaptic activity and reinforces abuse and hedonic behaviour.
EXPERIMENTAL APPROACH
Xenopus laevis oocytes expressing human (h) DAT were voltage-clamped and exposed to DA, R(-)AMPH, or S(+)AMPH.
KEY RESULTS
At -60mV, near neuronal resting potentials, S(+)AMPH induced a depolarizing current through hDAT, which after removing the drug, persisted for more than 30 min. This persistent leak in the absence of S(+)AMPH was in contrast to the currents induced by R(-)AMPH and DA, which returned to baseline immediately after their removal. Our data suggest that S(+)AMPH and Na+ carry the initial S(+)AMPH-induced current, whereas Na+ and Cl- carry the persistent leak current. We propose that the persistent current results from the internal action of S(+)AMPH on hDAT because the temporal effect was consistent with S(+)AMPH influx, and intracellular S(+)AMPH activated the effect. The persistent current was dependent on Na+ and was blocked by cocaine. Intracellular injection of S(+)AMPH also activated a DA-induced persistent leak current.
CONCLUSIONS AND IMPLICATIONS
We report a hitherto unknown action of S(+)AMPH on hDAT that potentially affects AMPH-induced DA release. We propose that internal S(+)AMPH acts as a molecular stent that holds the transporter open even after external S(+)AMPH is removed. Amphetamine-induced persistent leak currents are likely to influence dopaminergic signalling, DA release mechanisms, and amphetamine abuse.
Keywords: dopamine transporter, dopamine, amphetamine, electrophysiology, Xenopus oocyte expression
Introduction
Cocaine and amphetamine profoundly influence dopaminergic neurotransmission and therefore implicate the dopamine (DA) transporter (DAT). In response to prolonged exposure to these drugs, DAT reduces its capacity for DA transport (Iversen, 2006; Samuvel et al., 2008). In contrast to the DAT inhibitor, cocaine, amphetamine acts as a DAT substrate that flows into cells through the transporter (Volz et al., 2007; Erreger et al., 2008). Thus, cocaine and amphetamine both increase extracellular DA by decreasing its uptake, but they do so by entirely different mechanisms: whereas cocaine blocks DA uptake, amphetamine replaces DA as a substrate, and, in addition, amphetamine induces the release of DA into the synaptic cleft (Iversen, 2006). In striatal slices, as one example, amphetamine causes a gradual rise in extracellular DA that lasts for over 30 min in normal mice, whereas no analogous increase exists in −/− DAT mice (Giros et al., 1996). Although DAT is required for the DA releasing action of amphetamine, it is not needed for the vesicle-depleting action of amphetamine; furthermore, in the absence of amphetamine, cytoplasmic DA is considered to be insufficiently concentrated to reverse DAT, implying that AMPH releases DA from vesicular stores before DA efflux (Jones et al., 1998). In some cases, however, as in dendrodendritic autoinhibition, DAT block abolishes DA efflux in the absence of AMPH (Falkenburger et al., 2001).
Because amphetamine releases DA from terminals in the frontal lobe and limbic system, amphetamine may be used clinically to treat medical conditions such as attention-deficit hyperactivity disorder or narcolepsy (Burnette et al., 1996; Fleckenstein et al., 2007). Therapeutic drugs such as Adderall (dextro-amphetamine) are mixtures of racemic amphetamine and dextro-amphetamine salts. Clinical manifestations associated with the abuse of amphetamine or its precursors or derivatives, such as phenethylamine or methamphetamine, are well documented (Potkin et al., 1979; Romanelli and Smith, 2006; Winslow et al., 2007).
Amphetamine is a homologue of phenethylamine and the parent compound of a wide range of psychoactive derivatives, from the N-methylated methamphetamine (METH) to MDMA (3,4-methylenedioxy-N-methamphetamine), commonly known as ecstasy. Because DAT is the primary target for AMPH, the DAT is most frequently implicated in the reinforcing properties and abuse potential of AMPH (Sulzer et al., 1993; 1995; Seidel et al., 2005). The reward and addiction properties of AMPH apparently correlate with its ability to increase extracellular DA levels by mechanisms as yet only partially understood.
The principal mechanisms proposed for AMPH-induced increases in extracellular DA are the facilitated exchange model (Fischer and Cho, 1979), the DAT efflux channel and reverse transport model (Kahlig et al., 2005) and the vesicular depletion model, which is also called the weak-base model (Sulzer et al., 1993; 1995). Reverse transport depends on membrane depolarization, and thus the results reported here have implications for this model. Here we suggest a mechanism that results from our electrophysiological data and is based on a novel action of S(+)AMPH. S(+)AMPH transport and S(+)AMPH-induced current both depend on extracellular Na+, which also carries part of the current. In our model, hDAT transports S(+)AMPH into the cell where it becomes available to bind hDAT at an internally accessible site. S(+)AMPH binding to the internal site maintains hDAT in an open state long after the removal of external S(+)AMPH. This persistent open state defines a use-dependent ‘leak’ current, which may be related to the previously described substrate-independent leak (Sonders et al., 1997b), although this is speculative at the present time. The leak in oocytes is present in dopaminergic neurons from Caenorhabditis elegans (Carvelli et al., 2004; 2008), and possibly in dopaminergic neurons from rat (Ingram et al., 2002). Such leaks are a general phenomena in monoamine transporter (DeFelice and Goswami, 2007) and in other transport systems where they are called ‘slips’ and assigned a natural function in native cells (Nelson et al., 2002). Surprisingly, once hDAT has been exposed to S(+)AMPH, subsequent exposure to DA also results in a persistent leak. In an unclamped presynaptic terminal, the persistent leak would depolarize the presynaptic membrane and increase the likelihood of vesicular fusion and dopamine release.
Methods
Expression of the human DAT in Xenopus oocytes
Oocytes were harvested and prepared from adult Xenopus laevis females following standard procedures (Machaca and Hartzell, 1998; Iwamoto et al., 2006). We select stage V–VI oocytes for cRNA injection within 24 h of isolation. cRNA was transcribed in the pOTV vector (gift of Mark Sonders, Columbia University) using Ambion mMessage Machine T7 kit (Ambion Inc., Austin, TX). Each oocyte was injected with 50 nL of 1 µg·µL−1 hDAT cRNA (final amount 50 ng) (Nanoject AutoOocyteInjector, Drummond Scientific Co., Broomall, PA) and incubated at 18°C for 4–8 days in Ringer's solution supplemented with Na-pyruvate (550 µg·mL−1), streptomycin (100 µg·mL−1), tetracycline (50 µg·mL−1) and 5% dialyzed horse serum.
Two-electrode voltage clamp (TEVC)
Electrodes had resistances from 1–5 MΩ. Xenopus oocytes expressing hDAT were voltage-clamped to −60 mV (unless otherwise noted), and buffer was gently perfused until a stable baseline was obtained. Then the experimental substrates were perfused until stable currents were obtained for the time periods indicated. In order to compare data from different oocytes or after successive applications of a particular compound, we normalized data to an initial response to 10 µM DA.
Oocyte injection of S(+)AMPH
We injected a small volume of concentrated drug and calculated the final concentration by dilution in the oocyte volume. For example, 50 nL of 0.5 mM S(+)AMPH injected into an oocyte with an estimated volume of 1 µL (stage V–VI oocytes are 1–1.2 mm in diameter) gave a 25 µM final concentration. Repeated injections at the same concentration, or a single injection at a higher concentration produced a range of S(+)AMPH concentrations inside the oocyte from 0 to 180 µM.
Solutions
Extracellular (in mM): 120 NaCl, 7.5 HEPES, 5.4 K+ gluconate, 1.2 Ca2+ gluconate, pH 7.4 with KOH. For Na+-free solution, 120 NaCl is replaced with 120 mM N-methyl-D-glucamine (NMDG)-Cl. Intracellular: 3 M KCl.
Results
Structures of dopamine and amphetamine stereoisomers
Racemic amphetamine (AMPH) consists of equal amounts of S(+)AMPH and R(–)AMPH. The AMPH stereoisomers share similarity with the unique structure of dopamine (Figure 1). Although DA, S(+)AMPH and R(–)AMPH have similar structures, they have markedly distinct effects on the human DAT (hDAT) as regards inducing a current under voltage clamp conditions in hDAT-expressing oocytes (Figure 3A).
Figure 1.
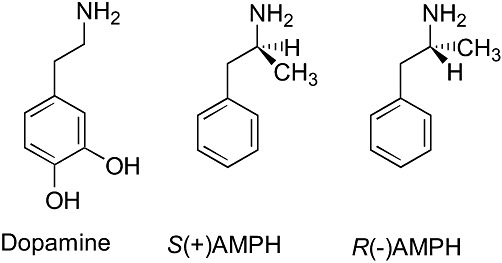
Structures of dopamine and enantiomers of amphetamine. Chemical composition of dopamine, S(+)amphetamine and R(–) amphetamine (labelled S(+)AMPH and R(–)AMPH, respectively), showing their structural similarity.
Figure 3.
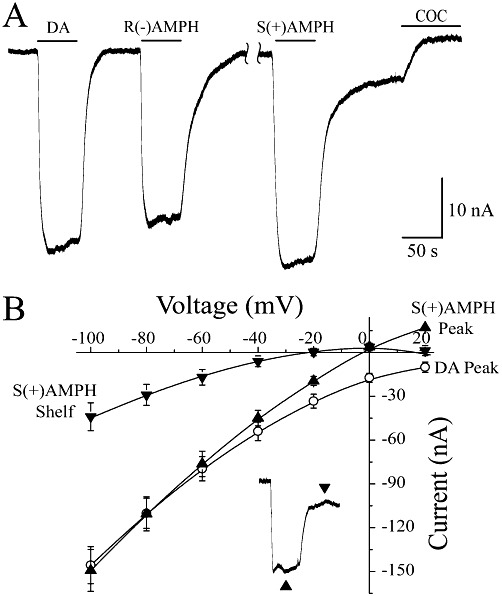
DA- and AMPH-induced currents. (A) At −60 mV, neither 10 µM DA nor 10 µM R(–)AMPH induced a shelf but the current always return to baseline after their removal. S(+)AMPH on the other hand induced a prominent shelf current that was blocked by cocaine (10 µM). The peak currents are approximately the same at −60 mV for 10 µM DA, R(–)AMPH, or S(+)AMPH. Note that in the presence of 10 µM cocaine the current returned to values positive to the initial baseline, indicating the presence of an endogenous leak current. (B) Baseline subtracted I(V) curves for DA, S(+)AMPH peak and S(+)AMPH shelf. The S(+)AMPH peak I(V) was shifted to the left above −60 mV compared with the DA peak, consistent with the conductance of both Na+ and S(+)AMPH through hDAT. The S(+)AMPH shelf I(V) was further shifted to the left, consistent with the absence of S(+)AMPH and presence of Cl- ions flowing through hDAT.
Dopamine and S(+)AMPH differentially affect hDAT
Xenopus oocytes expressing hDAT were exposed to DA or S(+)AMPH (10 µM, −60 mV) for time periods ranging from 20 to 200 s (Figure 2). Addition of 10 µM DA elicited an inward current from 10 to 70 nA at −60 mV, depending on the level of hDAT expression. We confined ourselves to expression levels in this range. When DA was removed, the DA-induced current returned to baseline (Figure 2A) regardless of exposure time. As with DA, brief exposures to 10 µM S(+)AMPH (30 s or less) elicited currents that returned to baseline following S(+)AMPH removal. However, for exposure times greater than 30 s, the current induced by 10 µM S(+)AMPH persisted despite removal of external S(+)AMPH. Furthermore, the amplitude of the persistent current depended on the length of exposure to S(+)AMPH (Figure 2B). S(+)AMPH-induced persistent currents, also referred to as ‘shelf’ currents, may last as long as 30 min (data not shown). We first discovered the shelf current using a racemic mixture of AMPH, which also induces a long-lasting persistent current after the racemate is removed (data not shown). The relationship between the amplitude of the shelf current and the duration of S(+)AMPH exposure (normalized to the initial hDAT-mediated peak current) shows that the shelf current amplitude saturates as a function of S(+)AMPH exposure time (Figure 2C). The existence of a shelf current after removal of S(+)AMPH is dependent not only on the duration of S(+)AMPH exposure but also on extracellular S(+)AMPH concentration. If the concentration of external S(+)AMPH was increased from 10 to 30 µM, an exposure time that was too brief to elicit a bona fide shelf current was now capable of doing so (Figure 2D).
Figure 2.
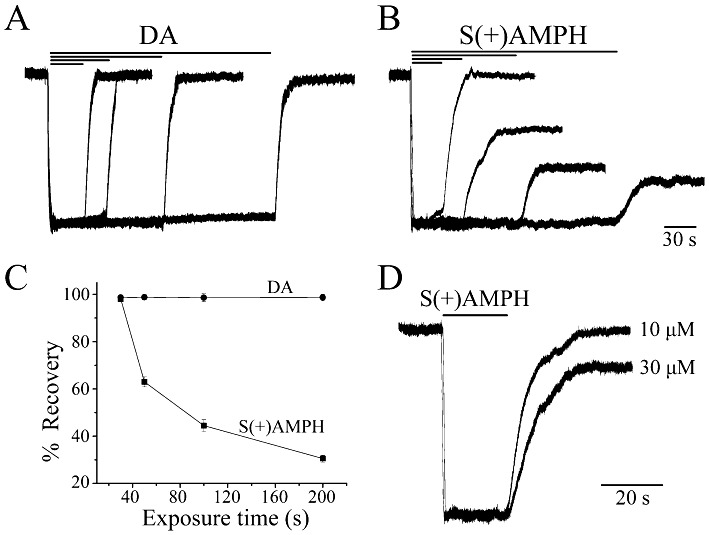
S(+)AMPH induces a persistent ‘shelf’ current through the human dopamine transporter (hDAT). (A) External DA (10 µM) induced a large inward ‘peak’ current at V =−60 mV that returned to baseline when DA was removed, regardless of DA exposure time. DA peak currents were normalized to the briefest exposure time. (B) S(+)AMPH (10 µM) induced a similar inward peak current for exposures less than 30 s; however, for longer exposure times, a current that we term ‘leak’ or ‘shelf’ remained long after S(+)AMPH had been removed, and the amplitude of the shelf was proportional to the length of exposure. S(+)AMPH peak currents were normalized to the briefest exposure time. (C) Amplitude of the shelf current relative to the initial peak current plotted against exposure time of external S(+)AMPH, compared with the corresponding DA currents (n= 4, ±SEM). (D) A relatively brief and initial exposure to S(+)AMPH (20 s.), which ordinarily would not produce a shelf current, did so if the concentration of S(+)AMPH was increased from 10 to 30 µM. For the same exposure range of times and concentrations, neither DA nor S(+)AMPH induced peak or shelf currents in mock-injected oocytes (data not shown).
The current–voltage relationship of currents induced by DA and S(+)AMPH
At −60 mV, 10 µM R(–)AMPH consistently induced a peak current slightly less in magnitude than that induced by 10 µM DA, whereas 10 µM S(+)AMPH induced a slightly larger current; these differences were less below −60 mV and more noticeable above −60 mV (Figure 3B). Strikingly, R(–)AMPH produced no authentic shelf current compared with S(+)AMPH but, at −60 mV, the current always returned to its pre-stimulus value (Figure 3A). However, after a sufficiently long exposure time, S(+)AMPH induced a prominent shelf current that was blocked by the hDAT inhibitor, cocaine (Figure 3A). Cocaine also blocked the peak currents for DA, R(–)AMPH and S(+)AMPH (not shown) and in all cases the current returned to positive values compared with the pre-stimulus baseline, as shown in Figure 3A. Thus, hDAT, like other co-transporters in this family, can produce cocaine-sensitive DA- and AMPH-induced currents and cocaine-sensitive leak currents (Sonders et al., 1997a; Amara and Sonders, 1998), to which we have now added the cocaine-sensitive shelf current. As Na+ has a major role in the transport of substrate by monoamine transporters (Nelson, 1998; Rudnick, 1998b), we investigated its effect on the currents induced by DA and S(+)AMPH. The peak currents induced by 10 µM DA, R(–)AMPH or S(+)AMPH were abolished when external Na+ was replaced with NMDG (data not shown), and the shelf current was dependent on Na+ (Figure 5). We generated I(V) curves for the initial current and shelf current induced by DA, S(+)AMPH peak and S(+)AMPH shelf (Figure 3B). The I(V) for DA bends downwards (towards greater negative currents) at more positive potentials. This is due to blockade of the endogenous leak current of the DAT (Sonders et al., 1997a; Ingram and Amara, 2000), also found in the 5-HT transporter (Galli et al., 1997) and NA transporter (Galli et al., 1998). When external S(+)AMPH was present, the I(V) curve shifted to the left (between −20 and 20 mV), possibly reflecting the co-conductance of Na+ and S(+)AMPH cations. The shelf I(V) was further shifted to the left, implying not only the absence of S(+)AMPH but also the probable presence of Cl- ions, which are known to carry current in DAT (Ingram et al., 2002; Carvelli et al., 2008)
Figure 5.
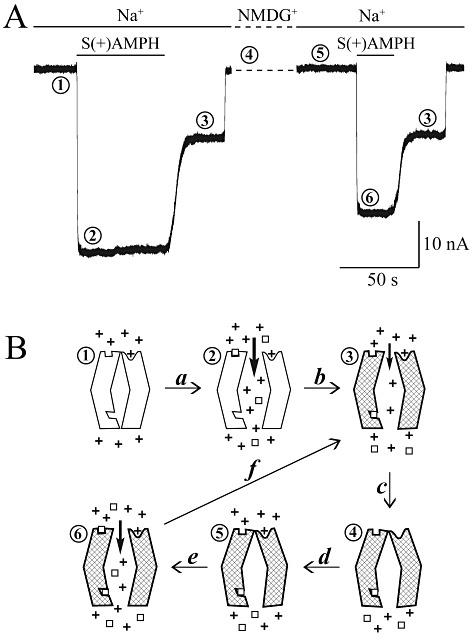
Model for S(+)AMPH-induced, hDAT-mediated peak and shelf currents. (A) Phase 1: the baseline current at V =−60 mV in external physiological solution containing Na+. Phase 2: the 10 µM S(+)AMPH-induced peak current in Na+, nominally between 10 and 25 nA. Phase 3: the shelf current (persistent leak current), which remains after removing external S(+)AMPH. At fixed voltage, the shelf amplitude depends on exposure time and concentration of external S(+)AMPH. Phase 4: replacing Na+ with NMDG introduces a shift in baseline that we removed from the figure (dashed line); however, re-introducing Na+ during phase 4 (phase 5) does not restore the shelf current but returns the current to the original baseline [S(+)AMPH has been transported into the cell during phase 2]. Phase 6: reintroducing external S(+)AMPH in the presence of Na+ induces a new peak current. Note that the second application of S(+)AMPH is normally too brief to elicit a shelf current; however, S(+)AMPH is already present inside the cell from the first application. The new peak is smaller than the first peak, possibly due to hDAT internalization. Finally, after removing S(+)AMPH for the second time, the shelf current again manifests itself. (B) The ‘+’ symbols stand for Na+ ions, and the ‘open squares’ stand for S(+)AMPH) or in some experiments DA. The hatched transporter indicates internal occupancy by S(+)AMPH and a long-lasting ‘molecular stent’ configuration. The numbers above each state of the transporter correspond to the traces in (A). Transition (a) opens the top and bottom gates, which for brief external S(+)AMPH exposures would close. Transition (b) occurs after longer exposures and S(+)AMPH has built up inside to the extent that the inner S(+)AMPH site remains occupied and holds both gates open (molecular stent hypothesis), even in the absence of external S(+)AMPH. Transition (c): removing external Na+ closes the outer gate, which does not reopen without external Na+ and S(+)AMPH both present (transitions d and e), rendering the transporter capable of (and indeed more prone to) forming the molecular stent (transition f), because internal S(+)AMPH is still present.
S(+)AMPH operates on hDAT from the inside
Until now, we have applied S(+)AMPH externally and have shown that an exposure time of 30–50 s or more (at 10 µM, −60mV) activates the shelf current. Because of this relatively long time to elicit an effect, and because increasing S(+)AMPH concentration reduces the time required to activate a shelf current (Figure 2D), we suspected that S(+)AMPH was being transported into the cell to exert its effect internally. To test this possibility, we injected S(+)AMPH into the oocyte to obtain a range of internal concentrations (see Methods) and correlated the internal concentration with the degree of persistent leak current. Whereas a brief application (10 s) of 10 µM S(+)AMPH externally was insufficient to induce a shelf current, injecting 10 µM S(+)AMPH into the oocyte induced a prominent shelf current (Figure 4A). Remarkably, using external DA instead of S(+)AMPH also resulted in a shelf current after the internal injection of S(+)AMPH (Figure 4B, centre trace), and increasing the internal S(+)AMPH (second injection in the same oocyte) generated a larger shelf current (Figure 4B, rightmost trace). Injecting DA into the oocyte did not have a similar effect on the external S(+)AMPH or DA (data not shown). These data suggest that S(+)AMPH is a use-dependent drug, and the ability to elicit the shelf current depends on its accessibility to hDAT from the inside. Thus, when hDAT has been previously exposed to S(+)AMPH, either external S(+)AMPH or external DA has the potential to generate persistent currents, though for the same external and internal concentrations, S(+)AMPH has a stronger effect (Figure 4C). Following the protocol in Figure 4B, we methodically titrated internal S(+)AMPH by repeated injections at the same concentration or higher concentrations in different oocytes (see Methods). These data are pooled in Figure 4D, which shows that for 10 µM external DA applied for 10 s, the greater the internal S(+)AMPH concentration, the greater the shelf current (the shelf current is plotted as a percentage of induced current that has returned to baseline after removing external DA). The DA-induced shelf current saturated at 80% full recovery in the presence of internal S(+)AMPH, with a Hill coefficient nH= 1.7 and k1/2= 37 µM.
Figure 4.
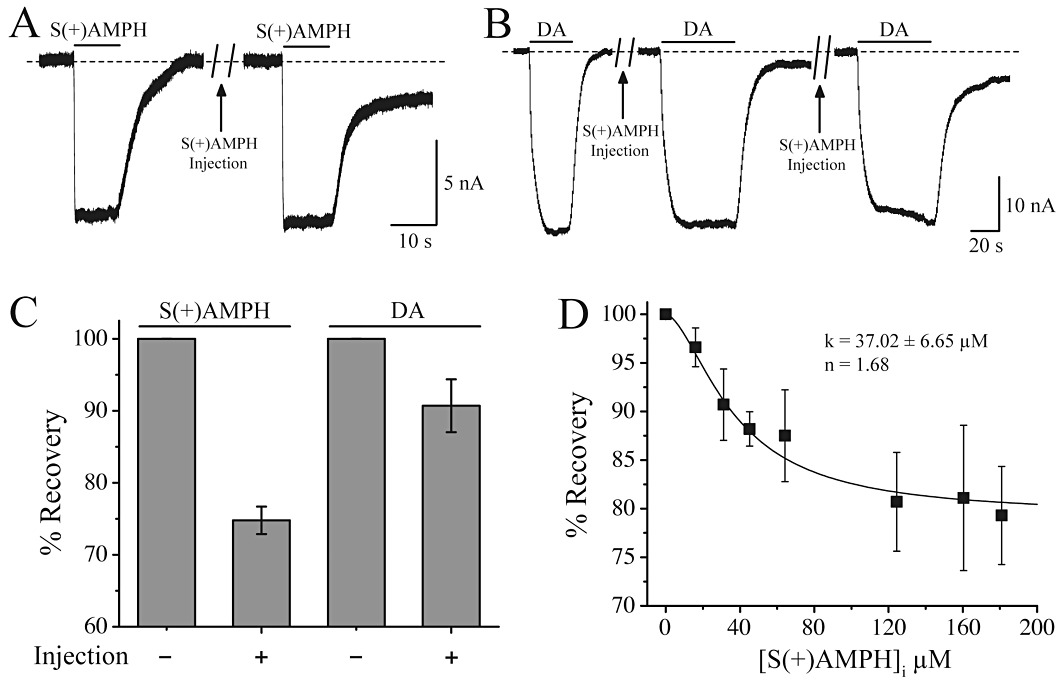
S(+)AMPH injection promotes the shelf current. (A) A brief application (10 s) of 10 µM external S(+)AMPH did not illicit a shelf current; however, after injecting sufficient S(+)AMPH into the cell to elevate the internal concentration to 25 µM (see Methods), the same challenge induced a prominent shelf current. (B) Dopamine, which ordinarily would not produce a shelf current (left trace), now induced a shelf current after its removal in added after a similar 25 µM injection of S(+)AMPH (centre trace). Doubling the internal S(+)AMPH concentration resulted in the generation of a larger shelf (right trace). Injecting DA into the oocyte had no similar effect on the responses to external S(+)AMPH or DA (data not shown). (C) For the same external and internal concentrations, S(+)AMPH generated a stronger shelf current than DA. Following the same protocol as in (B), we titrated internal S(+)AMPH by repeated injections into the same oocyte or single more concentrated injections in different oocytes (see Methods). (D) Pooled data for 10 µM external DA applied for 10 s: the greater the internal S(+)AMPH concentration the greater the DA-induced shelf current, that is, in the presence of internal S(+)AMPH, less of the DA-induced current was able to return to baseline after external DA removal. For a DA challenge, the shelf current saturates at 80% full recovery, with a Hill coefficient nH= 1.7 and k1/2= 37 µM.
Internal S(+)AMPH is silent without external S(+)AMPH and Na+
Based on our experiments, we formulated a model of the S(+)AMPH-induced persistent current based on two gates in the hDAT protein: an external gate operated by S(+)AMPH and Na+ and blocked by cocaine, and an internal gate operated by S(+)AMPH (Figure 5). Previous work suggests that Na+ plays a regulatory role at the internal face of hDAT (Khoshbouei et al., 2003); however, this possibility was not explicitly tested in the present study.
In stage 1 of Figure 5, we would expect hDAT to maintain a small leak current even in the absence of substrate. This endogenous leak, which can be revealed by cocaine, was present even without prior exposure to substrate (Figure 3A and Sonders and Amara, 1996); here we ignored this background leak and focused only on the substrate-induced currents. In our model, opening the outer gate requires both Na+ and S(+)AMPH. R(–)AMPH or DA also operate the outer gate. Opening the gate generates the peak (or steady state) current in phase 2, which is probably carried by Na+ and S(+)AMPH cations, although Cl- is also implicated (see Figure 3B and Ingram et al., 2002; Carvelli et al., 2008). As transport ensues, the inner gate becomes exposed to S(+)AMPH and, once occupied, remains open and holds the outer gate open, even though external S(+)AMPH has been removed. This allosteric action between the inner and outer gate acts as a molecular stent that holds the transporter in an open state 3. The molecular stent requires external Na+, and on removing Na+ the current returns to baseline 4. However, merely re-introducing Na+ does not restore the current 5; rather, S(+)AMPH and Na+ must both be present to regenerate the peak 6 and shelf 4 currents. Unless this dual condition is fulfilled, internal S(+)AMPH is silent.
Discussion
In this work, we present a novel finding for the action of S(+)AMPH on hDAT. Our results suggest a model in which S(+)AMPH is transported into the cell through hDAT, whereupon the drug has access to an internal site on the transporter that induces a persistent ‘shelf’ current. Bound to the internal site, S(+)AMPH induces a molecular stent in hDAT that holds the transporter open long after external amphetamine is removed. The internally accessibly site is in addition to an external site for (S+)AMPH, which initiates the transport of the drug to the inside. Structural and functional evidence argue for one centrally located substrate binding site for neurotransmitter transporters (Yamashita et al., 2005; Piscitelli et al., 2010), while other data (Shi et al., 2008; Shan et al., 2011; Zhao et al., 2011) support an additional site external to the central site (see also DeFelice and Goswami, 2007; De Felice, 2011). Figure 5 proposes two distinct functional sites for S(+)AMPH: an exterior site, which along with Na+ opens the pathway, and an internal site for S(+)AMPH (but not DA) that keeps the pathway open, even though the external site may be empty.
Regardless of the specific mechanism, the existence of the shelf current has physiological consequences for synaptic transmission. Previous results suggest the leak current would depolarize the presynaptic membrane (Ingram et al., 2002; Carvelli et al., 2004) and increase the probability of transmitter release. Furthermore, once it is exposed to S(+)AMPH, the presynaptic membrane would respond to DA abnormally for as long as S(+)AMPH remains inside the terminal (30 min) at a significant concentration (30 µM). S(+)AMPH is therefore a use-dependent drug that pre-conditions hDAT to generate a persistent leak when subsequently challenged with an endogenous transmitter, dopamine, or exogenous AMPH. The half-life of AMPH inside cells is not well known (Seiden et al., 1993), but it ranges from 2 h up to 12 h if measured from body fluids (Mofenson and Greensher, 1975; Verstraete, 2005; Verstraete and Heyden, 2005).
The persistent leak flows in the absence of external S(+)AMPH or DA and must therefore be entirely composed of co-transported ions, most likely Na+ and Cl-. We explicitly tested the dependence of S(+)AMPH-induced currents on external Na+, and the results are consistent with well-known criteria for coupled co-transport (Gu et al., 1998; Rudnick, 1998a,b; Petersen and DeFelice, 1999). Near the resting potential of most cells (−60 mV), the persistent leak current is constant as long as the voltage is held constant. In adult neurons, −60 mV is near the equilibrium potential of Cl-. A depolarizing leak could represent the entrance of Na+ or the exit of Cl-; however, near rest, the Na+ contribution would be larger than the Cl- contribution, resulting in net depolarization, whereby the Na+ leak would diminish and the Cl- leak would be enhanced. The net effect on actual neurons is difficult to predict. In two cases, however, a net depolarization was reported for dopaminergic neurons in tissue culture (Ingram et al., 2002; Carvelli et al., 2004).
It is already known that DA acting on DAT elicits inward currents through a channel-like mechanism that can depolarize dopaminergic neurons and increase their excitability (Ingram et al., 2002; Carvelli et al., 2004; 2008). Ingram and colleagues suggest that tonic activity excites dopaminergic neurons by activating an uncoupled Cl- conductance mediated by the DAT. In the Ingram et al. (2002) study, the effect of DA or amphetamine on voltage-clamped DAT was tested during external perfusion of the cells (corresponding to the peak or steady-state current in our model). In addition to the peak current, in which S(+)AMPH and DA both participate, we have uncovered a persistent leak current, which as discussed above would prolong depolarization long after amphetamine is removed. Prolonged depolarization of the presynaptic membrane would therefore further increase the excitability of neurons and the probability of neurotransmitter release as postulated by Ingram et al. A full understanding of this effect and its magnitude is at present unknown and would require a complete knowledge of current generating channels and receptors on the presynaptic membrane. In particular, D2 dopamine receptors are known to be involved in transmitter release (Schmitz et al., 2001). It has been suggested that released dopamine may feed back onto D2 autoreceptors to depress neuronal activity (Sulzer and Galli, 2003). In addition, AMPH may regulate hDAT indirectly by targeting or modulating proteins that then affect DAT function. These possible responses extend the range of synaptic states regulated by neurotransmitters, to which the newly discovered leak current will undoubtedly contribute to a more complete understanding of amphetamine action.
The density of the DAT would also be a contributing factor to the relative effect of DA- or AMPH-induced currents. Rapid treatment of rat striatal synaptosomes with low doses of amphetamine increases surface expression of the DAT. Either dopamine or amphetamine increase surface DAT within 10 s of substrate addition and steadily increase surface DAT until removal 2 min later. In these experiments, exocytosis of DAT was blocked with tetanus and botulinum neurotoxins. These data demonstrate that dopamine and amphetamine can rapidly increase surface DAT possibly to respond rapidly during dopamine secretion (Furman et al., 2009). However, it is also known that long-term exposure to amphetamine can decrease surface DAT expression (Saunders et al., 2000; Galici et al., 2003). Thus, the increase in surface DAT occurs within a minute and has a fairly short life of a few minutes, whereas longer treatments with AMPH, especially at doses equal to or greater than 10 mM, cause down-regulation of DAT. We believe that a decrease in surface hDAT is responsible for the consistently observed decreased response to a second application of S(+)AMPH, as seen in Figure 5A. Nevertheless, even brief exposure to amphetamine, normally too brief to elicit a shelf current, readily demonstrates a shelf current after the cell had been previously exposed to S(+)AMPH (pre-conditioning of the cell).
Persistent depolarizing currents following amphetamine exposure would be relevant to any model of presynaptic physiology and transmitter release. The resulting persistent depolarization of dopaminergic neurons caused in particular by S(+)AMPH, a component of street amphetamine and prescription drugs, could play an important role in behavioural effects, including craving, withdrawal and relapse, as well as the pleasurable effects of amphetamine, such as hyperactivity and euphoria.
Acknowledgments
We wish to recognize Prof Richard Glennon of VCU School of Pharmacy, Department of Medicinal Chemistry, and Dr Habibeh Khoshbouei of Meharry School of Medicine, Department of Pharmacology for contributions to this work. This research was supported by NIH 1RC1DA028112-01 (LJD) and DA026947-01A1 (HK).
Glossary
- AMPH
amphetamine
- DA
dopamine
- DAT
dopamine transporter
- hDAT
human DAT
- R(–)AMPH
levo (negative) amphetamine
- S(+)AMPH
dextro (plus) amphetamine
Conflict of interest
The authors of this article have no conflict of interest.
References
- Amara SG, Sonders MS. Neurotransmitter transporters as molecular targets for addictive drugs. Drug Alcohol Depend. 1998;51:87–96. doi: 10.1016/s0376-8716(98)00068-4. [DOI] [PubMed] [Google Scholar]
- Burnette WB, Bailey MD, Kukoyi S, Blakely RD, Trowbridge CG, Justice JB., Jr Human norepinephrine transporter kinetics using rotating disk electrode voltammetry. Anal Chem. 1996;68:2932–2938. doi: 10.1021/ac960022x. [DOI] [PubMed] [Google Scholar]
- Carvelli L, McDonald PW, Blakely RD, DeFelice LJ. Dopamine transporters depolarize neurons by a channel mechanism. Proc Natl Acad Sci USA. 2004;101:16046–16051. doi: 10.1073/pnas.0403299101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvelli L, Blakely RD, DeFelice LJ. Dopamine transporter/syntaxin 1A interactions regulate transporter channel activity and dopaminergic synaptic transmission. Proc Natl Acad Sci USA. 2008;105:14192–14197. doi: 10.1073/pnas.0802214105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice LJ. Evaluation of: Piscitelli CL et al. Nature 2010. 2011;468:1129–1132. doi: 10.1038/nature09581. F1000.com/7439956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelice LJ, Goswami T. Transporters as channels. Annu Rev Physiol. 2007;69:87–112. doi: 10.1146/annurev.physiol.69.031905.164816. [DOI] [PubMed] [Google Scholar]
- Erreger K, Grewer C, Javitch JA, Galli A. Currents in response to rapid concentration jumps of amphetamine uncover novel aspects of human dopamine transporter function. J Neurosci. 2008;28:976–989. doi: 10.1523/JNEUROSCI.2796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenburger BH, Barstow KL, Mintz IM. Dendrodendritic inhibition through reversal of dopamine transport. Science. 2001;293:2465–2470. doi: 10.1126/science.1060645. [DOI] [PubMed] [Google Scholar]
- Fischer JF, Cho AK. Chemical release of dopamine from striatal homogenates: evidence for an exchange diffusion model. J Pharmacol Exp Ther. 1979;208:203–209. [PubMed] [Google Scholar]
- Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol. 2007;47:681–698. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- Furman CA, Chen R, Guptaroy B, Zhang M, Holz RW, Gnegy M. Dopamine and amphetamine rapidly increase dopamine transporter trafficking to the surface: live-cell imaging using total internal reflection fluorescence microscopy. J Neurosci. 2009;29:3328–3336. doi: 10.1523/JNEUROSCI.5386-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galici R, Galli A, Jones DJ, Sanchez TA, Saunders C, Frazer A, et al. Selective decreases in amphetamine self-administration and regulation of dopamine transporter function in diabetic rats. Neuroendocrinology. 2003;77:132–140. doi: 10.1159/000068650. [DOI] [PubMed] [Google Scholar]
- Galli A, Petersen CI, deBlaquiere M, Blakely RD, DeFelice LJ. Drosophila serotonin transporters have voltage-dependent uptake coupled to a serotonin-gated ion channel. J Neurosci. 1997;17:3401–3411. doi: 10.1523/JNEUROSCI.17-10-03401.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli A, Blakely RD, DeFelice LJ. Patch-clamp and amperometric recordings from norepinephrine transporters: channel activity and voltage-dependent uptake. Proc Natl Acad Sci USA. 1998;95:13260–13265. doi: 10.1073/pnas.95.22.13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Gu H, Caplan MJ, Rudnick G. Cloned catecholamine transporters expressed in polarized epithelial cells: sorting, drug sensitivity, and ion-coupling stoichiometry. Adv Pharmacol. 1998;42:175–179. doi: 10.1016/s1054-3589(08)60721-8. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Amara SG. Arachidonic acid stimulates a novel cocaine-sensitive cation conductance associated with the human dopamine transporter. J Neurosci. 2000;20:550–557. doi: 10.1523/JNEUROSCI.20-02-00550.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram SL, Prasad BM, Amara SG. Dopamine transporter-mediated conductances increase excitability of midbrain dopamine neurons. Nat Neurosci. 2002;5:971–978. doi: 10.1038/nn920. [DOI] [PubMed] [Google Scholar]
- Iversen L. Neurotransmitter transporters and their impact on the development of psychopharmacology. Br J Pharmacol. 2006;147(Suppl. 1):S82–S88. doi: 10.1038/sj.bjp.0706428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto H, Blakely RD, DeFelice LJ. Na+, Cl–, and pH dependence of the human choline transporter (hCHT) in Xenopus oocytes: the proton inactivation hypothesis of hCHT in synaptic vesicles. J Neurosci. 2006;26:9851–9859. doi: 10.1523/JNEUROSCI.1862-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlig KM, Binda F, Khoshbouei H, Blakely RD, McMahon DG, Javitch JA, et al. Amphetamine induces dopamine efflux through a dopamine transporter channel. Proc Natl Acad Sci USA. 2005;102:3495–3500. doi: 10.1073/pnas.0407737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshbouei H, Wang H, Lechleiter JD, Javitch JA, Galli A. Amphetamine-induced dopamine efflux. A voltage-sensitive and intracellular Na+-dependent mechanism. J Biol Chem. 2003;278:12070–12077. doi: 10.1074/jbc.M212815200. [DOI] [PubMed] [Google Scholar]
- Machaca K, Hartzell H. Assymetric distribution of Ca-activated Cl channels in Xenopus oocytes. Biophys J. 1998;74:1286–1295. doi: 10.1016/S0006-3495(98)77842-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mofenson HC, Greensher J. Letter: physostigmine as an antidote: use with caution. J Pediatr. 1975;87(6 Pt 1):1011–1012. doi: 10.1016/s0022-3476(75)80946-2. [DOI] [PubMed] [Google Scholar]
- Nelson N. The family of Na+/Cl- neurotransmitter transporters. J Neurochem. 1998;71:1785–1803. doi: 10.1046/j.1471-4159.1998.71051785.x. [DOI] [PubMed] [Google Scholar]
- Nelson N, Sacher A, Nelson H. The significance of molecular slips in transport systems. Nat Rev Mol Cell Biol. 2002;3:876–881. doi: 10.1038/nrm955. [DOI] [PubMed] [Google Scholar]
- Petersen CI, DeFelice LJ. Ionic interactions in the Drosophila serotonin transporter identify it as a serotonin channel. Nat Neurosci. 1999;2:605–610. doi: 10.1038/10158. [DOI] [PubMed] [Google Scholar]
- Piscitelli CL, Krishnamurthy H, Gouaux E. Neurotransmitter/sodium symporter orthologue LeuT has a single high-affinity substrate site. Nature. 2010;468:1129–1132. doi: 10.1038/nature09581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potkin SG, Karoum F, Chuang LW, Cannon-Spoor HE, Phillips I, Wyatt RJ. Phenylethylamine in paranoid chronic schizophrenia. Science. 1979;206:470–471. doi: 10.1126/science.504988. [DOI] [PubMed] [Google Scholar]
- Romanelli F, Smith KM. Clinical effects and management of methamphetamine abuse. Pharmacotherapy. 2006;26:1148–1156. doi: 10.1592/phco.26.8.1148. [DOI] [PubMed] [Google Scholar]
- Rudnick G. Bioenergetics of neurotransmitter transport. J Bioenerg Biomembr. 1998a;30:173–185. doi: 10.1023/a:1020573325823. [DOI] [PubMed] [Google Scholar]
- Rudnick G. Ion-coupled neurotransmitter transport: thermodynamic vs. kinetic determinations of stoichiometry. Methods Enzymol. 1998b;296:233–247. doi: 10.1016/s0076-6879(98)96018-9. [DOI] [PubMed] [Google Scholar]
- Samuvel DJ, Jayanthi LD, Manohar S, Kaliyaperumal K, See RE, Ramamoorthy S. Dysregulation of dopamine transporter trafficking and function after abstinence from cocaine self-administration in rats: evidence for differential regulation in caudate putamen and nucleus accumbens. J Pharmacol Exp Ther. 2008;325:293–301. doi: 10.1124/jpet.107.130534. [DOI] [PubMed] [Google Scholar]
- Saunders C, Ferrer JV, Shi L, Chen J, Merrill G, Lamb ME, et al. Amphetamine-induced loss of human dopamine transporter activity: an internalization-dependent and cocaine-sensitive mechanism. Proc Natl Acad Sci USA. 2000;97:6850–6855. doi: 10.1073/pnas.110035297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel S, Singer EA, Just H, Farhan H, Scholze P, Kudlacek O, et al. Amphetamines take two to tango: an oligomer-based counter-transport model of neurotransmitter transport explores the amphetamine action. Mol Pharmacol. 2005;67:140–151. doi: 10.1124/mol.67.1.. [DOI] [PubMed] [Google Scholar]
- Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;33:639–677. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- Shan J, Javitch JA, Shi L, Weinstein H. The substrate-driven transition to an inward-facing conformation in the functional mechanism of the dopamine transporter. PLoS ONE. 2011;6:e16350. doi: 10.1371/journal.pone.0016350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter–inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell. 2008;30:667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonders MS, Amara SG. Channels in transporters. Curr Opin Neurobiol. 1996;6:294–302. doi: 10.1016/s0959-4388(96)80111-5. [DOI] [PubMed] [Google Scholar]
- Sonders MS, Zhu SJ, Zahniser NR, Kavanaugh MP, Amara SG. Multiple ionic conductances of the human dopamine transporter: the actions of dopamine and psychostimulants. J Neurosci. 1997a;17:960–974. doi: 10.1523/JNEUROSCI.17-03-00960.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonders MS, Zhu SJ, Zahniser NR, Kavanaugh MP, Amara SG. Multiple ionic conductances of the human dopamine transporter: the actions of dopamine and psychostimulants. J Neurosci. 1997b;17:960–974. doi: 10.1523/JNEUROSCI.17-03-00960.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Galli A. Dopamine transport currents are promoted from curiosity to physiology. Trends Neurosci. 2003;26:173–176. doi: 10.1016/S0166-2236(03)00063-8. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Maidment NT, Rayport S. Amphetamine and other weak bases act to promote reverse transport of dopamine in ventral midbrain neurons. J Neurochem. 1993;60:527–535. doi: 10.1111/j.1471-4159.1993.tb03181.x. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstraete AG. Oral fluid testing for driving under the influence of drugs: history, recent progress and remaining challenges. Forensic Sci Int. 2005;150:143–150. doi: 10.1016/j.forsciint.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Verstraete AG, Heyden FV. Comparison of the sensitivity and specificity of six immunoassays for the detection of amphetamines in urine. J Anal Toxicol. 2005;29:359–364. doi: 10.1093/jat/29.5.359. [DOI] [PubMed] [Google Scholar]
- Volz TJ, Hanson GR, Fleckenstein AE. The role of the plasmalemmal dopamine and vesicular monoamine transporters in methamphetamine-induced dopaminergic deficits. J Neurochem. 2007;101:883–888. doi: 10.1111/j.1471-4159.2006.04419.x. [DOI] [PubMed] [Google Scholar]
- Winslow BT, Voorhees KI, Pehl KA. Methamphetamine abuse. Am Fam Physician. 2007;76:1169–1174. [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl–dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Terry DS, Shi L, Quick M, Weinstein H, Blanchard SC, et al. Substrate-modulated gating dynamics in a Na(+)-coupled neurotransmitter transporter homologue. Nature. 2011;474:109–1013. doi: 10.1038/nature09971. [DOI] [PMC free article] [PubMed] [Google Scholar]