

Abstract
Structure-based design, synthesis, and biological evaluation of a series of dihydroquinazoline-derived β-secretase inhibitors incorporating thiazole and pyrazole-derived P2-ligands are described. We have identified inhibitor 4f which has shown potent enzyme inhibitory (Ki = 13 nM) and cellular (IC50 = 21 nM in neuroblastoma cells) assays. A model of 4f was created based upon the X-ray structure of 3a-bound β-Secretase. The model revealed critical interactions in the active site.
Keywords: β-Secretase, Alzheimer’s Diseas, Memapsin 2, Inhibitor, Design and Synthesis, Dihydroquinazoline
β-Secretase (BACE1, memapsin 2) is an important molecular target for the treatment of Alzheimer’s disease (AD).1 This membrane-bound aspartic protease catalyzes the cleavage of β-amyloid precursor protein (APP) leading to the formation of amyloid-β-peptide (Aβ) in the brain. The neurotoxicity of Aβ leads to brain inflammation, neuronal death, dementia, and AD.2,3 Early on, we designed a potent substrate-based transition-state inhibitor 1.4 An X-ray structure of 1-bound β-secretase led to the structure-based design of a series of BACE1 inhibitors.5 Over the years, we and others have designed a variety of BACE1 inhibitors incorporating novel peptidomimetic and nonpeptide scaffolds.6–8 A number of BACE1 inhibitors have been shown to reduce Aβ-levels in transgenic AD mice. Furthermore, we recently showed that administration of β-secretase inhibitor GRL8234 (2) rescued the decline of cognitive function in transgenic AD mice.9 While the development of a clinically effective inhibitor has not yet emerged, important progress has been made with small-molecule inhibitors belonging to different structural classes.1
In 2007, Baxter and co-workers reported an interesting class of 2-amino-dihydroquinazoline-derived BACE1 inhibitors.10 A representative example is inhibitor 3a (Figure 1) with reported a Ki of 11 nM. An X-ray structure of 3a-bound β-secretase provided the important molecular interactions responsible for its high affinity.10 Based upon this reported X-ray structure, we have designed a number of functionalities and heterocyclic scaffolds, to make specific interactions in the β-secretase active site to improve enzyme affinity and potency. Herein, we report the design, synthesis and evaluation of a series of potent 2-amino-dihydroquinazoline-derived inhibitors incorporating urethanes and heterocycles such as pyrazoles and thiazoles. A number of compounds exhibited potent β-secretase inhibitory activity and displayed good potency in cellular assays. To obtain molecular insight into the possible ligand-binding site of β-secretase, we have created an energy-minimized model of 4f based upon the 3a-bound β-secretase X-ray structure.
Figure 1.

Structures of inhibitors 1–3
The synthesis of various dihydroquinazoline derivatives is shown in Scheme 1. The Boc-protected (R)-cyclohexylglycine methyl ester 4 was reduced by DIBAL-H at −78 °C and the resulting aldehyde was reacted with (ethoxycarbonylmethylene)- triphenylphosphorane in a mixture of CH2Cl2 and methanol to provide the corresponding α,β-unsaturated ester. The ester was subjected to hydrogenation over Pd-C for 12 h to provide the corresponding saturated derivative. Saponification of the ester with aqueous LiOH afforded the acid 5 in 70% overall yield. For the synthesis of quinazoline derivative 3a, acid 5 was coupled with N-cyclohexylmethyl amine to provide amide 6 in 77% yield. Removal of the Boc -group was carried out by treatment with trifluoroacetic acid and the resulting amine was subjected to reductive amination with aldehyde 710,11 to afford nitro compound 8 in 62% yield. Hydrogenation of 8 over 10% Pd-C in ethyl acetate at 23 °C provided the corresponding amine. Reaction of this resulting amine with BrCN in ethanol at reflux furnished dihydroquinazoline derivative 3a in 84% yield. For the synthesis of inhibitor 3b, (R)-phenylglycinol derivative 9 was oxidized under Swern conditions and the resulting aldehyde was reacted with (ethoxycarbonylmethylene)triphenylphosphorane in THF to provide α,β-unsaturated ester 10. Hydrogenation of the ester over 10% Pd-C in ethyl acetate and saponification of the ester provided the corresponding acid which was coupled with N-cyclohexylmethyl amine to provide amide 11 in 53% overall yield. Amide 11 was converted to dihydroquinazoline derivative 3b by following the same sequence of reactions as 3a.
Scheme 1.

Synthesis of dihydroquinazoline-derived BACE1 inhibitors
We have investigated the potential of various urethane derivatives as BACE1 inhibitors. A representative synthesis of urethane derivative 3c is shown in Scheme 2. Phenylglycinol derivative 9 was reacted with 4-nitrophenylchloroformate to provide the corresponding mixed carbonate.12 Reaction of N-cyclohexylmethyl amine with this mixed carbonate afforded urethane derivative 12 in 90% yield in two steps. Removal of the Boc group with trifluoroacetic acid and reductive amination of the resulting amine with aldehyde 7 furnished nitro derivative 13 in 53% yield, in two steps. Hydrogenation of nitro compound 13 over 10% Pd-C in ethyl acetate followed by exposure of the resulting amine to BrCN afforded inhibitor 3c. Urethane derivatives 3d and 3e were prepared by an analogous procedure using Boc protected (R)-cyclohexyl glycinol as the starting material. For synthesis of urethane 3f, racemic amino ester 14 was prepared using a multicomponent reaction developed in our laboratory.13 Reaction of 14 with Boc2O in the presence of DMAP in CH3CN at 23 °C afforded the corresponding Boc derivative. Treatment of the resulting ester with magnesium powder in methanol under sonication afforded the corresponding Boc amino ester.14 Reduction of the resulting ester with LAH afforded the racemic alcohol 15 in 63% overall yield. This alcohol was converted to urethane derivative 3f and amide derivative 3g as described above.
Scheme 2.

Synthesis of urethane-derived inhibitors
Based upon the X-ray structure of 3a-bound β-secretase, we planned to incorporate various heterocyclic derivatives in place of the methyl group in 3a to interact with the β-secretase active site. We were particularly interested in designing inhibitors with thiazole and pyrazole derivatives since these heterocycles contain hydrogen bond donor and acceptor groups for interactions in the enzyme active site. Also, these heterocycles are inherent to numerous bioactive natural products and FDA approved therapeutic agents.15, 16 Accordingly we designed a number of inhibitors containing thiazole and pyrazole derivatives. The synthesis of various dihydroquinazoline derivatives are shown in Scheme 3. N-cyclohexyl derivative 16 was prepared by reductive amination of the corresponding 4-thiazolecarboxaldehyde17 and cyclohexyl amine. Similarly, amines 17–20 were prepared from the corresponding aldehydes.18–21 Various known pyrazole aldehydes22 were converted to amines 21–23 by reductive amination with cyclohexylamine. For synthesis of dihydroquinazoline 4b, acid 5 was coupled with amine 16 in the presence of EDC, and HOBt to provide amide 24. Removal of Boc and subsequent reductive amination with aldehyde 7 furnished nitro derivative 25 in 65% overall yield. This was converted to quinazoline 4b as described above. Other inhibitors 4c–4i were prepared following a similar protocol as 4b.
Scheme 3.

Synthesis of memapsin 2 inhibitors
The β-secretase inhibitory activity of various inhibitors was determined against recombinant β-secretase using our previously reported assay protocols.23 The results are shown in Table 1. As can be seen, our synthetic dihydroquinzoline derivative 3a has shown Ki value of 25 nM. The corresponding phenyl derivative 3b exhibited nearly a 6-fold lower enzyme inhibitory potency. We have then investigated the corresponding urethane derivative 3c. However, this inhibitor displayed nearly a 5-fold loss of potency compared to 3b. However, the corresponding cyclohexyl urethane derivative 3d, improved potency by 20-fold over 3c (entry 4). The presence of methyl group is important as the cyclohexyl urethane derivative 3e is less potent. We have also incorporated tetrahydropyran ring in place of the cyclohexyl group in 3d. As shown, racemic mixture (1:1) 3f has shown reduced potency over cyclohexyl derivative 3d. We have also examined the effect of a ring oxygen in carboxamide derivative 3g. This derivative too lost nearly 5-fold potency compared to cyclohexyl derivative 3a. We have also evaluated the cellular inhibition of β-secretase in neuroblastoma cells.24 Inhibitor 3a has shown an average cellular IC50 value of 71 nM. The corresponding phenyl derivative displayed an IC50 of 482 nM. The urethane derivative 3c was significantly less potent compared to inhibitor 3b. Similarly, urethane derivative 3f showed an IC50 value of 1·1 μM (entry 6).
Table 1.
Enzyme inhibitory and cellular activity of inhibitors
| Entry | Inhibitor | ki (nM) | IC50(nM)a,b |
|---|---|---|---|
| 1. |
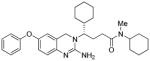 3a |
25 | 71 |
| 2. |
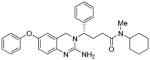 3b |
163.8 | 482 |
| 3. |
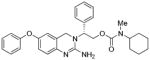 3c |
783 | 3400 |
| 4. |
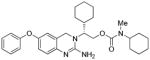 3d |
37.5 | nt |
| 5. |
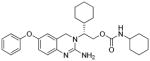 3e |
60 | nt |
| 6. |
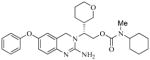 3f |
104 | 1100 |
| 7. |
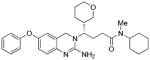 3g |
122 | nt |
IC50 was determined in neuroblastoma cells.
GRL-8234 exhibited Ki, = 1.8 nM, IC50 = 2.5 nM in this asay.9a
Based upon the reported X-ray structure of 3a-bound β-secretase, we have incorporated various thiazole and pyrazole-derived ligands in an effort to interact with residues in the β-secretase active site. As can be seen in Table 2, incorporation of (2-methylthiazol-4-yl)methyl substituent in place of the methyl group in 3d, resulted in nearly a 40-fold loss of enzyme inhibitory activity (entry 1). The corresponding amide derivative 4b also exhibited a loss of potency over 3a (entry 2). Interestingly, this compound displayed poor cellular β-secretase activity in neuroblastoma cells over 3a. Introduction of a methyl group on the methylene side chain in 4b resulted in 4c which showed a loss of potency (entry 3). Furthermore, chain elongation in 4d and incorporation of (2-isopropylthiazol-4-yl)methyl substituent in 4e did not improve potency (entries 4 and 5). Interestingly, incorporation of a methoxymethyl substituent on the thiazole side chain in 4f resulted in nearly a 6-fold improvement of enzyme activity over 4b. Furthermore, inhibitor 4f has shown very potent cellular inhibitory properties in neuroblastoma cells (entry 6). We have also investigated various substituted pyrazolylmethyl groups (entries 7–9). Methyl substituted pyrazole derivative 4g is more potent than the unsubstituted derivative 4h in both enzyme inhibitory and cellular assays. Incorporation of methyl group on the pyrazole ring in 4i did not improve potency over the alkylated or unalkylated derivatives.
To gain insight into specific ligand-binding site interactions, an energy minimized model structure of 4f was created in the active site of BACE1, based upon the crystal structure of 3a-bound β-secretase.10 The conformation of 4f was optimized using the CHARMM force field.25 As shown in Figure 2, 2-amino dihydroquinazoline functionality forms a unique hydrogen bonding network with the catalytic aspartic acids Asp32 and Asp228 and the (S)-cyclohexyl group nicely filled in the S1′-subsite as reported in the X-ray structure.10 The 2-methoxymethylthiazole moiety appears to fill in the hydrophobic pocket in the S2-subsite. Furthermore, the methoxy oxygen is within proximity to form a hydrogen bond with Thr232. This may explain a 6-fold enhancement of enzyme inhibitory potency over the 2-methylthiazole derivative 4b. The P1-cyclohexamide fits well into the S1-site of β-secretase.
Figure 2.

Stereoview of the model of inhibitor 4f (green carbon) with β-secretase. Possible hydrogen bonds between the inhibitor and β-secretase are shown in black dotted lines.
In summary, we have carried out structure-based modifications of 2-amino-3,4-dihydroquinazoline -derived β-secretase inhibitors. In particular, we have incorporated thiazole and pyrazole-based P2-ligands to make specific interactions in the S2-subsite. These efforts resulted in inhibitors with improved potency and cellular inhibitory properties compared to methyl-substituted inhibitor 3a. Inhibitor 4f has shown enhanced enzyme inhibitory activity as well as very good cellular inhibitory potency in neuroblastoma cells. A protein-ligand X-ray structure-based model of 4f-bound β-secretase has provided important molecular insight into the ligand-binding site interactions. Further design and improvement of inhibitor properties are currently in progress.
Table 2.
Enzyme inhibitory and cellular activity of inhibitors
| Entry | Inhibitor | ki (nM) | IC50(nM)a,b |
|---|---|---|---|
| 1. |
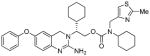 4a |
1463 | nt |
| 2. |
 4b |
79 | 390 |
| 3. |
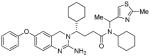 4c |
104 | nt |
| 4. |
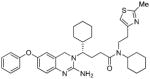 4d |
106 | nt |
| 5. |
 4e |
144 | nt |
| 6. |
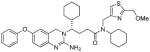 4f |
13 | 21 |
| 7. |
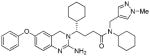 4g |
11 | 23 |
| 8. |
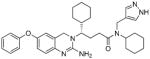 4h |
23 | 48 |
| 9. |
 4i |
39 | 51 |
IC50 was determined in neuroblastoma cells.
GRL-8234 exhibited Ki; = 1.8 nM, IC50 = 2.5 nM in this assay.9a
Acknowledgments
Financial support by the National Institutes of Health (AG 18933) is gratefully acknowledged. We would like to thank Professor D. Eric Walters (Rosalind Franklin University of Medicine and Science) for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Ghosh AK, Brindisi M, Tang J. J Neurochem. 2012;120:71–83. doi: 10.1111/j.1471-4159.2011.07476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Proc Natl Acad Sci, USA. 2000;97:1456–1460. doi: 10.1073/pnas.97.4.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vassar R, Bennettt BD, Babu-Khan S, Khan S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 3.(a) Selkoe DJ. Nature. 1999;399:A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]; (b) Selkoe D. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh AK, Shin D, Downs D, Koelsch G, Lin X, Ermolieff J, Tang J. J Am Chem Soc. 2000;122:3522–3523. doi: 10.1021/ja000300g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hong L, Koelsch G, Lin X, Wu S, Terzyan S, Ghosh AK, Zhang XC, Tang J. Scienc. 2000;290:150–153. doi: 10.1126/science.290.5489.150. [DOI] [PubMed] [Google Scholar]
- 6.Tang J, Hong L, Ghosh AK. In: Aspartic Acid Proteases as Therapeutic Targets. Ghosh AK, editor. Wiley-VCH Verlag GmbH & Co KGaA; Weinheim: 2010. pp. 413–440. [Google Scholar]
- 7.Iserloh U, Cumming JN. In: Aspartic Acid Proteases as Therapeutic Targets. Ghosh AK, editor. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2010. pp. 441–479. [Google Scholar]
- 8.Cole DC, Bursavich MG. In: Aspartic Acid Proteases as Therapeutic Targets. Ghosh AK, editor. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2010. pp. 481–509. [Google Scholar]
- 9.(a) Ghosh AK, Kumaragurubaran N, Hong L, Kulkarni S, Xu X, Miller HB, Reddy DS, Weerasena V, Turner R, Chang W, Koelsch G, Tang J. Bioorg Med Chem Lett. 2008;18:1031–1036. doi: 10.1016/j.bmcl.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chang WP, Huang X, Downs D, Cirrito JR, Koelsch G, Holtzman DM, Ghosh AK, Tang J. FASEB J. 2011;25:775–784. doi: 10.1096/fj.10-167213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baxter EW, Conway KA, Kennis L, Bischoff F, Mercken MH, De Winter HL, Reynolds CH, Tounge BA, Luo C, Scott MK, Huang Y, Braeken M, Pieters SMA, Berthelot DJC, Masure S, Bruinzeel WD, Jordan AD, Parker MH, Boyd RE, Qu J, Alexander RS, Brenneman DE, Reitz AB. J Med Chem. 2007;50:4261–4264. doi: 10.1021/jm0705408. [DOI] [PubMed] [Google Scholar]
- 11.Katritzky AR, Wang Z, Hall CD, Akhmedov NG. ARKIVOC. 2003;2:49–58. [Google Scholar]
- 12.Ghosh AK, Gemma S, Baldridge A, Wang Y-F, Kovalevsky AY, Koh Y, Weber IT, Mitsuya H. J Med Chem. 2008;51:6021–6033. doi: 10.1021/jm8004543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh AK, Xu CX, Kulkarni SS, Wink D. Org Lett. 2005;7:7–10. doi: 10.1021/ol048302j. [DOI] [PubMed] [Google Scholar]
- 14.Nyasse B, Grehn L, Ragnarsson U. Chem Commun. 1997:1017–1018. [Google Scholar]
- 15.(a) Ghosh AK, Kulkarni S. Org Lett. 2008;10:3907–3909. doi: 10.1021/ol8014623. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nicolaou KC, Roschangar F, Vourloumis D. Angew Chem Int Ed. 1998;37:2014–2045. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2014::AID-ANIE2014>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]; (c) Virgil SC. In: In Aspartic Acid Proteases as Therapeutic Targets. Ghosh AK, editor. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2010. pp. 147–152. and references cited therein. [Google Scholar]
- 16.(a) Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, Graneto MJ, Lee LF, Malecha JW, Miyashiro JM, Rogers RS, Rogier DJ, Yu SS, Anderson GD, Burton EG, Cogburn JN, Gregory SA, Koboldt CM, Perkins WE, Seibert K, Veenhuizen AW, Zhang YY, Isakson PC. J Med Chem. 1997;40:1347–1365. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]; (b) Terrett NK, Bell AS, Brown D, Ellis P. Bioorg Med Chem Lett. 1996;6:1819–1824. and references cited therein. [Google Scholar]
- 17.Sawada D, Shibasaki M. Angew Chem Int Ed. 2000;39:209–213. doi: 10.1002/(sici)1521-3773(20000103)39:1<209::aid-anie209>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 18.Brunjes M, Sourkouni-Agirusi G, Kirschning A. Adv Synth Catal. 2003;345:635–642. [Google Scholar]
- 19.Degoey David A, et al. From U.S. Pat. Appl. Publ., 20050131017. 2005 Jun 16;
- 20.Deady LW, Stanborough MS. Aust J Chem. 1981;34:1295–1302. [Google Scholar]
- 21.Hubbard RD, Bamaung NY, Palazzo F, Zhang Q, Kovar P, Osterling DJ, Hu X, Wilsbacher JL, Johnson EF, Bouska J, Wang J, Bell RL, Davidsen SK, Shappard GS. Bioorg Med Chem Lett. 2007;17:5406–5409. doi: 10.1016/j.bmcl.2007.07.037. [DOI] [PubMed] [Google Scholar]
- 22.(a) Frey RR, Curtin ML, Albert DH, Glaser KB, Pease LJ, Soni NB, Bouska JJ, Reuter D, Stewart KD, Marcotte P, Bukofzer G, Li J, Davidsen SK, Michaelides MR. J Med Chem. 2008;51:3777–3787. doi: 10.1021/jm701397k. [DOI] [PubMed] [Google Scholar]; (b) Taydakov IV, Krasnoselskiy SS, Dutova TY. Synth Commun. 2011;41:2430–2434. [Google Scholar]; (c) Baraldi PG, Cacciari B, Spalluto G, Romagnoli R, Braccioli G, Zaid AN, Pineda de las Infantas MJ. Synthesis. 1997:1140–1142. [Google Scholar]
- 23.Ermolieff J, Loy JA, Koelsch G, Tang J. Biochemistry. 2000;39:12450–12456. doi: 10.1021/bi001494f. [DOI] [PubMed] [Google Scholar]
- 24.Chang WP, Koelsch G, Wong S, Downs D, Da H, Weerasena V, Gordon B, Devasamudram T, Bilcer G, Ghosh AK, Tang J. J Neurochem. 2004;89:1409–1416. doi: 10.1111/j.1471-4159.2004.02452.x. [DOI] [PubMed] [Google Scholar]
- 25.Brooks BR, Brooks CL, III, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. J Comp Chem. 2009;30:1545–1615. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]