

Abstract
Angiotensin II (A-II) regulation of aldosterone secretion is initiated by inducing cell membrane depolarization, thereby increasing intracellular calcium and activating the calcium calmodulin/calmodulin kinase cascade. Mutations in the selectivity filter of the KCNJ5 gene coding for inward rectifying potassium channel (Kir)3.4 has been found in about one third of aldosterone-producing adenomas. These mutations result in loss of selectivity of the inward rectifying current for potassium, which causes membrane depolarization and opening of calcium channels and activation of the calcium calmodulin/calmodulin kinase cascade and results in an increase in aldosterone secretion. In this study we show that A-II and a calcium ionophore down-regulate the expression of KCNJ5 mRNA and protein. Activation of Kir3.4 by naringin inhibits A-II-stimulated membrane voltage and aldosterone secretion. Overexpression of KCNJ5 in the HAC15 cells using a lentivirus resulted in a decrease in membrane voltage, intracellular calcium, expression of steroidogenic acute regulatory protein, 3-β-hydroxysteroid dehydrogenase 3B2, cytochrome P450 11B1 and cytochrome P450 11B2 mRNA, and aldosterone synthesis. In conclusion, A-II appears to stimulate aldosterone secretion by depolarizing the membrane acting in part through the regulation of the expression and activity of Kir3.4.
Aldosterone secretion by the zona glomerulosa of the adrenal is regulated primarily by the renin-angiotensin system. Excessive or inappropriate aldosterone secretion results in hypertension and greater cardiac, vascular, and renal damage in patients with primary aldosteronism than those with essential hypertension of similar duration and severity (1, 2). Angiotensin II (A-II) stimulates aldosterone production by activating the Ca2+/CaMK (calmodulin kinase), MAPK, and cAMP cascade (3–5).
Primary aldosteronism, defined as the autonomous and excessive secretion of aldosterone, is most often due to aldosterone-producing adenomas (APA) or idiopathic hyperaldosteronism (1). Recent studies show that approximately 30–60% of patients with APA have somatic mutations of the KCNJ5 gene coding for an inwardly rectifying potassium channel (Kir3.4), and germline mutations in the KCNJ5 gene have been detected in some families with familial hyperaldosteronism type 3 (6–11). The KCNJ5 somatic mutations G151R, L168R, and T158A and deletion of amino acid 157 have been found in APA; T158A, G151E, and G151R germline mutations have been found in familial hyperaldosteronism type 3 families (6, 7, 11). Patients with the T158A and G151R germinal mutations have severe hyperaldosteronism and massive bilateral adrenal cortical hyperplasia with transitional zone characteristics (6, 11, 12), whereas germinal mutation G151E is associated with a mild phenotype (7, 11). We reported that the KCNJ5 T158A mutation increases aldosterone production by depolarizing the plasma membrane, thereby activating voltage-gated Ca2+ channels and Ca/calmodulin signaling in cultured human adrenocortical cells (13). Whereas we tend to focus on aberrant production, the rate of normal aldosterone synthesis must vary greatly depending on physiological and environmental conditions to maintain homeostasis. The fundamental role of Kir3.4 in regulating normal aldosterone production in adrenal zona glomerulosa cells has not been studied.
Inwardly rectifying potassium channels (Kir) transport potassium ions into and out of cells, and play a key role in a cell's ability to generate and transmit electrical signals (14, 15). In general, Kir channels tend to hyperpolarize the membrane potential in excitable cells such as neurons and cardiac myocytes, whereas they carry outward current in nonexcitable cells such as those in the anterior pituitary gland (14–16). The G protein-coupled inwardly rectifying potassium channel (known as “Kir3” or “GIRK”), is one of the seven Kir channel subfamilies denoted as “Kir1” to “Kir7.” Kir3 is gated by ligand-stimulated G protein-coupled receptors and activated by a large number of neurotransmitters, including acetylcholine, adenosine, ATP, dopamine, serotonin, and somatostatin,. The Kir3 gene family has four members including Kir3.1 to Kir3.4, with a wide tissue distribution, including the heart, neurons, neurosecretory cells, pancreas, pituitary gland (14–16), and adrenal gland. Their functions vary across cell types and have not been studied in the adrenal cortex.
We hypothesized that Kir3.4 plays a role in the stimulation of aldosterone production by A-II, a ligand for G protein-coupled receptors. To address this hypothesis, we investigated the effects of A-II, a Ca2+ ionophore and naringin, a Kir3 channel activator, on KCNJ5 expression and aldosterone production. We also overexpressed and knocked down KCNJ5 using lentivirus vectors in the HAC15 human adrenocortical cell line to determine the effect on the regulation of aldosterone synthesis.
Materials and Methods
Cell culture and materials
The HAC15 human adrenocortical carcinoma cell line, a subclone of the H295R, a human adrenocortical carcinoma cell (17, 18), was provided by Dr. William Rainey and cultured as described elsewhere (13). A-II, the calcium ionophore A23187, and the direct activator of Kir3 channel Naringin (19) were purchased from Sigma Aldrich Co. Ltd. (St. Louis, MO). The dye to detect membrane voltage, DiSBAC2(3) was purchased from AnaSpec (Fremont, CA). To detect intracellular Ca2+ concentration, Fluo-4 AM, was purchased from Invitrogen (Carlsbad, CA).
Plasmids
The full-length cDNA of KCNJ5 (pCR4-TOPO) was purchased from Open Biosystems (Huntsville, AL) and inserted into pENTR/d-TOPO for subsequent use in ligase reaction with the Gateway-compatible lentivector pLX303 plasmid (Addgene plasmid 25897; Addgene, Cambridge MA) (20) by the ligase reaction method (Invitrogen), resulting in pLX303-KCNJ5. The control plasmid employed was pLX303 without the KCNJ5 insert. Two lentiviral plasmids for short hairpin RNA (shRNA) of KCNJ5 (pGIPZ-shRNA-KCNJ5; a, no. V3LHS_310145 and b, no. V3LHS_310147) and pGIPZ empty plasmid were purchased from Open Biosystems. Construction of the pBM14-CYP11B2 promoter/gaussia to study promoter activity using a reporter gene was as previously described (13).
Lentiviral production and infection
Lentivirus production was performed in the 293TN cell line (System Biosciences, Mountain View, CA) using a lentiviral transfer vector, psPAX2, and pCMV-VSV-G, as previously described (13). Lentivirus transduction was performed 24 h after seeding HAC15, as we previously described (13, 21). To assess steroid production, mRNA expression, immunoblotting, membrane voltage, and intracellular Ca2+ concentration, 72 h after transduction the cells were serum deprived using DMEM-F12 supplemented with 0.1% cosmic calf serum for 24 h. The medium was replaced with fresh DMEM-F12 containing 0.1% cosmic calf serum with or without 10 nm A-II. After incubation for the indicated time, the supernatants were collected for steroid measurements, and cells were harvested for analysis of mRNA and protein.
Luciferase assay
We performed the gaussia luciferase assay to assess the CYP11B2 activity, as we previously reported (13). In brief, the cells stably transduced with pBM14-CYP11B2 promoter/gaussia luciferase were grown in 48-well plates and infected with pLX303 control or pLX KCNJ5 lentivirus. The supernatants were collected and placed in 96-well plates for luminescence detection using the gaussia luciferase substrate coelenterazine (Gold Biotechnology, St. Louis, MO), and read with a BMG FLUOstar Omega Reader (BMG Labtech, Ortenberg, Germany) (22).
RNA extraction and RT-PCR
Total RNA extraction, RT PCR, and the detection of CYP11B2, CYP11B1, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA expression by the Taqman Gene expression assay were performed as previously reported (13). Expression of mRNA for steroidogenic acute regulatory protein (STAR), cytochrome P450, family 11, subfamily A, polypeptide 1 (CYP11A1), 3β-hydroxysteroid dehydrogenase (HSD3B2), cytochrome P450, family 21, subfamily A, polypeptide 2 (CYP21A2), cytochrome P450, family 17, subfamily A, polypeptide 1 (CYP17A1), and GAPDH were detected using SYBR Green with PCR primers as previously reported (13). mRNA expression levels of KCNJ5 were also determined using SYBR Green using the real-time PCR primers: KCNJ5-forward, TGTGGTCATTCTAGAAGGGATGG; KCNJ5-reverse, TGAGGACTGGTGTGAATCGG. Real-time data were obtained during the extension phase, and critical threshold cycle values were calculated at the log phase of each gene amplification curve. Gene expression levels were analyzed as arbitrary units normalized against GAPDH mRNA expression. GAPDH expression in the experiments was unchanged by the different procedures employed.
Analysis of plasma membrane voltage and intracellular Ca2+ concentration
After serum starving as described above, the medium was replaced with HEPES buffer with or without 10 nm A-II or 100 μm Naringin. After incubation for the indicated times, 3 μm DiSBAC2(3) for membrane voltage or 3 μm Fluo-4 AM for intracellular Ca2+ concentration were added, the cells were washed three times with HEPES buffer, and fluorescence was detected with a BMG FLUOstar Omega Reader.
SDS-PAGE and immunoblotting analysis
Cells were lysed with RIPA buffer (50 mm Tris, 150 mm NaCl, 0.1% sodium dodecyl sulfate, 2 mm EDTA, 1% Triton X100, 1% sodium deoxycholate) supplemented with protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA). The cell lysates were centrifuged and the supernatant was mixed with Laemmli buffer (125 mm Tris, 4% sodium dodecyl sulfate, 20% glycerol, 10% 2-mercaptoethanol, and 0.01% bromophenol blue). Equal aliquots of cell lysates were subjected on 12.5% SDS-PAGE and transferred to polyvinylidene difluoride membranes using a wet technique. Immunoblotting analyses using anti-Kir3.4 (catalog no. AB9808; Millipore Corp., Billerica, MA) and anti-β-tubulin (catalog no. 6G7; Developmental Studies Monoclonal Bank, The University of Iowa, Iowa City, IA) were performed. Blots were developed with Super Signal West Pico Chemiluminescent substrate (Pierce Chemical Co., Rockford, IL) and exposed to autoradiography film. Films were scanned and quantified with a Kodak Image Station 440 using the one-dimensional (1D) Kodak image analysis software. Protein levels were analyzed as arbitrary units normalized against tubulin expression.
Steroid and protein assays
Aldosterone and cortisol levels were measured in cell culture supernatants by time-resolved fluorescence using the assay and primary antibodies developed in our laboratory as previously described (13, 23).
Statistical analysis
All results were expressed as mean ± se of at least three separate experiments in which each sample was assayed in triplicate or quadruplicate. Differences between two groups were analyzed for statistical significance by t test and multiple groups were analyzed by one-way ANOVA followed by Bonferroni comparisons. Time-response effects were tested by two-way ANOVA. The differences were considered to be significant at P < 0.05. Analyses were performed using SPSS for Windows (release 12.0; SPSS Inc., Chicago, IL).
Results
The effects of A-II on KCNJ5 expression
Stimulation with 10 nm A-II significantly suppressed KCNJ5 mRNA by 41.2% (Fig. 1A) and KCNJ5 protein (Kir3.4) by 52.7% compared with baseline (Fig. 1, B and 1C).
Fig. 1.
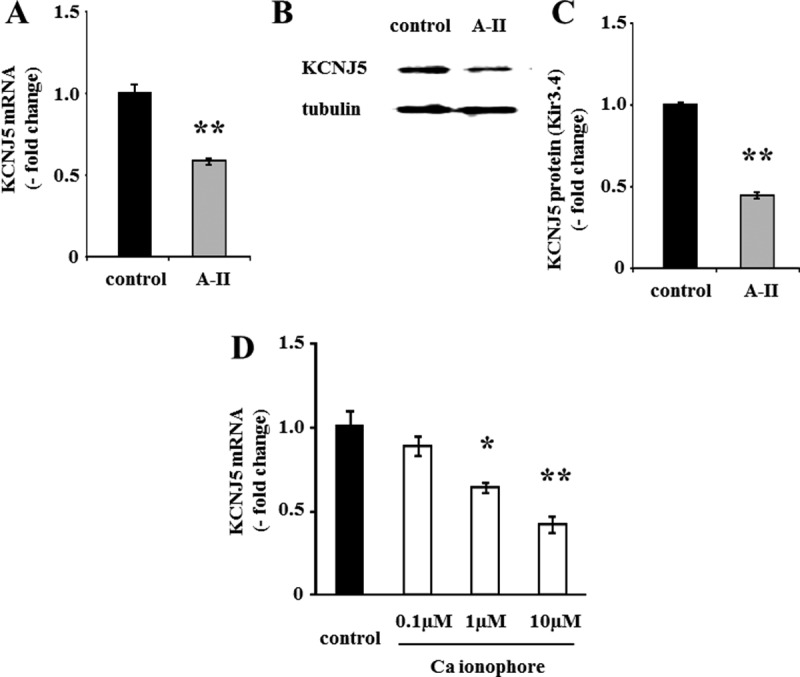
Effect of A-II and calcium ionophore on mRNA and protein expression of KCNJ5. A and D, Confluent cells were serum deprived in DMEM-F12 containing 0.1% cosmic calf serum for 24 h and then incubated with fresh media with 0.1% serum and no secretagogue, A-II (10 nm), or calcium ionophore A23187 for 3 h. After aspiration of media, the cells were collected for RNA extraction and real-time RT-PCR was performed. Results were normalized by GAPDH mRNA expression and expressed as fold change vs. control. B and C, Cells were serum deprived 72 h after infection in DMEM-F12 containing 0.1% cosmic calf serum for 24 h and then incubated with fresh media with 0.1% serum and no secretagogue or A-II (10 nm) for 8 h. After aspiration of media, the cells were harvested with protease inhibitor. The cell lysates were subjected to immunoblotting analysis using anti-KCNJ5 or antitubulin antibodies. The KCNJ5 band intensity of scanned images was adjusted by that of tubulin and expressed as fold change vs. control. *, P < 0.05 vs. control, n = 3; **, P < 0.01 vs. control, n = 3.
To examine the mechanism of A-II mediated KCNJ5 regulation, HAC15 cells were incubated with the calcium ionophore A23187. A23187 suppressed KCNJ5 mRNA levels in a concentration-dependent manner (Fig. 1D), suggesting that increased Ca2+ signaling down-regulates KCNJ5 expression.
Effects of naringin on membrane voltage, steroidogenesis factors, and aldosterone levels
We next investigated the roles of Kir3.4 in A-II-mediated aldosterone production by activating the Kir3.4 channel using naringin. Naringin is reported to directly activate Kir3 channels by binding tyrosines 148 and 150 of the receptor (19). As shown in Fig. 2A, naringin partially and significantly abrogated the A-II-mediated increase in membrane voltage (A-II without naringin, 1.43 ± 0.03; A-II with naringin, 1.17 ± 0.05; P = 0.016). In addition, the increase of CYP11B2 expression (A-II without naringin, 5.7 ± 0.4; A-II with naringin, 4.4 ± 0.2; P = 0.037) and aldosterone synthesis (A-II without naringin, 492.0 ± 17.5; A-II with naringin, 428.5 ± 19.1; P = 0.032) in response to A-II were also partly, but significantly, inhibited by naringin (Fig. 2, B and C). These results suggested that A-II-induced aldosterone production is mediated in part by Kir3.4.
Fig. 2.
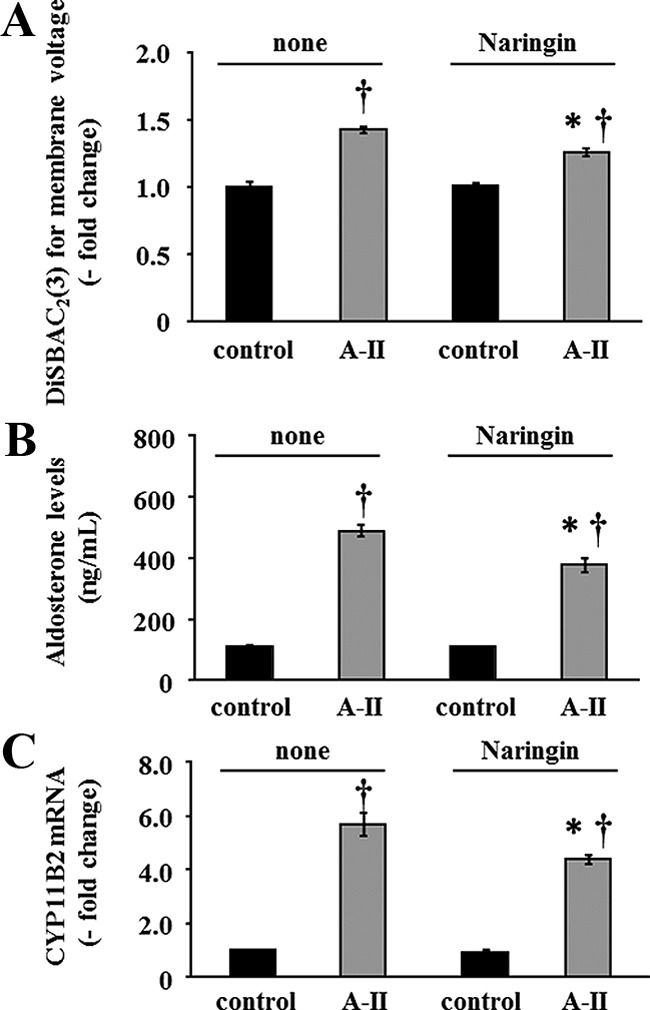
Effect of naringin on membrane voltage, CYP11B2 mRNA expression, and aldosterone production. Confluent cells were serum deprived in DMEM-F12 containing 0.1% cosmic calf serum for 24 h. Pretreatments with or without naringin for 30 min were performed for each experiment before adding secretagogue. A, HAC15 cells were incubated in HEPES buffer with 3 μm DiSBAC2(3) and no secretagogue or A-II (10 nm) for 5 min. Fluorescence was detected by a plate reader. Results were expressed as fold change vs. control cells. *, P < 0.05 comparison between with and without naringin after A-II stimulation, n = 4. †, P < 0.01 comparison with the respective control, n = 4. B, Cells were incubated with no secretagogue or with A-II (10 nm) and without and with naringin for 6 h, and aldosterone was measured in the media. *, P < 0.05 comparison between with and without naringin after A-II stimulation, n = 4. †, P < 0.01 comparison with the respective control, n = 4. C, After 3 h incubation with no secretagogue or A-II (10 nm) and without and with naringin, cells were harvested for RNA extraction, and real-time RT-PCR was performed. Results were normalized by GAPDH mRNA expression and expressed as fold change vs. control. *, P < 0.05 comparison between with and without naringin after A-II stimulation, n = 4. †, P < 0.01 comparison with the respective control, n = 4.
Regulation of membrane voltage and intracellular Ca2+ concentration by the modulation of KCNJ5 in HAC15 cells
Because A-II repressed the expression of Kir3.4, we investigated the effects of altering KCNJ5 expression on membrane voltage and intracellular Ca2+ concentration in HAC15 cells. Infection of HAC15 cells with two different lentiviruses delivering shRNA-KCNJ5 (line a and b) decreased KCNJ5 mRNA expression by 41.4% and 43.3%, respectively (Fig. 3A). As shown in Fig. 3, B and C, the protein levels of KCNJ5 also were decreased 60.9% (line a) and 70.7% (line b) by knockdown of KCNJ5 (KD-KCNJ5). Alteration of KCNJ5 expression would be expected to change membrane voltage and thereby increase or decrease intracellular Ca2+ concentration in HAC15 cells. We infected HAC15 cells with empty viral vectors as controls or with one of two different shRNA-KCNJ5 lentiviruses. Under basal conditions the KD-KCNJ5 cells transduced with either shRNA-KCNJ5 lentivirus showed no difference in membrane voltage or intracellular calcium concentration using DiSBAC2(3) and Fluo-4 AM from control cells before and after A-II stimulation (data not shown). We then investigated the relationship between overexpression of KCNJ5 (Supplemental Fig. 1 published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org) and membrane voltage and intracellular Ca2+ concentration, as detected by DiSBAC2(3) and Fluo-4 AM. The membrane voltages in HAC15 KCNJ5 cells were significantly lower than those in control cells before and after A-II stimulation (Fig. 4A), and KCNJ5 cells had lower intracellular Ca2+ concentration than control cells (Fig. 4B).
Fig. 3.
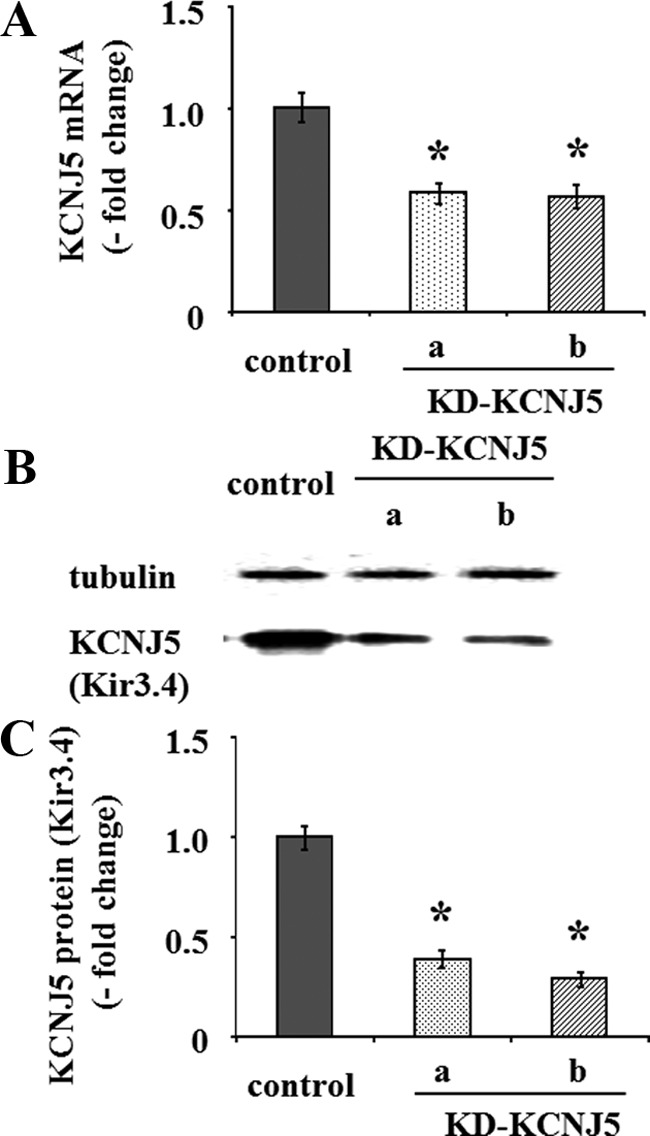
Effect of shRNA-KCNJ5 on mRNA and protein expression of KCNJ5. Cells were serum deprived 72 h after transduction with control or either shRNA-KCNJ5 lentiviruses as in Fig 1. A, After aspiration of media, the cells were collected for RNA as in Fig 1. B and C, After aspiration of media, the cells were harvested with protease inhibitor. The cell lysates were subjected to immunoblotting analysis using anti-KCNJ5 or antitubulin antibodies. The KCNJ5 band intensity of scanned images was adjusted by that of tubulin and expressed as fold change vs. control. *, P < 0.01 vs. control, n = 3.
Fig. 4.
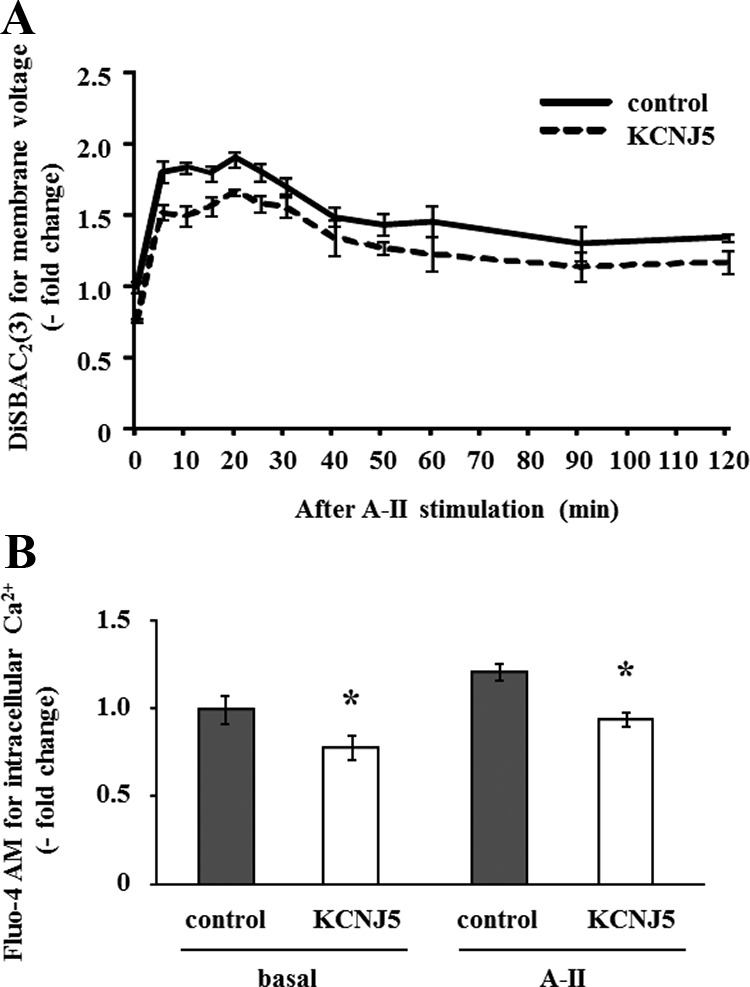
Effect of the KCNJ5 overexpression on membrane voltage and intracellular Ca2+ concentration in HAC15 cells transduced with control or KCNJ5 lentiviruses. Cells were serum deprived 72 h after infection in DMEM-F12 containing 0.1% cosmic calf serum for 24 h, and then incubated in HEPES buffer with 10 nm A-II and 3 μm DiSBAC2(3) for the indicated time (A) or 3 μm Fluo-4 AM for 10 min (B). Fluorescence was detected by a plate reader. Results were expressed as fold change vs. control cells. *, P < 0.05 vs. control, n = 4.
The effect of KCNJ5 suppression and overexpression on aldosterone production
To assess aldosterone production induced by KCNJ5 modulation, we measured aldosterone concentration in the medium in HAC15 cells transduced with control, shRNA-KCNJ5, or KCNJ5 lentiviral plasmid. Decreasing KCNJ5 expression had no effect on aldosterone (Fig. 5A) and cortisol production (data not shown). In contrast, KCNJ5 overexpression significantly repressed basal and A-II-induced aldosterone (Fig. 5B, basal, P = 0.018; and A-II, P < 0.01) and cortisol levels (Fig. 5C; basal, P < 0.01; and A-II, P < 0.01).
Fig. 5.

Basal and A-II-stimulated aldosterone and cortisol production by HAC15 transduced with control, shRNA-KCNJ5 (A), or KCNJ5 lentiviruses (B, C). Cells were serum deprived 72 h after infection in DMEM-F12 containing 0.1% cosmic calf serum for 24 h after which cells were incubated with no secretagogue or A-II (10 nm) for 24 h. *, P < 0.05 vs. control, n = 4, **, P < 0.01 vs. control, n = 4.
mRNA expression and reporter assay of steroid biosynthetic enzymes in cells overexpressing KCNJ5
Basal expression of StAR, HSD3B2, CYP11B1, and CYP11B2 mRNA in HAC15 cells overexpressing KCNJ5 was significantly suppressed by 51.0% (P < 0.001), 53.2% (P = 0.005), 65.1% (<0.001), and 30.2% (P = 0.018) in comparison with control cells at baseline, respectively (Fig. 6). In addition, KCNJ5+ cells had lower mRNA expression of StAR, HSD3B2, CYP11B1, and CYP11B2 by 27.3% (P = 0.036), 45.1% (P = 0.097), 44.1% (P = 0.005), and 54.4% (P < 0.001) compared with control cells after A-II stimulation, respectively (Fig. 6). No differences of other steroidogenic enzymes were observed between control and KCNJ5+ cells (Fig. 6) at baseline or after A-II stimulation.
Fig. 6.

Effect of the KCNJ5 overexpression on mRNA expression of the key proteins for adrenal steroid synthesis such as StAR, CYP11A1, HSD3B2, CYP21A2, CYP17A1, CYP11B1, and CYP11B2. Cells were serum deprived 72 h after infection in DMEM-F12 containing 0.1% cosmic calf serum for 24 h and then incubated with fresh media with 0.1% serum for 3 h. After aspiration of media, the cells were collected for RNA extraction and real-time RT-PCR was performed. Results were normalized by GAPDH mRNA expression and expressed as fold change vs. control. *, P < 0.05 vs. control, n = 3, **, P < 0.01 vs. control, n = 3.
We investigated the transcriptional regulation of CYP11B2 analyzed by luciferase assays using HAC15 cells stably infected with each reporter construct. As shown in Fig. 7, basal CYP11B2 transcriptional activity in KCNJ5+ and control HAC15 cells was not significantly different; however, A-II stimulated CYP11B2 transcriptional activity was 37.2% in the KCNJ5 overexpressing cells compared with that in control cells.
Fig. 7.
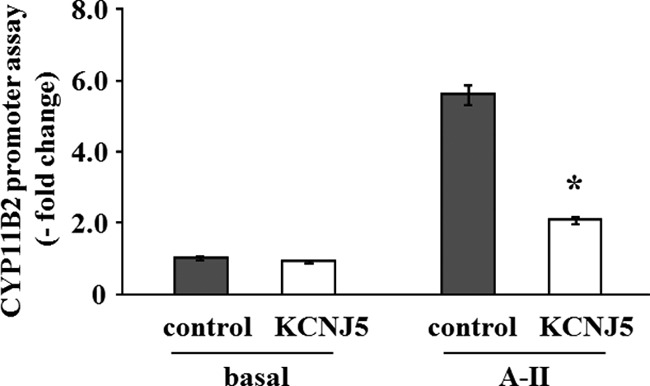
Effect of the KCNJ5 overexpression on CYP11B2 reporter gene expression. HAC15 cells stably infected with pBM14-CYP11B2 promoter/gaussia were infected with control or KCNJ5 lentiviruses. After 72 h incubation, cells were serum deprived in DMEM-F12 containing 0.1% cosmic calf serum for 24 h and incubated in fresh media with 0.1% serum for 24 h, and supernatants were collected to assess luciferase activity. Results are shown as fold change vs. control cells. *, P < 0.01 vs. control, n = 3.
Discussion
The Kir3.4 channel, product of the KCNJ5 gene, regulates potassium flow and the membrane voltage and is gated by G protein-coupled receptors (14–16). Our results indicate that A-II stimulates aldosterone production in part through Kir3.4. A-II decreased the expression of Kir3.4 at the mRNA and protein level. Overexpression of KCNJ5 decreased plasma membrane voltage and intracellular Ca2+ concentration, mRNA expression of steroidogenic enzymes, and aldosterone production in HAC15 cells under basal conditions and with A-II stimulation. The Kir3.4-selective activator naringin did not affect basal membrane voltage but inhibited depolarization by A-II. These results suggest that Kir3.4 channels transfer potassium from inside to out of HAC15 cells continuously, a function that A-II interrupts, thereby depolarizing the membrane, as summarized in Fig. 8. Overexpression of Kir3.4 and naringin treatment suppressed the response of the HAC15 cells to A-II, consistent with a report that activation of Kir3.4 by dopamine led to hyperpolarization of the lactotrope membrane and inhibition of prolactin secretion (24).
Fig. 8.

A possible mechanism by which A-II regulates aldosterone production via Kir3.4. A-II stimulates aldosterone synthesis (CYP11B2) by increasing intracellular Ca2+ concentration through inhibition of Kir3.4 receptor and activation of voltage-gated Ca2+ channel. The increase of intracellular Ca2+ suppresses Kir3.4 expression.
Knocking down KCNJ5 using the shRNA did not alter basal aldosterone synthesis in HAC15 cells, and although the results are surprising, we can only suggest that there must be redundant mechanisms maintaining proper membrane polarization. Kir3 channels consist of homo- or heterotetramer with KCNJ5, KCNJ3, KCNJ6, and KCNJ9 products (14–16); other Kir3 channels may increase and compensate for the loss of Kir3.4 in the KD-KCNJ5 cells, maintaining basal membrane voltage and the sequential cascade for aldosterone production. In addition, TASK1 and BK channels, two other potassium channels have roles in the regulation of membrane voltage and aldosterone production in adrenocortical cells (25, 26) and may help maintain normal membrane voltage in the KD-KCNJ5 cells. Our finding that activation of the channel by naringin results in only a slight suppression of membrane voltage and aldosterone secretion in response to A-II suggests the participation of other potassium channels in the regulation of membrane voltage and aldosterone production.
In our study, CYP11B2 mRNA expression was suppressed by KCNJ5 overexpression both at baseline and after A-II stimulation. Overexpression is expected to result in membrane hyperpolarization, decreased intracellular calcium mobilization, and reduced Ca2+/CaMK signal cascade, resulting in less transcription of the CYP11B2 gene. Inhibitors of Ca2+/CaMK signaling similarly decrease the expression of CYP11B2 induced by A-II (3, 27). These data are concordant with our recent report that transduction of the mutant KCNJ5 T158A in HAC15 adrenocortical cells increased membrane depolarization and Ca2+ signaling, and enhanced CYP11B2 expression (13). The regulation of CYP11B2 mRNA expression appears to be dependent upon the Ca2+ cascade.
The HAC15 and its H295 predecessor adrenocarcinoma cell lines behave as pluripotential adrenal cells with zona glomerulosa, zona fasciculate, and zona reticularis characteristics. In the HAC15 subclone, the zona fasciculata enzyme 17α-hydroxylase is expressed and the steroid, cortisol, is synthesized and regulated as if they were from the zona glomerulosa. The transcription factors involved in defining the zones of the adrenal cortex remain unknown and are of great interest.
In conclusion, we demonstrated one of the mechanisms of A-II-mediated aldosterone production is by regulation of Kir3.4. This inwardly rectifying potassium channel appears to transfer potassium from inside to outside of the human adrenocortical cells to continuously regulate membrane voltage. Suppression of Kir3.4 by A-II results in the depolarization of the plasma membrane, an increase in intracellular Ca2+ concentration, and enhanced expression of steroidogenic factors.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants HL27255 and HL105383, and by Award 1018X007080 from the Biomedical Laboratory Research & Development Service of the Veterans Affairs Office of Research and Development (to E.G.S.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- A-II
- Angiotensin II
- APA
- aldosterone-producing adenomas
- CaMK
- calmodulin kinase
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- HSD3B2
- 3β-hydroxysteroid dehydrogenase
- Kir
- inwardly rectifying potassium channel
- shRNA
- short hairpin RNA
- STAR
- steroidogenic acute regulatory protein.
References
- 1. Funder JW, Carey RM, Fardella C, Gomez-Sanchez CE, Mantero F, Stowasser M, Young WF, Jr, Montori VM. 2008. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 93:3266–3281 [DOI] [PubMed] [Google Scholar]
- 2. Milliez P, Girerd X, Plouin PF, Blacher J, Safar ME, Mourad JJ. 2005. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol 45:1243–1248 [DOI] [PubMed] [Google Scholar]
- 3. Condon JC, Pezzi V, Drummond BM, Yin S, Rainey WE. 2002. Calmodulin-dependent kinase I regulates adrenal cell expression of aldosterone synthase. Endocrinology 143:3651–3657 [DOI] [PubMed] [Google Scholar]
- 4. Otis M, Gallo-Payet N. 2006. Differential involvement of cytoskeleton and rho-guanosine 5′-triphosphatases in growth-promoting effects of angiotensin II in rat adrenal glomerulosa cells. Endocrinology 147:5460–5469 [DOI] [PubMed] [Google Scholar]
- 5. Nogueira EF, Rainey WE. 2010. Regulation of aldosterone synthase by activator transcription factor/cAMP response element-binding protein family members. Endocrinology 151:1060–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Choi M, Scholl UI, Yue P, Björklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ, Lolis E, Wisgerhof MV, Geller DS, Mane S, Hellman P, Westin G, Åkerström G, Wang W, Carling T, Lifton RP. 2011. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 331:768–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mulatero P, Tauber P, Zennaro MC, Monticone S, Lang K, Beuschlein F, Fischer E, Tizzani D, Pallauf A, Viola A, Amar L, Williams TA, Strom TM, Graf E, Bandulik S, Penton D, Plouin PF, Warth R, Allolio B, Jeunemaitre X, Veglio F, Reincke M. 2012. KCNJ5 Mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension 59:235–240 [DOI] [PubMed] [Google Scholar]
- 8. Azizan EA, Murthy M, Stowasser M, Gordon R, Kowalski B, Xu S, Brown MJ, O'Shaughnessy KM. 2012. Somatic mutations affecting the selectivity filter of KCNJ5 are frequent in 2 large unselected collections of adrenal aldosteronomas. Hypertension 59:587–591 [DOI] [PubMed] [Google Scholar]
- 9. Boulkroun S, Beuschlein F, Rossi GP, Golib-Dzib JF, Fischer E, Amar L, Mulatero P, Samson-Couterie B, Hahner S, Quinkler M, Fallo F, Letizia C, Allolio B, Ceolotto G, Cicala MV, Lang K, Lefebvre H, Lenzini L, Maniero C, Monticone S, Perrocheau M, Pilon C, Plouin PF, Rayes N, Seccia TM, et al. 2012. Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension 59:592–598 [DOI] [PubMed] [Google Scholar]
- 10. Taguchi R, Yamada M, Nakajima Y, Satoh T, Hashimoto K, Shibusawa N, Ozawa A, Okada S, Rokutanda N, Takata D, Koibuchi Y, Horiguchi J, Oyama T, Takeyoshi I, Mori M. 2012. Expression and mutations of KCNJ5 mRNA in Japanese patients with aldosterone-producing adenomas. J Clin Endocrinol Metab 97:1311–1319 [DOI] [PubMed] [Google Scholar]
- 11. Scholla UI, Nelson-Williamsa C, Yuec P, Grekind R, Wyatt RJ, Dillon MJ, RC, Hammer LK, Harley FL, Farhia A, Wang W-H, Lifton RP. 2012. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci USA 109:2533–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geller DS, Zhang J, Wisgerhof MV, Shackleton C, Kashgarian M, Lifton RP. 2008. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab 93:3117–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oki K, Plonczynski MW, Luis Lam M, Gomez-Sanchez EP, Gomez-Sanchez CE. 2012. Potassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesis. Endocrinology 153:1774–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. 2010. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90:291–366 [DOI] [PubMed] [Google Scholar]
- 15. Lüscher C, Slesinger PA. 2010. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci 11:301–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morishige K, Inanobe A, Yoshimoto Y, Kurachi H, Murata Y, Tokunaga Y, Maeda T, Maruyama Y, Kurachi Y. 1999. Secretagogue-induced exocytosis recruits G protein-gated K+ channels to plasma membrane in endocrine cells. J Biol Chem 274:7969–7974 [DOI] [PubMed] [Google Scholar]
- 17. Parmar J, Key RE, Rainey WE. 2008. Development of an adrenocorticotropin-responsive human adrenocortical carcinoma cell line. J Clin Endocrinol Metab 93:4542–4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang T, Rainey WE. 2012. Human adrenocortical carcinoma cell lines. Mol Cell Endocrinol 351:58–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yow TT, Pera E, Absalom N, Heblinski M, Johnston GA, Hanrahan JR, Chebib M. 2011. Naringin directly activates inwardly rectifying potassium channels at an overlapping binding site to tertiapin-Q. Br J Pharmacol 163:1017–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, Green TM, Johannessen CM, Silver SJ, Nguyen C, Murray RR, Hieronymus H, Balcha D, Fan C, Lin C, Ghamsari L, Vidal M, Hahn WC, Hill DE, Root DE. 2011. A public genome-scale lentiviral expression library of human ORFs. Nat Methods 8:659–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Romero DG, Zhou MY, Yanes LL, Plonczynski MW, Washington TR, Gomez-Sanchez CE, Gomez-Sanchez EP. 2007. Regulators of G-protein signaling 4 in adrenal gland: localization, regulation, and role in aldosterone secretion. J Endocrinol 194:429–440 [DOI] [PubMed] [Google Scholar]
- 22. Tannous BA. 2009. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat Protoc 4:582–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gomez-Sanchez CE, Foecking MF, Ferris MW, Chavarri MR, Uribe L, Gomez-Sanchez EP. 1987. The production of monoclonal antibodies against aldosterone. Steroids 49:581–587 [DOI] [PubMed] [Google Scholar]
- 24. Gregerson KA, Flagg TP, O'Neill TJ, Anderson M, Lauring O, Horel JS, Welling PA. 2001. Identification of G protein-coupled, inward rectifier potassium channel gene products from the rat anterior pituitary gland. Endocrinology 142:2820–2832 [DOI] [PubMed] [Google Scholar]
- 25. Nogueira EF, Gerry D, Mantero F, Mariniello B, Rainey WE. 2010. The role of TASK1 in aldosterone production and its expression in normal adrenal and aldosterone-producing adenomas. Clin Endocrinol (Oxf) 73:22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grimm PR, Irsik DL, Settles DC, Holtzclaw JD, Sansom SC. 2009. Hypertension of Kcnmb1−/− is linked to deficient K secretion and aldosteronism. Proc Natl Acad Sci USA 106:11800–11805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pezzi V, Clyne CD, Ando S, Mathis JM, Rainey WE. 1997. Ca2+-regulated expression of aldosterone synthase is mediated by calmodulin and calmodulin-dependent protein kinases. Endocrinology 138:835–838 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.