

Abstract
Background
Testicular germ cell tumors (TGCTs) are the most frequent malignances in young adult men. The two main histological forms, seminomas and nonseminomas, differ biologically and clinically. pRB protein and its immediate upstream regulator p16INK4a are involved in the RB pathway which is deregulated in most TGCTs. The objective of this study was to evaluate the occurrence of loss of heterozygosity (LOH) of the CDKN2A (p16INK4a) and RB1 tumor suppressor genes in TGCTs.
Materials and methods.
Forty TGCTs (18 seminomas and 22 nonseminomas) were analyzed by polymerase chain reaction using the restriction fragment length polymorphism or the nucleotide repeat polymorphism method.
Results
LOH of the CDKN2A was found in two (6%) out of 34 (85%) informative cases of our total TGCT sample. The observed changes were assigned to two (11%) nonseminomas out of 18 (82%) informative samples. Furthermore, LOH of the RB1 was detected in two (6%) out of 34 (85%) informative cases of our total TGCT sample. Once again, the observed changes were assigned to two (10.5%) nonseminomas out of 19 (86%) informative samples. Both LOHs of the CDKN2A were found in nonseminomas with a yolk sac tumor component, and both LOHs of the RB1 were found in nonseminomas with an embryonal carcinoma component.
Conclusions
The higher incidence of observed LOH in nonseminomas may provide a clue to their invasive behavior.
Keywords: loss of heterozygosity, CDKN2A, RB1, seminomas, nonseminomas
Introduction
Testicular germ cell tumor (TGCT) is diagnosed mainly after puberty and is the most frequent malignancy in young adult men1, however, it is also not rare in childhood.2 The two main histological forms, seminomas and nonseminomas, differ biologically and clinically. About 50% of TGCTs are pure seminomas and 40% pure or mixed nonseminomas. The remaining 10% containing both seminoma and nonseminoma components are classified as being nonseminoma according to the World Health Organization (WHO) classification system.3 The genetic alterations underlying the development of these neoplasms have not been understood fully, although much has been done to elucidate them.4,5
The cell cycle regulatory pathway deregulated in almost all human tumors appears to be the G1 phase-controlling mechanism centered around the pRB protein. Different cancers seem to have altered different key components of that mechanism, which may be connected with gene activity patterns in different target cells.6 The mechanism involves pRB and its immediate upstream regulators, the cyclin dependent kinases (CDK4 and CDK6), their catalytic partners (cyclin D1, cyclin D2 and cyclin D3), and the members of the INK4 family of CDK inhibitors (p16INK4a, p15INK4b, p18INK4c and p19INK4d). This mechanism seems to be a common point for various signaling pathways, serving as a growth factor dependent cell cycle switch. Deregulation of the RB pathway may be an obligatory step in oncogenesis, making tumor cells less dependent on growth stimuli.6,7
The pRB is essential in cell cycle regulation and its function is regulated by phosphorylation. In G0 and the early G1 phase, hypophosphorylated pRB is complexed with the transcription factor E2F.8 In late G1, a significant hyperphosphorylation of the pRB by CDK4 and CDK6 in complex with D cyclins (D1, D2, D3) occurs.9
The CDKN2 locus at chromosomal region 9p21 encodes p16INK4a tumor suppressor protein involved in the RB cell cycle control pathway.10 p16INK4a functions as a regulator of G1/S phase transition by inhibiting the activity of CDK4 and CDK6. Thus, by inhibiting pRB phosphorylation, p16INK4a can promote the formation of a pRB-E2F repressive transcriptional complex, which blocks cell cycle progression past G1/S restriction point.11
In diverse types of cancer the RB pathway becomes deregulated through alterations in one or more of its components. The most common defects of the RB pathway are mutations or deletions of RB1 and inactivating mutations or promoter methylation of the CDKN2A (p16INK4a) tumor suppressor gene, as well as the overexpression of the cyclin D2/CDK4 complex.6,12,13
The objective of this study was to evaluate the occurrence of the loss of heterozygosity (LOH) of the CDKN2A and RB1 tumor suppressor genes in TGCTs.
Materials and methods
Patients and tumor material
Fourty TGCT samples (18 seminomas and 22 nonseminomas) were collected from Sisters of Mercy University Hospital and University Hospital Center, Zagreb, Croatia. The samples were formalin-fixed and paraffin-embedded. Clinical and pathological data for 40 TGCTs according to the WHO 2004 classification are shown in Table 1.
TABLE 1.
Clinical and pathological data for 40 testicular germ cell tumor cases
| Patient no.* | Age | pTNM | Histology |
|---|---|---|---|
| 1 | 26 | pT1NXMX | ITGCN, S |
| 2 | 26 | pT1NXMX | ITGCN, S |
| 3 | 37 | pT1NXMX | S |
| 4 | 33 | pT1NXMX | ITGCN, S |
| 5 | 31 | pT1NXMX | ITGCN, S |
| 6 | 29 | pT1NXMX | ITGCN, S |
| 7 | 39 | pT1NXMX | ITGCN, S |
| 8 | 27 | pT3NXMX | S |
| 9 | 41 | pT1NXMX | ITGCN, S |
| 10 | 48 | pT1NXMX | S |
| 11 | 48 | pT2NXMX | S |
| 12 | 34 | pT1NXMX | ITGCN, S |
| 13 | 60 | pT1NXMX | ITGCN, S |
| 14 | 29 | pT1NXMX | ITGCN, S |
| 15 | 60 | pT1NXMX | S |
| 16 | 29 | pT1NXMX | ITGCN, S |
| 17 | 28 | pT1NXMX | ITGCN, S |
| 18 | 32 | pT1NXMX | ITGCN, S |
| 19 | 37 | pT1NXMX | EC |
| 20 | 18 | pT2NXMX | EC, IT, MT, S |
| 21 | 24 | pT1NXMX | EC, ITGCN, S |
| 22 | 22 | pT2NXMX | EC, YST |
| 23 | 37 | pT1NXMX | EC, ITGCN, S |
| 24 | 28 | pT2NXMX | C, EC, IT, MT |
| 25 | 17 | pT2NXMX | EC, MT |
| 26 | 34 | pT2NXMX | EC |
| 27 | 19 | pT1NXMX | EC, ITGCN, MT, YST |
| 28 | 39 | pT1NXMX | MT, YST |
| 29 | 21 | pT2NXMX | EC, MT, YST |
| 30 | 23 | pT2NXMX | EC, IT, MT |
| 31 | 22 | pT1NXMX | MT, YST |
| 32 | 25 | pT3NXMX | EC |
| 33 | 45 | pT2NXMX | EC, ITGCN, S, YST |
| 34 | NK | pT2NXMX | C, EC, ITGCN, S, YST |
| 35 | 23 | pT2NXMX | EC, IT, ITGCN, MT, YST |
| 36 | 39 | pT1NXMX | EC, ITGCN, S, YST |
| 37 | 24 | pT2NXMX | EC, ITGCN, YST |
| 38 | 30 | pT1NXMX | EC, ITGCN, YST |
| 39 | 36 | pT1NXMX | EC, ITGCN, MT, YST |
| 40 | 58 | pT2NXMX | EC, ITGCN, YST |
seminomas, patients no. 1–18; nonseminomas, pateints no. 19–40
C = choriocarcinoma; EC = embryonal carcinoma; IT = immature teratoma; ITGCN = intratubular germ cell neoplasia; MT = mature teratoma; S = seminoma; YST = yolk sac tumor; NK = not known
DNA extraction
For each specimen, 20 μm paraffin-embedded section was prepared for DNA extraction. In addition, 4 μm section was stained with hematoxylin-eosin to identify the tumor and normal tissue areas which were removed separately from the microscopic slide, transferred to microtubes and extracted using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany).
LOH analysis of CDKN2A gene
A previously described polymorphic microsatellite marker hMp16α-I1 consisting of a mononucleotide tract of (A)23 located close to intron 1 of the CDKN2A gene was analyzed in this study.14 Primers used for polymerase chain reaction (PCR) amplifications were 5’-CAATTACCACATTCTGCGCTT-3’ and 5’-CAGGCAGAGAGCACTGTGAG-3’, which produced 190–210 bp fragments. PCR amplifications were performed in 25 μl reaction volume with a final concentration 0.2 mM of each dNTP, 3 mM MgCl2, 0.2 μM of each primer (Sigma-Aldrich, St. Louis, MI, USA), 1x Flexi buffer (Promega, Madison, WI, USA) and 0.5 U of GoTaq® Hot Start Polymerase (Promega, Madison, WI, USA). One hundred nanograms of DNA were used in each PCR reaction. PCR amplifications were carried out in a Eppendorf Mastercycler Personal (Hamburg, Germany), with cycling times of 96ºC for 5 min (one cycle), then 45 cycles of 96ºC for 30 s, 57ºC for 45 s, and 72ºC for 30 + 1 s. The final step was incubation at 72º C for 10 min. Amplified DNA fragments were analyzed on silver-stained 15% polyacrylamide gels. LOH of CDKN2A was considered to had occured if one out of two alleles (heterozygous samples) of a gene marker was missing or significantly reduced in comparison to alleles from adjacent normal tissue.
LOH analysis of RB1 gene
LOH of RB1 was detected using polymerase chain reaction-restriction fragment length polymorphism method (PCR-RFLP). Amplification with RB1 primers 5′-TCCCACCTCAGCCTCCTTAG-3’ and 5′-GTAGGCCAAGAGTGGCAGCT-3′ used in our study produced a 190 bp segment of intron 17.15 PCR amplifications were performed under conditions mentioned above. To generate the RFLP pattern for LOH analysis, 10 μl of PCR product were digested with 5 U of XbaI restriction enzyme (Fermentas, Vilnius, Lithuania) in a total volume of 25 μl for 12 h. The restriction digestion resulted in fragments of 75 and 115 bp. DNA fragments were analyzed on silver-stained 15% polyacrylamide gels. LOH was recognized as a partial or complete loss of either the uncleaved (190 bp) or the cleaved (75 + 115 bp) allele.
Results
In this study 40 TGCTs, 18 seminomas and 22 nonseminomas, were analyzed. First, we searched for LOH of the intragenic polymorphic microsatellite marker hMp16α-I1 in the CDKN2A gene. From 40 TGCTs, 34 (85%) tumors were informative for this polymorphism, 16 (89%) seminomas and 18 (82%) nonseminomas. Our analysis revealed that two (6%) samples showed LOH of hMp16α-I1 marker. The observed changes were assigned to two nonseminomas (11%, patients no. 31 and 34, Table 2). In both tumor cases, one out of two allels of gene marker was missing in comparison to alleles from the adjacent normal tissue (Figure 1). In addition, both LOHs of the CDKN2A were found in nonseminomas with a yolk sac tumor component. LOH of the CDKN2A gene was not observed among seminomas.
TABLE 2.
A) Observed loss of heterozygosity (LOH) and B) distribution of observed LOH of CDKN2A and RB1 genes in testicular germ cell tumors
|
A) observed LOH
| ||
| Patient no. | CDKN2A | RB1 |
|
| ||
| 20 | LOH | |
| 25 | LOH | |
| 31 | LOH | NI |
| 34 | LOH | I |
|
B) distribution of observed LOH
| ||
| Tumor | CDKN2A | RB1 |
|
| ||
| Seminoma, Σ 18 | 0% (0/16) | 0% (0/15) |
| Nonseminoma, Σ 22 | 11% (2/18) | 10.5% (2/19) |
I = informative (heterozygous); NI = not informative (homozygous)
Numbers in parentheses: the number of tumors demonstrating LOH over the number of informative tumors.
Figure 1.
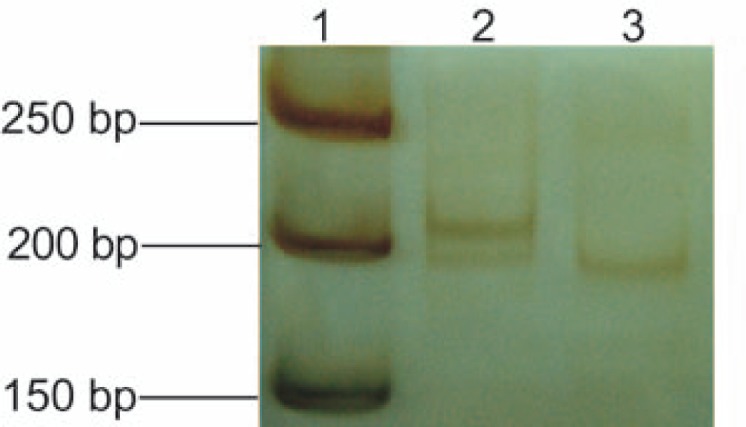
Loss of heterozygosity (LOH) of the CDKN2A gene at polymorphic microsatellite marker hMp16α-I1. Silver-stained 15% polyacrylamide gel. Lane 1: 50-bp DNA ladder (Fermentas, Vilnius, Lithuania); lane 2: heterozygous normal testis tissue; lane 3: LOH in the corresponding testicular germ cell tumor (nonseminoma, patient no. 31).
The analysis of intragenic polymorphic restriction marker of the RB1 gene showed that 34 (85%) of total TGCTs were heterozygous for this polymorphism; 15 (83%) seminomas and 19 (86%) nonseminomas. LOH was observed in two (6%) samples when looking at the total TGCTs analyzed. Once again the observed allelic losses were assigned to nonseminomas: two samples (10.5%, patients no. 20 and 25, Table 2) had one of the alleles missing in comparison to bands from the adjacent normal testis tissue. These nonseminoma samples showed loss of the cleaved allele (75- and 115-bp fragments), as the single uncleaved allele (190-bp fragment) appeared on the silver stained 15% polyacrylamide gel (Figure 2). Furthermore, both LOHs of the RB1 were found in nonseminomas with an embryonal carcinoma component. None of the seminomas demonstrated LOH of the RB1 gene.
Figure 2.

Loss of heterozygosity (LOH) of the RB1 gene at intron 17 (XbaI restriction polymorphism). Silver-stained 15% polyacrylamide gel. Lane 1: 50-bp DNA ladder (Fermentas, Vilnius, Lithuania); lane 2: heterozygous normal testis tissue; lane 3: LOH in the corresponding testicular germ cell tumor (nonseminoma, patient no. 25).
No statistically relevant correlation between the occurrence of LOH, form of TGCT, histological type of contained components and tumor stage according to TNM classification could be determined by Fisher’s exact test.
Discussion
TGCT is associated with characteristic abnormalities in the RB pathway including upregulation of cyclin D2, and downregulation of pRB and the CDK inhibitors such as p16INK4a.7
The inactivation of the CDKN2A gene, which encodes an inhibitor of CDK4 and CDK6, is one of the most common molecular events in human neoplasms. The major mechanisms contributing to CDKN2A silencing are promoter methylation, gene mutations and hemizygous or homozygous deletions. When one CDKN2A allele is mutated or methylated, the second allele is often deleted.16
The analysis of the expression of INK4 family has pointed to a down-regulation of CDKN2A in testicular neoplasms.7,12 Honorio et al.17 demonstrated that promoter hypermethylation of that gene is not involved in the decrease of p16INK4a protein expression. In contrast, some studies have found promoter mutation, a half of analyzed TGCTs had de novo promoter methylation and approximately half of TGCTs showed hypermethylation of CDKN2A exon 1α. All that correlated with a decreased level of CDKN2A mRNA expression.1,18 However, Chaubert et al.18 have not detected any CDKN2A mutations and observed LOH of the CDKN2A gene in only one of 29 TGCTs with a yolk sac tumor component, using seven different markers. These observations indicate that CDKN2A gene inactivation might be an important mechanism leading to cell deregulation in TGCTs.
Despite of promoter methylation and mutations being the most common ways of inactivating CDKN2A in TGCTs, various studies detected LOH at the position of the CDKN2A gene, varying from as low as 5.5% to as high as 42%. The LOHs of CDKN2A were reported mostly in nonseminomas.5,19 Genomic region containing CDKN2A (9p21) is reported to be the most commonly deleted region early in the development of nonseminomas, which may be implicated in their ability to differentiate into various types, for various markers located within this region.20
In our study only nonseminomas demonstrated LOH (Table 2). Both LOHs of the CDKN2A were found in nonseminomas with a yolk sac tumor component, one sample also having an embryonal carcinoma component. Furthermore, one nonseminoma with the LOH of CDKN2A demonstrated LOH of TP53 gene, and the other showed LOH of the CDH1 gene.21
The RB1 gene is often deleted or mutated to an inactive form in a variety of human tumors. Cells of embryonal testes and intratubular germ cell neoplasia (ITGCN) show no expression of pRB, whereas it is expressed in healthy testes during spermatogenesis. The lack of pRB in most TGCTs may, therefore, reflect its deregulation by normal mechanisms in testicular germ cells. However, the lack of pRB may facilitate the transition of those cells to tumor cells of ITGCN and thus contribute to molecular pathogenesis of TGCTs.7,12 Lowered levels of pRB mRNA compared with normal testis did not reflect a grossly altered structure of the DNA coding regions, but instead relates to a potentially reversible transcriptional modulation through the promoter methylation. The pRB appears to be differentially expressed according to the differentiation status of the tumor, more differentiated cells of teratocarcinoma show positive immunohistochemical staining, less differentiated forms of TGCT such as embryonal carcinoma are stained negatively.12,22,23
In contrast, deletions of RB1 gene are, along with its mutations, also reported as one of the most common alterations of the RB pathway. Various studies revealed deletions of the RB1 gene region in testicular cancer.5 For example, Peng et al.24 used short variable number of tandem repeats in RB1 introns 16 and 20, and found LOH in 5% of seminomas and 28% of nonseminomas analyzed within 93% of informative TGCT cases. The location of the RB1 gene is reported to be one of the most commonly involved in allelic imbalance within TGCTs.4 The exact alterations of the RB1 in various forms of TGCTs needs to be further elucidated in more detail. Studies also revealed a different pattern of LOH in different histological types of nonseminomas for markers located within the genomic region containing the RB1 gene (13q14), varying from 0% in yolk sac tumor component to 50% in choriocarcinoma.25
In our study, LOH of the RB1 gene was found in nonseminomas with an embryonal carcinoma component, and both nonseminomas with LOH of RB1 also demonstrated LOH of the TP53 gene.21 Interestingly, the amount of embryonal carcinoma component in TGCT, along with vascular invasion, has been proved so far to be the only clinically valid prognostic factor for the development of stage II metastatic testicular cancer.26
LOH of CDKN2A, RB1, TP53 and CDH1 in TGCTs may increase their tumorigenic potential by the increased proliferation capacity due to RB1 loss and decreased rate of apoptosis due to TP53 alteration.19,21,27 It has been shown that TP53 is abundant but inactive in cells of TGCTs. In healthy testes such reversibly inactivated TP53 may play a role in switching between proliferation and apoptosis in cells undergone meiosis.27 It was reported that, in cells that sustained lesion in the RB pathway, there was a strong selection for the loss or inactivation of wild type TP53. Alterations of RB1 are often seen together with alterations of TP53 in variety of different cancers.6,10,15 It is possible that the inactivation of both RB1 and TP53 genes in a cell produces a synergistic effect, which imposes a stronger selective pressure for the cellular transformation. This may also help to explain the high proliferation rate and/or invasiveness of TGCTs with embryonal carcinoma and yolk sac tumor component. A higher incidence of LOH in nonseminomas may provide a clue to their invasive behavior, because for some of the nonseminoma types there seem to be a region of preferential loss (3q27–3q28 in embryonal carcinoma), and all of the TGCTs show gain of 12p11–12p12 sequences.20 Knowing the exact nature of genetic alterations associated with these tumors may provide novel treatment strategies.28
However, the low frequency of observed LOHs in this study could be a consequence of genomic instability in above mentioned nonseminomas, rather than the main cause of CDKN2A and RB1 inactivation.24
Acknowledgments
This work was supported by Grant 058-0582261-2246 from Ministry of Science and Technology, Republic of Croatia.
References
- 1.Fombonne J, Devouassoux-Shisheboran M, Bouvier R, Droz J-P, Benahmed M, Krantic S. Analysis of p16INK4A gene promoter in male germ-cell tumors: identification of a new point mutation. Cancer Detect Prev. 2005;29:1–7. doi: 10.1016/j.cdp.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Kachanov DY, Dobrenkov KV, Shamanskaya TV, Abdullaev RT, Inushkina EV, Savkova RF. Solid tumors in young children in Moscow Region of Russian Federation. Radiol Oncol. 2008;42:39–44. [Google Scholar]
- 3.Eble JN, Sauter G, Epstein JI, Sesterhenn IA. Tumours of the testis and paratesticular tissue. In: Kleihues P, Sobin LH, editors. World Health Organization Classification of Tumour. Lyon: IARC Press; 2004. pp. 217–78. [Google Scholar]
- 4.Bergthorsson JT, Agnarsson BA, Gudbjartsson T, Magnusson K, Thoroddsen A, Palsson B, et al. A genome-wide study of allelic imbalance in human testicular germ cell tumors using microsatellite markers. Cancer Genet Cytogenet. 2006;164:1–9. doi: 10.1016/j.cancergencyto.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 5.von Eyben FE. Chromosomes, genes, and development of testicular germ cell tumors. Cancer Genet Cytogenet. 2004;151:93–138. doi: 10.1016/j.cancergencyto.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Bartek J, Bartkova J, Lukas J. The retinoblastoma protein pathway in cell cycle control and cancer. Exp Cell Res. 1997;237:1–6. doi: 10.1006/excr.1997.3776. [DOI] [PubMed] [Google Scholar]
- 7.Bartkova J, Rajpert-De Meyts E, Skakkebæk NE, Lukas J, Bartek J. Deregulation of the G1/S-phase control in human testicular germ cell tumours. APMIS. 2003;111:252–66. doi: 10.1034/j.1600-0463.2003.1110129.x. [DOI] [PubMed] [Google Scholar]
- 8.Adams PD, Li X, Sellers WR, Baher KB, Leng X, Harper JW, et al. Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin-cdk complex. Mol Cell Biol. 1999;19:1068–80. doi: 10.1128/mcb.19.2.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parry D, Mahony D, Willis K, Lees E. Cyclin D-CDK subunit arrangement is dependent on the availability of competing INK4 and p21 class inhibitors. Mol Cell Biol. 1999;19:1775–83. doi: 10.1128/mcb.19.3.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–77. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 11.Zhang HS, Postigo AA, Dean DC. Active transcriptional repression by the Rb-E2F complex mediates G1 arrest triggered by p16INK4a, TGFβ, and contact inhibition. Cell. 1999;97:53–61. doi: 10.1016/s0092-8674(00)80714-x. [DOI] [PubMed] [Google Scholar]
- 12.Bartkova J, Lukas C, Sørensen CS, Rajpert-De Meyts E, Skakkebæk NE, Lukas J, et al. Deregulation of the RB pathway in human testicular germ cell tumours. J Pathol. 2003;200:149–56. doi: 10.1002/path.1353. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt BA, Rose A, Steinhoff C, Strohmeyer T, Hartman M, Ackermann R. Up-regulation of cyclin-dependent kinase 4/cyclin D2 expression but down-regulation of cyclin-dependent kinase 2/cyclin E in testicular germ cell tumors. Cancer Res. 2001;61:4214–21. [PubMed] [Google Scholar]
- 14.Herranz M, Urioste M, Santos J, Rivas C, Martinez B, Benitez J, et al. Analysis of the INK4a/ARF locus in non-Hodkin’s lymphomas using two new internal microsatellite markers. Leukemia. 1999;13:808–10. doi: 10.1038/sj.leu.2401409. [DOI] [PubMed] [Google Scholar]
- 15.Xing EP, Yang G-Y, Wang L-D, Shi ST, Yang CS. Loss of heterozygosity of the Rb gene correlates with pRb protein expression and associates with p53 alteration in human esophageal cancer. Clin Cancer Res. 1999;5:1231–40. [PubMed] [Google Scholar]
- 16.Jones PA, Laird PW. Cancer epigenetics comes of age. Nature Genet. 1999;21:163–67. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 17.Honorio S, Agathanggelou A, Wernert N, Rothe M, Maher ER, Latif F. Frequent epigenetic inactivation of the RASSF1A tumour suppressor gene in testicular tumours and distinct methylation profiles of seminoma and nonseminoma testicular germ cell tumours. Oncogene. 2003;22:461–66. doi: 10.1038/sj.onc.1206119. [DOI] [PubMed] [Google Scholar]
- 18.Chaubert P, Guillou L, Kurt A-M, Bertholet M-M, Metthez G, Leisinger H-J. Frequent p16INK4 (MTS1) gene inactivation in testicular germ cell tumors. Am J Pathol. 1997;151:859–65. [PMC free article] [PubMed] [Google Scholar]
- 19.Heidenreich A, Gaddipati JP, Moul JW, Srivastava S. Molecular analysis of P16Ink4/CDKN2 and P15 Ink4B/MTS2 genes in primary human testicular germ cell tumors. J Urol. 1998;159:1725–30. doi: 10.1097/00005392-199805000-00101. [DOI] [PubMed] [Google Scholar]
- 20.Faulkner SW, Leigh DA, Oosterhuis JW, Roelofs H, Looijenga LHJ, Friedlander ML. Allelic losses in carcinoma in situ and testicular germ cell tumours of adolescents and adults: evidence suggestive of the linear progression model. Br J Cancer. 2000;83:729–36. doi: 10.1054/bjoc.2000.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vladušić T, Hrašćan R, Vrhovac I, Krušlin B, Gamulin M, Grgić M, et al. Loss of heterozygosity of selected tumor suppressor genes in human testicular germ cell tumors. Pathol Res Pract. 2010;206:163–7. doi: 10.1016/j.prp.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Jones RH, Vasey PA. New directions in testicular cancer; molecular determinants of oncogenesis and treatment success. Eur J Cancer. 2003;39:147–56. doi: 10.1016/s0959-8049(02)00612-3. [DOI] [PubMed] [Google Scholar]
- 23.Strohmeyer T, Reissmann P, Cordon-Cardo C, Hartmann M, Ackermann R, Slamon D. Correlation between retinoblastoma gene expression and differentiation in human testicular tumors. Proc Natl Acad Sci. 1991;88:6662–6. doi: 10.1073/pnas.88.15.6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng H-Q, Bailey D, Bronson D, Goss PE, Hogg D. Loss of heterozygosity of tumor suppressor genes in testis cancer. Cancer Res. 1995;55:2871–75. [PubMed] [Google Scholar]
- 25.Rothe M, Albers P, Wernert N. Loss of heterozygosity, differentiation, and clonality in microdissected male germ cell tumours. J Pathol. 1999;188:389–94. doi: 10.1002/(SICI)1096-9896(199908)188:4<389::AID-PATH364>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 26.Heidenreich A, Sesterhenn IA, Mostof FK, Moul JW. Prognostic risk factors that identify patients with clinical stage I nonseminomatous germ cell tumors at low risk and high risk for metastasis. Cancer. 1998;83:1002–11. [PubMed] [Google Scholar]
- 27.Bartkova J, Falck J, Rajpert-De Meyts E, Skakkebæk NE, Lukas J, Bartek J. Chk2 tumor suppressor protein in human spermatogenesis and testicular germ-cell tumours. Oncogene. 2001;20:5897–902. doi: 10.1038/sj.onc.1204746. [DOI] [PubMed] [Google Scholar]
- 28.Kamensek U, Sersa G. Targeted gene therapy in radiotherapy. Radiol Oncol. 2008;42:115–35. [Google Scholar]