

Abstract
Background and Aims
Wild soybean (Glycine soja), a native species of East Asia, is the closest wild relative of the cultivated soybean (G. max) and supplies valuable genetic resources for cultivar breeding. Analyses of the genetic variation and population structure of wild soybean are fundamental for effective conservation studies and utilization of this valuable genetic resource.
Methods
In this study, 40 wild soybean populations from China were genotyped with 20 microsatellites to investigate the natural population structure and genetic diversity. These results were integrated with previous microsatellite analyses for 231 representative individuals from East Asia to investigate the genetic relationships of wild soybeans from China.
Key Results
Analysis of molecular variance (AMOVA) revealed that 43·92 % of the molecular variance occurred within populations, although relatively low genetic diversity was detected for natural wild soybean populations. Most of the populations exhibited significant effects of a genetic bottleneck. Principal co-ordinate analysis, construction of a Neighbor–Joining tree and Bayesian clustering indicated two main genotypic clusters of wild soybean from China. The wild soybean populations, which are distributed in north-east and south China, separated by the Huang-Huai Valley, displayed similar genotypes, whereas those populations from the Huang-Huai Valley were different.
Conclusions
The previously unknown population structure of the natural populations of wild soybean distributed throughout China was determined. Two evolutionarily significant units were defined and further analysed by combining genetic diversity and structure analyses from Chinese populations with representative samples from Eastern Asia. The study suggests that during the glacial period there may have been an expansion route between south-east and north-east China, via the temperate forests in the East China Sea Land Bridge, which resulted in similar genotypes of wild soybean populations from these regions. Genetic diversity and bottleneck analysis supports that both extensive collection of germplasm resources and habitat management strategies should be undertaken for effective conservation studies of these important wild soybean resources.
Keywords: Wild soybean, Glycine soja, microsatellites, genetic diversity, population structure
INTRODUCTION
Crop wild relatives (CWRs) have been recognized as valuable genetic resources for crop improvement (Prescott-Allen and Prescott-Allen, 1986, 1988; Feuillet et al., 2008). They are also important for both applied and basic research as a means of understanding the biology of crop plants (Tanksley and McCouch, 1997; Damania, 2008; Feuillet et al., 2008). However, global climate change and the destruction of the ecological balance have sped up the extinction rate of these species (Saunders et al., 1991; Thomas et al., 2004). More attention should be paid to the effective conservation of plant biodiversity, especially for wild relatives that have potential for the genetic improvement of cultivars (Myers et al., 2000; Rao and Hodgkin, 2002). Therefore, comprehensive and extensive investigation of the population genetic structure and the phylogenetic relationship of CWRs is a requisite for identifying conservation units and developing in situ/ex situ conservation priorities for CWRs (Heywood et al., 2007).
Wild soybean (Glycine soja) is well known as the closest wild relative of the cultivated soybean (G. max). It is endemic over a wide range of areas of East Asia including China, the Russian Far East, the Korean Peninsula and Japan. A long history of domestication, cultivation and breeding has narrowed the genetic basis of cultivated soybean, limiting further improvement of crop yield and quality. In contrast, wild soybeans, which inhabit a wide range of eco-geographic regions in East Asia, have diverse genetic variability in pest and disease resistance genes and other useful agricultural and ecological characteristics (Hajjar and Hodgkin, 2007; Chung and Singh, 2008). Thus wild soybeans have been explored as a very important genetic resource for cultivated soybean improvement in response to global climate change (Chung and Singh, 2008).
Previous genetic diversity analyses showed a high level of genetic variation in East Asia, especially in China (Kuroda et al., 2006; Lee et al., 2008; Li et al., 2009; Wen et al., 2009). Wild soybeans occur in most provinces of China except the Xinjiang, Qinghai and Hainan Provinces (Li, 1993; Dong et al., 2001). The possible distribution centre of genetic diversity of wild soybean in China is debated. For example, based on different developmental responses to photo-thermo effects in wild soybean, Xu et al. (1987) proposed seven wild soybean ecotypes in China. Dong et al. (2001) suggested three genetic diversity centres of wild soybean in China based on morphological traits and suggested that the north-east centre was the primary centre. More recent research also reported three geographically distinct genetic groups of wild soybean in China using microsatellites (Li et al., 2009; Wen et al., 2009). Xu et al. (1999) detected the greatest genetic diversity of wild soybeans in southern China from the analysis of isozymes and restriction fragment length polymorphisms (RFLPs) of cytoplasmic DNA collected from different ecological regions. Furthermore, analysis of nuclear and cytoplasmic genomic polymorphism suggests that the primary diversity centre of wild soybean is in southern China (Shimamoto et al., 1998; Wen et al., 2009).
Despite increasing interest in the analysis of genetic diversity of wild soybeans, most reports have used representative individuals or populations limited to narrow geographic regions. Understanding genetic variation within and between natural populations is critical for sustainable utilization and conservation of wild soybean. Detailed studies on the genetic structure and phylogeographic history of this important wild relative endemic to East Asia are thus necessary to provide new opportunities for the improvement of soybean breeding (Hajjar and Hodgkin, 2007).
Genetic structure in natural populations is mainly shaped by their mating systems, life cycles and the historical demography related to geological and climate changes (Avise et al., 1987; Slatkin, 1987; Hewitt, 2000, 2004). In the present study, 20 microsatellite loci from the 20 chromosomes of soybean were employed to investigate the genetic diversity and population structure of 40 natural wild soybean populations at a regional scale in China. Furthermore, by integrating previous genotype data of representative samples from East Asia (Guo et al., 2010), the genetic structure of wild soybean covering the whole distribution area will be discussed.
MATERIALS AND METHODS
Plant materials and genotyping
A total of 712 individuals from 40 natural populations of wild soybean (Glycine soja Siebold & Zucc.) were sampled, covering the major geographical distribution regions in China (Fig. 1 and Table 1). For each population, mature seeds were collected from each individual, with an interval of >5 m between individuals. Two to 20 individuals were collected for each population. Seeds are preserved in the Wuhan Botanical Garden of the Chinese Academy of Sciences, and the Institute of Crop Science of the Chinese Academy of Agricultural Sciences. Two individuals of G. tomentella were sampled as outgroups.
Fig. 1.

Geographical distribution of wild soybean populations in China. The dotted lines separate the three eco-regions in China based on the population genetic structure. The image at bottom-left shows the distribution area of wild soybean in East Asia, including China, the Russian Far East, the Korean Peninsula and Japan.
Table 1.
Genetic diversity of 40 wild soybean populations and three inferred eco-regions
| Eco-region | Population | n | NE | HE | FIS | IAM | TPM |
|---|---|---|---|---|---|---|---|
| NE (north-east China) | HWK | 20 | 2·3 | 0·460 | 0·920 | 9NS/9NS | 11NS/7NS |
| HHN | 20 | 2·6 | 0·567 | 0·967 | 3NS/17*** | 4NS/16** | |
| HYS | 20 | 2·5 | 0·544 | 0·959 | 4NS/16*** | 7NS/13* | |
| HHL | 20 | 2·5 | 0·515 | 0·915 | 2NS/16*** | 6NS/12* | |
| JTY | 20 | 1·9 | 0·329 | 0·756 | 15***/5NS | 17***/3NS | |
| JLJ | 20 | 3·1 | 0·643 | 0·895 | 2NS/18*** | 3NS/17** | |
| LKY | 20 | 2·1 | 0·463 | 0·897 | 2NS/16*** | 3NS/15** | |
| LDD | 20 | 2·4 | 0·508 | 0·845 | 4NS/16*** | 7NS/13NS | |
| LCY | 20 | 1·8 | 0·379 | 0·983 | 1NS/14*** | 1NS/14*** | |
| HCD | 20 | 2·7 | 0·543 | 0·954 | 5NS/13** | 7NS/11NS | |
| NTL | 20 | 1·9 | 0·390 | 0·785 | 2NS/15*** | 4NS/13** | |
| Overall | 220 | 5·6 | 0·759 | – | – | – | |
| NC (the Huang-Huai Valley) | BBJ | 20 | 2·5 | 0·513 | 0·979 | 5NS/13** | 7NS/11NS |
| HXT | 18 | 1·0 | 0·016 | 1·000 | 2NS/0NS | 2NS/0NS | |
| SWS | 20 | 1·9 | 0·336 | 1·000 | 3NS/11** | 4NS/10* | |
| SQY | 20 | 1·1 | 0·019 | 0·815 | 3NS/0NS | 3NS/0NS | |
| HZZ | 20 | 1·4 | 0·146 | 1·000 | 5NS/5NS | 5NS/5NS | |
| HXH | 15 | 1·6 | 0·195 | 1·000 | 15***/2NS | 17***/0NS | |
| HNX | 20 | 1·0 | 0·005 | 1·000 | 1NS/0NS | 1NS/0NS | |
| SKL | 20 | 2·1 | 0·422 | 0·965 | 3NS/15** | 4NS/14* | |
| JYC | 20 | 2·5 | 0·521 | 0·868 | 1NS/16*** | 6NS/11* | |
| AWH | 18 | 2·6 | 0·558 | 0·865 | 5NS/14** | 8NS/11NS | |
| SXA | 20 | 1·5 | 0·212 | 1·000 | 1NS/12*** | 2NS/11* | |
| NYC | 12 | 1·9 | 0·367 | 0·939 | 0NS/14*** | 0NS/14*** | |
| GQS | 12 | 2·1 | 0·416 | 0·942 | 1NS/17*** | 1NS/17*** | |
| Overall | 235 | 4·7 | 0·721 | – | – | – | |
| SC (south China) | CKX | 20 | 2·1 | 0·358 | 0·885 | 8NS/9NS | 10NS/7NS |
| HYC | 20 | 1·6 | 0·193 | 0·977 | 14**/3NS | 15***/2NS | |
| HWH | 20 | 2·6 | 0·558 | 0·949 | 2NS/16*** | 3NS/15*** | |
| HTB | 17 | 2·4 | 0·501 | 0·995 | 6NS/14** | 6NS/14NS | |
| ACH | 20 | 1·7 | 0·242 | 0·947 | 9*/6NS | 11***/4NS | |
| HFH | 11 | 2·2 | 0·477 | 1·000 | 2NS/17*** | 3NS/16*** | |
| HAH | 16 | 1·6 | 0·279 | 0·989 | 1NS/10*** | 1NS/10*** | |
| HHS | 8 | 1·5 | 0·195 | 1·000 | 15***/0NS | 15***/0NS | |
| GRY | 7 | 2·2 | 0·458 | 0·810 | 4NS/13** | 8NS/9* | |
| GXA | 20 | 2·0 | 0·390 | 0·953 | 5NS/13* | 8NS/10NS | |
| JJA | 20 | 2·2 | 0·450 | 0·943 | 1NS/15*** | 1NS/15*** | |
| JNC | 20 | 1·9 | 0·400 | 0·953 | 2NS/14*** | 3NS/13*** | |
| ZQY | 16 | 1·4 | 0·139 | 1·000 | 12**/2NS | 12***/2NS | |
| FXP | 20 | 1·1 | 0·053 | 0·693 | 8**/1NS | 8**/1NS | |
| FJY | 20 | 2·2 | 0·454 | 0·960 | 7NS/12NS | 9NS/10NS | |
| FLC | 2 | 1·0 | 0·000 | – | – | – | |
| Overall | 257 | 6·2 | 0·785 | ||||
| Total | 712 | – | 0·813 | 0·929 | – | – |
n, number of samples; NE, number of effective alleles, HE, expected heterozygosity; FIS, fixation index.
Demographic analysis was carried out under two models: IAM model, infinite allele model; TPM model, two-phased model of mutation which proposes 30 % multistep changes. The ratio indicates the number of loci with heterozygosity deficiency/number of loci with heterozygosity excess. NS, not significant; *P < 0·05; **P < 0·01; ***P < 0·001.
The seeds of each individual were used for germination. After germination, the leaf tissue was collected and used for DNA extraction following the cetyltrimethylammonium bromide (CTAB) protocol (Doyle and Doyle, 1987). Twenty unlinked microsatellites from the 20 soybean chromosomes were selected for genotyping (Supplementary Data Table S1) (Cregan et al., 1999; Song et al., 2004). The PCR protocol was based on the description in Cregan et al. (1999). Briefly, approx. 10 µg of template DNA, 2·5 µmol of forward and reverse primers, and 1 U of Taq polymerase were used in each 10 µL mixture. PCR products were separated by 6 % PAGE, then visualized by silver staining and scored according to a 25 bp DNA ladder (Promega, Madison, WI, USA).
Genetic diversity analysis
Genetic diversity statistics for each locus, population and eco-region identified by STRUCTURE 2·2 (Pritchard et al., 2000) were assessed by calculating the expected heterozygosity (HE), number of detected alleles (NA), effective allele number (NE) and fixation index (FIS) using GENALEX V6 software (Peakall and Smouse, 2006). The outcrossing rate was calculated from the fixation index using the equation t = (1 – FIS)/(1 + FIS) (Weir, 1996).
Demographic history
The program BOTTLENECK (Cornuet and Luikart, 1996) was implemented to detect deviation from mutation–drift equilibrium, which deduces historical changes in population size for each population. The program is based on the principle that a population that had experienced recent variation in their effective population size would present a related variation in allele number and genetic diversity. For example, populations that experienced a recent bottleneck should exhibit a faster reduction of allele number than of genetic diversity under a supposed mutation model at mutation–drift equilibrium. In contrast, populations with recent expansion should exhibit a faster increase in allele numbers relative to genetic diversity (Cornuet and Luikart, 1996). Two mutation models were performed in our study for the test. These were the infinite allele model (IAM), which proposes that the microsatellite may evolve to infinite alleles, and the two-phased model (TPM), which proposes 30 % multistep changes (Gladieux et al., 2008). A one-tailed Wilcoxon sign-rank test (Luikart et al., 1998) was conducted to determine whether a population exhibited a significant number of loci with an excess or deficiency of diversity.
Genetic structure analysis
Principal co-ordinate analysis (PCA), which used multivariate techniques to detect patterns of variation in complex data sets, was performed based on the Nei's genetic distance matrix (Nei, 1978) by the GENALEX V6 program (Peakall and Smouse, 2006).
Model-based Bayesian analysis was carried out using STRUCTURE 2·2 (Pritchard et al., 2000) to detect the genetic structure of wild soybean populations. This algorithm assumes that each individual has admixture ancestral genotypes from more than one cluster regardless of sampling location or taxonomy. A series of K = 1–40 was used to estimate the number of clusters under the admixture model with allele frequencies correlated. For each K, at least five independent runs of 100 000 iterations were processed following a burn-in period of 50 000 iterations. The ad hoc statistic, ΔK, which was calculated based on the rate of change of the log-likelihood for the present K value, was employed to identify the optimal number of populations present in the data set (Evanno et al., 2005). The optimal genetic structure (the maximum value of ΔK) was graphically displayed using DISTRUCT (Rosenberg, 2004).
Analysis of molecular variance (AMOVA) was conducted using the ARLEQUIN V3·0 software (Excoffier et al., 2005) to examine the distribution of genetic variation within populations, among populations within eco-regions and among eco-regions as identified by STRUCTURE 2·2 (Pritchard et al., 2000). Genetic differentiation and a test for significance between population pairs were further assessed by the F-statistics estimator (FST) using FSTAT V2·9·3 (Goudet, 2001). Gene flow among populations was estimated by calculating the number of migrants (Nm) based on F-statistics with the equation Nm = (1 – FST)/FST (Slatkin and Barton, 1989).
Phylogenetic relationship
Pairwise genetic distances between populations were calculated using the Cavalli-Sforza and Edwards chord distance by MICRSAT V1·5 with 10 000 replications (Cavalli-Sforza and Edwards, 1967; Minch et al., 1996). We used the PHYLIP V3·67 software package (Felsenstein, 2004) to construct a Neighbor–Joining (NJ) tree of wild soybean populations. Glycine tomentella was used as the outgroup to root the tree.
Combined analysis with previous microsatellite data from representative individuals
In order to explain further the phylogeographical structure of wild soybeans in China, we incorporated our data with our previous data from representative individual samples from the whole distribution area in East Asia (Guo et al., 2010). These included 216 individuals from China, five individuals from the Russian Far East, five individuals from South Korea and five individuals from Japan, which were genotyped with 56 microsatellites (Guo et al., 2010). The genetic structure of 216 individuals was investigated using the model-based analysis by STRUCTURE 2·2 (Pritchard et al., 2000) with K ranging from 1 to 10 and parameters as described above.
RESULTS
Genetic diversity and differentiation
In this study, 40 naturally occurring wild soybean populations evenly distributed throughout the whole distribution area of China were analysed for genetic diversity and population structure using 20 unlinked microsatellite loci. A total of 317 alleles were detected across the 20 microsatellite loci in 712 individuals (Supplementary Data Table S1). At the species scale, wild soybean showed a high level of genetic diversity with HE = 0·813 (Table 1). For each population analysed, the highest level of genetic diversity was found in the JLJ population (NE = 3·1, HE = 0·643) (Table 1), but no genetic diversity was found in the FLC population due to the small number of individuals (n = 2) sampled.
The global FIS was extremely high (FIS = 0·929), suggesting a low outcrossing rate in wild soybean populations. At the population level, the FIS value varied from 0·693 to 1, with an average outcrossing rate of 3·7 % (Table 1). The low outcrossing rates are most probably due to the predominantly self-pollinating nature of wild soybean. These low outcrossing rates resulted in high overall genetic differentiation (FST = 0·561) and low gene flow (Nm = 0·783) among populations.
Bottleneck analysis
Demographic analysis of the 39 polymorphic populations (excluding the monomorphic FLC population) revealed that most populations had an expected heterozygosity excess under both the IAM (27 populations) and TPM models (26 populations). The one-tailed Wilcoxon sign-rank test predicted that 25 and 19 populations had a heterozygosity excess significantly derived from the mutation–drift equilibrium under the IAM and TPM models, respectively (Table 1). This suggested that a recent population bottleneck had occurred in most of the natural populations.
Population structure
The factor plate of PCA analysis projecting scatter plots of the first two principal components is shown in Fig. 2. The first two principal components 1 and 2 accounted for 48·5 % of the total variation and roughly grouped the 40 populations into two main groups: the populations from central China including most of the area of the Huang-Huai Valley (NC) formed one group (Group A in Fig. 2), while the populations from north-east China (NE) and south China (SC) formed another group (Group B in Fig. 2).
Fig. 2.
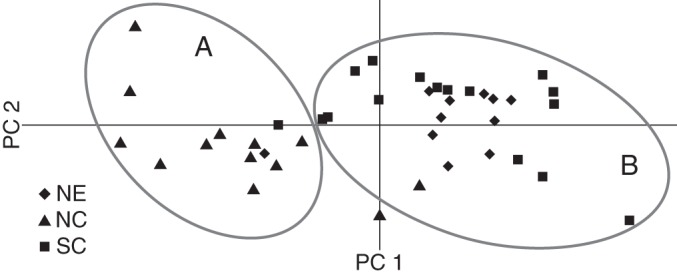
Principal co-ordinate analysis (PCA) of wild soybean populations from the genotype of microsatellites. Populations in group A are mainly from Huang-Huai Valley. Populations in group B are mainly from north-east China and south China.
Based on ΔK statistics (Evanno et al., 2005) in the Bayesian analysis using STRUCTURE 2·2, the highest likelihood for K was 2 (Supplementary Data Fig. S1a). When K = 2, two main groups were identified (Fig. 3A): populations from north-east China (NE) and south China (SC) showed a similar ancestral genotypic origin, while populations from the Huang-Huai Valley (NC) were independent. This was consistent with the result of the PCA. Further analysis based on K = 3 revealed that most of the populations from north-east and south China were genetically separated (Fig. 3B). All 40 populations could be divided into three clusters corresponding to three eco-regions of wild soybean previously defined in China: the north-east China eco-region (NE), the Huang-Huai Valley eco-region (NC) and the south China eco-region (SC) (Figs 1 and 3B).
Fig. 3.

STRUCTURE estimation of the wild soybean genetic structure. NE, wild soybean from north-east China; NC, wild soybean from the Huang-Huai Valley; SC, wild soybean from south China; R, wild soybean from the Russian Far East; K, wild soybean from South Korea; J, wild soybean from Japan. (A) Genetic structure of 40 populations covering most of the distribution area in China based on 20 microsatellites, with K = 2 and (B) K = 3. (C) Genetic structure of 231 representative individuals covering the whole distribution area in East Asia based on 56 microsatellites, with K = 2. (D) Genetic structure of representative individuals from East Asia except the Huang-Huai Valley based on 56 microsatellites, with K = 3.
High pairwise differentiations between populations were observed (0·277–0·725 with an average of 0·547), suggesting significant genetic differentiation and limited gene flow between populations. The overall AMOVA revealed that 56·08 % of the molecular variances were found among populations and 43·92 % were found within populations, indicating great genetic diversity within populations (Table 2). AMOVA was used for further detection of the genetic differentiation between different eco-regions identified by STRUCTURE 2·2. As shown in Table 2, the greatest genetic differentiation occurred between the NC and NE regions, with 8·16 % of the variation among groups, while the NE and SC regions exhibited the lowest genetic differentiation, with 3·09 % of the variation among groups (Table 2). This was consistent with the results from the PCA and STRUCTURE analysis, which both revealed a closer genetic relationship between NE and SC than between NC and SC, or between NC and NE.
Table 2.
Hierarchical analyses of molecular variance (AMOVA) within and among three wild soybean eco-regions
| Variance component (%) |
|||||
|---|---|---|---|---|---|
| Grouping structure | No. of groups | No. of populations | Within populations | Among populations within groups | Among groups |
| Overall | 1 | 40 | 43·92 | 56·08 | – |
| NE and NC | 2 | 24 | 44·73 | 47·11 | 8·16 |
| NE and SC | 2 | 27 | 48·6 | 48·31 | 3·09 |
| NC and SC | 2 | 29 | 36·35 | 56·5 | 7·15 |
Phylogenetic analysis
Relationships among wild soybean populations based on the 20 microsatellites were visualized in an NJ tree. Two main clusters were identified (Fig. 4) as per the PCA. Cluster I included populations mainly from the Huang-Huai Valley (NC) along with three populations from south-west China (SC) including populations CKX, HFH and HYC. Cluster II contained populations from north-east China (NE) and most of the populations from south China. For the three eco-regions identified by STRUCTURE 2·2, the populations from south China are distributed across both clusters, while those from north-east China and the Huang-Huai Valley clustered independently from each other in different clusters.
Fig. 4.

Neighbor–Joining tree with the chord distance of wild soybean populations based on 20 microsatellites. Cluster I includes populations mainly from the Huang-Huai Valley. Cluster II includes populations from north-east China and south China.
DISCUSSION
Genetic diversity of G. soja
The present study detected high overall genetic diversity of wild soybean populations in China. This is consistent with previous studies (Li et al., 2009; Wen et al., 2009; Guo et al., 2010). The expected heterozygosity detected in wild soybean was much higher than that reported for cultivated soybean (Kuroda et al., 2006; Guo et al., 2010). Wild soybean from China could provide abundant genetic resources for cultivar improvement.
Mating system and the life history cycle play central roles in shaping the distribution of the population genetic diversity of plants (Hamrick and Godt, 1996; Nybom, 2004). The overall level of genetic diversity is high in wild soybean (Shimamoto et al., 1998; Xu et al., 1999; Dong et al., 2001; Xu et al., 2002; Li et al., 2009; Wen et al., 2009; Li et al., 2010), but the expected heterozygosity of each natural population is relatively low (with average HE = 0·345, Table 1). Though higher than that detected in wild soybean populations from Japan (HE = 0·228) (Kuroda et al., 2006), it is much lower than the observed genetic diversity of many flowering plants based on microsatellites (an average HE of 0·410 for selfing species and 0·460 for annual species) (Nybom, 2004). First, as an annual selfing species, limited gene flow between populations might account for the relatively low genetic diversity found in wild soybean relative to other flowering plants. A high selfing rate (81·9–100 %) was detected in the natural populations of wild soybean. The average outcrossing rate of the natural populations is 3·7 %, which is congruent with a rate of 3·4 % for wild soybeans from Japan (Kuroda et al., 2006). Secondly, changes in climate and the environment are likely to influence the distribution and propagation of wild soybean. Sakai et al. (2003) used four intergenic spacer regions of chloroplast DNA to analyse the genus Glycine, and suggested that wild soybean rapidly expanded its distribution in East Asia. Thus, relatively low genetic diversity in natural populations might be exacerbated due to genetic drift (Sakai et al., 2003). This is consistent with the BOTTLENECK analysis in this study, which indicated that most natural populations have experienced a population bottleneck. The genetic diversity analysis revealed that the eco-region from south China had the greatest genetic diversity measured by either NE or HE (Table 1). Similarly, previous studies based on simple sequence repeats (SSRs), amplified fragment length polymorphisms (AFLPs) and RFLPs (Shimamoto et al., 1998; Wen et al., 2009; Xu et al., 1999; Guo et al., 2010) found genetic diversity to be highest in south China and suggest that a genetic diversity centre of wild soybean exists in south China.
Strong genetic differentiation (FST = 0·76) among populations of wild soybean from Japan has been reported in a previous study (Kuroda et al., 2006). It is much higher than that detected in wild soybean populations from China (FST = 0·561). AMOVA revealed that 43·92 % of the genetic diversity occurred within populations in this study. Relatively larger population size (most populations have 20 individuals) might account for the molecular variance within and among populations, as larger populations may encompass more of the available genetic diversity of the natural population. Therefore, both the distribution range and plentiful sampling are equally important for effective conservation studies of genetic resources.
Population structure of G. soja
Several studies have discussed the discrimination of sub-populations for wild soybean based on the geographic distribution of representative individuals from China (Xu et al., 1987; Dong et al., 2001; Li et al., 2009; Wen et al., 2009; Li et al., 2010). However, with genetic information of the natural population, we were able to detect novel population genetic structures of wild soybeans in the whole distribution area of China.
The Bayesian analysis using STRUCTURE 2·2 detected two genotypic groups of wild soybean from China: (1) the SC and NE group; and (2) the NC group (Fig. 3A). The genetic distinctiveness of NC was also evident in PCA (Fig. 2) and the NJ tree (Fig. 4). Both analyses revealed that most of the populations from NE and SC (Fig. 2B; Fig.4 cluster II) clustered together, while NC (Fig. 2A; Fig. 4 cluster I) clustered independently. The population structure was also evidenced by the hierarchical AMOVA (Table 2), whereby genetic differentiation between NE and SC (3·09 %) was lower than that between NE and NC (8·16 %) and between NC and SC (7·15 %). This suggests two evolutionarily significant units in China, which is different from the seven ecotypes defined based on differences in the photo-thermo effects on the development of wild soybeans by Xu et al. (1987), and the three geographic genetic differentiation groups reported previously (Dong et al., 2001; Wang et al., 2001; Wen et al., 2009; Li et al., 2010).
This study relied on neutral microsatellite loci distributed in the 20 linkage groups of soybean and analysed across natural soybean populations evenly distributed throughout China, which highlighted the population structure of wild soybean from heredity. The two evolutionarily significant units of wild soybean detected in the present study agree with findings of our previous study (Guo et al., 2010) based on representative individuals from East Asia, which demonstrated that individuals from the Huang-Huai Valley were clustered independently from other regions, including south China (SC), north-east China (NE), the Russian Far East (R), South Korea (K) and Japan (J) (Fig. 3C).
It was surprising that wild soybeans from south China (SC) and those from north-east China (NE) showed similar genotypes, because these two regions are geographically separated by the Huang-Huai Valley (NC). We employed the genotype data from our previous study to explain further the existing population structure in China. Sub-division structure analysis of representative individuals from East Asia excluding those from the Huang-Huai Valley (NC) gave an optimal population number of K = 3 (Supplementary Data Fig. S1), which showed that individuals sampled from South Korea and Japan shared most of the genotypes with individuals from the south-east coast of China (Fig. 3D). Previous investigations on the vegetation of East Asia based on fossil pollen evidence suggested that during the Last Glacial Maximum, the south-east coast of China, the Korean Peninsula and Japan might have been connected by temperate forest in the East China Sea Land Bridge (Harrison et al., 2001). It was likely that there was an expansion event during the glacial age, which resulted in similar genotypes of wild soybeans in these regions. Thus it is proposed that insufficient time for lineage sorting and differentiation after expansion resulted in similar genotypes in north-east and south China despite their geographical separation. In addition, phylogenetic analysis revealed that wild soybean populations from the Huang-Huai Valley clustered with some populations from the south-west of China, where there is considered to be an important biodiversity hotspot (Myers et al., 2000). Relatively low genetic diversity in the Huang-Huai Valley implies that there may have been a rapid expansion of wild soybean from southwest China.
Concluding remarks and implications for conservation
Wild soybeans are distributed across East Asia, covering the mainland of China, Taiwan, the Korean Peninsula, Japan and the Russian Far East. As wild species can be important genetic resources for cultivar improvement, several groups have reported the genetic structure of wild soybean from East Asia (Xu et al., 2002; Lee et al., 2008; Kuroda et al., 2009; Wen et al., 2009; Li et al., 2010). We have detected two evolutionarily significant units of wild soybean populations from China by using unlinked microsatellite loci to analyse the population structure of natural wild soybean populations from the whole distribution area. Further analysis of polymorphic sequences, such as chloroplast or nuclear sequences, across more representative population samples from the whole distribution area in East Asia are needed to elucidate the population structure and phylogeographical expansion routes of G. soja in more depth. This would also enable prediction of a time frame of expansion for wild soybean in East Asia to support population diffusion or interaction via the East China Sea Land Bridge.
Although most of the genotypes were shared between north-east and south China, morphological diversity and investigation have shown that the north-east was a very important diversity centre (Dong et al., 2001; Wen et al., 2009). North-east China, the Huang-Huai Valley and south China regions with different predominant genotypes could be considered as independent gene pools for cultivated soybean improvement, especially in regards to native adaptive characteristics for cultivation in these regions.
Populations with low genetic diversity might be at increased risk for extinction due to limited adaptive potential and fixation of deleterious alleles (Frankham, 1995; Keller and Waller, 2002). In this study, although the overall genetic diversity of wild soybean was high due to extensive geographic distribution, the genetic diversity within populations was relatively low (Table 1). BOTTLENECK analysis further showed that most populations deviated significantly from mutation–drift equilibrium, suggesting a recent population bottleneck. Extensive collection of germplasm resources is recommended for comprehensive and long-term conservation of these important genetic resources ex situ. In addition, habitat management and the monitoring of population dynamics for wild soybean should be undertaken to maintain the dynamic evolutionary potential for the two evolutionarily significant units discovered, especially those from south China, which is considered a genetic diversity centre of wild soybean.
SUPPLEMENTARY DATA
ACKNOWLEDGMENTS
The project was partially supported by The National Natural Science Foundation of China (30670158), the State Key Basic Research and Development Plan of China (973) (G1998010203), CAS/SAFEA International Partnership Program for Creative Research Teams Project and the 100 Talents Program of the Chinese Academy of Sciences. Special thanks to Professor Xinan Zhou, Professor Dianxiang Zhang, the Chinese Academy of Agricultural Sciences and South China Botanical Garden for their assistance in sample collection. The authors thank Drs Ting Wang, Alice Hayward, Jing Wang, Xiaohong Yao and Chi Song for suggestions on the data analysis and manuscript preparation.
LITERATURE CITED
- Avise JC, Arnold J, Ball RM, et al. Intraspecific phylogeography: the mitochondrial DNA bridge between population genetics and systematics. Annual Review of Ecology and Systematics. 1987;18:489–522. [Google Scholar]
- Cavalli-Sforza L, Edwards A. Phylogenetic analysis: models and estimation procedures. Evolution. 1967;21:550–570. doi: 10.1111/j.1558-5646.1967.tb03411.x. [DOI] [PubMed] [Google Scholar]
- Chung G, Singh RJ. Broadening the genetic base of soybean: a multidisciplinary approach. Plant Science. 2008;27:295–341. [Google Scholar]
- Cornuet JM, Luikart G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics. 1996;144:2001–2014. doi: 10.1093/genetics/144.4.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan PB, Jarvik T, Bush AL, et al. An integrated genetic linkage map of the soybean genome. Crop Science. 1999;39:1464–1490. [Google Scholar]
- Damania AB. History, achievements, and current status of genetic resources conservation. Agronomy Journal. 2008;100:9–21. [Google Scholar]
- Dong YS, Zhuang BC, Zhao LM, Sun H, He MY. The genetic diversity of annual wild soybeans grown in China. Theoretical and Applied Genetics. 2001;103:98–103. [Google Scholar]
- Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin. 1987;19:11–15. [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schneider S. Arlequin version 3·0: an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP (Phylogeny Inference Package), version 3·6. 2004 http://evolution.genetics.washington.edu/phylip.html . [Google Scholar]
- Feuillet C, Langridge P, Waugh R. Cereal breeding takes a walk on the wild side. Trends in Genetics. 2008;24:24–32. doi: 10.1016/j.tig.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Frankham R. Conservation genetics. Annual Review of Genetics. 1995;29:305–327. doi: 10.1146/annurev.ge.29.120195.001513. [DOI] [PubMed] [Google Scholar]
- Gladieux P, Zhang XG, Afoufa-Bastien D, Sanhueza RMV, Sbaghi M, Le Cam B. On the origin and spread of the scab disease of apple: out of central Asia. PLoS ONE. 2008;3:e1455. doi: 10.1371/journal.pone.0001455. http://dx.doi.org/10.1371/journal.pone.0001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet J. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2·9. 3) 2001 http://www2.unil.ch/popgen/softwares/fstat.htm . [Google Scholar]
- Guo J, Wang Y, Song C, et al. A single origin and moderate bottleneck during domestication of soybean (Glycine max): implications from microsatellites and nucleotide sequences. Annals of Botany. 2010;106:505–514. doi: 10.1093/aob/mcq125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar R, Hodgkin T. The use of wild relatives in crop improvement: a survey of developments over the last 20 years. Euphytica. 2007;156:1–13. [Google Scholar]
- Hamrick JL, Godt MJW. Effects of life history traits on genetic diversity in plant species. Philosophical Transactions of the Royal Society B: Biological Sciences. 1996;351:1291–1298. [Google Scholar]
- Harrison SP, Yu G, Takahar H, Prentice IC. Diversity of temperate plants in East Asia. Nature. 2001;413:129–130. doi: 10.1038/35093166. [DOI] [PubMed] [Google Scholar]
- Hewitt GM. The genetic legacy of the Quaternary ice ages. Nature. 2000;405:907–913. doi: 10.1038/35016000. [DOI] [PubMed] [Google Scholar]
- Hewitt GM. Genetic consequences of climatic oscillations in the Quaternary. Philosophical Transactions of the Royal Society B: Biological Sciences. 2004;358:183–195. doi: 10.1098/rstb.2003.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heywood V, Casas A, Ford-Lloyd B, Kell S, Maxted N. Conservation and sustainable use of crop wild relatives. Agriculture Ecosystems and Environment. 2007;121:245–255. [Google Scholar]
- Keller LF, Waller DM. Inbreeding effects in wild populations. Trends in Ecology and Evolution. 2002;17:230–241. [Google Scholar]
- Kuroda Y, Kaga A, Tomooka N, Vaughan DA. Population genetic structure of Japanese wild soybean (Glycine soja) based on microsatellite variation. Molecular Ecology. 2006;15:959–974. doi: 10.1111/j.1365-294X.2006.02854.x. [DOI] [PubMed] [Google Scholar]
- Kuroda Y, Tomooka N, Kaga A, Wanigadeva S, Vaughan D. Genetic diversity of wild soybean (Glycine soja Sieb. et Zucc.) and Japanese cultivated soybeans [G. max (L.) Merr.] based on microsatellite (SSR) analysis and the selection of a core collection. Genetic Resources and Crop Evolution. 2009;56:1045–1055. [Google Scholar]
- Lee JD, Yu JK, Hwang YH, et al. Genetic diversity of wild soybean (Glycine soja Sieb. and Zucc.) accessions from South Korea and other countries. Crop Science. 2008;48:606–616. [Google Scholar]
- Li FS. Studies on the ecological and geographical distribution of the Chinese resources of wild soybean (G. soja) Scientia Agriculture Sinica. 1993;26:47–55. [Google Scholar]
- Li XH, Wang KJ, Jia JZ. Genetic diversity and differentiation of Chinese wild soybean germplasm (G. soja Sieb. & Zucc.) in geographical scale revealed by SSR markers. Plant Breeding. 2009;128:658–664. [Google Scholar]
- Li Y-H, Li W, Zhang C, et al. Genetic diversity in domesticated soybean (Glycine max) and its wild progenitor (Glycine soja) for simple sequence repeat and single-nucleotide polymorphism loci. New Phytologist. 2010;188:242–253. doi: 10.1111/j.1469-8137.2010.03344.x. [DOI] [PubMed] [Google Scholar]
- Luikart G, Allendorf FW, Cornuet JM, Sherwin WB. Distortion of allele frequency distributions provides a test for recent population bottlenecks. Journal of Heredity. 1998;89:238–247. doi: 10.1093/jhered/89.3.238. [DOI] [PubMed] [Google Scholar]
- Minch E, Ruiz-Linares A, Goldstein D, Feldman M, Cavalli-Sforza LL. Microsat (version 1·5): a computer program for calculating various statistics on microsatellite allele data. Stanford, CA: Stanford University Medical Center; 1996. [Google Scholar]
- Myers N, Mittermeier RA, Mittermeier CG, da Fonseca GAB, Kent J. Biodiversity hotspots for conservation priorities. Nature. 2000;403:853–858. doi: 10.1038/35002501. [DOI] [PubMed] [Google Scholar]
- Nei M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics. 1978;89:583–590. doi: 10.1093/genetics/89.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nybom H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Molecular Ecology. 2004;13:1143–1155. doi: 10.1111/j.1365-294X.2004.02141.x. [DOI] [PubMed] [Google Scholar]
- Peakall ROD, Smouse PE. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes. 2006;6:288–295. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott-Allen C, Prescott-Allen R. The first resource: wild species in the North American economy. New Haven, CT: Yale University Press; 1986. [Google Scholar]
- Prescott-Allen R, Prescott-Allen C. Using wild genetic resources for food and raw materials. London: Earthscan Publications; 1988. [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RV, Hodgkin T. Genetic diversity and conservation and utilization of plant genetic resources. Plant Cell, Tissue and Organ Culture. 2002;68:1–19. [Google Scholar]
- Rosenberg NA. DISTRUCT: a program for the graphical display of population structure. Molecular Ecology Notes. 2004;4:137–138. [Google Scholar]
- Sakai M, Kanazawa A, Fujii A, Thseng FS, Abe J, Shimamoto Y. Phylogenetic relationships of the chloroplast genomes in the genus Glycine inferred from four intergenic spacer sequences. Plant Systematics and Evolution. 2003;239:29–54. [Google Scholar]
- Saunders DA, Hobbs RJ, Margules CR. Biological consequences of ecosystem fragmentation: a review. Conservation Biology. 1991;5:18–32. [Google Scholar]
- Shimamoto Y, Fukushi H, Abe J, et al. RFLPs of chloroplast and mitochondrial DNA in wild soybean, Glycine soja, growing in China. Genetic Resources and Crop Evolution. 1998;45:433–439. [Google Scholar]
- Slatkin M. Gene flow and the geographic structure of natural populations. Science. 1987;236:787–792. doi: 10.1126/science.3576198. [DOI] [PubMed] [Google Scholar]
- Slatkin M, Barton NH. A comparison of three indirect methods for estimating average levels of gene flow. Evolution. 1989;43:1349–1368. doi: 10.1111/j.1558-5646.1989.tb02587.x. [DOI] [PubMed] [Google Scholar]
- Song QJ, Marek LF, Shoemaker RC, et al. A new integrated genetic linkage map of the soybean. Theoretical and Applied Genetics. 2004;109:122–128. doi: 10.1007/s00122-004-1602-3. [DOI] [PubMed] [Google Scholar]
- Tanksley SD, McCouch SR. Seed banks and molecular maps: unlocking genetic potential from the wild. Science. 1997;277:1063–1666. doi: 10.1126/science.277.5329.1063. [DOI] [PubMed] [Google Scholar]
- Thomas CD, Williams SE, Cameron A, et al. Biodiversity conservation: uncertainty in predictions of extinction risk/Effects of changes in climate and land use/Climate change and extinction risk (reply) Nature. 2004;430:1–2. [Google Scholar]
- Weir BS. Genetic data analysis II. Sunderland, MA: Sinauer Associates; 1996. [Google Scholar]
- Wen ZX, Ding YL, Zhao TJ, Gai JY. Genetic diversity and peculiarity of annual wild soybean (G. soja Sieb.et Zucc.) from various eco-regions in China. Theoretical and Applied Genetics. 2009;119:371–381. doi: 10.1007/s00122-009-1045-y. [DOI] [PubMed] [Google Scholar]
- Xu B, Lu QH, Zhuang BC. Analysis of ecotypes of wild soybean (G. soja) in China. Scientia Agriculture Sinica. 1987;20:29–35. [Google Scholar]
- Xu DH, Gao Z, Tian QZ, et al. Genetic diversity of the annual wild soybean (Glycine soja) in China. Chinese Journal of Applied and Environmental Biology. 1999;5:439–443. [Google Scholar]
- Xu DX, Abe JA, Gai JG, Shimamoto YS. Diversity of chloroplast DNA SSRs in wild and cultivated soybeans: evidence for multiple origins of cultivated soybean. Theoretical and Applied Genetics. 2002;105:645–653. doi: 10.1007/s00122-002-0972-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.