

Membrane protein structure and function, especially for small membrane proteins, can be highly sensitive to the membrane mimetic environment used for structural characterization, as exemplified by the M2 protein from influenza A virus that has been characterized in liquid crystalline lipid bilayers, detergent micelles and in detergent based crystals.[3–8] Various transmembrane (TM) helical tilt angles, different drug binding sites and amphipathic helix interactions, as well as a lack of consensus on the sidechain geometry for the functionally critical residues is apparent from this set of structures. Many of these structural differences can be explained based on the influence of the protein's environment. Hydrophobic thickness influences the helical tilt; detergent penetration into the helical bundle and crystal contacts influence the packing and hence tilt of the helices, while the highly curved surface of micelles destabilize the interactions of amphipathic helices with what would be the bilayer interface.[9] These structural perturbations can influence functional properties such as the binding of the antiviral drug to the protein and our understanding of the proton channel functional mechanism. Exactly how well the native membrane needs to be modeled to achieve a native membrane protein structure is explored here, where we aim to validate the structure of the tetrameric M2 conductance domain (M2CD; residues 22–62; PDB #2L0J) that has been structurally characterized in synthetic lipid bilayers. We have set out to do this by observing the full length protein in synthetic bilayers, as well as in native E. coli membranes. For the first time we report on structural insights from the full length M2 (M2FL) protein using magic angle spinning solid state NMR (ssNMR) and we present spectra of the protein as it is inserted into the E. coli membranes by the cellular apparatus without ever being exposed to a detergent environment. These results validate the earlier structural results obtained from the M2CD observed in a liquid crystalline bilayer envionment.
In addition to its proton channel activity, M2 facilitates viral budding that localizes the protein to this site on the surface of an infected cell suggesting that M2 is not imbedded in the high cholesterol and sphingomyelin environment that dominates the viral particle.[10, 11] Instead, M2 appears to be preferentially localized to the periphery of the raft-like lipid domains from which the viral particle buds and where the amphipathic helices of the M2 protein can induce membrane curvature. For this reason and because it has recently been shown that raft like lipids do not support an M2 conformation compatible with amantadine (the antiviral drug that targets M2) binding[12] nor do these lipids support a conformation having pKas for the His37 tetramer (the critical residues for proton transit) consistent with high proton affinity,[13] consequently, we have used liquid crystalline lipid environments for M2 that support both of these functional properties.[14] The synthetic lipid bilayer we have used is the same as that used for the structural study of M2CD,[7] and the second is another liquid crystalline domain, that of the E. coli plasma membrane, an environment that models the lipid and protein complexity of the environment surrounding raft-like domains. Previously, several solid state NMR in situ studies of other membrane proteins in cellular membranes have been reported.[15–20] Here, we use an in situ preparation to validate a structure of a proven drug target for Influenza A. Such validation is necessitated by the sensitivity of this protein's structure to its membrane mimetic environment used for structural studies.
The M2 proton channel is a tetramer of a 97 residue protein having a single (TM) helix[21] including the H37xxxW41 signature sequence for a proton channel.[22] The functional mechanism involves shuttling protons across the central energy barrier facilitated by these four His37 residues, while the Val27 and Trp41 tetramers act as gates at the entrance and exit of the pore, respectively.[6, 7, 23–26] 13C-13C correlation spectra of uniformly [13C, 15N]-valine (Fig. 1A,B,E) and uniformly [13C, 15N]-alanine (Fig. 1C,D,F) labeled M2FL in reconstituted liposomes and in E. coli membranes are very similar in these two environments. The liposome samples represent protein that has been isolated and purified using detergents and then reconstituted into lipid bilayers (SI). The in situ samples were prepared by simply isolating the E. coli cellular membranes from an isotopically labeled culture expressing M2FL (SI).
Figure 1.

Comparison of 13C-13C correlation spectra using 50 ms mixing time at 243K of uniformly [13C,15N]-valine (A,B,E,) and uniformly [13C,15N]-alanine (C,D,F,) labeled M2FL in reconstituted liposomes at pH 7.5 (blue) and in E. coli membranes at pH 8.0 (red). (A,D) Cα, Cβ, Cγ1, and Cγ2 correlations with C' for valine and Cα, Cβ correlations with C' for alanine labeled samples. (B,C) Correlations between Cα, Cβ, Cγ1, and Cγ2 for valine and Cα-Cβ correlations for alanine labeled samples. (E) Spectral slices at 67.3 ppm through the valine resonances from M2CD (black), M2FL reconstituted (blue), and M2FL in situ (red) samples. Spectral slices at 55.5 ppm through the alanine resonances from M2CD (black), M2FL reconstituted (blue), and M2FL in situ (red) samples.
There are 7 valine and 5 alanine residues in M2FL: Val−14 and Ala−1 in the N-terminal purification tag, Val7 in the N-terminal domain on the viral exterior, Val27,28 and Ala29,30 in the TM helix, and Val68,84,92 and Ala83,86 in the C-terminal domain on the viral interior. While there is a lack of resolved resonances, there is excellent secondary structure spectral dispersion especially for the C', Cα, and Cβ sites of alanine and valine, but also significant spectral dispersion for the Cγ1 and Cγ2 resonances of valine. The conformation dependent frequencies span characteristic frequencies for helix, coil and sheet motifs. The resonance frequencies observed for the M2CD, Val27,28 Ala29,30 are indicated on the spectra and in Table S1.[27]
In Fig. 1E slices through both full length spectra and the spectrum of UL 15N-13C labeled M2CD at the Cα frequency for the Val27 and Val28 residues in the TM helix are presented. In Fig. 1F similar slices through the alanine spectra and the M2CD spectrum at the Cα frequency for the Ala29 and Ala30 residues in the TM helix are presented. The spectral slices show that the resonance frequencies are nearly identical across all three samples for these sites in the TM helix suggesting that the proton conducting pore has the same or nearly the same structure in all three of these samples. However, the resonance linewidths are somewhat broader for the full length protein samples, especially for the in situ sample even for the TM helix resonances. Outside of the TM helix the resonances as seen in the 2D spectra in both M2FL preparations are substantially broader and hence the resonances are less intense. However, there is a remarkable overlap of the spectral intensity for all of the valine and alanine spectral intensity from these samples suggesting that the similarity in structure may extend beyond the TM helices to the rest of the protein.
13C-13C correlation spectra of uniformly [13C,15N]-labeled M2CD (Fig. 2A,E) and M2FL (Fig. 2C,D) both reconstituted in the same liposome environment as well as uniformly [13C,15N]-labeled M2 (Fig. 2B) in situ show numerous similarities. In the aliphatic-aliphatic regions of M2FL there are broad resonance intensities underlying the narrower resonances. These latter resonances are mostly consistent with the resonances from M2CD, but the former very broad resonances, as seen in Figure 1, are from those regions outside of the conductance domain and hence not present in the spectrum of M2CD. Also as stated previously the resonances of the TM helix appear to be somewhat broader in the M2FL protein than in the M2CD spectra. Even for M2CD the resonance patterns for each of the amino acid types - valine, alanine, leucine, and isoleucine overlap severely making sequence specific assignments very challenging. Furthermore, even if sequence specific assignments were achieved identifying cross peaks between the aliphatic resonances would be most difficult to assign to specific distances without some form of limited labeling strategy. Here, we focus on the comparison of isotropic chemical shift frequencies for the more unique residues in the TM helix (Pro25, Ser31, Gly34, His37, Trp41, Asp44 and Arg45). To this end we have shown in Figure 2 the aliphatic-aromatic regions of the M2CD (Fig. 2E) and M2FL (Fig. 2D) samples in reconstituted liposomes. The resonances of the unique residues as well as those for Val27,28 and Ala29,30 have been uniquely assigned for M2CD.[27] Most of these resonances can also be readily recognized in the M2FL spectra by their resonance pattern and nearly identical frequencies (RMSD for 45 resonances is 0.2 ppm) and the resonance frequencies are given in Table S1.
Figure 2.

13C-13C correlation spectra of uniformly [15N,13C]-labeled M2CD (A,E) and M2FL (C,D) both in reconstituted liposomes, and M2FL (B) in situ. (A,B,C) Aliphatic-aliphatic regions displaying spectra obtained with 10 (red) and 50 (blue) ms mixing times. (D,E) Aliphatic-aromatic regions displaying spectra obtained with 20 (red) and 50 (blue) ms mixing times. Lines drawn between resonances identify similar chemical shifts in the various spectra for which the sequence specific assignments have been previously achieved for M2CD. All spectra were obtained with 13C-optimized 1H/13C/15N 3.2-mm NHMFL biosolids MAS probe utilizing Low-E coil technology.[1, 2]
The M2FL sample reconstituted in liposomes at pH 6.6 exhibits a clear doubling of the His37 and Trp41 Cα-Cβ crosspeaks (Fig. 2C) similar to that for M2CD at pH 7.5 (Fig. 2A).[27, 28] Such doubling reflects the +2 charged state for the histidine tetrad that has two pKas of 8.2 forming imidazole-imidazolium (HisA-HisB) dimers.[14] The M2FL in situ sample at pH 8.0 does not display so clearly the doubling of the His37 and Trp41 Cα-Cβ crosspeaks, probably due to the high pH of this sample (pH 8.0) and that it is in the midst of the titration for these two pKas. As shown in Fig. S1 the spectrum of the M2FL reconstituted in liposomes displays a very similar resonance pattern at high pH.
Surprisingly, these histidines near the middle of the lipid bilayer have an affinity for protons that is nearly two orders of magnitude greater than histidines exposed to a polar aqueous environment. In addition, these charges are close together and would be expected to destabilize the tetrameric structure via charge repulsion. Instead, the formation of two interhelical hydrogen bonds (N2ε - H - Nδ1) between pairs of His37 residues distributes the charge reducing the charge repulsion while stabilizing the tetrameric structure (Fig. 3). Indeed, these hydrogen bonds appear to account for the three orders of magnitude increase in structural stability observed between pH 9 and 6.5.[29] The unique chemistry of the histidine tetrad, actually a dimer of dimers in this charged state, is responsible for shuttling protons across a dehydrated zone in the M2 pore. The +2 charged state observed in the M2CD and M2FL reconstituted liposome samples is known as the histidine locked state, an inactive state. This protein is activated at low pH, presumably by the addition of a third proton to the histidine tetrad leading to proton transport once the Trp41 gate opens.[7] While each of the imidazoles share a charge in the +2 state they do not share the charge equally. A comparison of these resonance frequencies with those of the free amino acid [30]suggests that the shared protons of the imidazole-imidazolium dimers have a preferred location on the Nε2 site creating more charge density on this imidazole than on its partner that is contributing an Nδ1 site for the hydrogen bond. The histidine residues with the highest charge density have lower Cα and Cβ resonance frequencies and higher Cδ2 resonance frequencies than the pair of histidine residues with lower charge density.
Figure 3.
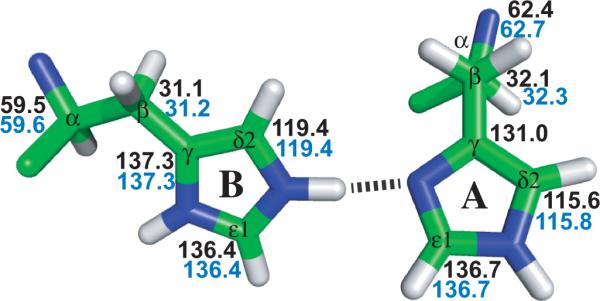
One of the two pair of His37 dimers from the M2CD PDB structure #2L0J, each of which share a charge at pH 6.6 (M2FL sample) and pH 7.5 (M2CD sample). The chemical shifts for these two samples are almost identical: M2FL (blue) and M2CD (black).
The numerous nearly identical chemical shifts for M2FL and M2CD throughout the TM helix, in particular the structurally and chemically sensitive chemical shifts of the His37 tetrad, suggest that the structure of the M2 pore is essentially identical for the M2CD as for the M2FL protein, thereby validating the conductance domain construct as an appropriate construct for assessing the structure of the M2 protein channel. Furthermore, the uniform overlap of the valine and alanine resonances from M2FL in synthetic lipids and in situ in E. coli membranes along with multiple chemical shifts from the TM helix observed in situ in uniform [15N,13C] labeled M2FL confirms that the synthetic bilayers represent an adequate environment for supporting a native-like structure. This is particularly significant since the in situ sample has been inserted into the E. coli membrane by the cellular apparatus and the protein was observed directly in this environment along with the complex mixture of lipids and native E. coli membrane proteins. Therefore, the structure of the M2CD (PDB #2L0J) is validated along with the unique histidine tetrad chemistry responsible for the conductance mechanism.
Supplementary Material
Justification for Angewandte Chemie.
The proton channel/transporter formed by a tetramer of the M2 protein from Influenza A is a that is a proven drug target and for which there has been a renewed and massive effort to develop new drugs - nearly 40 papers on drug development against M2 in the past 2 years. This manuscript represents the first structural restraints on the M2 protein obtained for the protein as it is inserted into the E. coli plasma membrane by the cellular apparatus during protein expression. The multiple structures of M2 protein fragments obtained by crystallography, solution and solid state NMR have been is serious conflict with each other illustrating that the membrane mimetic environment can seriously perturb the protein structure. Here we validate the structure obtained for the M2 proton conductance domain as determined by solid state NMR in liquid crystalline lipid bilayers. In this process we have confirmed the unique histidine chemistry of this terameric protein that is responsible for proton conductance. These results clearly demonstrate the need to characterize the structures of membrane proteins in native-like environments and that a liquid crystalline lipid bilayer appears to be an adequate environment to stabilize the native-like membrane protein structure.
Acknowledgments
This work was supported in part by the National Institutes of Health Grants AI23007 and AI074805. Some of the research was conducted at the National High Magnetic Field Laboratory supported by Cooperative Agreement 0654118 between the Division of Materials Research of the National Science Foundation and the State of Florida.
References
- [1].Gor'kov PL, Chekmenev EY, Li C, Cotten M, Buffy JJ, Traaseth NJ, Veglia G, Brey WW. J Magn Reson. 2007;185:77. doi: 10.1016/j.jmr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- [2].McNeill SA, Gor'kov PL, Shetty K, Brey WW, Long JR. J Magn Reson. 2009;197:135. doi: 10.1016/j.jmr.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schnell JR, Chou JJ. Nature. 2008;451:591. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cross TA, Sharma M, Yi M, Zhou HX. Trends Biochem Sci. 2011;36:117. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pielak RM, Chou JJ. Biochem Biophys Res Commun. 2010;401:58. doi: 10.1016/j.bbrc.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Acharya R, Carnevale V, Fiorin G, Levine BG, Polishchuk AL, Balannik V, Samish I, Lamb RA, Pinto LH, DeGrado WF, Klein ML. Proc Natl Acad Sci U S A. 2010;107:15075. doi: 10.1073/pnas.1007071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou HX, Cross TA. Science. 2010;330:509. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Nature. 2008;451:596. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cross TA, Dong H, Sharma M, Busath DD, Zhou HX. Curr Opin Virol. 2012;2:128. doi: 10.1016/j.coviro.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rossman JS, Jing X, Leser GP, Lamb RA. Cell. 2010;142:902. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rossman JS, Lamb RA. Virology. 2011;411:229. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cady S, Wang T, Hong M. J Am Chem Soc. 2011;133:11572. doi: 10.1021/ja202051n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hu F, Schmidt-Rohr K, Hong M. J Am Chem Soc. 2011;134:3703. doi: 10.1021/ja2081185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hu J, Fu R, Nishimura K, Zhang L, Zhou HX, Busath DD, Vijayvergiya V, Cross TA. Proc Natl Acad Sci U S A. 2006;103:6865. doi: 10.1073/pnas.0601944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fu R, Wang X, Li C, Miranda AN, Pielak GJ, Tian F. J Am Chem Soc. 2011;133:12370. doi: 10.1021/ja204062v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Renault M, Tommassen-van Boxtel R, Bos MP, Post JA, Tommassen J, Baldus M. Proc Natl Acad Sci U S A. 2012;109:4863. doi: 10.1073/pnas.1116478109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jacso T, Franks WT, Rose H, Fink U, Broecker J, Keller S, Oschkinat H, Reif B. Angew Chem Int Ed Engl. 2011;51:432. doi: 10.1002/anie.201104987. [DOI] [PubMed] [Google Scholar]
- [18].Sivertsen AC, Bayro MJ, Belenky M, Griffin RG, Herzfeld J. J Mol Biol. 2009;387:1032. doi: 10.1016/j.jmb.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lewis BA, Harbison GS, Herzfeld J, Griffin RG. Biochemistry. 1985;24:4671. doi: 10.1021/bi00338a029. [DOI] [PubMed] [Google Scholar]
- [20].Kamihira M, Vosegaard T, Mason AJ, Straus SK, Nielsen NC, Watts A. J Struct Biol. 2005;149:7. doi: 10.1016/j.jsb.2004.10.002. [DOI] [PubMed] [Google Scholar]
- [21].Lamb RA, Zebedee SL, Richardson CD. Cell. 1985;40:627. doi: 10.1016/0092-8674(85)90211-9. [DOI] [PubMed] [Google Scholar]
- [22].Venkatamaran P, Lamb RA, Pinto LH. J. Biol. Chem. 2005;280:21463. doi: 10.1074/jbc.M412406200. [DOI] [PubMed] [Google Scholar]
- [23].Tang Y, Zaitseva F, Lamb RA, Pinto LH. J Biol Chem. 2002;277:39880. doi: 10.1074/jbc.M206582200. [DOI] [PubMed] [Google Scholar]
- [24].Yi M, Cross TA, Zhou HX. J Phys Chem B. 2008;112:7977. doi: 10.1021/jp800171m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hu F, Luo W, Hong M. Science. 2010;330:505. doi: 10.1126/science.1191714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhou HX. Biophys J. 2011;100:912. doi: 10.1016/j.bpj.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Can TV, Sharma M, Hung I, Gor'kov P, Brey W, Cross TA. J Am Chem Soc. 2012;134:9022. doi: 10.1021/ja3004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Andreas LB, Eddy MT, Pielak RM, Chou J, Griffin RG. J Am Chem Soc. 2010;132:10958. doi: 10.1021/ja101537p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ma C, Polishchuk AL, Ohigashi Y, Stouffer AL, Schon A, Magavern E, Jing X, Lear JD, Freire E, Lamb RA, DeGrado WF, Pinto LH. Proc Natl Acad Sci U S A. 2009;106:12283. doi: 10.1073/pnas.0905726106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li S, Hong M. J Am Chem Soc. 2011;133:1534. doi: 10.1021/ja108943n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.