

Abstract
HIV infection is associated with increased risk of cardiovascular complications, the underlying mechanism of which remains unclear. Plasma levels of the coagulation biomarker D-dimer (DD) correlate with increased mortality and cardiovascular events in HIV-infected patients. We compared the incidence of cardiovascular lesions and the levels of the coagulation markers DD and thrombin antithrombin in pathogenic SIV infections of rhesus and pigtailed macaques (PTMs) and in nonpathogenic SIV infection of African green monkeys (AGMs) and sooty mangabeys. Hypercoagulability and cardiovascular pathology were only observed in pathogenic SIV infections. In PTMs infected with SIV from AGMs (SIVagm), DD levels were highly indicative of AIDS progression and increased mortality and were associated with cardiovascular lesions, pointing to SIVagm-infected PTMs as an ideal animal model for the study of HIV-associated cardiovascular disease. In pathogenic SIV infection, DD increased early after infection, was strongly correlated with markers of immune activation/inflammation and microbial translocation (MT), and was only peripherally associated with viral loads. Endotoxin administration to SIVagm-infected AGMs (which lack chronic SIV-induced MT and immune activation) resulted in significant increases of DD. Our results demonstrate that hypercoagulation and cardiovascular pathology are at least in part a consequence of excessive immune activation and MT in SIV infection.
Introduction
There are increasing data demonstrating the significant impact of non-AIDS–defining comorbidities on the outcome of HIV infection. Cardiovascular disease (CVD) emerged as a major comorbidity during the era of highly active antiretroviral therapy (HAART),1–3 especially in urban African-American and Hispanic populations, which are disproportionately affected by HIV and are also at high risk for CVD. CVD may be intrinsically related to HIV infection2 or to long-term HAART, which may increase CVD risk by adverse metabolic effects (lipid changes) or vascular toxic effects.3 To date, however, our understanding of the relationship among HIV infection, HAART, and CVD risk is unclear and incomplete, and this is a critical barrier preventing interventions that may reduce CVD mortality in these patients.
CVD complications in HIV-infected subjects include thrombotic micrangiopathy (TMA), arteriopathy, dilated cardiomyopathy, abnormal coronary artery pathology (including atherothrombotic disease), and myocarditis.4–12 Abnormally high levels of coagulation markers, endothelial activation markers, and platelet activation markers have also been documented in HIV-infected patients.13–15 The SMART (Strategies for Management of Anti-Retroviral Therapy) study confirmed that a mild-to-moderate hypercoagulable state exists in HIV infection.2 This study also showed that increased levels of the coagulation biomarker D-dimer (DD) are associated with increases in inflammation markers (ie, IL-6), both being strongly linked to the loss of HIV/SIV control, disease progression, and death,2 suggesting a causal relationship between immune activation/inflammation and CVD in HIV-infected patients.
It is widely accepted that immune activation levels more accurately predict HIV disease progression to AIDS and death than do levels of viral replication or CD4+ T cells.16–18 This paradigm is supported by data from our group and others showing that natural hosts of SIV such as African green monkeys (AGMs), mandrills and sooty mangabeys (SMs) generally do not progress to AIDS despite having levels of viral replication similar to or higher than those of untreated HIV-1–infected patients.19,20 This suggests that the ability of natural hosts to maintain low levels of immune activation in the face of SIV infection and high viral load (VL) may be a major determinant of their resistance to disease progression.19,21–23
Immune activation is a multifactorial process.24 Although viral replication is a key factor driving immune activation during acute HIV/SIV infection,24 this association is not evident during chronic infection. HIV-infected patients with similar VLs may have different levels of immune activation and different rates of disease progression.16 HIV patients who receive HAART and exhibit supressed viremia but who continue to exhibit increased levels of immune activation have poor T-cell restoration and prognosis.25,26 Natural hosts of SIV maintain chronically high levels of VLs that are independent of immune activation and disease progression.27 Therefore, factors in addition to viral replication were proposed to drive the excessive immune activation characteristic of HIV infection, most notably the translocation of gut microbial products into the bloodstream.28,29 This mechanism is supported by data from natural hosts of SIV in which normal levels of immune activation during chronic SIV infection are associated with lack of microbial translocation (MT).21,22,29,30
A correlation between the levels of DD and sCD14, a product of bacterial lipopolysaccharide (LPS)–activated monocytes, was recently established in HIV-infected patients.31,32 These cross-cutting correlations between cardiovascular biomarkers and inflammatory biomarkers and between inflammatory biomarkers and MT biomarkers led us to hypothesize that the MT associated with HIV/SIV infection activates inflammatory mediators that in turn go on to play a role in the onset of CVD.
In the present study, we used 4 animal models of SIV infection to: (1) identify the most relevant animal model to study SIV-related CVD, (2) compare CVD-related biomarkers between progressive and nonprogressive SIV infections, (3) determine the timing and the pathways of CVD development during SIV infection, and (4) test directly the hypothesis that MT associated with HIV/SIV infection plays a role in the development of CVD observed in HIV-1–infected patients.
We report herein that CVD pathology is present in pathogenic SIV infections and is associated with disease progression but is absent in natural, nonprogressive SIV infections. We identified pigtailed macaques (PTMs) infected with SIV from AGMs (SIVagm) as the most appropriate animal model for the study of SIV-related CVD pathology. In this model, hypercoagulability was correlated with immune activation and MT. LPS administration to chronically SIVagm-infected AGMs (a species that lack MT during SIV infection22) significantly affected coagulability, strongly supporting MT as a major cause of CVD in progressive SIV infections.
Methods
Animals and infections
We used 4 nonhuman primate (NHP) species, 2 of which are representative of progressive SIV infections (rhesus macaques [RMs] and PTMs) and 2 of nonprogressive SIV infections (AGMs and SMs). The following study groups were included:
Group 1 consisted of 8 uninfected and 8 RMs infected with the reference strain SIVmac239, 13 uninfected and 13 SIVagm-infected PTMs, 15 uninfected and 26 SIVagm-infected AGMs, and 4 uninfected and 21 SMs infected with SIV from SMs (SIVsmm). The goal was to compare and contrast the changes in the levels of coagulation biomarkers between progressive and nonprogressive SIV infections and to assess the differences in the levels of coagulation biomarkers between uninfected and chronically SIV-infected monkeys within the same species to identify an NHP model for HIV-related hypercoagulability.
Group 2 consisted of 9 PTMs (6 males and 3 females 6-13 years of age) and 8 RMs (males 5-15 years of age). These monkeys were IV infected with 300 tissue-culture–infectious doses of SIVagm strain92018 and SIVmac239, respectively. The follow-up for these animals was until progression to AIDS: 1-2 years after inoculation for PTMs and 4-18 months after inoculation for RMs. The goal was to determine the timing of biomarker changes and to establish correlations between coagulation biomarkers and other virologic/immunologic biomarkers at different stages of SIV infection to unravel the pathways responsible for coagulation abnormalities.
Group 3 consisted of 6 adult AGM males 6-11 years of age experimentally inoculated with 300 tissue-culture–infectious doses of SIVagm92018. During chronic infection (starting from day 200 after inoculation) these animals received LPS (an endotoxin) intravenously every 2 days for a period of 3 weeks. Based on a preliminary study,21 the initial LPS dose was of 18 IU/kg. To minimize tolerance to the LPS, the dose was increased by 5 IU every week. The goal was to assess directly the role of MT in the genesis of hypercoagulation in SIV-infected NHPs.
All RMs and SMs, 8 PTMs, and 22 AGMs were housed and handled at the Tulane National Research Primate Center; 5 PTMs and 4 AGMs were housed and handled at the Regional Industrial Development Corporation NHP facility of the University of Pittsburgh. Housing and handling of all animals were in accordance with American Association for Accreditation of Laboratory Animal Care's Guide for the Care and Use of Laboratory Animals (US Public Health Service)33 and the Animal Welfare Act. All animal procedures were approved by the institutional animal care and usage committees of the Tulane University and University of Pittsburgh.
Tissue sampling
Blood and intestinal biopsies were collected as described previously.22 Plasma and PBMCs and mononuclear cells from the intestinal biopsies were isolated as described previously.22,34 At the necropsy, kidneys, hearts, aortas, coronary arteries, intestines, lungs, and brains were collected from SIVagm-infected PTMs and AGMs and fixed in 10% buffered formalin.
Viral quantification
Plasma VLs were quantified by specific real-time PCR for SIVagmSab92018 and SIVmac239, as described previously.34
Abs and flow cytometry
Whole blood and mononuclear cells isolated from intestinal biopsies were stained for flow cytometry as described previously22 to assess changes in the levels of major T-cell populations and their immune activation status. The mAbs used were: CD3-Pacific Blue, CD4-allophycocyanin, CD8-Texas Red, HLA-DR-allophycocyanin-Cy7, Ki-67–FITC (BD Biosciences). All Abs were validated and titrated using PBMCs from AGMs, RMs, and PTMs. Samples were stained for Ki-67 using the Ki-67/FITC–conjugated mouse anti–human mAb set (BD Pharmingen) as per the manufacturer's instructions. Stained cells were analyzed with an LSRII flow cytometer (BD Biosciences) and FlowJo Version 7.6 software (TreeStar). CD4+ and CD8+ T-cell percentages were obtained by first gating on lymphocytes, then on CD3+ T cells. Activation markers were determined by gating on lymphocytes, then on CD3+ T cells, and finally on CD4+CD3+ or CD8+CD3+ T cells.
Dynamics of inflammatory cytokines and chemokines
Cytokine testing in plasma was done using the human cytokine 25-Plex system (Biosource International) as per the manufacturer's instructions. Results were read by the Bio-Plex reader (Bio-Rad), which uses Luminex technology (Luminex Corporation). The analysis was focused on IL-6, because plasma levels of this cytokine have a well-known association with CVD35 and may be produced as a result of bacterial exposure. In HIV-infected patients, increases in IL-6 and DD have been shown to be strongly associated with disease progression and death.2
MT
Plasma-soluble CD14 (sCD14) levels were measured by ELISA (Quantikine Human sCD14 Immunoassay; R&D Systems). The analytical coeficient of variation ranged from 7.19%-10.9%.
sCD163
Plasma-soluble CD163 (sCD163) levels were measured by ELISA (Trillium Diagnostics). The analytical coeficient of variation ranged from 4.6%-13.6%.
Coagulation markers
Coagulation status was estimated by determining the plasma levels of DD and the thrombin–antithrombin complex (TAT).
DD is a terminal product of plasmin acting on a fibrin clot. It increases during the activation states of coagulation, disseminated intravascular coagulation, and deep vein thrombosis.36 DD was measured using a STAR automated coagulation analyzer, (Diagnostica Stago) and an immunoturbidimetric assay (Liatest D-DI; Diagnostica Stago). The analytical coeficient of variation ranged from 5%-14%.
TAT measures thrombin production and subsequent inhibition by antithrombin; antithrombin being in excess, TAT primarily reflects recent thrombin production. TAT was measured by ELISA (Enzygnost TAT micro; Siemens). The analytical coeficient of variation ranged from 7.3%-17.43%.
Histological assays
Hearts, aortas, kidneys, brains, intestines, and lungs were collected at the necropsy, fixed in 10% buffered formalin, and embedded in paraffin. We routinely checked for pathology similar to that seen in HIV-infected patients, including renal TMA, arteriopathy, myocardial hypertrophy and fibrosis, atherosclerosis (ATS), infarction, and myocarditis.4–12 Four-micron paraffin sections were stained with H&E for routine histopathology diagnosis and with Masson trichrome for collagen detection.
Immunohistochemical staining was performed on the formalin-fixed, paraffin-embedded tissues using an avidin-biotin complex HRP technique (Vectastain Elite ABC kit; Vector Laboratories) and a rabbit polyclonal antifibrinogen Ab (DAKO) as a primary Ab.
Statistical analysis
A paired t test and the Mann-Whitney U test were used to compare variables before and after infection using Prism Version 5.03 software (GraphPad). We used general estimating equations to test the relationship between DD and the other variables (ie, immune activation, inflammation, and VLs) in pathogenic infection. We present the slope of these estimates and, as a crude measure of the fraction of variance explained by the model (analogous to a correlation coefficient, r2), we calculated f = 1 − s2model/s2DD, where smodel is the scale parameter of the model and s2DD is the variance of the DD data, as described previously.37 For these tests, it was assumed that the full covariate conditional mean assumption was verified.38 These statistical tests were performed in R (Comprehensive R-Archive Network, http://CRAN.R-project.org/). A significance level of P = .05 was used throughout.
Results
Chronic pathogenic SIV infections, but not nonpathogenic SIV infections, are characterized by a procoagulant state
A procoagulant state was reported to occur in HIV-1–infected patients.2,31,39 To determine whether these findings could be reproduced in the NHP models, we compared the levels of DD and TAT in a variety of uninfected and chronically SIV-infected NHPs. Two models of progressive SIV infection (SIVmac239-infected RMs and SIVagm-infected PTMs) and 2 models of natural, nonprogressive SIV infection (SIVagm-infected AGMs and SIVsm-infected SMs) were compared.
Similar to HIV-infected patients, DD levels significantly increased during chronic pathogenic SIV infections in RMs and PTMs (Figure 1A). The average DD levels were 2.021 ± 0.26 μg/mL in chronically infected RMs, significantly higher (P = .0002) than in uninfected RMs (average, 0.38 ± 0.058 μg/mL). Similar increases were observed in chronically SIVagm-infected PTMs (2.3 ± 0.16 μg/mL vs 0.25 ± 0.05 μg/mL, P < .0001). Conversely, no significant increase in DD levels was observed in either chronically SIV infected AGMs or SMs (0.18 ± 0.03 μg/mL vs 0.23 ± 0.05 μg/mL, P > .05, in AGMs and 0.2 ± 0.05 μg/mL vs 0.26 ± 0.05 μg/mL, P > .05, in SMs; Figure 1A).
Figure 1.
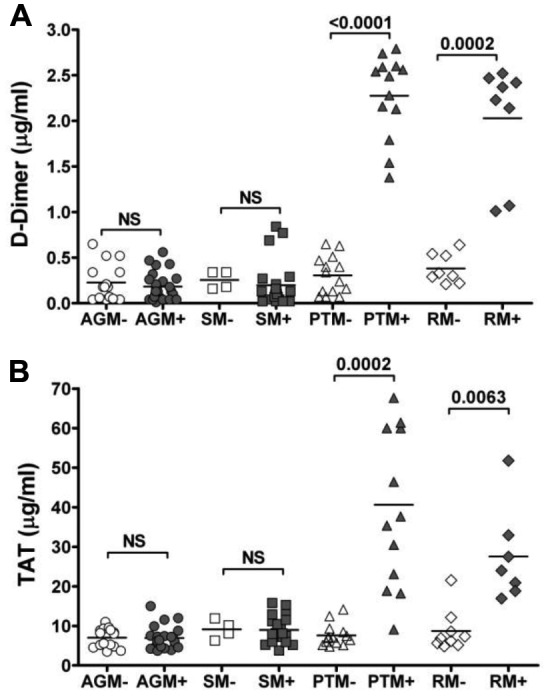
Differences in coagulation status of natural, nonprogressive SIV infections of AGMs and SMs and pathogenic SIV infections of RMs and PTMs. (A) Levels of DD are unchanged between uninfected and SIV-infected African NHPs, whereas they were increased significantly in chronically SIV-infected RMs and PTMs. (B) Similar changes observed by testing a second coagulation marker, TAT. P values were calculated with the Mann-Whitney U test.
Similarly, TAT levels were significantly increased (P = .0002 and P = .0063) in chronic pathogenic SIV infections in PTMs and RMs, respectively, but not in chronically infected AGMs and SMs (P > .05; Figure 1B).
Cross-sectional testing of DD and TAT revealed for the first time that DD is a reliable marker for measuring coagulation status in SIV infection and that hypercoagulation is characteristic of progressive SIV infections in macaques (similar to HIV-1 infection in humans), whereas SIV-infected nonprogressive hosts maintain normal coagulation markers.
The coagulation biomarkers DD and TAT increase early after SIV infection and are excellent predictors of SIV disease progression
To assess the timing of the SIV-related coagulation biomarker disturbances and their ability to serve as predictive markers of HIV/SIV disease progression, we assessed prospectively changes in DD and TAT levels at key time points in the 2 models of pathogenic SIV infection (SIVagm/PTMs and SIVmac/RMs; Figure 2). DD and TAT levels were measured throughout the acute and chronic stages of experimental infection, including end-stage AIDS. This comparative study showed that DD levels were significantly elevated in both species during acute SIV infection (0.24 ± 0.21 μg/mL vs 2.02 ± 0.58, P < .0001, in PTMs and 0.38 ± 0.16 μg/mL vs 1.2 ± 0.7 μg/mL, P = .0281, in RMs; Figure 2), with PTMs exhibiting the greatest increase (Figure 2A). With the transition to chronic infection, differences between the 2 models were uncovered: in SIVagm-infected PTMs, the levels of DD remained significantly increased throughout infection (Figure 2A). Furthermore, DD levels increased significantly in PTMs with the progression to AIDS (P = .0227; Figure 2A). Conversely, in SIVmac239-infected RMs, DD levels transiently decreased around the set point and during the early chronic infection (Figure 2B), but did not return to baseline levels. With the progression to AIDS, DD levels rebounded in RMs, but, differently from PTMs, the increase was not statistically different from chronic infection (P = .072; Figure 2B).
Figure 2.

Changes in the levels of 2DD and TAT assessed at critical time points of SIV infection. (A) SIVmac239-infected RMs. (B) SIVagm-infected PTMs. SP indicates set point. P values were calculated with the paired t test.
TAT had similar dynamics to DD during the course of acute and chronic SIV infection in both RM and PTMs, confirming the existence, timing, and predictive value for disease progression of the SIV-related coagulation disorder (Figure 2).
The dynamics of coagulation markers in progressive SIV infections showed that hypercoagulability occurs very early after SIV infection and that, similar to HIV-infected patients, increases in DD arising in SIVagm-infected PTMs are strongly correlated with AIDS progression and death. Overall, these results suggest that the SIVagm-infected PTM is a better model for investigating HIV-related CVD hypercoagulability than the SIVmac-infected RM.
Histological analyses reveal a high incidence of cardiovascular lesions in pathogenic SIVagm/PTM compared with nonpathogenic SIVagm/AGM infections
To confirm that the SIVagm/PTM model reliably reproduces the broad spectrum of cardiovascular abnormalities reported in HIV patients,4–12 we investigated numerous tissues collected at necropsy from 9 chronically SIVagm-infected PTMs. Cardiovascular pathology has already been reported in persistent progressive SIVmac/HIV-2–infected RMs and PTMs.40–43 However, the spectrum of cardiovascular lesions in SIVagm-infected PTMs is currently unknown. We also compared tissues from SIVagm-infected PTMs with SIVagm-infected AGMs to assess whether differences in the incidence of cardiovascular lesions exist between progressive and nonprogressive hosts infected with the same virus strain.
In the present study, we report that SIVagm-infected PTMs develop pathologic evidence of CVD. Similar to the renal TMA described in HIV-infected patients, numerous thrombi were present in the glomerular capillary loops and small arteries in the kidneys (Figure 3A) in 7 of 9 SIVagm-infected PTMs. The presence of thrombi in the kidney was confirmed by immunohistochemistry for fibrinogen (Figure 3B). TMA was also detected in the small blood vessels of the intestine (data not shown), lung (Figure 3C), and brain (Figure 3D) in SIVagm/PTMs, which is consistent with the frequent neurologic disease observed in this species. TMA appear to be specifically associated with SIV infection, because it was reported previously to be absent in uninfected PTMs.41
Figure 3.

Diverse spectrum of cardiovascular lesions in pathogenic SIVagm infection of PTMs. Thrombotic microangiopathy (TMA) characterized by numerous thrombi in capillaries and small arteries was detected in multiple organs. (A) Kidney (H&E staining). (B) Immunohistochemistry for fibrinogen (brown). (C) Lung (H&E staining). (D) Brain (H&E staining). (E) Glomerulopathy characterized by either focal and segmental glomerulosclerosis or collapse and fibrosis of the entire glomerular tuft was observed in SIVagm-infected PTMs (H&E staining). (F) Arteritis, characterized by significantly thickened wall (double arrow) and lumen occlusion (arrow) was detected in the kidney (H&E staining). (G) Heart hypertrophy characterized by enlarged myocytes with irregular nuclei (arrow) was observed (H&E staiing). (H) Fibrosis of the heart characterized by increased diffuse deposition of collagen replacing drop-out or lysed myocytes (blue), or (I) focal large area of fibrosis replacing areas of infarction was detected using Gomori 1-step trichrome staining (nuclei-black, muscle-red, collagen-blue). (J) Myocarditis characterized by severe infiltration with mononuclear cells (arrows; Gomori staining) and (K) extensive myocyte cytolysis (arrows; Gomori staining) was also diagnosed. (L) Incipient lesion of atherosclerosis (fatty streaks) were found in the aorta of SIVagm-infectd PTMs, characterized by accumulation of foamy macrophages (inset) in the tunica intima, under the aortic endothelium (H&E staining). Slides were visualized with a Carl Zeiss Axio Imager M1 microscope using the following objectives: 10×/0.3 DICI Plan Neofluar, 20×/0.8 DICII Plan Apochromat, and 40×/0.75 DICII Plan Neofluar. Micrographs were taken with a Carl Zeiss AxioCam MRc5. Images were acquired and analyzed using Carl Zeiss Axio Vision SE64 Release 4.8.2. The original magnification of all the pictures was 400×. Images were cropped with Adobe Photoshop CS6.
Five of 9 SIVagm-infected PTMs also had histological lesions similar to those described in HIV-associated nephropathy; these included focal and segmental glomerulosclerosis and collapsing glomerulopathy (Figure 3A,E) and HIV arteriopathy (in 2 of 9; ie, thickened-wall arteries and vessel occlusion; Figure 3F). Numerous myocardial lesions similar to those identified in HIV patients were also identified in SIVagm-infected PTMs; these included: myocardial hypertrophy (in 7 of 9 PTMs, enlarged myocytes with irregular nuclei; Figure 3G); fibrosis (in 3 of 9 PTMs) with increased collagen deposition replacing either small groups of drop-out myocytes (in 1 of 9 PTMs; Figure 3H) or larger areas of infarction (in 2 of 9; Figure 3I); and myocarditis (in one of 9 PTMs) with mononuclear cell infiltration (Figure 3J) and myocytolysis (Figure 3K). Finally, although ATS lesions are very rare in young adult SIV-negative macaques,44 fatty streaks (the first visible lesions in the development of ATS) composed of foamy macrophages (Figure 3L inset) were found to accumulate in the tunica intima and under the aortic endothelium in 2 of 9 SIVagm-infected PTMs (Figure 3L).
Analysis of tissues collected from 4 PTMs euthanized during acute SIVagmSab infection (data not shown) did not reveal the cardiovascular lesions observed in chronically infected animals, which is consistent with previous studies reporting that dilative cardiomyopathy, fibrosis, and arteriopathy are present in chronically but not acutely infected macaques.42,43 These findings, in conjunction with the lack of TMA in uninfected PTMs, demonstrates that microangiopathy and cardiovascular lesions observed are clearly the result of the infection and are not simply due to a background increase in cardiovascular lesions in PTMs.
Finally, histological examination of the kidneys (Figure 4A), brains (Figure 4B) lungs (Figure 4C), intestines (Figure 4D), aortas (Figure 4E), and hearts (Figure 4F) collected at necropsy from 9 chronically infected AGMs failed to identify any of the lesions described in SIVagm-infected PTMs.
Figure 4.

Lack of cardiovascular pathology in SIVagm-infected AGMs. Histological investigations of kidney (A), brain (B), lung (C), intestine (D), aorta (E), and heart (F) in chronically SIVagm-infected AGMs failed to identify cardiovascular lesions. Slides were visualized with a Carl Zeiss Axio Imager M1 microscope using the following objectives: 10×/0.3 DICI Plan Neofluar, 20×/0.8 DICII Plan Apochromat, and 40×/0.75 DICII Plan Neofluar. Micrographs were taken with a Carl Zeiss AxioCam MRc5. Images were aquired and analyzed by using Carl Zeiss Axio Vision SE64 Release 4.8.2. The original magnification of all the pictures was 400×. Images were cropped with Adobe Photoshop CS6.
Histological analyses confirmed that the SIVagm/PTM has a high incidence of tissue cardiovascular lesions in addition to hypercoagulability. Furthermore, the nonprogressive natural hosts do not develop cardiovascular lesions during chronic SIV infection.
sCD163 increases in SIVagm-infected PTMs and is correlated with DD levels
CD163 was reported to be a biomarker of coronary ATS based on its significant increases in both HIV-uninfected patients with coronary artery disease45 and HIV patients with noncalcified ATS plaques.46 Therefore, we measured sCD163 in SIVagm-infected PTMs and detected significant increases of this monocyte/macrophage activation marker during all stages of SIV infection (Figure 5). sCD163 was increased in all PTMs, the highest levels being observed in the 2 animals with incipient ATS lesions. We found a strong correlation between the levels of sCD163 and DD (P = .0016). Our results therefore support a possible role of monocyte/macrophage activation in the development of CVD.
Figure 5.
Significant increase of sCD163 levels during acute and chronic SIVagm infection in PTMs.
Our data suggest that the SIVagm-infected PTM is a relevant NHP model for the study of HIV-related CVD mechanisms and for developing therapeutic interventions to reduce the risk of HIV-related CVD, because it has a high incidence of cardiovascular lesions, hypercoagulability during SIV infection, and increased markers of monocyte/macrophage activation.
Procoagulant state is correlated with MT and immune activation biomarkers
In Figure 6, we show the SIV associated changes in: (1) viremia (panel A); (2) CD4+ T-cell depletion (panel B); (3) T-cell immune activation (measured as the fraction of CD4+ and CD8+ T cells expressing Ki-67 [panels C-D] and HLA-DR [data not shown]); (4) systemic inflammation (measured by plasma levels of IL-6; panel E); and (5) MT (estimated by plasma levels of sCD14; panel F).
Figure 6.

Dynamics of virologic and immunologic parameters of pathogenic SIVagm infection in PTMs. (A) Viral replication. (B) Mucosal CD4+ T cells. (C) Levels of immune activation (Ki-67) of CD4+ T cells. (D) Levels of immune activation (Ki-67) of CD8+ T cells. (E) Dynamics of inflammation as assessed by changes in the levels of IL-6. (F) Prospective assessment of microbial translocation based on testing of the surrogate marker sCD14. Two PTMs (□) died with peritonitis because of surgery complications. In these monkeys, the coagulation markers paralleled the levels of sepsis and inflammation and not viral replication, which was controlled. Index of CD4+ T cells is defined by the percentage of depletion from the baseline.
To identify candidate mechanisms for the HIV/SIV–related procoagulant state in the SIVagm-infected PTM model of CVD, we correlated DD and those disease parameters. DD was strongly correlated with levels of immune activation (for Ki-67+ CD8+ T cells, P = .01, slope = 0.062, and f = 0.73; for Ki-67+ CD4+ T cells, P < .000 01, slope = 0.102, and f = 0.9) and inflammation (for IL-6, P = .0002, slope = 0.504, and f = 0.76) as the PTMs progressed from uninfected to chronic infection and then to AIDS.
A strong correlation was also observed between DD and sCD14 (P = .000 02, slope = 2.23, and f = 0.75). This correlation was reinforced by 2 cases of peritonitis that showed high levels of sCD14, immune activation, and DD despite exquisite control of viral replication (Figure 6).
We could not formally test the association between DD and VL because there is no preinfection viral replication. However, cross-sectional correlations between DD and viremia showed that the 2 parameters were not correlated during chronic SIV infection (P = .261 and r2 = 0.18). The lack of correlation between DD and viremia was reinforced by the animals that strongly controlled chronic virus replication but maintained increased DD levels. These findings are in agreement with the data from nonprogressive models showing that viral replication was very high, whereas DD and TAT remained at preinfection levels during chronic infection47 (Figure 1).
Therefore, our data point to MT and immune activation as potential factors responsible for increased HIV/SIV–related cardiovascular risk and suggest that viral replication has a limited impact on HIV-associated hypercoagulation.
LPS administration induces immune activation and a procoagulant state in chronically SIV-infected AGMs
To test directly the hypothesis that HIV/SIV-associated MT is responsible for increased immune activation and the procoagulant status of SIV-infected NHPs, 6 chronically SIVagm-infected AGMs were treated with LPS for 3 weeks. The choice of SIVagm-infected AGMs for this study is justified by their lack of MT and their normal levels of immune activation during chronic SIVagm infection despite high levels of virus replication.22 Because of the remarkable stability of the VLs in this system (high levels of chronic viremia maintained for decades) and the exquisite control of immune activation and MT, the SIVagm/AGM system allows accurate detection of discrete alterations in immune activation and coagulation biomarkers after therapeutic interventions.
LPS administration induced increased sCD14 levels (Figure 7A), demonstrating increased activation of macrophages due to bacterial products and validating sCD14 as a surrogate marker with which to assess accurately the magnitude of MT during HIV infection or other clinical conditions.
Figure 7.

LPS administration to chronically SIVagm-infected AGMs. Shown are macrophage activation (sCD14; A) and DD (B) and viral (C) loads.
The levels of DD also increased significantly after LPS administration (Figure 7B). Finally, VLs showed moderate but sustained increases after LPS administration (Figure 7C) that were statistically significant (P = .0313). Our study therefore established a direct causal relationship between MT and hypercoagulation in SIVagm-infected AGMs, providing proof-of-concept data for the mechanism of the procoagulant status in HIV-infected patients.
Discussion
There is a critical need to identify a relevant animal model with which to study the mechanisms underlying HIV-associated CVD and to design therapies to improve clinical outcomes. CVD pathology is a major non-AIDS–defining comorbidity that affects the outcome of HIV infection significantly.1 However, a systematic approach to comprehensively define the mechanisms of HIV-related CVD is difficult in HIV-infected patients for several reasons: (1) the long duration of HIV infection in humans; (2) late HIV diagnosis, which often focuses investigation on the chronic infection only; (3) interference of multiple confounding factors such as smoking, diet, and diverse HAART regimens that may affect the development of cardiovascular lesions3; and (4) ethical limitations that prevent invasive tissue sampling for the assessment of cardiovascular lesions or interventions aimed at reducing the frequency of these lesions. As a result, most of the published HIV-associated CVD studies have been cross-sectional, limited to the chronic phase of infection, and correlative.2,13–15
The use of animal models that reproduce HIV-associated CVD permits circumvention of these limitations, exploration of the mechanisms of CVD occurring in HIV-1 infection and design, and testing of therapies aimed at controlling CVD and improving the clinical outcome of chronic HIV infection.
By comparing and contrasting changes in coagulation biomarkers and CVD tissue lesions in NHP models representing pathogenic and nonpathogenic SIV infections, we report that CVD occurs with high frequency and is specifically associated with pathogenic SIV infection. The SIVagm-infected PTM appears to be the model of choice for the study of SIV-associated CVD based on its similarities to HIV-1 infection. These similarities include significant increases in immune activation and coagulation biomarkers, specifically those predictive of disease progression and mortality in humans,2 a high incidence of cardiovascular lesions and significant increases in sCD163 (a marker of activated monocytes/macrophages associated with coronary disease in HIV patients4–11,46).
We identified DD as the best predictive marker of SIV disease progression in SIVagm-infected PTMs. During chronic infection, progression to AIDS was associated with significant increases in DD, whereas neither VLs nor CD4+ T-cell counts changed significantly (Figure 6). The mechanistic basis for this observation is that, unlike HIV infection, the highly pathogenic SIV infection of PTMs is characterized by a very poor control of viral replication and virtually no CD4+ T-cell restoration during early chronic infection. Therefore, during transition to AIDS, neither the VL nor CD4+ T-cell loss increases significantly,48 and this is a major limitation of the studies in NHP models. Our observation that coagulation biomarkers predict disease progression and death in SIVagm-infected PTMs is therefore a major achievement to the field and may improve the follow-up of SIV-infected macaques significantly.
DD can be nonspecific as a biomarker of SIV-related hypercoagulability because it may be elevated in the presence of infectious disease processes other than SIV, which may account for false-positive results of DD testing. To validate the DD testing, we measured TAT as a second marker associated with the hypercoagulable status. The results of the present study strongly suggest that DD level increases are directly related to the stage of SIV infection, because DD and TAT gave similar results in both cross-sectional and prospective studies. Furthermore, increases in these 2 markers were associated with the presence of TMA in several tissues, which confirmed the usefulness of DD in assessing HIV/SIV-associated coagulopathy.
Our study of the SIVagm/PTM model has identified several important features of cardiovascular disease in SIV-infected monkeys. First, changes in coagulation biomarker levels occurred in the absence of HAART, suggesting that, similar to SIV-infected NHPs, the hypercoagulability observed in HIV-infected patients is directly related to viral infection and not only to HAART. Furthermore, CVD occurred in SIV-infected PTMs in the absence of risk factors specific for human subjects, such as a high cholesterol diet and smoking, pointing to a direct role of lentiviral infection in inducing cardiovascular comorbidity. A direct comparison between PTMs and AGMs infected with the same viral strain (SIVagmSab92018) demonstrated that the SIV-associated CVD is host dependent and not virus related. Furthermore, our prospective studies showed that significant changes of coagulation biomarkers occur early in SIV infection. Finally, we identified a very diverse pattern of CVD presentation in SIVagm-infected PTMs. Because this plethora of cardiovascular lesions was observed in a relatively small number of monkeys, we concluded that the incidence of CVD is very high in SIVagm/PTMs. This may also indicate that the incidence of CVD is underestimated in HIV-infected patients because of limited access to tissues.
To identify mechanisms underlying the hypercoagulable state in SIV-infected NHPs, we compared increases in coagulation biomarkers with other major parameters of pathogenic SIV infection. We report that during chronic infection, increases in coagulation markers are associated specifically with immune activation and MT and not with VLs. Thus, both PTMs and AGMs have high VLs during chronic infection, but experience divergent levels of immune activation, MT, and different incidences of coagulopathy. Two PTMs had low VLs during chronic infection, but died with peritonitis and associated high levels of MT and INFL. These 2 animals had high levels of DD despite their ability to control viral replication. Similarly, occasional progression of HIV infection occurring in elite controllers associates high immune activation levels and abnormally high ATS despite viral control.49,50
Conversely, during acute infection, massive increases in coagulation biomarkers were associated with both increased immune activation and MT and high VLs. To reconcile these findings, we propose that, during chronic infection, MT drives the levels of immune activation and cardiovascular pathology. Conversely, during acute SIV infection, additional factors such as viral replication affect immune activation and subsequent coagulopathy.
Such a dichotomy was observed for other pathogenic parameters of HIV infection, leading Hunt and Deeks to propose that acute and chronic HIV infection are 2 different diseases.25 In this context, during acute infection, immune activation responses and the consequent changes in the coagulation markers limit HIV/SIV replication and establish a steady-state infection that limits perturbation of CD4+ T-cell homeostasis and maintains the optimal function of the immune system.25 During chronic infection, immune activation results from persistent damage of the mucosal barrier, leading to persistent bacteremia, which may give rise to altered cardiovascular function and subsequent CVD.
We tested the hypothesis that MT may be the main determinant of the CVD observed in SIV-infected NHPs and HIV-infected patients in 2 sets of experiments. First, in the cross-sectional study, we showed that both AGMs and SMs, which maintain immune system homeostasis during chronic SIV infection,19,20,23 also maintain a healthy coagulation status during chronic SIV infection. Moreover, no cardiovascular lesions could be identified in chronically SIVagm-infected AGMs. In natural hosts, the mucosal barrier is preserved21,22 and, therefore, the levels of MT and immune activation22,23 during chronic infection are similar to the levels in uninfected AGMs and SMs.22,29,30 Our results strongly suggest that CVD may be due to the endotoxemia related to immune dysfunction observed in pathogenic HIV/SIV infection. However, like studies performed in humans, such observations made in both cross-sectional and/or longitudinal analyses are intrinsically correlative. Therefore, to identify the mechanisms of CVD in HIV infection and to confirm our hypothesis that MT associated with pathogenic HIV/SIV infection is at the origin of CVD, we administered LPS to chronically SIVagm-infected AGMs. The rationale of performing this study in AGMs is that, in this animal model, high VLs, mucosal barrier integrity, and control of MT and immune activation are maintained at stable levels for decades, thus permitting easy identification of any minor perturbation of the system.
LPS administration affected the virus/host equilibrium in chronic SIVagm-infected AGMs by activating macrophages significantly (measured by the levels of sCD14) and by elevating the levels of viral replication. This experiment demonstrated that the maintenance of sCD14 at baseline levels in chronically infected AGMs is not because of an intrinsic tolerance to the LPS or LPS neutralization. More importantly, LPS administration resulted in a significant increase in the levels of coagulation biomarkers, confirming our hypothesis that MT observed in chronically HIV-infected patients and SIV-infected macaques is at the origin of hypercoagulation. The significance of this experiment is that it was performed in a stable system. The administration of LPS to chronically infected macaques would likely not have a similar predictive value, because it would be performed in the context of a damaged mucosal barrier, persistent MT, and preexisting coagulopathy and CVD. Therefore, our study provides for the first time proof-of-concept data that experimental modeling of MT observed in chronically HIV-infected patients results in an altered coagulation status.
At this stage, we cannot conclude that hypercoagulability is the only pathophysiologic mechanism driving CVD in HIV-infected patients. Interventions aimed at normalizing coagulation in SIV/HIV-infected patients and macaques should be performed to address this question. Alternative mechanistic pathways driven by inflammation/immune activation, which are increased in SIV/HIV infection, are equally possible. It is plausible that CVD results from the interaction of multiple factors triggered by MT.
In conclusion, in the present study, we have identified an animal model that, because of its high incidence of CV lesions, hypercoagulability, and increased markers of monocyte/macrophage activation during SIV infection, is appropriate for the study of CVD complications of HIV/SIV infection. This model permits establishing the pathogenic determinants of the HIV/SIV-related CVD, which may affect significantly the management of HIV-infected patients. This model also allows validation of biomarkers that can be used to identify those HIV-infected patients who, despite similar presentations (ie, similar VLs in the absence or the presence of therapy), may have a variety of underlying pathobiologies requiring individualized therapeutic interventions, such as anti-inflammatory therapy and/or anticoagulant or anti-ATS drugs. The use of NHP models allowed us to test directly the hypothesis that MT is the root cause of CVD comorbidities in HIV-infected patients. Therefore, our results indicate that interventions aimed at reducing gut inflammation and MT have the potential to reduce CVD risk in HIV-infected patients. These findings can be readily translated to clinical practice to improve the management of HIV-infected patients. Preventing, diagnosing, and treating HIV-related cardiovascular complications, which represent a major cause of morbidity and mortality in HIV-infected patients, may affect the outcome of HIV infection significantly, as HAART did 15 years ago.
Acknowledgments
The authors thank Drs Jason Brenchley and Daniel Douek for helpful discussions and technical assistance and the Division of Veterinary Medicine of Tulane National Primate Research Center and Division of Laboratory Animal Resources of the University of Pittsburgh for animal care.
This work was supported by National Institutes of Health/National Institute of Allergy and Infectious Diseases/National Center for Research Resources grants RO1 AI064066 (to I.P.), R01 AI065325 (to C.A.), R01 RR025781 (to C.A. and I.P.), and RR-00168 (to the Tulane National Research Primate Center). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
There is an Inside Blood commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: I.P., C.W., C.A., A.L., and R.T. designed the study; I.P., E.C., D.M., J.K., C.X., and C.A. performed the experiments; G.S.H.-R. and A.T. were in charge of the animal studies; I.P., R.M.R., and C.A. analyzed the data; and I.P., C.W., C.A., A.L., and R.T. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ivona Pandrea, MD, PhD, Center for Vaccine Research, 9045 BST3, 3501 Fifth Ave, Pittsburgh, PA 15261; e-mail: pandrea@pitt.edu.
References
- 1.Palella FJ, Jr, Phair JP. Cardiovascular disease in HIV infection. Curr Opin HIV AIDS. 2011;6(4):266–271. doi: 10.1097/COH.0b013e328347876c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuller LH, Tracy R, Belloso W, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5(10):e203. doi: 10.1371/journal.pmed.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friis-Møller N, Reiss P, Sabin CA, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med. 2007;356(17):1723–1735. doi: 10.1056/NEJMoa062744. [DOI] [PubMed] [Google Scholar]
- 4.Barbaro G, Di Lorenzo G, Grisorio B, Barbarini G. Incidence of dilated cardiomyopathy and detection of HIV in myocardial cells of HIV-positive patients. Gruppo Italiano per lo Studio Cardiologico dei Pazienti Affetti da AIDS. N Engl J Med. 1998;339(16):1093–1099. doi: 10.1056/NEJM199810153391601. [DOI] [PubMed] [Google Scholar]
- 5.Barbaro G, Di Lorenzo G, Grisorio B, Barbarini G. Cardiac involvement in the acquired immunodeficiency syndrome: a multicenter clinical-pathological study. Gruppo Italiano per lo Studio Cardiologico dei pazienti affetti da AIDS Investigators. AIDS Res Hum Retroviruses. 1998;14(12):1071–1077. doi: 10.1089/aid.1998.14.1071. [DOI] [PubMed] [Google Scholar]
- 6.Blanchard DG, Hagenhoff C, Chow LC, McCann HA, Dittrich HC. Reversibility of cardiac abnormalities in human immunodeficiency virus (HIV)-infected individuals: a serial echocardiographic study. J Am Coll Cardiol. 1991;17(6):1270–1276. doi: 10.1016/s0735-1097(10)80134-2. [DOI] [PubMed] [Google Scholar]
- 7.Butt AA, Chang CC, Kuller L, et al. Risk of heart failure with human immunodeficiency virus in the absence of prior diagnosis of coronary heart disease. Arch Intern Med. 2011;171(8):737–743. doi: 10.1001/archinternmed.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen IS, Anderson DW, Virmani R, et al. Congestive cardiomyopathy in association with the acquired immunodeficiency syndrome. N Engl J Med. 1986;315(10):628–630. doi: 10.1056/NEJM198609043151007. [DOI] [PubMed] [Google Scholar]
- 9.De Castro S, d'Amati G, Gallo P, et al. Frequency of development of acute global left ventricular dysfunction in human immunodeficiency virus infection. J Am Coll Cardiol. 1994;24(4):1018–1024. doi: 10.1016/0735-1097(94)90864-8. [DOI] [PubMed] [Google Scholar]
- 10.Jacob AJ, Sutherland GR, Bird AG, et al. Myocardial dysfunction in patients infected with HIV: prevalence and risk factors. Br Heart J. 1992;68(6):549–553. doi: 10.1136/hrt.68.12.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joshi VV, Pawel B, Connor E, et al. Arteriopathy in children with acquired immune deficiency syndrome. Pediatr Pathol. 1987;7(3):261–275. doi: 10.1080/15513818709177129. [DOI] [PubMed] [Google Scholar]
- 12.Lipshultz SE, Chanock S, Sanders SP, Colan SD, Perez-Atayde A, McIntosh K. Cardiovascular manifestations of human immunodeficiency virus infection in infants and children. Am J Cardiol. 1989;63(20):1489–1497. doi: 10.1016/0002-9149(89)90014-3. [DOI] [PubMed] [Google Scholar]
- 13.de Larrañaga GF, Petroni A, Deluchi G, Alonso BS, Benetucci JA. Viral load and disease progression as responsible for endothelial activation and/or injury in human immunodeficiency virus-1-infected patients. Blood Coagul Fibrinolysis. 2003;14(1):15–18. doi: 10.1097/00001721-200301000-00004. [DOI] [PubMed] [Google Scholar]
- 14.Solages A, Vita JA, Thornton DJ, et al. Endothelial function in HIV-infected persons. Clin Infect Dis. 2006;42(9):1325–1332. doi: 10.1086/503261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torriani FJ, Komarow L, Parker RA, et al. Endothelial function in human immunodeficiency virus-infected antiretroviral-naive subjects before and after starting potent antiretroviral therapy: the ACTG (AIDS Clinical Trials Group) study 5152s. J Am Coll Cardiol. 2008;52(7):569–576. doi: 10.1016/j.jacc.2008.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giorgi JV, Hultin LE, McKeating JA, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis. 1999;179(4):859–870. doi: 10.1086/314660. [DOI] [PubMed] [Google Scholar]
- 17.Giorgi JV, Lyles RH, Matud JL, et al. Predictive value of immunologic and virologic markers after long or short duration of HIV-1 infection. J Acquir Immune Defic Syndr. 2002;29(4):346–355. doi: 10.1097/00126334-200204010-00004. [DOI] [PubMed] [Google Scholar]
- 18.Rodríguez B, Sethi AK, Cheruvu VK, et al. Predictive value of plasma HIV RNA level on rate of CD4 T-cell decline in untreated HIV infection. JAMA. 2006;296(12):1498–1506. doi: 10.1001/jama.296.12.1498. [DOI] [PubMed] [Google Scholar]
- 19.Pandrea I, Sodora DL, Silvestri G, Apetrei C. Into the wild: simian immunodeficiency virus (SIV) infection in natural hosts. Trends Immunol. 2008;29(9):419–428. doi: 10.1016/j.it.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pandrea I, Apetrei C. Where the wild things are: pathogenesis of SIV infection in African nonhuman primate hosts. Curr HIV/AIDS Rep. 2010;7(1):28–36. doi: 10.1007/s11904-009-0034-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pandrea I, Gaufin T, Brenchley JM, et al. Cutting edge: Experimentally induced immune activation in natural hosts of simian immunodeficiency virus induces significant increases in viral replication and CD4+ T cell depletion. J Immunol. 2008;181(10):6687–6691. doi: 10.4049/jimmunol.181.10.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pandrea IV, Gautam R, Ribeiro RM, et al. Acute loss of intestinal CD4+ T cells is not predictive of simian immunodeficiency virus virulence. J Immunol. 2007;179(5):3035–3046. doi: 10.4049/jimmunol.179.5.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silvestri G, Paiardini M, Pandrea I, Lederman MM, Sodora DL. Understanding the benign nature of SIV infection in natural hosts. J Clin Invest. 2007;117(11):3148–3154. doi: 10.1172/JCI33034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sodora DL, Silvestri G. Immune activation and AIDS pathogenesis. AIDS. 2008;22(4):439–446. doi: 10.1097/QAD.0b013e3282f2dbe7. [DOI] [PubMed] [Google Scholar]
- 25.Hunt PW, Martin JN, Sinclair E, et al. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J Infect Dis. 2003;187(10):1534–1543. doi: 10.1086/374786. [DOI] [PubMed] [Google Scholar]
- 26.Lederman MM, Calabrese L, Funderburg NT, et al. Immunologic failure despite suppressive antiretroviral therapy is related to activation and turnover of memory CD4 cells. J Infect Dis. 2011;204(8):1217–1226. doi: 10.1093/infdis/jir507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pandrea I, Onanga R, Kornfeld C, et al. High levels of SIVmnd-1 replication in chronically infected Mandrillus sphinx. Virology. 2003;317(1):119–127. doi: 10.1016/j.virol.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 28.Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nat Immunol. 2006;7(3):235–239. doi: 10.1038/ni1316. [DOI] [PubMed] [Google Scholar]
- 29.Brenchley JM, Price DA, Schacker TW, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12(12):1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 30.Gordon SN, Klatt NR, Bosinger SE, et al. Severe depletion of mucosal CD4+ T cells in AIDS-free simian immunodeficiency virus-infected sooty mangabeys. J Immunol. 2007;179(5):3026–3034. doi: 10.4049/jimmunol.179.5.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Funderburg NT, Mayne E, Sieg SF, et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood. 2010;115(2):161–167. doi: 10.1182/blood-2009-03-210179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang W, Lederman MM, Hunt P, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis. 2009;199(8):1177–1185. doi: 10.1086/597476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.National Research Council. Guide for the Care and Use of Laboratory Animals. Washington, DC: National Academy Press; 1996. [Google Scholar]
- 34.Pandrea I, Apetrei C, Dufour J, et al. Simian immunodeficiency virus SIVagm.sab infection of Caribbean African green monkeys: a new model for the study of SIV pathogenesis in natural hosts. J Virol. 2006;80(10):4858–4867. doi: 10.1128/JVI.80.10.4858-4867.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harris TB, Ferrucci L, Tracy RP, et al. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am J Med. 1999;106(5):506–512. doi: 10.1016/s0002-9343(99)00066-2. [DOI] [PubMed] [Google Scholar]
- 36.Levy G. Value and limitations of the assay of a fibrin degradation product: D dimer. [article in French]. Sem Hop (Paris) 1987;63(25):2061–2064. [Google Scholar]
- 37.Twisk JWR. Applied Longitudinal Data Analysis for Epidemiology. Cambridge, United Kingdom: Cambridge University Press; 2003. [Google Scholar]
- 38.Diggle PJ, Heagerty P, Laing K-Y, Zeger SL. Analysis of Longitudinal Data. 2nd Ed. Oxford, United Kingdom: Oxford University Press; 2002. [Google Scholar]
- 39.Boulware DR, Hullsiek KH, Puronen CE, et al. Higher levels of CRP, D-dimer, IL-6, and hyaluronic acid before initiation of antiretroviral therapy (ART) are associated with increased risk of AIDS or death. J Infect Dis. 2011;203(11):1637–1646. doi: 10.1093/infdis/jir134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chalifoux LV, Simon MA, Pauley DR, MacKey JJ, Wyand MS, Ringler DJ. Arteriopathy in macaques infected with simian immunodeficiency virus. Lab Invest. 1992;67(3):338–349. [PubMed] [Google Scholar]
- 41.Eitner F, Cui Y, Hudkins KL, et al. Thrombotic microangiopathy in the HIV-2-infected macaque. Am J Pathol. 1999;155(2):649–661. doi: 10.1016/S0002-9440(10)65161-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shannon RP, Simon MA, Mathier MA, Geng YJ, Mankad S, Lackner AA. Dilated cardiomyopathy associated with simian AIDS in nonhuman primates. Circulation. 2000;101(2):185–193. doi: 10.1161/01.cir.101.2.185. [DOI] [PubMed] [Google Scholar]
- 43.Yanai T, Lackner AA, Sakai H, Masegi T, Simon MA. Systemic arteriopathy in SIV-infected rhesus macaques (Macaca mulatta). J Med Primatol. 2006;35(2):106–112. doi: 10.1111/j.1600-0684.2005.00145.x. [DOI] [PubMed] [Google Scholar]
- 44.Chawla KK, Murthy CD, Chakravarti RN, Chhuttani PN. Arteriosclerosis and thrombosis in wild rhesus monkeys. Am Heart J. 1967;73(1):85–91. doi: 10.1016/0002-8703(67)90312-2. [DOI] [PubMed] [Google Scholar]
- 45.Aristoteli LP, Moller HJ, Bailey B, Moestrup SK, Kritharides L. The monocytic lineage specific soluble CD163 is a plasma marker of coronary atherosclerosis. Atherosclerosis. 2006;184(2):342–347. doi: 10.1016/j.atherosclerosis.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 46.Burdo TH, Lo J, Abbara S, et al. Soluble CD163, a novel marker of activated macrophages, is elevated and associated with noncalcified coronary plaque in HIV-infected patients. J Infect Dis. 2011;204(8):1227–1236. doi: 10.1093/infdis/jir520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pandrea I, Silvestri G, Onanga R, et al. Simian immunodeficiency viruses replication dynamics in African NHP hosts: common patterns and species-specific differences. J Med Primatol. 2006;35(4-5):194–201. doi: 10.1111/j.1600-0684.2006.00168.x. [DOI] [PubMed] [Google Scholar]
- 48.Picker LJ, Hagen SI, Lum R, et al. Insufficient production and tissue delivery of CD4+ memory T cells in rapidly progressive simian immunodeficiency virus infection. J Exp Med. 2004;200(10):1299–1314. doi: 10.1084/jem.20041049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsue PY, Hunt PW, Schnell A, et al. Role of viral replication, antiretroviral therapy, and immunodeficiency in HIV-associated atherosclerosis. AIDS. 2009;23(9):1059–1067. doi: 10.1097/QAD.0b013e32832b514b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hunt PW, Brenchley J, Sinclair E, et al. Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J Infect Dis. 2008;197(1):126–133. doi: 10.1086/524143. [DOI] [PMC free article] [PubMed] [Google Scholar]