

Abstract
Background
Non-small cell lung cancers (NSLC) are aggressive cancers that are insensitive to chemotherapies and accounts for nearly 33% of all cancer deaths in the United States. Two hallmarks of cancer that allow cells to invade and metastasize are sustained proliferation and enhanced motility. In this study we investigate the relationship between urokinase plasminogen activator (uPA)/uPA receptor (uPAR) signaling and Na+/H+ exchanger isoform 1 (NHE1) expression and activity.
Methods and Results
The addition of 10nM uPA increased the carcinogenic potential of three NSCLC cell lines, NCI-H358, NCI-H460, and NCI-H1299. This included an increase in the rate of cell proliferation 1.6 to 1.9 fold; an increase in the percentage of cells displaying stress fibers 3.05 to 3.17 fold; and an increase in anchorage-independent growth from 1.64 to 2.0 fold. In each of these cases the increase was blocked when the experiments were performed with NHE1 inhibited by 10 μM EIPA (ethylisopropyl amiloride). To further evaluate the role of uPA/uPAR and NHE1 in tumor progression we assessed signaling events using full-length uPA compared to the uPA amino terminal fragment (ATF). Comparing uPA and ATF signaling in H460 cells, we found that both uPA and ATF increased stress fiber formation approximately 2 fold, while uPA increased matrix metalloproteinase 9 (MMP9) activity 5.44 fold compared to 2.81 fold for ATF. To expand this signaling study, two new cell lines were generated, one with reduced NHE1 expression (H460 NHE1 K/D) and one with reduced uPAR expression (H460 uPAR K/D). Using the K/D cell lines we found that neither uPA nor ATF could stimulate stress fiber formation or MMP9 activity in cells with dramatically decreased NHE1 or uPAR expression. Finally, using in vivo tumor formation studies in athymic mice we found that when mice were injected with H460 cells 80% of mice formed tumors with an average volume of 390 mm3. This was compared to 20% of H460 uPAR K/D injected mice forming tumors with an average volume of 15 mm3 and 10% of H460 NHE1 K/D injected mice forming tumors with an average volume of 5 mm3.
Conclusion
Taken together, these data demonstrate that uPA/uPAR-mediated tumor progression and metastasis requires NHE1 in NSCLC cells and suggests a potential therapeutic approach to blocking cancer progression.
1 Introduction
Non-small cell lung cancer (NSCLC) is an aggressive malignancy, is insensitive to many chemotherapy or other adjuvant approaches and accounts for nearly 1/3rd of all cancer deaths [1]. Due to its insidious nature, most NSCLC is not diagnosed until after the disease has advanced and metastasized. Two distinct hallmarks of cancer that allow cells to invade and metastasize include sustained proliferation and enhanced cellular motility [2]. A number of reports find that increased activity or expression of urokinase plasminogen activator (uPA) or its cognate receptor (uPAR) is associated with elevated proliferation, increased cell motility and poor patient outcomes with NSCLC [3–10]. uPA is an extracellular serine protease with several distinct domains. The uPA receptor-binding domain found on the amino terminal fragment of uPA (ATF – also used in recombinant form to bind to the uPA receptor without protease activity) is separate from its serine protease catalytic domain and the clotting blotting associated-kringle domain [6, 11, 12]. The mechanism of action of uPA is diverse and cell-type dependent. In addition to proteolytic activation of plasmin, uPA can act as a classical agonist, binding to its receptor to activate cell proliferation and migration through a number of signaling pathways in a mechanism that may be independent of proteolysis [7, 8, 13]. While inhibition of uPA-uPAR function has been reported to be critical for blocking invasion and migration in several cancer types [14] the signaling mechanism in tumor cells has yet to be fully defined.
One of the permissive steps in proliferation and in cell migration is the alkalinization of the intracellular pH (pHi) and the resulting acidification of the extracellular environment [15–18]. The sodium hydrogen exchanger, isoform 1 (NHE1) exchanges an intracellular proton for an extracellular sodium ion and coordinates cellular motility through protein interaction with a host of intracellular proteins [16, 17, 19]. Increases in pHi induced by NHE1 are critical for increased DNA synthesis and cell cycle progression. The resulting acidification of the extra cellular pH (pHe) supports the degradation of the ECM by proteases including MMP9 promoting invasion. Furthermore, NHE1 acts as a modulator of directed cell motility as it is localized to the leading edge of migrating tumor cells supporting the polarization of invading cells. Regulation of cytoskeletal proteins such as filamentous actin by NHE1 aids in the proper development of focal adhesion complexes in invadapodia and other cellular structures. Loss of either proton transport or the ability to bind to cytoskeletal proteins results in a loss of directed motility and tumor invasion in a range of cancer cell types [18, 20, 21].
Like NHE1, uPAR is localized to the leading edge of migrating tumor cells after binding uPA [7, 8]. While the effects of uPAR are both intra-and extracellular, the signaling mechanism is cell type specific [6]. Both NHE1 and uPAR have been shown to be involved in a variety of cancer cells and are thought to be important drivers of survival and tumor motility. Inhibition of NHE1 blocks cell proliferation and induces apoptosis in small cell lung cancer [22]. We found that inhibition of NHE1 in H358 NSCLC cells significantly reduced the ability of the lung cancer cells to invade through the ECM [21] while similar effects on lung cancer development have been reported for uPA. Therefore, it is interesting that the uPA-uPAR complex is spatially and temporally located with NHE1 and that uPAR also activates many of the signaling intermediates that activate NHE1. However mechanism through which NHE1 and uPA-uPAR functions in this system has not been identified.
This study aims to identify the effect of NHE1 in NSCLC and its relationship to uPA/uPAR signaling. The key parameters necessary to link these two protein systems in a common signaling pathway leading to cancer are to show that NHE1 is critical for uPAR mediated cellular migration and invasion. Our data demonstrate that human NSCLC cells utilize a non-proteolytic uPAR mediated pathway to activate NHE1 and that NHE1 is critical in uPA induced cell motility events. Pharmacological inhibition of NHE1 and shRNAi knockdown of NHE1 expression blocks both MMP9 activity and the formation of stress fibers as well as inhibits the ability of uPA to induce NSCLC tumor formation in vitro and in vivo. The results presented here suggest that both uPA-uPAR and NHE1 contribute to lung cancer cell invasion and progression. Moreover, the results provide a potential novel therapeutic approach to treating a cancer whose current five-year survival rate is very low.
2 Materials & methods
2.1 Reagents, cell lines and transfections
Anti-uPA receptor, anti ERK and anti-phospho ERK were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-NHE1 and the colormetric uPA activity assay were obtained from Millipore (Billerica, MA). Viability/cytotoxicity fluorescent assay, BCECF, and phalloidin-Alexa Fluor 488 were from Invitrogen (Carlsbad, CA).
The invasive human non-small cell lung cancer cell lines NCI-H358 (H358: bronchioalveolar carcinoma), NCI-H460 (H460: large cell carcinoma) and NCI-H1299 (H1299: carcinoma isolated from lymph node) were obtained from American Type Culture Collection, Rockville MD. All three cell lines were grown in RPMI supplemented with 10% fetal calf serum without antibiotics and incubated at 37 °C in 5% CO2. In some experiments, cells were treated with 10 μM of the specific NHE1 inhibitor 5-(N-ethyl-N-isopropyl)-amiloride (EIPA: Sigma Aldrich), alone or in combination with either 10 nM uPA (a mixture of high and low molecular weight urokinase, Millipore Billerica, MA) or 10 nM of recombinant human urokinase amino terminal fragment (ATF - Molecular Innovations, Novi MI).
H460 stable knockdown cells against NHE isoform 1 (NHE1 K/D) were generated using short hairpin RNAs (shRNA 19mer target sequence cctcatgaagataggtttc) or scramble controls constructed in vector psi-nH1 (GeneCopia, Rockville MD). Cells were transfected with shRNA containing plasmid using JetPei transfection reagent (New York, NY) and incubated in 5.0 μg/ml puromycin for three passages to select for stable transfected cells. Cells which lack NHE1 activity were further selected using a H+ suicide technique [23]. This method uses the reversible nature of the exchanger where Li+ is used in place of Na+ to generate toxic intracellular conditions in cells possessing NHE1 activity. Trypsinized cells were resuspended and incubated for two hours in LiCl saline solution (130 mM LiCl, 5 mM KCl, 1 mM MgSO4, 2 mM CaCl2, 5 mM glucose and 20 mM Hepes-Tris pH 7.4), washed and subjected to a one-hour acid loading incubation in choline chloride solution (130 mM choline-Cl, 5 mM KCl, 1 mM MgSO4, 2 mM CaCl2, 20 mM MES-Tris pH 5.5). The cells were then allowed to recover in RMPI media containing 10% FBS for 2–4 days before being subjected to a second acid load/H+ suicide treatment.
H460 stable knockdown cells directed against the urokinase plasminogen receptor (uPAR K/D) with a non-silencing control sequence were generated using Lentiviral constructs expressing shRNA targeting uPAR (Santa Cruz Biotechnology, CA). The cells were transfected with 20000 transducing particles per milliliter of shRNA in DMEM with 5 μg/ml Polybrene for two days before selecting stable clones in 5.0 μg/ml puromycin. Both NHE1 K/D and uPAR K/D were subsequently cultured in 1.0 μg/ml puromycin to maintain selection.
2.3 Proliferation assays
Cell proliferation was performed by the reduction of XTT (2.3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxyanalide) from Trevigen (Gaithersburg, MD). Cells were plated at 1 × 103 cells/well in a 96 well plate and incubated overnight before making the cells quiescent by incubating the cells for 16 hours in low serum (0.5% FBS) containing medium. The cells were then incubated in low serum medium containing treatments as indicated for 24 hours. XTT reagent was added to each culture well to attain a final concentration 0.3 mg/ml and absorbance read at 490 nm with a reference of 630 nm.
2.4 Stress fiber formation
Treated and control cells grown on glass coverslips were fixed with 3% paraformaldehyde at 4°C for 30 min and permeabilized with 0.4% Triton X-100 for 10 min. Actin stress fibers were stained with 0.5 μg/ml phalloidin-Alexa Fluor 488 for 60 min. The cells were treated with prolong anti-fade prior to mounting the glass slides and analyzed on an inverted Olympus IX70 microscope in fluorescence mode using a 100X objective. Cells displaying significant and strong stress fibers (well formed and prominent fibers which are organized and stretched through the majority of the cell) were counted in five random fields for each slide. Percent of cells displaying stress fibers was determined as the total number of cells with stress fibers vs. the total number of cells evaluated in the observed fields.
2.5 Anchorage independent soft agar colony formation
1×104 cells were seeded in 0.6% agar in RPMI medium containing 0.5% serum with the indicated agonists or inhibitors (Cytoselect Cell Transformation Assay, Cell Biolabs, San Diego CA). The plate was incubated at 37°C for 9 days with a change of overlay medium (with agonists and inhibitors) at days 3 and 6. Agar was solubilized and relative cell density measured with by observing CyQuant fluorescence at 485/520 nm.
2.6 Intracellular pH determination
Single cell steady state intracellular pH was measured using the pH-sensitive fluorescent dye, 2′,7′-bis-(carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF). Cells were cultured in bicarbonate free complete medium or bicarbonate free medium with minimal FBS (0.5% FBS) for 24 hours prior to assay. The cells were incubated with 10 μM of the uncharged acetoxymethyl ester form of BCECF (BCECF-AM, Molecular Probes) for 20 min at 25 °C in sodium buffer (Na buffer–20 mM HEPES pH 7.40 at 37 °C, 145 mM NaCl, 3 mM KCl, 1 mM MgCl2, 1 mM CaCl2, and 5 mM Glucose). The fluorescence intensity of emission was measured in glass bottom dishes on an Olympus ix70 at 525 nm while the dye was excited alternately at the pH-sensitive wavelength of 502 nm (556 msec) and the pH-insensitive isoexcitation wavelength of 439 nm (334 msec). Fluorescence intensity for each wavelength was corrected for background fluorescence by subtracting intensities from adjacent areas in each field. An average of 40 cells were identified for each measurement. pHi was determined by the fluorescence intensity ratio method with a calibration curve generated using the K+-Nigericin method [24].
Initial rate of alkalinization after an acid load was determined in each cell line using the ammonium chloride pre-pulse technique [25]. During acid loading, pulsing the cells with NH4+ induces an alkalinization of the cytosol and the intracellular pH acidifies upon removal of NH4+ in a sodium free solution. The ability of cells to recover the pHi is determined in the presence of sodium. Dye-loaded cells were treated with 2 mM NH4Cl and incubated for 10 min. After incubation, the ammonium chloride solution was removed, sodium buffer was added, and the rate of recovery from the ensuing acid load was measured for 15 min. dpHi/min was determined by calculating the slope of the first 30 to 60 s of the recovery.
2.7 RhoA-GTPase assay
G-Protein activation was determined by a pull-down assay using GST-Rhotekin (CellBio Labs, San Diego, CA). H460 cells were cultured in cell culture dishes and starved in serum-free medium for 12 h. Cells were then exposed to UPA or ATF for 15 min, quickly washed with ice-cold Tris-buffered saline, and lysed in 500 μl of lysis buffer containing 20 mM Tris-HCl, pH 8.0, 137 mM NaCl, 10% glycerol, 1% Nonidet P-40, 50 mMβ-glycerophosphate, 10 mM NaF, 1 mMNa3VO4, 1 mM phenylmethylsulfonyl fluoride, 2 μg/ml aprotinin, and 2 μg/ml leupeptin. Cell lysates were immediately centrifuged at 8000 rpm at 4 °C for 5 min and 50 μg of lysate was incubated with 30 μg GST-fusion protein beads for 1 h at 4 °C. The beads were washed with wash buffer, and bound small G-protein was eluted by boiling each sample in Laemmli sample buffer. Eluted samples from the beads and total cell lysate were then electrophoresed on 14% SDS–polyacrylamide gel electrophoresis gels, transferred to nitrocellulose, and analyzed by Western blotting using a monoclonal anti-RhoA antibody.
2.8 Gelatin zymography
Conditioned medium was collected following a 24 hour treatement with indicated agonists or inhibitors and then analyzed for MMP9 activity by zymography. The samples were mixed one to one with loading buffer (0.125 M Tris–Cl pH 7.2, 5% SDS, 40% sucrose, and 0.1% bromophenol blue). The protein samples were resolved by 10% SDS–PAGE in the presence of 1 mg/ml gelatin (Biorad, Hercules, CA). The resulting gels were washed in 2% Triton-X100 for 60 min and then incubated for 24 h at 37 °C in zymogen gel buffer (0.1 M Tris–Cl pH 8.0, 1 mM CaCl2, and 2% Triton-X100). The gels were then stained using Coomassie brilliant blue R-250, gelatinase activity was identified as clear bands. Band intensities were analyzed using the BioRad Image Station and the BioRad Quantitiy One Program.
2.9 Semi-quantitative RT-PCR analysis
Total RNA was extracted from cells using the RNAeasy kit according to the manufacturer’s protocol (Qiagen, Germantown, MD) and previously published [26]. cDNA was prepared by reverse transcription from 1 μg of total RNA after genomic DNA elimination using the RT2 First Strand from SABiosciences (Frederick MD). NHE1 and uPA Receptor in wild-type and shRNA expressing cells were analyzed by real time qPCR. Wells without template RNA was included as a negative control and β-actin was used as a normalizing gene. Each experiment was carried out four times, and the ΔΔCt values were calculated by normalizing the gene expression levels to β-actin, and the relative expression level was expressed as a fold change.
2.10 Ectopic and Orthotopic tumor studies in nude mice
Six week-old athymic female nude mice nu/nu (Harlan, Indianapolis, IN) were used conforming to guidelines laid down by the National Research Council and approved by Minnesota State University Moorhead Institutional Animal Care and Use Committee. Mice were kept in laminar-flow cages in standardized environmental conditions. Sterilized food (Harlan, Indianapolis, IN) and water were supplied ad libitum.
Ectopic model
Cells were harvested at near confluence with trypsin/EDTA solution and each animal received subcutaneous injections (s.c.) of the indicated cells (5 × 106) suspended in 100 μl of sterile saline into their right flank. Tumor growth was periodically monitored, tumor volume was calculated using the formula: V (mm3) = (D × d2)/2, where d (mm) and D (mm) are the smallest and largest perpendicular tumor diameter, respectively. Animals receiving intraperitoneal injections (i.p.) were given either vehicle (1% DMSO) or 10 mg/kg EIPA in 200 μl aliquots every three days. Mice injected with NHE1 K/D or uPAR K/D cells were not treated by i.p. Animals were euthanized and tumors examined six weeks post-injection.
Orthotopic model
To investigate the pathogenesis of tumor growth and metastasis 5 × 106, cells in 100 μl were injected into the animal’s bloodstream via retro-orbital injection. Mice were monitored daily for behavior and health. Once an animal appeared morbid which included tumors over 2 cm, hind limb paralysis or cachexia, they were sacrificed.
2.11 Differential gene expression profile
The mRNA from H460 and NHE1 K/D cells was isolated and converted to cDNA as described for semi-quantitative RT-PCR with an additional on-column DNAase treatment to ensure high quality total RNA and as previously described [26]. RNA concentration and quality were determined by UV spectrophotometry and ribosomal integrity determined after electrophoresis and analysis of the 18S and 28S rRNA bands. The cDNA template was mixed with RT2 Real-Time SYBR Green/Rox Master Mix, and RNAse-free water (SuperArray Biosciences, Frederick, MD). A final reaction volume of 25 μl was added to each well of the Tumor Metastasis PCR Array PAHS-028 (SuperArray Biosciences) before amplification on a Stratagene MX3000P following the manufacturer’s protocol. Fold changes between wild-type and NHE1 null expressing H460 cells were analyzed using SABiosciences webportal software. A fold change from wild-type cells greater than 2.0 was selected to be a significant.
3 RESULTS
3.1 NSCLC Cell Lines Secrete uPA and Express the uPA Receptor
To evaluate the role of uPA stimulation of NHE1 in the progression of NSCLC we studied three cell lines. The NCI-H358 (H358) cell line is a bronchioalveolar carcinoma derived from a tumor in the bronchus, NCI-H1299 (H1299) is a carcinoma cell line derived from a lymph node metastatic site, and NCI-H460 (H460) is a carcinoma line derived from pleural effusion. We first examined the ability of each cell line to produce uPA and for the presence of the uPA receptor. The conditioned media was collected at 24 hrs and uPA activity determined using a colorometric assay. H1299 cells had the highest uPA activity at 6.99 +/− 0.10 μmol substrate/min with the H358 and H460 cells generating 5.4 +/−0.09 and 5.02 +/− 0.026 μmol substrate/min (Fig. 1A, n=6). To assess the potential role of uPA in the regulation of cancer progression in NSCLC, we investigated whether H358, H460, and H1299 cells express uPAR by western blot analysis. Figure 1A shows that all three cell lines express a protein of approximately 55 to 60 kDA that was identified using rabbit anti-uPAR antibody. This result is in agreement with previous finings in H460 cells for the expression of uPA and its receptor [10, 27]
Fig. 1.
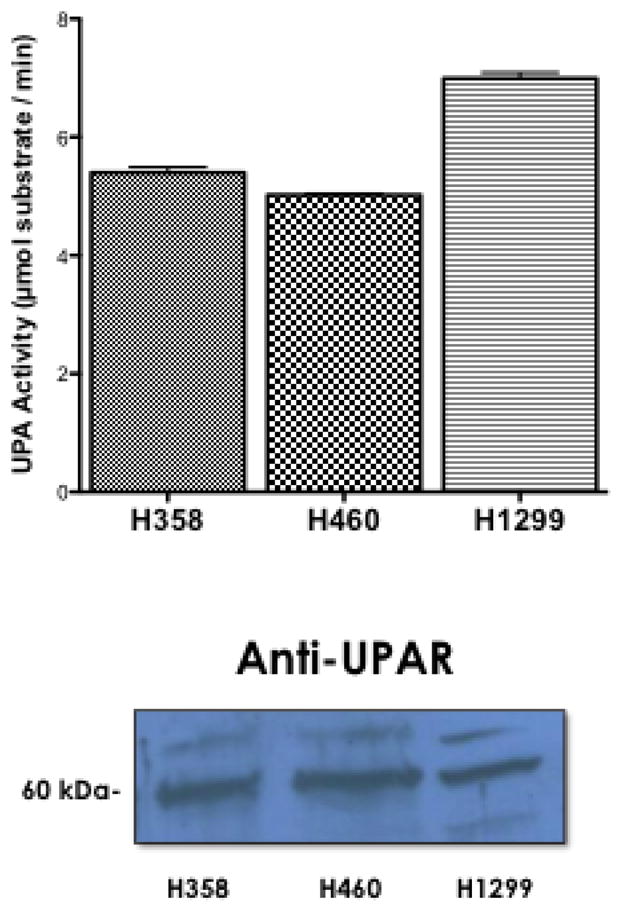

uPA activity and uPAR expression in NSCLC. A. (Upper Panel) Conditioned media from each cell line was collected and assayed for uPA activity. n=6, no statistical difference among all three groups was determined using student a one way anova. (Lower Panel) Whole cell lysates from each cell line were collected and 50 μg of each lysate was subjected to SDS-PAGE. Expression of uPA receptor was determined with anti-UPAR antibodies. B. Recombinant purified uPA was incubated with vehicle (0.1% DMSO) or the indicated concentration of EIPA in 0.1% DMSO for 2 hours (white columns). Cultures of H460 were treated with EIPA (10μM) for 24 hours before collecting conditioned medium (grey columns). uPA activity was determined using a tripeptide substrate conjugated with a pNA. Statistical analysis was determined using a one tailed type one student T-test vs control, n=3.
It has been reported that both amiloride and EIPA (a selective NHE inhibitor derivative of amiloride) directly inhibit the protease activity of uPA [28, 29]. To ensure that data collected on cell behavior was due to inhibition of NHE1 and not uPA, we assessed the impact of EIPA on uPA activity using recombinant purified uPA and uPA secreated into conditioned media. Figure 1B shows that the activity of purified uPA was not altered by EIPA. The reaction rate in the absence of inhibitor was 0.98+/− 0.024 μmol substrate/min. This was unchanged in the presence of either 1 μM EIPA (0.98 +/− 0.015) or 10 μM EIPA (1.04 +/− 0.014). Likewise, in conditioned media control samples without EIPA the uPA activity was 5.89 +/− 0.34 μmol substrate/min while in the presence of 10 μM EIPA, the uPA activity was not significantly different (6.13 +/− 0.17 μmol substrate/min). These data indicate that under the experimental conditions displayed here, EIPA does not inhibit uPA protease activity.
3.2 uPA Enhances tumor-like behavior in NSCLC
When cells receive a growth stimulus, one of the first physiological responses is an increase in pHi typically mediated by NHE1. Binding of uPA to its receptor increases cell growth and tumor formation in a wide range of normal and transformed cell lines and enhances invasive potential of lung cancer cells [30–33]. One of the hallmark capabilities of cancer is the fundamental ability to sustain chronic proliferation [2]. To assess the ability of uPA to alter cell proliferation in an NHE1-dependent manner, the growth rate of the three cell lines H358, H460, H1299 was studied. Figure 2A shows that in each cell type, 10 nM uPA increased cell proliferation rates 1.87, 1.76, and 1.63 fold respectively. This enhanced rate of proliferation was returned to control levels or below by the presence of 10 μM EIPA. To ensure that prolonged exposure of cells to EIPA was not simply leading to cell death instead of blocking cell proliferation we performed a live/dead assay on cells treated with either vehicle or EIPA for 24 hrs. (Fig. 2A inset). In each case, greater than 98% of cells were living at the end of the 24 hr. period.
Fig. 2.



uPA and NHE1 influence cell proliferation, motility and tumor growth. Where indicated cells were treated with 10 nM uPA in the absence or presence of 10 nM EIPA. A. Cell proliferation of three cell lines was determined using the XTT assay. Cells (1 × 103/well) were seeded, allowed to attach for one day and then incubated overnight in low serum medium alone or in the presence of the indicated agonist or inhibitor for 24 hours. All data are presented as the mean +/− SEM (n ≥ 8). Inset – a live-dead assay was performed in parallel with a high dose (50 μM EIPA). Green cells indicated living cells while red cells identify apoptotic cells. B. Cells were grown to 20–30% confluency on glass coverslips and cultured 12–16 hours in media containing 0.5% serum. The cells were then deprived of serum for 1 h. Agonist alone or in combination with EIPA was added and cells were incubated at room temperature for 30 min and stained for actin polymerization. Five random fields were counted for each coverslip. The average percent of cells displaying stress fiber formation was determined for each field in each experiment. Representative micrographs were obtained by using an Olympus IX70 fluorescence microscope. C. Cells were suspended in 0.6% agar saturated with RPMI medium containing 0.5 FBS. Cell plugs were incubated at 37° C for 9 days with the indicated agonist or inhibitor. Following the incubation period, the agar was suspended, cells solubilized and quantitated by CyQuant-DNA analysis. Data are presented as the mean ± SEM (n=4). Statistical analysis was performed using a non-paired one-way Anova (p>0.001) as compared to each cell type control sample group.
In addition to supporting cell growth, activation of NHE1 is a central requirement for the formation of actin filaments in a variety of cell lines. NHE1 is found at the leading edge of migrating cells where it coordinates actin filament organization, contributing to directional cell migration and thus enhances metastasis [15, 17, 19]. A number of cell types including keratinocytes and fibroblasts utilize stress fibers to enhance cell spreading and increase the filipodial and invadapodial formation critical for cell motility [20, 34, 35]. The formation of actin filaments (stress fiber formation), was assessed by treating cells with 10 nM uPA for 15 min in the presence or absence of 10 μM EIPA (Fig. 2B). In H358 cells, uPA stimulation increased the percentage of cells displaying stress fibers from 12% to 38% (3.17 fold increase) with EIPA inhibition returning cells with stress fibers to basal levels (9%). In H460 cells, the percentage of cells displaying stress fibers increased from 19% to 59% (3.05 fold increase) with EIPA inhibition decreasing cells displaying stress fibers to 3%. Finally, in H1299 cells uPA stimulation increased the percentage of cells displaying stress fibers from 11% to 34% (3.09 fold increase) with EIPA inhibition decreasing stress fiber forming cells to 6%.
Increased cell proliferation and stress fiber formation both contribute to the tumorigenic potential of a cell line. Soft agar or anchorage independent cell growth is another hallmark characteristic of cellular transformation and uncontrolled cell growth. To evaluate the ability of uPA to alter neoplastic transformation in NSCLC cell lines and the requirement for NHE1 in this process we performed a soft agar assay to assess changes in the ability to proliferate and form tumors in a 3D environment. In each cell line, performing the assay in the presence of 10 nM uPA substantially increased colony formation in soft agar compared to basal low serum conditions (Fig. 2C). In H358, H460, and H1299 cells, uPA increased anchorage-independent growth 2.0, 1.84, and 1.64 fold respectively. For each cell line, uPA induced increase in soft agar colony formation was significantly blocked by addition of 10 μM EIPA. Collectively these data demonstrate that uPA can stimulate the hallmarks of cancer cell growth in NSCLC and that inhibition of NHE1 abrogates the cellular response to uPA.
3.4 NHE1 transport in H460 cells
Because pharmacological inhibition of NHE1 showed a dramatic inhibition of tumor development in all three NSCLC cell types, we next investigated the invasive and metastatic H460 cell line for its NHE1 related proton transport activity. Cells were incubated in 0.5% FBS containing media for 12–16 hours and the rate of recovery from an NH4+ pre-pulse acid load measured in nominally HCO3− free buffer was used as a measure of NHE1 transport. The rate of recovery for cells pretreated with EIPA was significantly lower than control cells (0.105 +/− 0.025 dpH/min for control vs −0.035 +/− 0.031dpHi/min for EIPA treated cells, Fig. 3) indicating the recovery observed in control cells was due to an active NHE1 transporter. Surprisingly, when uPA was incubated with the cells prior to, during and following the NH4+ pre-pulse, there was no significant effect on transport rate. This suggests that either uPA does not enhance NHE1 transport or that the signaling pathway, which regulates the NHE1 set point, is already activated (Fig. 3). NHE1 activation is complex involving many covalent and non-covalent modifiers activated by a wide range of cell signaling pathways. Both growth factor pathways and small G protein RhoA signaling are known upstream regulators of NHE1. Therefore, to investigate the lack of NHE1 transport response to uPA, we assessed the level of activation of both ERK and RhoA in resting and in uPA-stimulated cells. The level of ERK activation in quiescent-serum deprived cells was significant and did not increase upon 10 nM uPA stimulation or addition of the uPA amino-terminal receptor domain, ATF (Fig. 4A). Extended serum-deprivation (up to 48 hours – data not shown) did not decrease baseline ERK phosphorylation. These results were not surprising as H460 cells contain K-Ras mutations leaving these cells with a constitutively active ERK signaling pathway [53]. Unlike the high-basal level of ERK phosphorylation found in resting cells, there was low but detectable levels of GTP-RhoA in un-stimulated cells. Whereas addition of either uPA or ATF doubled the amount of GTP-RhoA detected in the cells (Fig. 4B). While we cannot discount the contribution of endogenous uPA secreted by H460 cells (Fig. 1) use of the NHE1 inhibitor provide evidence that exogenously added uPA stimulates or enhances several biological processes in these NSCLC cells in a NHE1 depend manner. Furthermore, that ERK whose basal activity is high, combined with an inducible RhoA activation suggests that NHE1 imparts its effect in these lung tumor cells through cytoskeleton regulation and other non-growth factor regulation of NHE1.
Fig. 3.

NHE1 activity in H460 cells. Mean proton efflux rate in the absence or presence of 10 nM uPA or 10 μM EIPA. n = >6 recordings with each recording consisting of an average of 30–40 individual cell recoveries. *** p< 0.001 as compared to the control group.
Fig. 4.
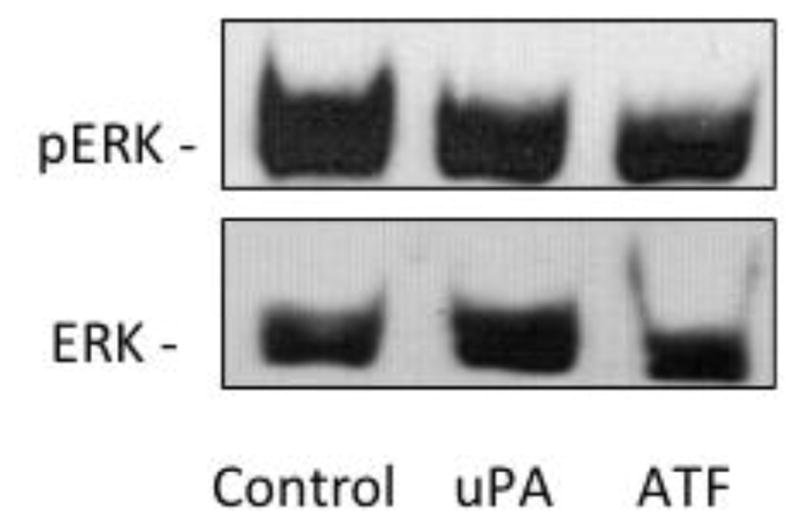

Effects of uPAR on ERK and RhoA in H460 cells. A. ERK activation was determined in quiescent, serum-deprived cells incubated with 10 nM of the indicated agonist or for 15 min Following treatment, cells were rinsed with PBS and lysed in Laemmli sample buffer. A total of 20 μg of each sample was resolved on a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and total ERK and phosphorylation state of ERK was determined by immunoanalysis. Data are representative of three separate experiments. B. (Top Panel) GTP bound RhoA was determined by affinity pulldown performed with GST-RBD following agonist treatment. Samples were blotted with an antibody against RhoA. Blot is representative of four blots. The normalized relative density of each blot was averaged (Lower Panel).
3.5 uPA signaling through uPAR in H460 cells
As uPA has its own extracellular capacity to signal and because of the diversity of uPAR signaling, we next evaluated the differences in uPA signaling based upon the requirement for receptor involvement versus protease activity. The amino terminal fragment of uPA (ATF) consists of the amino acids 1 to 135 of the amino terminus of uPA and includes the uPAR binding domain but not the protease domain. In H460 cells, 17% of untreated cells displayed stress fiber formation (Fig. 5). This value was decreased to 3% in the presence of 10μM EIPA. When H460 cells are treated with either 10 nM uPA or 10 nM ATF stress fiber formation increased to 34% and 33% respectively. In both cases, adding uPA or ATF in the presence of 10 μM EIPA blocked stress fiber formation maintaining the percentage of cells with stress fibers below control levels.
Fig. 5.

H460 Stress Fiber Formation. H460s were grown to a 30–50% confluency on a glass cover slip. After being placed in 0.5% serum overnight H460 cells were serum deprived and stimulated 10nM uPA or ATF without and with 10 μM EIPA 15 minutes prior to stimulation. Five random fields were examined for each cover slip and the average percent of cells expressing stress fibers determined for each field. Data indicated with a *** were significantly different from the control data using a one-way Anova with Tukey’s Multiple Comparison with a 99.9% confidence interval (P-value of < 0.001). Data indicated with ††† and ¥¥¥ were compared for the corresponding agonist alone vs. the agonist and inhibitor treatment.
We previously found that inhibition of NHE1 blocked MMP9 activity and NSCLC invasion [20, 21]. Furthermore, uPAR activation of MMP9 has been established although the mechanism remains unclear [4, 5, 36]. Therefore, we next evaluated the differences between uPA and ATF signaling on MMP9 activation in H460 cells using zymography. MMP9 activity in the growth media was evaluated following 24 hour incubation under the different experimental conditions. Figure 6 shows that addition of 10 nM uPA increased MMP9 activity 5.44 fold +/− 1.08 SEM. This increase was only partially inhibited by the addition of 10 μM EIPA. Comparatively, addition of 10 nM ATF increased MMP9 activity 2.81 fold +/− 0.48 SEM and addition of 10 μM EIPA completely abrogated the activation. It should be noted that EIPA has a diminished effect on the ATF-induced stress fiver than uPA while the NHE1 inhibitor had a greater effect on MMP9 activity induced by ATF than uPA (Fig 5 and 6). Together, these results suggest an intracellular signaling role for uPA-uPAR and that NHE1 transport and scaffolding activity has a significant but not exclusive role in determining cell motility.
Fig. 6.

uPAR stimulation of MMP9 activity requires NHE1. Serum-starved H460 cells were stimulated in the absence or presence of either 10nM uPA or ATF for 16 hours. Where indicated, cells were treated with the NHE1 inhibitor EIPA for the duration of the experiment. After 24 hours, the conditioned medium was collected and analyzed for MMP9 activity by the hydrolysis of gelatin using a zymogen gel assay. Zymography is a representative gel. Bars represent the average of n=6 +/− SEM. Data were analyzed with a one-way Anova using Tukey’s Multiple Comparison with a 99.9% confidence interval between indicated groups (P-value of < 0.001).
3.6 NHE1 or uPAR Knockdown Alters H460 Cells Responses
To further clarify the role of NHE1 and uPAR in NSCLC tumor progression, we prepared two sets of knockdown cell lines. The H460 uPAR K/D cell line was prepared using lentil virus infection using shRNA to create a stable cell line with substantially decreased uPAR expression. Figure 7A shows both western blot analysis and qPCR analysis of uPAR expression in H460 versus the H460 uPAR K/D cell lines. As can be seen, uPAR K/D cells show virtually no uPAR expression by western blot analysis and qPCR indicates a greater than 75% reduction in uPAR mRNA. H460 NHE1 K/D cells were prepared using shRNA plasmid transfection to generate a stable cell with dramatically reduced NHE1 expression. Figure 7B shows western blot analysis of NHE1 expression with virtually no NHE1 present and qPCR results demonstrating a greater than 86% reduction in NHE1 mRNA between the H460 and the H460 NHE1 K/D cells. Control experiments using nonsense scramble sequences showed no difference from wild type cells for either the uPAR or NHE1.
Fig. 7.


Generation of uPAR and NHE1 H460 knockdown cell lines. Western blot and qPCR were performed to confirm protein knockdown uPAR (A) and NHE1 (B). qPCR data (n = 6) were analyzed using a Student two tailed T test with confidence intervals of 99%.
Acid load recovery of NHE1 K/D was significantly diminished compared to wild-type H460 cells and the NHE1 K/D transport rates were similar to the recovery rates observed with EIPA treated H460 cells (rates of recovery H460: 0.12 +/− 0.06 dpH/min, H460 NHE1 K/D: 0.01 +/− 0.07 dpH/min) indicating the loss of NHE1 expression correlated with an inability to recover from acid load in nominal bicarbonate conditions. Interestingly, steady state pHi for control cells was 0.22 pH units higher than either knock down cell lines whether cultured in normal or serum deprived conditions (Fig 8) indicating that loss of expression of NHE1 or uPAR activation of NHE1 leads to an intracellular acidification. Serum deprivation is typically used to reduce growth factor pathway activation and to mimic the in vivo tumor conditions of a solid tumor [37] and did not shift the intracellular pH in any of the three cells lines (Fig 8). Further evidence of sustained growth factor pathway activation was found as ERK phosphorylation levels remained high even after 48 hours of serum deprivation most likely due to Ras mutation (data not shown).
Fig. 8.

NHE1 activity in NHE1 and uPAR knockdown H460 cells. Resting pHi in cells cultured for 24 hours in RPMI medium containing 10% FBS or 0.5% FBS in HCO− free RPMI buffer and cells calibrated using the Nigericin method.
Using these knockdown cells, we once again investigated cell proliferation, stress fiber formation, and MMP9 activity. In the wild-type H460 cells addition of 10 nM uPA or 10 nM ATF increased the rate of cell proliferation by 24 and 18% respectively, whereas both the uPAR K/D and NHE1 K/D cells showed a slower level of basal proliferation and were not responsive to either uPA or ATF addition. Addition of uPA or ATF to the knock down cell lines had no impact on the number of cells displaying stress fiber over their respective controls (Fig. 9B) in contrast to the greater than 3.5 fold increase in stress fiber formation observed in uPA or ATF stimulated wild-type cells. In both knockdown cell lines, neither uPA nor ATF addition was able to stimulate stress fiber formation. NHE1 K/D cells did not form stress fibers in any condition. This pattern continued in our examination of MMP9 activation (Fig. 9C). Neither of the knockdown cell lines, MMP9 showed activity was equal to or greater than the wild-type cells (Fig. 6 and 9C). Neither uPA nor ATF stimulated MMP9 activation in the H460 NHE1 K/D and H460 uPAR K/D cell lines. As can be seen in zymogen assays, multiple cleared bands were observed suggesting an incomplete processing of MMP9. The requirement of NHE1 expression and function for proper maturation of MMP9 was observed in fibroblast NHE1 null cells or fibroblast expressing a non-proton transport functioning NHE1 [20]. These findings further establish a role for uPA-receptor signaling in cell motility and proliferative events and that this signaling is independent of the proteolytic activity of uPA. This work also provides evidence showing the requirement for NHE1 in NSCLC tumorigenicity.
Fig. 9.
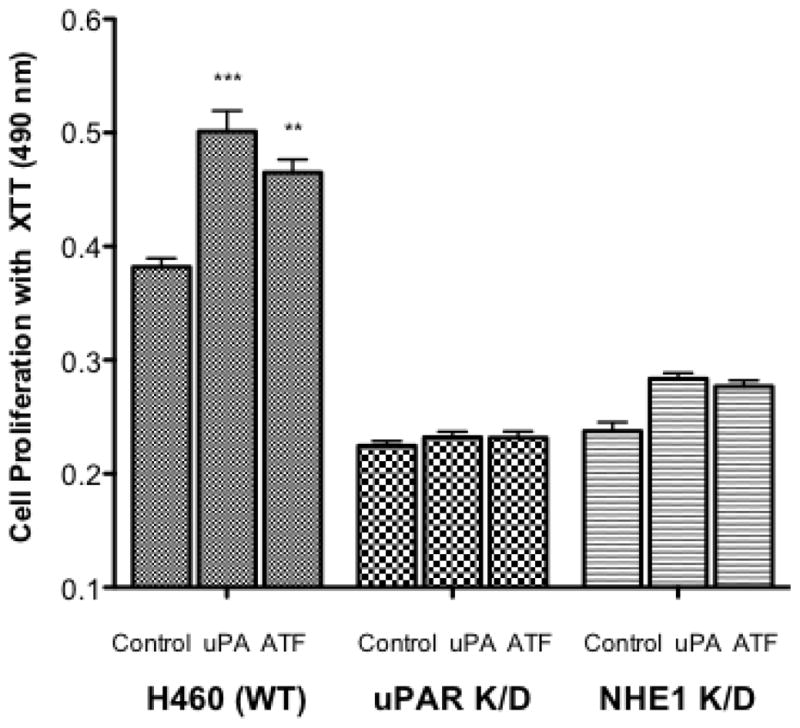
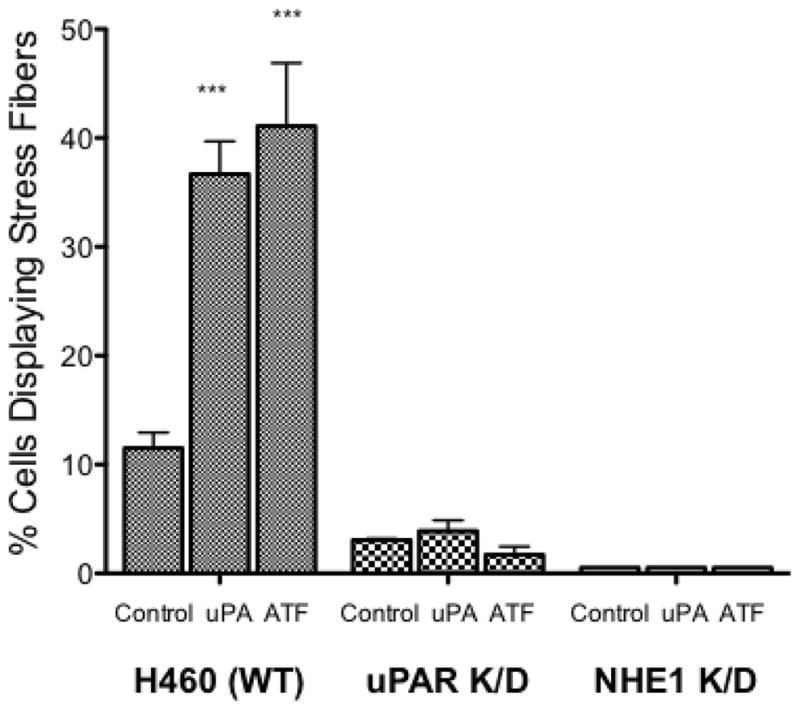
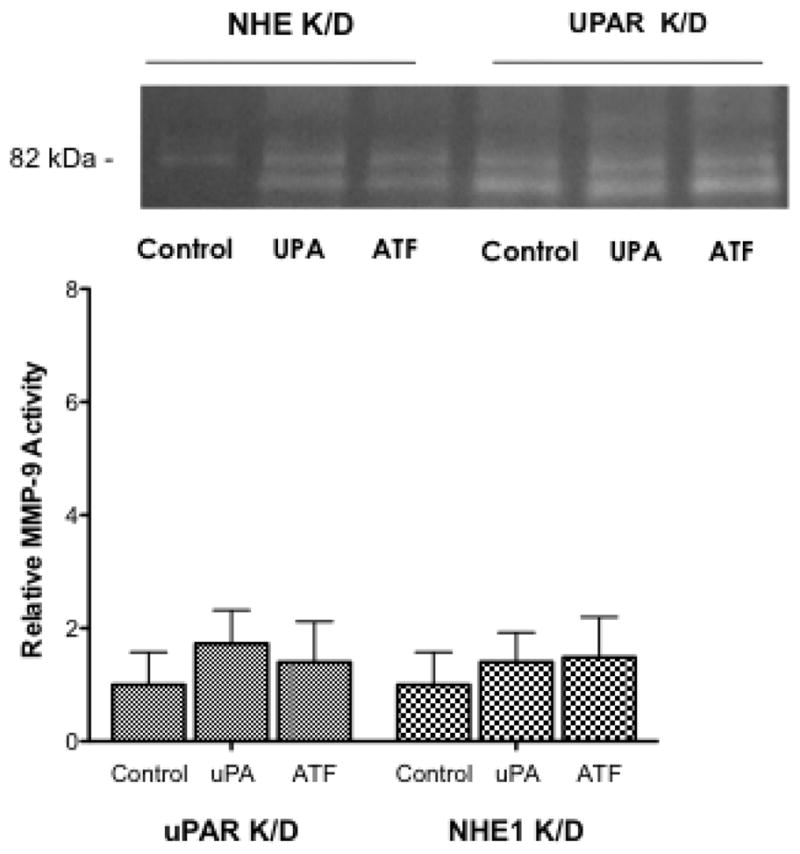
Impact of uPAR and NHE1 knockdown expression on H460 tumor behavior. A. Cell proliferation was measured for 24 hours after the addition of uPA or ATF. All data are presented as the mean +/− SEM (n ≥ 16). B. Stress fiber formation for each cell line (n > 6) was determined 15 min following uPA or ATF addition. C. MMP9 activity was measured in 24 hour conditioned medium. Low activity for each knockdown cell line was normalized against wild-type H460 cells (bottom panel). n = 4, data were analyzed using a one-way Anova (p>0.05) for significance.
3.7 In vivo in H460 cell tumor formation
To better understand the impact of NHE1 and uPAR expression in NSCL tumor development, we next evaluated the ability of the H460 wild-type, NHE1 K/D, and uPAR K/D cell lines to form tumors in athymic nude mice. We conducted two different sets of experiments in which wild-type and knock down cells were added either subcutaneously or intravenously. The first involved subcutaneous injections into four sets of 10 animals. The groups included H460 injected, H460 injected treated with IP EIPA, NHE1 K/D injected, and uPAR K/D injected. The results are shown in Figure 10A. In the group of animals injected with H460 wild-type cells, 8 of 10 mice formed tumors with an average tumor volume of 390 mm3. In the group injected with H460 wild-type cells and then treated with EIPA, 6 of 10 animals formed tumors with an average tumor volume of 80 mm3. Only 2 of 10 animals injected with uPAR K/D cells formed tumor with an average tumor volume of 15 mm3. Finally, in the NHE1 K/D injected group, only 1 of 10 animals formed tumors with a tumor volume of 5 mm3. In the second series of experiments the animals were given retro-orbital injections of the three different cell constructs and followed for survival outcomes over a 40 day interval (Fig 10B). In this experiment 100% of the mice injected with H460 wild type cells displayed morbidity within 20 days post-injection. For animal injected with uPAR K/D cells, 40% died by day 20, 50% died by day 25, and 40% remained alive at the end of the 40 day period. Finally, in the animals injected with NHE1 K/D cells, 20% of the animals died by 32 days following injection and 80% were still alive at 40 days. Necroscopy of the orthotopic injected animals identified tumors invading the thoracic cavity with significant metastasis to the spleen and lung. This is in agreement with earlier studies with H460 cells implanted in the pleural cavity [38]. Both sets of animal studies indicate that inhibition or loss of expression of NHE1 or uPAR are both crucial for NCSLC tumor growth and invasion in an in vivo murine model.
Fig. 10.
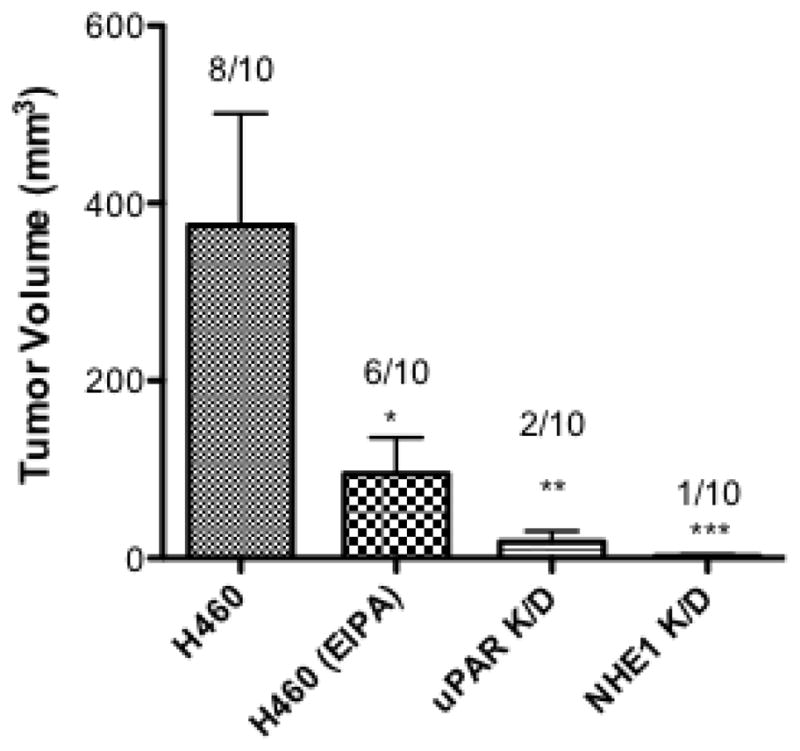

Both uPAR and NHE1 are critical for tumor formation and metastasis in nude mice. A Athymic, nude mice were injected subcutaneously with 5 × 106 of the indicated cells. Control mice (H460 injected) received vehicle, 1% DMSO intraperitonealy whereas animal group indicated with H460 (EIPA) were injected with 10 μg/kg every three days. Animals injected with uPAR or NHE1 knockdown cells did not receive injections. Six weeks post-injection, animals were euthanized and tumor volume determined. Data represent groups of 10 animals. Numbers above bars represent the number of animals out of ten which displayed tumors. A one way Anova with Bonferroni’s Multiple Comparison test with a p<0.001 was used to analyze the data. B. Six week, male athymic nude mice were injected with 5 × 106 cells retro orbitaly and monitored for 40 days post injection.
3.8 NHE1 deficiency differentially regulates gene expression of genes involved in tumor metastasis in H460 non-small cell lung cancer cells
To determine whether the changes observed in tumor progression and metastasis in in vitro and in vivo work, total RNA from H460 wild-type and NHE1 K/D cells was extracted and analyzed by real-time quantitative PCR using a Human Tumor Metastasis RT2 Profiler™ PCR Array. The number of cycles needed to reach the defined threshold was normalized to the β-actin signal to obtain the corrected cycle numbers. The array encodes 84 genes known to be involved in metastasis. Comparison of the array analysis of genes expressed in NHE1 K/D and H460 wild-type cells identified nine genes with differential expression. Five genes displayed a two-fold or greater expression and four genes showed a decreased expression further highlighting the impact of NHE1 on gene expression and on the potential of NHE1 to modulate a range of cellular functions (Table I).
Table I.
Mean values for fold expression
| GeneBank | Symbol | Description | Fold Expression |
|---|---|---|---|
| NM_006914 | RORB | RAR-Related Orphan Receptor B | 12.50 |
| NM_003810 | TNFS10 | Tumor Necrosis Factor Ligand Superfamily Member 10 | 6.85 |
| NM_000576 | IL1B | Interleukin 1-Beta Proprotein | 6.40 |
| NM_000369 | TSHR | Thyroid Stimulating Hormone Receptor | 2.88 |
| NM_002422 | MMP3 | Matrix Metalloproteinase Isoform 3 | 2.33 |
| NM_003255 | TIMP2 | TIMP Metallopeptidase Inhibitor 2 | −2.63 |
| NM_001797 | CDH11 | Cadherin 11, Type 2 | −3.33 |
| NM_003177 | SYK | Spleen Tyrosine Kinase | −11.56 |
| NM_002425 | MMP10 | Matrix Metalloproteinase Isoform 10 | −13.10 |
4 Discussion
The uPA-uPAR system is a complex and diverse mechanism that regulates cell migration, proliferation and tumor development. Once uPA is bound to uPAR, the pair can form a signaling complex with a number of partially defined partner receptors to coordinate cellular functions. The role of uPAR in regulating tumor behavior makes it an attractive therapeutic target, however the multiple modalities of action of uPA have made finding a successful treatment elusive. Activation of the uPA receptor induces its localization to the leading edge of migrating cells where NHE1 is located [7, 15, 17, 39]. The polarization of migrating cells by both NHE1 and uPAR leads to enhanced tumor invasion and migration, extracellular acidification, and coordination of cellular cytoskeletal dynamics. While numerous works have been published showing both uPAR and NHE1 are critical in tumor behavior, little evidence links the two. There are however, a number of studies which show a dependence of NHE1 transport on protease activity including cathepsin B, MMP2 and MT1-MMP[40–43]. However, while this work shows a relationship between pHi/NHE1 and tumor cell function, the work does not involve uPAR and is done in non-lung tissues and cell types. In an earlier study, we found that inhibition of NHE1 blocked agonist-induced invasion in H358 lung cancer cells [21]. These studies led us to hypothesize that NHE1 was critical in uPAR regulation of metastasis and tumor development. Here we demonstrate that uPAR signals through NHE1 without proteolysis to stimulate H460 lung cancer cell growth and motility. Using both NHE1 and uPAR knockdown cells we provide evidence for the requirement of both to induce cell growth and survivability in vivo. We also identified genes involved with tumor metastasis whose expression is altered after loss of NHE1 expression. These data may provide evidence for an additional and novel therapeutic approach to treating lung cancer.
uPA is secreted as an inactive zymogen, its expression and activation is tightly controlled in healthy tissue, and its protease activity plays a critical role in tumor progression. Expression patterns for uPAR are correlated with tumor progression and poor survival [43]. We show that both uPA and its cognate receptor are expressed in each of the cell lines examined, indicating potential autocrine activation in H1299, H358 and H460 cells. These findings parallel previous observations where uPAR expression was detected in H460 cells [44] and in other work reporting low levels of secreted uPA [10]. Furthermore, the expression pattern of both uPA and uPAR mirrors that of aggressively growing tumor cells, including H460 cells [32, 45]. While there is some basal level of uPA secretion, each cell line was able to increase its tumorigenic behavior in the presence of exogenous uPA (Fig. 2). It is interesting that lung tumor-associated macrophages express both uPA and MMP9 and the conditioned media from these cells increase the migration and invasion of H460 cells in a manner that required both proteases [46]. This hints at the potential for stromal or other cells to impact lung tumor development in a manner similar to that observed here. Proliferation requires the extrusion of the excess intracellular acid produced in metabolically active tumor cells. We show that exogenously added uPA increased proliferation in all three cell types while inhibiting proton transport blocked the intracellular alkalization critical for cell cycle progression [17, 19] suggesting that uPA signals through the exchanger. Similar patterns were observed with the formation of stress fibers (Fig. 2B). Stress fibers play a critical role in the epithelial-mesenchymal transition of lung cancer cells as they form structural networks driving the protrusion of lamellipodium and fillipodia [4, 47, 48]. Consistent with this, we found that EIPA blocked the formation of stress fiber formation in all three cell types (Fig. 2b). The ability of NHE1 inhibition to significantly decrease uPA stimulated anchorage-independent tumor growth demonstrates both the impact of uPA signaling on tumor progression and supports NHE1 as a critical player mediating these responses (Fig. 2c).
Because the tumor progression of each cell line was abrogated when NHE1 was inhibited and because uPA has both a receptor and non-receptor mechanism, we determined the signaling mechanism for uPA to NHE1 in H460 cells. In several studies, uPA signaling takes place through Ras-ERK signaling pathways, small G proteins or a combination of the two [7, 49,50]. H460 cells are moderately aggressive, metastatic adenocarcinoma cells with mutant K-ras [51,56]. Thus it was not surprising to find high levels of active ERK in resting or serum deprived H460 cells (Fig. 4A). However, the lack of uPA stimulation of NHE1 mediated acid load recovery was somewhat unexpected (Fig. 3). NHE1 contributes to cell migration by two distinct mechanisms effecting proton transport and by serving as a plasma membrane anchor for the cytoskeleton. The basal pHi in resting or serum deprived H460 cells was higher than NHE1 knockdown cells. This elevated transport set-point for pHi, (Fig. 8) indicates that there is a chronic elevation of proton transport from cells likely due to NHE1. This correlates with the high proliferation rate observed for wild-type H460 cells versus cells where NHE1 expression was diminished or the transport inhibited. However, while there were observable levels of GTP-RhoA in control cells, addition of uPA or ATF increased the small G protein-signaling pathway two-fold (Fig. 4B). Thus, it is possible that elevated ERK signaling is primarily activating the proton transport function of NHE1 explaining the lack of NHE1 transport response to the agonist. Meanwhile, the RhoA-Rock pathway, which phosphorylates distinct residues of NHE1 from ERK or ERKs downstream kinase RSK, [17,48] could be responsible for the uPA mediated effects in H460 cells in the presence of the elevated pHi caused by ERK activation.
Both uPA and uPAR can promote cell invasion and migration independent of proteolysis by binding to other cell surface proteins such as vitronectin and integrins at the leading edge of migrating cells. Evidence for this non-traditional receptor-based signaling includes inhibition of receptor protease activity with neutralizing antibodies [32] or the several studies showing an uPA-uPAR signals via integrin interaction [8]. We confirmed this observation using the uPAR binding domain of uPA which does not include the proteolytic domain. Here we show that ATF was sufficient to signal to RhoA, induce stress fiber formation, increase cell proliferation and enhance MMP9 activity. Studies in NHE1 and uPAR knockdown H460 cells confirm that uPA induction of tumor progression requires uPAR association and that the impact on uPA is independent of uPA proteolytic activity. Collectively these results indicate that uPA receptor occupancy is sufficient to enhance the signaling pathways altering NHE1 function and tumor development.
Increased levels of both MMP9 and uPA have been identified in NSCLC cancer patients and have been sought as a therapeutic target to treat lung cancer [14, 36, 52–54]. The relationship between the two proteases is less clear and is tissue and condition dependent. However, there are uPA induced bronchial epithelial cell migration is mediated by the plasmin system and requires MMP9 [52] and the complex of uPA and its receptor can signal to MMP9 without degrading extracellular matrix products [4, 31]. Here we report that the non-proteolytic domain of uPA was sufficient to enhance active MMP9 in conditioned media (Fig. 6). Our observations demonstrating that both uPA and ATF increase the level of active MMP9 found in conditioned media further supports the relationship between the proteases. Specifically that uPA signals through its receptor via NHE1 to stimulate MMP9 activity in H460 cells. Furthermore, our studies show that uPAR knockdown nearly blocks all uPA or ATF-stimulated MMP9 activity (Fig. 9C). In earlier work, we found that MMP9 activation in another NSCLC cell line, H358 was not only dependent on NHE1 expression, but that MMP9 maturation depended on both proton transport and cytoskeletal binding [21]. This is in agreement with work shown here demonstrating that NHE1 expression is critical for uPA-MMP9 activation. Interestingly, invadapodia, actin rich membrane extensions which are involved in the degradation of extracellular matrix contain multiple proteases including MMP9 and uPAR [55]. The work of Reshkin has identified thatNHE1 is critical to promote acidification and proteolytic activity necessary for tumor cell invasion through the matrix [56]. Thus it is possible that uPA coordinates tumor cell microenvironment leading to the corrosion of the extracellular matrix via NHE1. These results suggest that tissue remodeling by MMP9 required for invasion and migration is dependent on uPA stimulation and NHE1 however the structural mechanism for this activity is yet to be discovered.
Reduction of uPA activity or uPAR expression has been reported to inhibit tumor growth and invasion in a number of lung carcinoma models [3, 31, 53]. Our observations showing that uPAR knockdown abrogated tumor formation in vivo further supported these findings. The morbidity study (Fig. 10B) shows a dramatic decrease in tumor progression when cancer cells were delivered into the circulatory system. While 40% of the mice injected with H460 uPAR K/D cells did not survive, the life span of the mice was increased implying therapeutic value. One limitation in the study might be the loss of shRNA targeting uPAR implying these cells may have escaped from knockdown although a similar pattern was observed in other uPAR orthotopic studies [31]. The work presented where NHE1 was pharmacologically inhibited (Fig 10A) or in NHE1 K/D cells (Figs. 10A and 10B) both relates the importance of NHE1 in lung cancer tumor burden and metastasis. This work is consistent with several previous reports from other groups using other cell lines. Using injected karatinocytes, 75% of the tumor burden was blocked when using cells deficient in NHE1 expression or when NHE1 was chemically inhibited [37]. While these studies do not distinguish between the importance of uPAR and NHE1 in tissue remodeling or explain the mechanism of action, the work parallels the in vitro assays shown here and highlights the potential of both uPA-uPAR and NHE1 in lung cancer therapy.
The impact of NHE1 expression or function have been conducted in mouse muscle fibroblast cells as well as in brains of NHE1 null mice [57, 58]. It is not surprising that there is no common set of altered proteins between the limited set of genes examine here in NSCLC and either muscle or brain cells. Here we demonstrate a number of genes whose expression is altered in lung cancer cells when NHE1 expression is diminished (Table I). Three genes that displayed a decrease in expression in NHE1 knockdown cells (TIMP2, CDH11 and MMP10) are typically enhanced in aggressive cell lines and have been associated with tumor aggressiveness and reoccurrence. Loss of NHE1 expression increased tumor necrosis factor 10 which could result in an increase in apoptosis although none was observed in the cultures. Two of the other genes up regulated after loss of NHE1 expression, are associated with promoting cell growth and ECM remodeling. While two of the genes with increased expression, TOTB and TSHR, both function as nuclear receptors. It is possible that either a subtle shift in pHi due to NHE1 expression loss drives the transcriptional change or that the altered gene expression pattern is due to cellular compensation to adjust for the loss of proton transport. However, the pattern of some genes associated with tumor aggressiveness is consistent with the other reports of gene profiling and with the results found in vivo and in vitro reported here.
Tumor invasion and metastasis are associated with tumor progression and cancer deaths. Our work demonstrates that uPA/uPAR-mediated tumor progression and metastasis requires NHE1 in NSCLC cells and suggests a potential therapeutic approach to inhibiting invasion and metastasis. These data suggest that inhibition of NHE1 blocks uPA signaling through the uPA receptor and involves several functions involved in invasion and cellular motility. The signaling of the uPA recptor involves the ERK and RhoA pathway and influences the activity of MMP9. Loss of functioning NHE1 and the resulting pH microenviroment changes alters the ability of tumor cells to grow and impacts the death rate observed in our mouse model. Thus the altered intra and extra-cellular pH may be a mechanism supporting invasion and metastasis in NSCLC and provide a novel way to prevent and treat chemo refractive NSCLC.
Acknowledgments
This work was supported by National Institutes of Health Grant 1 R15 HL074924-01A1
References
- 1.Johnson DHBW, Carbone DP, Johnson DH, Blot WJ, Carbone DP, et al. Clinical Oncology. In: Abeloff AJMD, Niederhuber JE, Kastan MB, McKena WG, editors. Cancer of the Lung: non-small cell lung cancer and small cell lung cancer. 4. Chapter 76 Philadelphia: Churchill Livingstone Elsevier; 2008. [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Carriero MV, et al. Structure-based design of an urokinase-type plasminogen activator receptor-derived peptide inhibiting cell migration and lung metastasis. Molecular cancer therapeutics. 2009;8(9):2708–17. doi: 10.1158/1535-7163.MCT-09-0174. [DOI] [PubMed] [Google Scholar]
- 4.Rao JS, et al. Inhibition of invasion, angiogenesis, tumor growth, and metastasis by adenovirus-mediated transfer of antisense uPAR and MMP-9 in non-small cell lung cancer cells. Mol Cancer Ther. 2005;4(9):1399–408. doi: 10.1158/1535-7163.MCT-05-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nalla AK, et al. Targeting MMP-9, uPAR, and cathepsin B inhibits invasion, migration and activates apoptosis in prostate cancer cells. Cancer gene therapy. 2010;17(9):599–613. doi: 10.1038/cgt.2010.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nature reviews Molecular cell biology. 2002;3(12):932–43. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 7.Blasi F, Sidenius N. The urokinase receptor: focused cell surface proteolysis, cell adhesion and signaling. FEBS letters. 2010;584(9):1923–30. doi: 10.1016/j.febslet.2009.12.039. [DOI] [PubMed] [Google Scholar]
- 8.Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nature reviews Molecular cell biology. 2010;11(1):23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- 9.Veale D, Needham G, Harris AL. Urokinase receptors in lung cancer and normal lung. Anticancer research. 1990;10(2A):417–21. [PubMed] [Google Scholar]
- 10.Liu G, Shuman MA, Cohen RL. Co-expression of urokinase, urokinase receptor and PAI-1 is necessary for optimum invasiveness of cultured lung cancer cells. International journal of cancer Journal international du cancer. 1995;60(4):501–6. doi: 10.1002/ijc.2910600413. [DOI] [PubMed] [Google Scholar]
- 11.Dear AE, Medcalf RL. The urokinase-type-plasminogen-activator receptor (CD87) is a pleiotropic molecule. European journal of biochemistry/FEBS. 1998;252(2):185–93. doi: 10.1046/j.1432-1327.1998.2520185.x. [DOI] [PubMed] [Google Scholar]
- 12.Steffens GJ, et al. The complete amino acid sequence of low molecular mass urokinase from human urine. Hoppe-Seyler’s Zeitschrift fur physiologische Chemie. 1982;363(9):1043–58. doi: 10.1515/bchm2.1982.363.2.1043. [DOI] [PubMed] [Google Scholar]
- 13.Ossowski L, Aguirre-Ghiso JA. Urokinase receptor and integrin partnership: coordination of signaling for cell adhesion, migration and growth. Current opinion in cell biology. 2000;12(5):613–20. doi: 10.1016/s0955-0674(00)00140-x. [DOI] [PubMed] [Google Scholar]
- 14.Dass K, et al. Evolving role of uPA/uPAR system in human cancers. Cancer treatment reviews. 2008;34(2):122–36. doi: 10.1016/j.ctrv.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Martin C, et al. Intracellular pH gradients in migrating cells. American journal of physiology Cell physiology. 2011;300(3):C490–5. doi: 10.1152/ajpcell.00280.2010. [DOI] [PubMed] [Google Scholar]
- 16.Parks SK, Chiche J, Pouyssegur J. pH control mechanisms of tumor survival and growth. Journal of cellular physiology. 2011;226(2):299–308. doi: 10.1002/jcp.22400. [DOI] [PubMed] [Google Scholar]
- 17.Meima ME, Mackley JR, Barber DL. Beyond ion translocation: structural functions of the sodium-hydrogen exchanger isoform-1. Curr Opin Nephrol Hypertens. 2007;16(4):365–72. doi: 10.1097/MNH.0b013e3281bd888d. [DOI] [PubMed] [Google Scholar]
- 18.Stock C, Schwab A. Protons make tumor cells move like clockwork. Pflugers Archiv : European journal of physiology. 2009;458(5):981–92. doi: 10.1007/s00424-009-0677-8. [DOI] [PubMed] [Google Scholar]
- 19.Webb BA, et al. Dysregulated pH: a perfect storm for cancer progression. Nature reviews Cancer. 2011 doi: 10.1038/nrc3110. [DOI] [PubMed] [Google Scholar]
- 20.Wallert M, et al. alpha-(1) adrenergic receptor-induced cytoskeletal organization and cell motility in CCL39 fibroblasts requires Phospholipase D1. Journal of cellular biochemistry. 2011 doi: 10.1002/jcb.23227. [DOI] [PubMed] [Google Scholar]
- 21.Taves J, et al. Sodium hydrogen exchanger and phospholipase D are required for alpha1-adrenergic receptor stimulation of metalloproteinase-9 and cellular invasion in CCL39 fibroblasts. Arch Biochem Biophys. 2008;477(1):60–6. doi: 10.1016/j.abb.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 22.Li S, et al. Inhibition of proliferation and apoptosis induced by a Na+/H+ exchanger-1 (NHE-1) antisense gene on drug-resistant human small cell lung cancer cells. Oncology reports. 2009;21(5):1243–9. doi: 10.3892/or_00000347. [DOI] [PubMed] [Google Scholar]
- 23.Pouyssegur J, et al. A specific mutation abolishing Na+/H+ antiport activity in hamster fibroblasts precludes growth at neutral and acidic pH. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(15):4833–7. doi: 10.1073/pnas.81.15.4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas JA, et al. Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry. 1979;18(11):2210–8. doi: 10.1021/bi00578a012. [DOI] [PubMed] [Google Scholar]
- 25.Wallert MA, Frohlich O. Na+-H+ exchange in isolated myocytes from adult rat heart. Am J Physiol. 1989;257(2 Pt 1):C207–13. doi: 10.1152/ajpcell.1989.257.2.C207. [DOI] [PubMed] [Google Scholar]
- 26.Vomhof-DeKrey EE, Haring JS, Dorsam GP. Vasoactive intestinal peptide receptor 1 is downregulated during expansion of antigen-specific CD8 T cells following primary and secondary Listeria monocytogenes infections. Journal of neuroimmunology. 2011;234(1–2):40–8. doi: 10.1016/j.jneuroim.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crowley CW, et al. Prevention of metastasis by inhibition of the urokinase receptor. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(11):5021–5. doi: 10.1073/pnas.90.11.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen YX, O’Brien ER. Ethyl isopropyl amiloride inhibits smooth muscle cell proliferation and migration by inducing apoptosis and antagonizing urokinase plasminogen activator activity. Can J Physiol Pharmacol. 2003;81(7):730–9. doi: 10.1139/y03-066. [DOI] [PubMed] [Google Scholar]
- 29.Matthews H, Ranson M, Kelso MJ. Anti-tumour/metastasis effects of the potassium-sparing diuretic amiloride: An orally active anti-cancer drug waiting for its call-of-duty? International journal of cancer Journal international du cancer. 2011 doi: 10.1002/ijc.26156. [DOI] [PubMed] [Google Scholar]
- 30.Ghosh S, et al. Functional relevance of urinary-type plasminogen activator receptor-alpha3beta1 integrin association in proteinase regulatory pathways. The Journal of biological chemistry. 2006;281(19):13021–9. doi: 10.1074/jbc.M508526200. [DOI] [PubMed] [Google Scholar]
- 31.Tang CH, et al. Signaling through urokinase and urokinase receptor in lung cancer cells requires interactions with beta1 integrins. Journal of cell science. 2008;121(Pt 22):3747–56. doi: 10.1242/jcs.029769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duriseti S, et al. Antagonistic anti-urokinase plasminogen activator receptor (uPAR) antibodies significantly inhibit uPAR-mediated cellular signaling and migration. The Journal of biological chemistry. 2010;285(35):26878–88. doi: 10.1074/jbc.M109.077677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei Y, et al. Urokinase receptors are required for alpha 5 beta 1 integrin-mediated signaling in tumor cells. The Journal of biological chemistry. 2007;282(6):3929–39. doi: 10.1074/jbc.M607989200. [DOI] [PubMed] [Google Scholar]
- 34.Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265(1):23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Pellegrin S, Mellor H. Actin stress fibres. Journal of cell science. 2007;120(Pt 20):3491–9. doi: 10.1242/jcs.018473. [DOI] [PubMed] [Google Scholar]
- 36.Yang SF, et al. Increased plasma levels of urokinase plasminogen activator and matrix metalloproteinase-9 in nonsmall cell lung cancer patients. Clin Chim Acta. 2005;354(1–2):91–9. doi: 10.1016/j.cccn.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 37.Reshkin SJ, et al. Phosphoinositide 3-kinase is involved in the tumor-specific activation of human breast cancer cell Na(+)/H(+) exchange, motility, and invasion induced by serum deprivation. J Biol Chem. 2000;275(8):5361–9. doi: 10.1074/jbc.275.8.5361. [DOI] [PubMed] [Google Scholar]
- 38.Kraus-Berthier L, et al. Histology and sensitivity to anticancer drugs of two human non-small cell lung carcinomas implanted in the pleural cavity of nude mice. Clinical cancer research : an official journal of the American Association for Cancer Research. 2000;6(1):297–304. [PubMed] [Google Scholar]
- 39.Estreicher A, et al. The receptor for urokinase type plasminogen activator polarizes expression of the protease to the leading edge of migrating monocytes and promotes degradation of enzyme inhibitor complexes. The Journal of cell biology. 1990;111(2):783–92. doi: 10.1083/jcb.111.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bourguignon LY, et al. CD44 interaction with Na+-H+ exchanger (NHE1) creates acidic microenvironments leading to haluronidase-2 and cathepsin B activation and breast tumor cell invasion. J Biol Chem. 2004;279(26):16991–7007. doi: 10.1074/jbc.M311838200. [DOI] [PubMed] [Google Scholar]
- 41.Yang W, et al. Inhibition of Na(+)/H(+) exchanger 1 by 5-(N-ethyl-N-isopropyl) amiloride reduces hypoxia-induced hepatocellular carcinoma invasioin and motility. Cancer Lett. 2010;295(2):198–204. doi: 10.1016/j.canlet.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Lin Y, et al. NHE1 mediates MDA-MB-231 cells invasion through the regulation of MT1-MMP. Exp Cell Res. 2011;317(14):2031–40. doi: 10.1016/j.yexcr.2011.05.026. [DOI] [PubMed] [Google Scholar]
- 43.Allgayer H. Translational research on u-PAR. European journal of cancer. 2010;46(7):1241–51. doi: 10.1016/j.ejca.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 44.Nikolova DA, et al. Cetuximab attenuates metastasis and u-PAR expression in non-small cell lung cancer: u-PAR and E-cadherin are novel biomarkers of cetuximab sensitivity. Cancer research. 2009;69(6):2461–70. doi: 10.1158/0008-5472.CAN-08-3236. [DOI] [PubMed] [Google Scholar]
- 45.Shetty S, Idell S. Posttranscriptional regulation of urokinase receptor gene expression in human lung carcinoma and mesothelioma cells in vitro. Molecular and cellular biochemistry. 1999;199(1–2):189–200. doi: 10.1023/a:1006914800447. [DOI] [PubMed] [Google Scholar]
- 46.Wang R, et al. Tumor-associated macrophages provide a suitable microenvironment for non-small lung cancer invasion and progression. Lung cancer. 2011 doi: 10.1016/j.lungcan.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 47.Wyckoff JB, et al. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Current biology : CB. 2006;16(15):1515–23. doi: 10.1016/j.cub.2006.05.065. [DOI] [PubMed] [Google Scholar]
- 48.Baumgartner M, Patel H, Barber DL. Na(+)/H(+) exchanger NHE1 as plasma membrane scaffold in the assembly of signaling complexes. Am J Physiol Cell Physiol. 2004;287(4):C844–50. doi: 10.1152/ajpcell.00094.2004. [DOI] [PubMed] [Google Scholar]
- 49.Kunigal S, et al. SPARC-induced migration of glioblastoma cell lines via uPA-uPAR signaling and activation of small GTPase RhoA. International journal of oncology. 2006;29(6):1349–57. [PMC free article] [PubMed] [Google Scholar]
- 50.Jo M, et al. Cooperativity between the Ras-ERK and Rho-Rho kinase pathways in urokinase-type plasminogen activator-stimulated cell migration. The Journal of biological chemistry. 2002;277(14):12479–85. doi: 10.1074/jbc.M111147200. [DOI] [PubMed] [Google Scholar]
- 51.Mitsudomi T, et al. Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene. 1991;6(8):1353–62. [PubMed] [Google Scholar]
- 52.Legrand C, et al. uPA/plasmin system-mediated MMP-9 activation is implicated in bronchial epithelial cell migration. Experimental cell research. 2001;264(2):326–36. doi: 10.1006/excr.2000.5125. [DOI] [PubMed] [Google Scholar]
- 53.Henneke I, et al. Inhibition of urokinase activity reduces primary tumor growth and metastasis formation in a murine lung carcinoma model. American journal of respiratory and critical care medicine. 2010;181(6):611–9. doi: 10.1164/rccm.200903-0342OC. [DOI] [PubMed] [Google Scholar]
- 54.Duffy MJ. The urokinase plasminogen activator system: role in malignancy. Current pharmaceutical design. 2004;10(1):39–49. doi: 10.2174/1381612043453559. [DOI] [PubMed] [Google Scholar]
- 55.Linder S. The matrix corroded: podosomes and invadopodia in extracellular matrix degredation. Trends in Cell Biology. 2007;17(3):107–117. doi: 10.1016/j.tcb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 56.Busco G, et al. NHE1 promotes invadopodial ECM proteolysis through acidification of the peri-invadopodial space. The FASEB Journal. 24(10):3903–3915. doi: 10.1096/fj.09-149518. [DOI] [PubMed] [Google Scholar]
- 57.Putney LK, Barber DL. Expression profile of genes regulated by activity of the Na-H exchanger NHE1. BMC Genomics. 2004;5(1):46. doi: 10.1186/1471-2164-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou D, et al. Na+/H+ exchanger 1 deficiency alters gene expression in mouse brain. Physiological genomics. 2004;18(3):331–9. doi: 10.1152/physiolgenomics.00076.2004. [DOI] [PubMed] [Google Scholar]
- 59.Zhong M, et al. MicroRNAs reduce tumor growth and contribute to enhance cytotoxicity induced by gefitinib in non-small cell lung cancer. Chem Biol Interact. 2010;184(3):431–8. doi: 10.1016/j.cbi.2010.01.025. [DOI] [PubMed] [Google Scholar]