

Abstract
Diagnosis of hereditary colorectal cancer syndromes requires clinical suspicion and knowledge of such syndromes. Lynch syndrome is the most common cause of hereditary colorectal cancer. Other less common causes include familial adenomatous polyposis (FAP), Peutz-Jeghers syndrome (PJS), juvenile polyposis syndrome, and others. There have been a growing number of clinical and molecular tools used to screen and test at risk individuals. Screening tools include diagnostic clinical criteria, family history, genetic prediction models, and tumor testing. Patients who are high risk based on screening should be referred for genetic testing.
Keywords: Lynch syndrome, familial adenomatous polyposis, Peutz-Jeghers syndrome
Objectives: After completion of this article, the reader should be able to identify the clinical characteristics and genetic basis of multiple hereditary syndromes that are a cause of hereditary colorectal cancer.
Colorectal cancer (CRC) is the second leading cause of cancer deaths in men and women, with over 1 million cases estimated worldwide in 2009.1 There were over 140,000 de novo colorectal cancers (CRCs) diagnosed in the United States in 2010. Most of these cancers are thought to be sporadic; however, heredity is a significant factor in 10 to 15% of cases.2,3 Given the significant burden of this disease, prevention by identifying patients at high risk of developing CRC remains an important clinical goal.
In ~5% of all cases, CRC is associated with a highly penetrant dominantly inherited syndrome.4 Lynch syndrome is the most common, accounting for 2 to 5% of all CRC cases in the United States.5 Nonsyndromic familial presentations, defined as ≥ 2 first-degree relatives with CRC,6 make up an additional 20% of cases. Less than 1% is caused by FAP and other rare genetic syndromes. A considerable number of patients with CRC are therefore born with a predisposition to developing the disease, and emerging genomic research suggests that an even greater percentage than previously thought may carry a germline predisposition for CRC. This knowledge offers a screening and disease prevention opportunity to considerably affect the established natural history of CRC; an essential first step is the identification of individuals at increased risk.7
Several studies have shown that surveillance of Lynch syndrome families reduces the development of CRC by 60% and also decreases mortality.8,9 Although the effect of surveillance in familial colorectal cancer on mortality is unknown, various studies have reported a high yield of advanced adenomas in these families when compared with surveillance of average risk individuals.10 These findings suggest that the increased relative risk of developing CRC may outweigh the possible disadvantages of surveillance in persons suspected of having a familial CRC syndrome. Individuals at high risk are identified based on clinical suspicion and appropriate screening and genetic testing can be used to confirm the diagnosis. This review will focus on identification of common CRC syndromes. For many years, most of these syndromes were identified by the use of clinical findings; however, in the age of molecular diagnosis, sequencing plays a greater role.
Family History
There are several clinical characteristics that suggest that an individual may be at higher risk of a familial CRC syndrome. Some of these factors include a strong family history of CRC or early age at diagnosis. Patients who present with synchronous or metachronous colorectal cancers are also more likely to have a familial CRC syndrome. Patients with multiple polyps (more than three) and extraintestinal manifestations of known CRC syndromes (see Table 1) should be screened appropriately.
Table 1. Colorectal Cancer Predisposition Syndromes.
| Syndrome | Colon Presentation | Lifetime Colon Cancer Risk | Extracolonic Manifestations |
|---|---|---|---|
| FAP | Over 100 adenomatous colorectal polyps (average age of polyposis onset is 16 years) | Nearly 100% | Duodenal and periampullary cancers (3–5% risk), childhood hepatoblastoma, other cancers: pancreatic, thyroid, gastric, brain (all rare); desmoid tumors (20% risk); Gardner syndrome: osteomas (often of the jaw), epidermoid cysts, CHRPE, dental anomalies; Turcot syndrome: medulloblastoma |
| Lynch syndrome | Colon cancer (often early onset, average age of onset 44–61 years) | 50–80% | Endometrial cancer (40–60% risk), ovarian cancer (9–14% risk); other cancers: stomach, renal, ureter, small intestine, biliary (all 10% or less); Muir-Torre syndrome: cutaneous keratoacanthomas, sebaceous gland tumors; Turcot syndrome: glioblastoma |
| AFAP | 10–100 adenomatous colorectal polyps with a tendency toward polyps in the right side of the colon (average age of polyposis onset is 26) | 80% | Similar to FAP |
| MAP | 10 to over 100 colorectal polyps | Undefined, but increased over the general population | Not generally seen; some reports of patients with CHRPE, osteomas, dental cysts, duodenal adenomas, and/or gastric cancer |
| HMPS | Colorectal polyposis with polyps of different histologies (adenomas–classic, serrated, tubular; hyperplastic; juvenile; mixed juvenile-adenomatous or hyperplastic adenomatous) | Undefined | Rare extracolonic cancers including pancreatic, breast, thyroid, and kidney |
| HPS | Colorectal polyposis featuring large, hyperplastic polyps and some adenomas/serrated adenomas | Undefined | None reported |
| Peutz-Jeghers | Colorectal polyposis involving characteristic hamartomatous polyps | 39% | Blue/brown pigmentation (starting in childhood around the mouth, nose, and/or eyes and on the buccal mucosa and fingers; spots fade with age); upper GI polyposis (particularly small intestine); other cancers: breast, ovarian, pancreatic, small intestine, gastric, esophageal, cervical (adenoma malignum); sex cord tumors, Sertoli cell tumors |
| JPS | Colorectal polyposis involving juvenile polyps | 17–22% by age 35, ~ 68% by age 60 |
Gastric polyps (if present, 21% risk of gastric adenocarcinoma), other cancers: pancreatic and small intestine |
| Cowden syndrome | Colorectal hamartomas | Unclear, around 9% | Breast cancer (30% risk in women), thyroid cancer (10%), upper GI hamartomas; macrocephaly, fibrocystic breasts, dermatologic features (80% of affected individuals) including oral papillomas, trichilemmomas, keratoses of the hands and feet, and lipomas |
GI, Gastrointestinal; FAP, familial adenomatous polyposis; AFAP, attenuated familial adenomatous polyposis; MAP, MYH-associated polyposis; HMPS, hereditary mixed polyposis syndrome; HPS, hereditary polyposis syndrome; JPS, juvenile polyposis; CHRPE, congenital hypertrophy of retinal pigment epithelium.
From Gammon A, Kohlmann W, Burt R. Can we identify the high-risk patients to be screened? A genetic approach. Digestion 2007;76(1):7–19. Copyright: S. Karger AG, Basel, Switzerland.
Family history is the most important tool for the identification of hereditary CRC (Fig. 1). It should therefore be assessed during all primary contacts between doctors and patients. The minimum information that should be collected is as follows:
Figure 1.
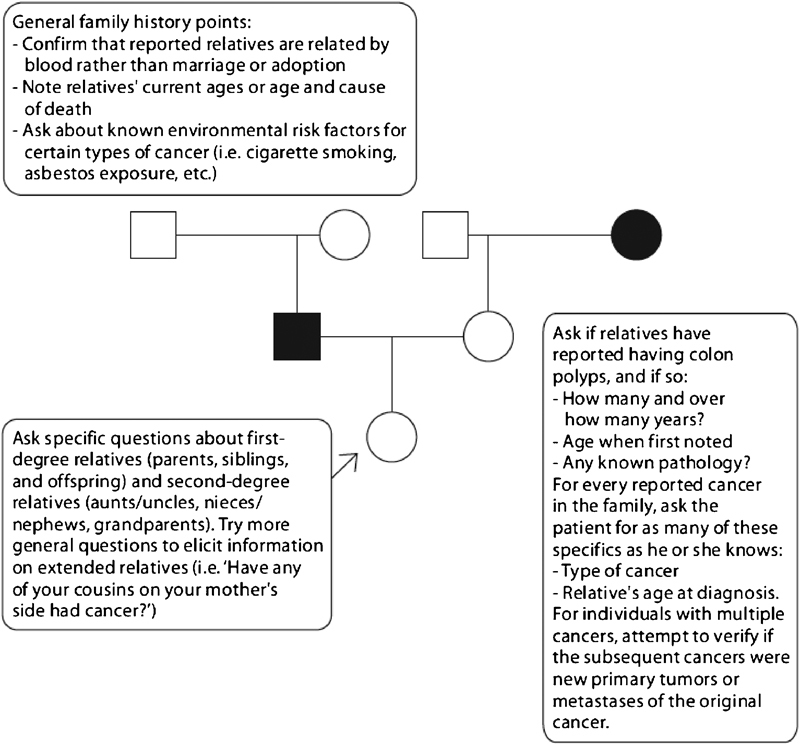
Family history gathering directed at identifying patients at high risk for a colorectal cancer predisposition syndrome. From: Gammon A, Kohlmann W, Burt R. Can we identify the high-risk patients to be screened? A genetic approach. Digestion 2007;76(1):7–19. Copyright: S. Karger AG, Basel, Switzerland.
The size of the family
The number of first-degree relatives (and second-degree relatives if a first-degree relative is affected) with cancer
Type of cancer
Age at diagnosis11
Identification of Individuals at High Risk for Lynch Syndrome (including Muir-Torre syndrome and Turcot syndrome) and Familial Colorectal Cancer Type X
Lynch syndrome is an autosomal dominant condition caused by a mutation in one of several DNA mismatch repair genes12,13 that maintain the fidelity of DNA replication. These genes encode proteins that form a multimeric DNA mismatch repair (MMR) complex that corrects the small insertions or deletions that frequently occur during somatic replication.14,15,16 Defective MMR proteins lead to the so-called mutator or replication error phenotype where a markedly increased rate of mutation, inevitably involving cell-cycle regulation, increases the potential for malignancy.17 The average age of CRC diagnosis in Lynch syndrome is ~44 years, versus 64 years in sporadic cancer, although individuals with mutations in MSH6 have a mean age at CRC diagnosis of 55 to 57 years.18 The lifetime risk for developing CRC is 80%, although evidence of differing patterns of penetrance are emerging for each gene,19,20,21 with CRC occurring earlier in male MLH1-carriers than female. Lynch syndrome accounts for ~3% to 5% of all CRC22,23 and 2% of endometrial cancer.24 It is the commonest inherited colon cancer syndrome.
Families who meet clinical diagnostic criteria (Amsterdam criteria, see below) with intact MMR have been classified as familial colorectal cancer type X.25,26,27,28,29,30 It is probable that there are unidentified genes that are associated with this phenotype. There is a trend to only refer to Lynch syndrome in patients with a known MMR gene defect31; the term hereditary nonpolyposis colorectal cancer (HNPCC) remains as an umbrella term including broadly all those who fulfill clinical diagnostic criteria.
Clinical Suspicion and Diagnosis
Patients with Lynch syndrome may have synchronous and metachronous CRC with a predilection for right-sided cancer, proximal to the splenic flexure. There are also several extracolonic manifestations of this disease including cancers of the small intestine, endometrium pancreas and biliary tract, brain, and transitional cell carcinoma of the ureter and renal pelvis.32,33,34,35 The most common extracolonic cancer is endometrial adenocarcinoma, which affects at least one female in about half of all Lynch syndrome families with mean age at diagnosis also in the fifth decade.36 The lifetime risk for endometrial cancer in women may be 21 to 71% at age 7037; risk varies with the underlying gene involved. For example, MSH2 mutation carriers have higher endometrial cancer risk than do carriers of MLH1 mutations. Associated endometrial cancer subtypes include endometrioid, clear cell carcinoma, uterine papillary serous carcinoma, and malignant mixed Mullerian tumors.38
Clinical Criteria
In 1991, the Amsterdam criteria (Table 2) were proposed to identify individuals who were likely to be mutation carriers. They required the presence of young-onset CRC, in addition to a family history of three CRCs involving two successive generations.39 The Amsterdam II criteria include other Lynch-associated malignancies,40 and therefore have a higher sensitivity than the Amsterdam criteria. With the introduction of tumor molecular analysis for Lynch syndrome, the Bethesda guidelines were proposed to help identify patients for additional tumor and genetic testing.41 Studies evaluating the performance of clinical criteria in populations at high-risk for Lynch syndrome have shown that the Bethesda guidelines have a higher sensitivity than the Amsterdam I and II criteria.42
Table 2. Amsterdam I Criteria (1991)*.
| All criteria must be met: |
| 1. One member diagnosed with colorectal cancer before age 50 |
| 2. Two affected generations |
| 3. Three affected relatives, one of them a first-degree relative of the other two |
| 4. Familial adenomatous polyposis excluded |
| 5. Tumors verified by pathologic examination. |
| Amsterdam II criteria (1999)† |
| Identical to the Amsterdam I criteria except in broadening the third criterion, it still requires at least three affected relatives, but now also requires any of the following recognized Lynch syndrome-related cancers: colorectal, endometrial, small bowel, ureter, or renal pelvis. |
Data from Vasen H, Watson P, Mecklin J, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC. Gastroenterology 1999; 116(6):1453–1456.
Data from LIor X, Pons E, Xicola R, et al. Differential features of colorectal cancers fulfilling Amsterdam criteria without involvement of the mutator pathway. Clin Cancer Res 2005; 11(20):7304–7310.
Recently, revised Bethesda guidelines were proposed to improve the accuracy of identifying patients with Lynch syndrome (see Table 3).43 Hampel et al44 highlighted the limitations of these criteria, noting that in a population-based cohort of 1066 patients with CRC, 5 of 23 mutation carriers did not meet the Bethesda or revised Bethesda criteria and would otherwise have been missed if genetic evaluation was limited to individuals who met these criteria. In light of these limitations, an alternative strategy involving universal tumor testing for microsatellite instability (MSI) and/or immunohistochemistry (IHC) of all individuals with CRC was proposed. Even if this strategy were found to be cost effective, it may still fail to identify cases in which MMR mutations disrupt MMR function but do not result in MSI, as seen with MSH6 mutations or when IHC results are normal despite a nonfunctional MMR protein.45
Table 3. Revised Bethesda Criteria (2004).
| Any one criterion would support MSI testing: |
| 1. One member diagnosed with CRC before age 50 |
| 2. Presence of synchronous, metachronous CRC or other Lynch syndrome-associated tumor* in an individual regardless of age |
| 3. CRC with MSI-H pathologic features diagnosed in an individual younger than 60 years (presence of tumor-infiltrating lymphocytes, Crohn-like lymphocytic reaction, mucinous/ signet-ring differentiation or medullary growth pattern) |
| 4. CRC or Lynch syndrome-associated tumor* in at least one first-degree relative younger than 50 |
| 5. CRC or Lynch syndrome-associated tumor* diagnosed in two |
CRC, Colorectal cancer; MSI, microsatellite instability.
Endometrial, stomach, ovarian, pancreas, small bowel, biliary tract, ureter or renal pelvis, brain, sebaceous gland adenoma or keratoacanthoma.
Data from Valle L, Perea J, Carbonell P, et al. Clinicopathologic and pedigree differences in Amsterdam I-positive hereditary nonpolyposis colorectal cancer families according to tumor microsatellite instability status. J Clin Oncol 2007; 25(7):781–786.
Microsatellite Instability and Immunohistochemistry
Various tumor-testing strategies exist in patients suspected of having Lynch syndrome. This includes testing for microsatellite instability (MSI) and/or immunohistochemistry (IHC). MSI refers to a mutation in small DNA segments that are 100 to 200 base pairs in length. Due to defects in MMR gene mutations in these sequences, there are changes in the length of individual microsatellites. This change in the length of the strands is referred to as microsatellite instability. A standard panel of five microsatellites is recommended for tumor testing by the National Cancer Institute. Tumors are reported as MSI-high (MSI-H), MSI-low (MSI-L), or MSI-stable (MSS). MSI-H tumors demonstrate changes in two or more of the markers, whereas MSI-L have only one positive marker. MSS refer to tumors with no abnormal markers.
Approximately 90% of Lynch syndrome-associated CRCs will have MSI-H making this analysis very sensitive. The specificity is much lower, however, as ~15% of sporadic CRCs are also MSI-H. Sporadic MSI-H CRCs are the result of somatic hypermethylation of the hMLH1 promoter region, as opposed to Lynch syndrome tumors, which are the result of a germline gene mutation.
Tumor testing with IHC utilizes four monoclonal antibodies specific for hMLH1, hMSH2, hMSH6, and hPMS2 proteins to evaluate tumors for MMR deficiency. The sensitivity of IHC is comparable to that of MSI analysis. However, IHC analysis can direct genetic testing to the appropriate MMR gene when loss of MMR protein expression is identified. Additional tumor testing, including BRAF mutation and hMLH1 promoter methylation analyses, can be helpful in differentiating sporadic versus Lynch syndrome-associated CRCs. Tumor testing in endometrial cancers has proven to be equally as effective at identifying Lynch syndrome.46 Other Lynch syndrome-associated tumors also frequently display MSI-H and loss of MMR protein expression, although their sensitivity and specificity in the clinical setting is not well established.4
Genetic Prediction Models
Prediction models have been developed to help identify individuals at risk for Lynch syndrome. The Barnetson et al47 model, the PREMM,48,49,50 and the MMRpro model are often cited as examples of this approach.51 Barnetson et al47 analyzed a population-based cohort of 870 patients diagnosed with CRC before 55 years of age. They developed a model to predict MLH1, MSH2, and MSH6 mutations using multivariable regression analysis. The model included patient age, gender, tumor location, presence of synchronous and metachronous CRCs, family history of endometrial cancer and CRC, age of the youngest relative with CRC, tumor MSI and IHC results. The model was then validated in 155 patients with CRC diagnosed before 45 years of age. The model had a sensitivity of 62%, specificity of 97%, and positive predictive value of 80%. The performance of the model exceeded that of the Bethesda and Amsterdam criteria. The ability of the model to separate mutation carriers from those without an MMR mutation (model discrimination) was similar between the derivation and validation cohorts. However, this model was developed and validated in patients with young-onset CRC and did not include Lynch-associated cancers other than endometrial cancer.
The PREMM48,49 model (prediction of mutations in MLH1 and MSH2) was developed using a cohort of 1914 individuals at moderate risk for Lynch syndrome.50 Clinical data from 898 probands were used for model derivation. The model was then validated in a separate large cohort of probands. The final multivariable logistic regression model included diagnosis of CRC, colonic adenomas, extracolonic Lynch-associated cancers, and a family history of Lynch cancers. The PREMM48,49 model showed good discrimination with an area under the receiver operating curve of 0.80. It has also been validated in a large population-based cohort.52 Strengths of the model include its ability to incorporate extracolonic Lynch-associated neoplasms and provide individualized risk prediction using an easy-to-use web-based calculator (Fig. 2). However, the PREMM48,49 model does not consider family size or unaffected family members.
Figure 2.
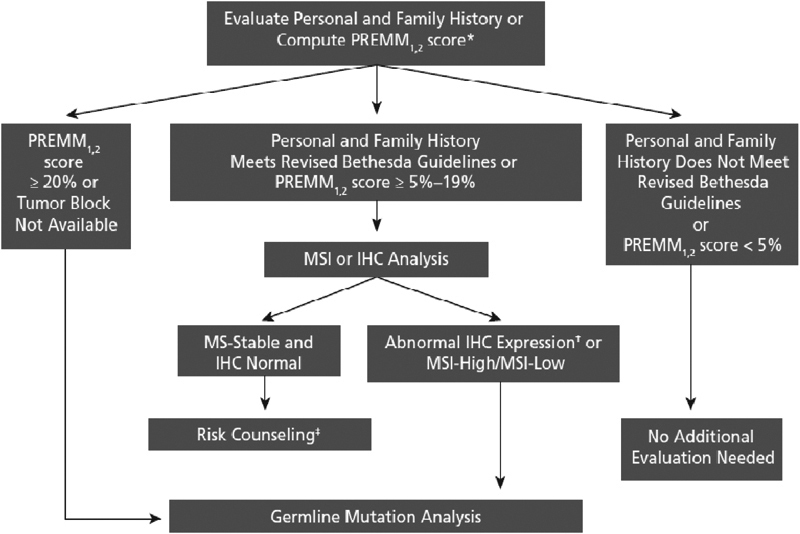
Algorithm for genetic evaluation of individuals with colorectal cancer based on revised Bethesda guidelines and PREMM score. IHC, immunohistochemistry; MSI, microsatellite instability.*Other models may be used. Each model has its own prespecified cutoff (PREMM1,2: http://www.dana-farber.org/pat/cancer/gastrointestinal/crc-calculator/default.asp; Barnetson: https://hnpccpredict.hgu.mrc.ac.uk/; MMR pro: http://astor.som.jhmi.edu/Bayes-Mendel/). †If loss of MLH1 expression, BRAF analysis should be performed. ‡Surveillance recommendations based on personal and family history. From Grover S, Syngal S. Genetic testing in gastroenterology: Lynch syndrome. Best Pract Res Clin Gastroentrol 2009; 23:185–196. Reprinted with kind permission of Elsevier.
The MMRpro model uses estimates of mutation prevalence and the penetrance of MMR genes to estimate the probability of carrying a clinically significant mutation in MMR genes.51 The model can also estimate the probability of developing CRC or endometrial cancer in unaffected relatives. The MMRpro model proved to have a higher sensitivity than the Bethesda guidelines, although it slightly overpredicted the number of carriers. Advantages of the MMRpro model include its ability to account for family size by including unaffected relatives and to incorporate MSI data. For individuals with indeterminate or uninformative genetic testing results, the MMRpro model can provide postsequencing probability of a deleterious mutation. These estimates are particularly valuable given that genetic testing has limitations in sensitivity and that uninformative results may lead to false reassurance and poor adherence to recommended cancer screening.53
Muir-Torre and Turcot Syndromes
Muir-Torre syndrome is a variant of Lynch syndrome that combines colorectal tumors with multiple cutaneous adnexal neoplasms (sebaceous adenomas and carcinomas and keratoacanthomas) and tumors in endometrium, kidney, ovaries, stomach, and small intestine. Mutations in MSH2 account for most of Muir-Torre syndrome.54,55,56 Turcot syndrome represents patients with a Lynch syndrome CRC in association with a glioblastoma. This entity should not be confused with medulloblastoma in familial adenomatous polyposis (FAP), also called Turcot syndrome.
Identification of Individuals at High Risk for Familial Adenomatous Polyposis (including Gardner Syndrome) and Attenuated Familial Adenomatous Polyposis (AFAP)
Familial adenomatous polyposis (FAP) is an autosomal dominant syndrome caused by germline mutations of the APC (adenomatous polyposis coli) gene.57 This gene is located on chromosome 5. FAP has a frequency of 1 in 5000 to 10,000 live births and affects males and females equally.58 It accounts for 1% of all CRC.59 FAP is the result of an inactivating mutation in APC and clinical presentation may be associated with the site of mutation, although it may also be clinically heterogeneous even within the same family. This suggests a role for modifier genes and/or environmental factors in modulating disease expression.60
Clinical Suspicion and Diagnosis
Classic FAP
Colorectal polyposis, numbering from hundreds to thousands, is nearly pathognomic of FAP. Classic FAP is suspected when an individual presents with >100 colorectal adenomas or has multiple adenomas and is a first-degree family member of a patient diagnosed with FAP. The presence of extracolonic manifestations reinforces this diagnosis.61
Polyps are generally less than 1 cm and occur throughout the colorectum with a predilection for sigmoid colon and rectum.62 They may be sessile or pedunculated with histology varying from tubular to villous adenoma. The age at onset of colorectal adenomas is variable, being present in only 15% of FAP gene carriers at age 10 years, 75% by age 20, and 90% by 30 years if untreated.63,64 In a review of more than 180 families and 922 affected individuals, the mean age at presentation was 27 and mean age at colectomy was 29.65
FAP has multiple extracolonic manifestations (Table 4). All three primordial germ layers may have associated abnormalities in an FAP patient. Endodermal lesions include gastric and small bowel polyps and carcinomas. Mesodermal abnormalities include desmoid tumors, osteomas, and dental abnormalities. Ectodermal lesions can affect the eye, brain, and skin.
Table 4. Extracolonic Tumor Risks in Familial Adenomatous Polyposis.
| Tumor | Relative Risk | Absolute Lifetime Risk (%) |
|---|---|---|
| Desmoid | 852.0 | 15.0 |
| Duodenum | 330.8 | 3.0–5.0 |
| Thyroid | 7.6 | 2.0 (<12% in women) |
| Brain | 7.0 | 2.0 |
| Ampullary | 123.7 | 1.7 |
| Pancreas | 4.5 | 1.7 |
| Hepatoblastoma | 847.0 | 1.6 |
| Gastric | – | 0.6* |
The Leeds Castle Polyposis Group.
From the National Cancer Institute, Genetics of Colorectal Cancer. Available at: http://www.cancer.gov/cancertopics/pdq/genetics/colorectal/health Professional/Table 4. Accessed November 30, 2009.
From Shah NB, Lindor NM. Lower gastrointestinal tract cancer predisposition syndromes. Hematol Oncol Clin N Am 2010;24:1229–1252. Reprinted with kind permission of Elsevier.
Desmoid tumors are histologically benign clonal neoplasms composed of fibrous tissue. They arise as mostly intraabdominal soft tissue tumors66 and occur in ~10 to 25% of patients with FAP.67 Trauma has been suggested to be an inciting factor, as 84% of FAP-associated desmoids developed within 5 years of abdominal surgery in one series.68 Desmoid tumors do not metastasize, but they can be locally invasive and can cause significant mass effect, obstruction, pain, and death. Desmoid tumors may also occur sporadically or in a hereditary manner without colon findings,69,70 but in cases of families with desmoid tumors or individuals with two or more desmoids, attempts should be made to exclude APC mutation. Gardner syndrome refers to the association of FAP with desmoids tumors. Gardner syndrome is not genetically distinct from FAP.
Osteomas may occur in any bone, but often localize to the face or skull. Dental abnormalities affect 70% of patients with FAP and include supernumerary teeth, congenitally absent teeth, fused roots, and osteomas of the jaw.62 Depending on the location, they can lead to symptoms and identification of FAP. Congenital hypertrophy of retinal pigment epithelium (CHRPE) is an asymptomatic hamartoma of the retinal epithelium occurring in 66 to 92% of patients with FAP.71 Detection of CHRPE on funduscopic exam used to be a significant tool for screening patients for FAP. In the era of molecular diagnosis, this technique is no longer useful.
Extracolonic tumors (Table 4) cause significant morbidity in FAP. Desmoid tumors and duodenal cancers being the second and third commonest causes of death after CRC.72 In one series, 88% of patients with FAP developed duodenal polyps, often near the ampulla and papilla,73 with a lifetime risk of duodenal carcinoma of 4 to 12%.74 Duodenal polyps may be associated with different germline APC mutations than those with severe colonic polyposis.75 Gastric cystic fundic gland polyps may develop in up to 33% of FAP patients. Gastric carcinoma is rare in FAP, but may be higher in Asian populations.76,77 Hepatoblastoma occurs in an estimated 0.6% of children before 6 years, but is rare thereafter.78 Thyroid carcinoma may affect 12% of patients with FAP,79 but carries a good prognosis. They are predominantly well-differentiated papillary cancers affecting young women. Before suspicion of FAP is confirmed, studies to identify possible extracolonic manifestations must be completed.61 The combination of CRC and brain tumors was referred to as Turcot syndrome. However, molecular studies have shown that although the combination of colonic polyposis and medulloblastoma is associated with APC mutations, the combination of CRC and glioblastoma is associated with defective mismatch repair genes and is also called Turcot syndrome.80
Attenuated FAP
Attenuated FAP (AFAP) is defined as the presence of fewer than 100 synchronous colorectal adenomas. This presentation of FAP shows a right-sided colonic predilection and presents at a later age.81
Genetic Diagnosis
Classic FAP
APC is a tumor suppressor gene consisting of 15 exons and encodes a protein of 2843 amino acids82 that is involved in cell adhesion, signal transduction, transcription regulation, cell cycle control, apoptosis, and maintenance of the fidelity of chromosomal segregation. As part of a scaffolding protein complex, it negatively regulates Wnt signaling.82,83 APC inactivation is the hallmark of the chromosomal instability pathway (CIN) phenotype that occurs in most CRCs. Increasing size, number, and worsening histology of polyps reflect the linear process of carcinogenesis along the CIN pathway. More than 800 different APC germline mutations were reported84 through 2007.57 APC mutations are not distributed evenly, with “hotspots” at codons 1061 and 1309 accounting for ~11% and 17%, respectively, of germline mutations. Most lie in the mutation cluster region (MCR) between codons 1250 and 1464 in the 50 region of exon 15.84
Mutation analysis can identify sequence changes in up to 95% of classic FAP cases. However, the early development of adenomas raises special considerations relating to genetic testing of children. Genetic consultation is recommended for newly diagnosed FAP families as this can determine whether genetic testing would be informative for at risk relatives. A negative test within a family with a known APC mutation allows colorectal screening to revert to that recommended to the population with background cancer risk, i.e., colonoscopy or equivalent test starting at age 50.
Management can be affected by genotype, as severity of disease and extracolonic tumors may correlate with the location of APC mutations. Mutations between codons 1250 and 1464, especially codon 1309, often lead to profuse polyposis with earlier presentations.85,86,87,88
Attenuated FAP
AFAP has been linked to mutations in exons 1 to 4, 30 regions of APC distal to codon 1580, and the alternatively spliced site of exon 9.57,84,89,90 However, some patients with this phenotype and no identified APC mutation have been shown to have compound heterozygous mutations in the base excision repair gene MYH,91 leaving open the possibility that cases of AFAP may be related to MYH-associated polyposis (MAP). If germline APC mutation testing is negative in suspected AFAP, testing for MYH mutations may be indicated.
If negative or uncertain results are obtained, an analysis of the APC gene and MSI should be performed to evaluate the possible presence of Lynch syndrome. As with classic FAP, if a mutation in APC is identified, the study should be offered to direct relatives. If the mutation is in MYH, which is recessively inherited, it is necessary to recommend the study to first-degree relatives and to the spouse of the affected individual.61
Identification of Individuals at High Risk for MYH-Associated Polyposis (MAP)
Mutations in the MYH (or MUTYH) gene on 1p32.1-p34.3 cause an autosomal recessive CRC predisposition syndrome associated with multiple colonic polyps. It may be indistinguishable from classical or attenuated FAP.91,92 Establishing the correct genetic diagnosis will direct cancer surveillance for family members. Classical and attenuated FAP are dominantly inherited with risk for successive generations, whereas only a single generation is at risk for recessively inherited.31
MYH is a base-excision repair (BER) gene that repairs mutations caused by reactive oxygen species.93 It codes for a DNA glycosylase that identifies and removes adenine residues that have been incorrectly paired with 8-oxo-7, 8-dihydro-20-deoxyguanosine (8-oxodG).94 Failure to correct this causes an increase in G:C / T:A transversions, particularly at GAA sequences, which leads to a stop codon, TAA. The APC gene is a major downstream target of MYH mutations.92 MAP tumors are generally MSS. More than 80 germline variants have been reported. Most are missense, but also reported are six truncating mutations, splice-site mutations, and several small insertion/ deletions.95 The commonest mutations in whites are Y179C and G396D (formerly called Y165C and G382D, respectively) accounting for 53% and 32% of all mutations, respectively. The Y179C mutation is more deleterious than the G396D mutation.96,97 Approximately 1% of the general population is heterozygous for an MYH mutation. MYH carriers could acquire a somatic mutation (a “second hit”) in the wild-type allele and develop CRC; however, somatic MYH mutations are infrequent in CRC.98 Moreover, the role of somatic mutations in MYH in the development of nonfamilial CRC is yet to be understood. It is notable that MYH mutations have not yet been implicated in nongastrointestinal cancers in which reactive oxygen species are thought to play a role in carcinogenesis, including lung, breast, kidney, liver, and prostate.99,100,101,102,103
Clinical Suspicion and Diagnosis
MAP tends to present later than classical FAP. In two major series, the mean ages at presentation were 46 and 51 with a range of 13 to 70 and the presenting feature in 50% of cases was CRC.104,105 Jenkins and colleagues106 reported cumulative risk to age 70 of 80%, which is a 50-fold increased risk of CRC compared with the general population. There was also a threefold increase in risk in monoallelic carriers (8% cumulative risk to age 70), but other data show no appreciable increase risk in monoallelic MYH carriers.96,107,108,109,110
Colorectal polyps in MAP range in number from a few to more than 500 and tend to be mainly small tubular or tubulovillous adenomas with mild dysplasia with occasional hyperplastic polyps. Cancer can arise anywhere in the colorectum, but the adenomas may show a right colonic predilection.31
Extracolonic manifestations of MAP include duodenal adenomas, gastric fundic gland polyps, CHRPE, osteomas and dental anomalies, and desmoid tumors. These findings were previously hallmarks of FAP, but have now been reported in MAP. Duodenal adenomas with or without duodenal adenocarcinoma have been reported in ~5%.97,111
Identification of Individuals at High Risk for Hamartomatous Polyposis Syndromes
Peutz-Jeghers Syndrome
Peutz-Jeghers syndrome (PJS) is a rare, highly penetrant, autosomal dominant disorder characterized by hamartomatous polyposis and mucocutaneous pigmentation. The incidence of PJS is estimated to be 1 in 8300 to 1 in 200,000 live births112; 25% of cases appear to be nonfamilial. PJS has been reported worldwide113,114 and occurs in males and females equally. There is variability in both the severity of disease as well as age of onset of symptoms.
PJS carries an increased risk for multiple benign and malignant tumors. The cumulative risk of any cancer is 67 to 85% by age 70 and the cumulative risk for CRC is 3% (40 years), 5% (50 years), 15% (60 years), and 39% (70 years).115,116 The risk to age 70 for cancers of the pancreas, uterus/ovary/cervix, breast, and lung were 11%, 18%, 45%, and 17%, in the same series, respectively. An increased risk of primitive biliary cancer was reported in PJS.117 No correlation has been found between risk of cancer and severity of polyposis or presence of pigmentation.118
Clinical Suspicion and Diagnosis
Polyps may occur anywhere along the gastrointestinal tract, but occur most consistently in the jejunum.31 Multiple adenomas can occur in the colon. Classical PJS intestinal polyps are hamartomatous and experienced pathologists are capable of distinguishing PJS polyps from juvenile polyps. PJS polyps manifest characteristic arborization and hypertrophy or hyperplasia of the smooth-muscle layer.119
Extraintestinal sites of PJS polyps include kidney, ureter, gallbladder, bronchus, and nasal passages. About one-third of patients will develop polyp-related symptoms by age 10 years and close to two-thirds by age 20 years.120 The hyperpigmented macules of PJS develop in 95% of affected individuals and arise most commonly in the perioral region, around the eyes and nostrils, on the buccal mucosa, the perianal area, and on the digits of hands and feet. They usually appear by the end of the first year of life and are almost always present by age 5 years.119 Macules may be dark blue to dark brown, vary in size from 1 to 5 mm, and may fade in puberty and adulthood, and are not precancerous.
PJS is associated with mutations in STK11. STK11 encodes for a 433-amino acid protein that is ubiquitously expressed and present primarily in the cytoplasm and to a lesser extent in the nucleus.121,122 STK11 is the only tyrosine kinase known to function as a tumor suppressor by physically associating with TP53 to regulate TP53-dependent apoptosis pathways.123,124 STK11 also interacts with PTEN, which is responsible for other hereditary hamartoma syndromes and also plays a role in the vascular endothelial growth factor (VEGF) pathway and cellular polarity. Inactivation of STK11 is a critical early event in the development of hamartomas and adenocarcinomas.125 Adenocarcinomas in Peutz-Jeghers syndrome demonstrate altered TP53 expression and loss of heterozygosity (LOH) in 17p and 18q. Microsatellite instability, LOH near the APC gene, or KRAS mutations have been identified in some tumors,125 with indications that tumorigenic potential of STK11 mutations is mediated through alternative mechanisms in different tissues, especially those in which hamartoma development is not a feature. Hamartomatous polyps have generally been considered to have a very low malignant potential; it was uncertain that PJS-associated hamartomas were the premalignant lesions in PJS. However, molecular and histologic studies have confirmed that hamartomatous polyps can undergo malignant transformation in PJS.126 It is not known whether inactivation of both STK11 alleles is necessary for carcinogenesis, or if a 50% decrease in protein expression is sufficient (haploinsufficiency). Data from studies in lkb1 +/− and lkb1 −/− mice (LKB1, also called STK 11, knockout mice) support both possibilities.127,128
Genetic Diagnosis
Clinical genetic testing for PJS is available. If a disease-causing mutation has been identified, it is appropriate to offer genetic testing to at risk relatives and, if positive, surveillance is indicated. If no disease-causing mutation is found in an individual with PJS, then first-degree relatives must be advised that they may still be at risk for PJS and that PJS cancer surveillance is advisable.31 Germline mutations in STK11 encoding a tyrosine kinase on chromosome 19p13.3 have been identified in nearly all PJS families,129,130 and 94% of patients with PJS overall.131
Juvenile Polyposis Syndrome
Juvenile polyposis (JP) is an autosomal dominant disorder characterized by multiple (5–200) hamartomatous polyps of the gastrointestinal tract.132 It is the most common of the hamartomatous polyp syndromes. Population prevalence is estimated to be between 1 in 16,000 and 1 in 100,000.133 Twenty percent to 50% of cases are inherited. Solitary juvenile polyps may be seen in ~2% of healthy children, but these are seldom dysplastic and are not associated with increased malignancy or extracolonic manifestations.131,134 In JP, “juvenile” refers to the type of polyp (resembling sporadic inflammatory hamartomatous polyps of childhood) rather than the age of onset, although most affected individuals have some polyps by age 20 years. The hamartomas of JP have a frondlike growth pattern, fewer stroma, and dilated glands with more proliferative smaller glands compared with solitary, sporadic juvenile polyps.135
Most juvenile polyps are benign, but malignant transformation may occur resulting in increased lifetime risk for cancers of the colon (10–40%), stomach (21%), and less commonly involving the small bowel and pancreas. The lifetime risk of cancers has been hard to define and may vary with underlying genetic cause; it is likely reduced by screening polypectomies. Malignant transformation is suspected to follow a traditional adenomatous polyp to cancer transformation sequence.136,137 However, additional work is required to determine if individuals with JP are also predisposed to malignancy separately from the predisposition to polyps.
JP may be misdiagnosed, as it shares clinical features with several other colonic hamartomatous polyp syndromes (Cowden, Bannayan-Riley-Ruvalcaba, Peutz-Jeghers, basal cell nevus/Gorlin). Because of this overlap, JP remains a diagnosis of exclusion. Physical examination, family history, and molecular testing may help differentiate between these possibilities.138,139
Clinical Suspicion and Diagnosis
The clinical criteria for diagnosis of JP include five juvenile polyps in the colon/rectum, juvenile polyps throughout the gastrointestinal tract, or any number of juvenile polyps with a family history of JP.131
Although only five polyps have been proposed as the minimum number for diagnosis, some individuals will have more than 100 polyps. In a review of 272 individuals with JP of undefined genetic subtype, 98% had involvement of the colorectum, 14% of the stomach, 7% of the jejunum and ileum, and 2% of the duodenum.140 Polyps usually range from 5 to 50 mm in size, can be single or multilobulated, are spherical in shape, and commonly show surface erosion. Clinical symptoms of JP may include bleeding, diarrhea, abdominal pain, intussusceptions, rectal prolapse, and even protein-losing enteropathy. Digital clubbing has been noted, perhaps owing to the overlap with hereditary hemorrhagic telangiectasia and arteriovenous shunting in these patients.141
Genetic Diagnosis
JP is clinically and genetically heterogeneous. Three genes, SMAD4, BMPR1A, and ENG, have been implicated so far. Each encodes proteins of either transforming growth factor- (TGF-) b or bone morphogenetic protein- (BMP-) signaling pathways. The low combined mutation detection rate has prompted a search for other candidate genes/proteins within these pathways. About 20% of individuals with JP have a mutation of SMAD (also known as MADH4 or DPC).139,142,143 SMAD4 is part of the TGF-b signal transduction pathway. The SMAD gene family is on chromosome 18q21.1, adjacent to DCC (deleted in colon cancer). SMAD4 complexes combine with other members of the SMAD family of proteins to transmit the TGF-b growth-suppressing signal from the cell surface receptor to nuclear downstream targets, mediating apoptosis and growth inhibition.
It has been postulated that the abundant stroma in JP may create an abnormal microenvironment, disrupting TGF-b signaling.144,145 This theory is supported by the fact that as hamartomatous polyps enlarge and mesenchymal component expands, they take on a serrated or villous-type configuration associated with epithelial dysplasia. Mutations in BMPR1A (ALK3) at 10q22.3, are found in ~20% to 25% of individuals with JP.139,142,143 BMPR1A is a serine-threonine kinase type I receptor of the TGF-b superfamily, which when activated leads to phosphorylation of SMAD4. A reduced number of gastric polyps have been observed in BMPR1A mutation-positive patients compared with SMAD4 mutation-positive patients.142,146,147
Mutations in ENG on chromosome 9q34.1 have been reported in very-early-onset JP.148 ENG encodes endoglin, an accessory receptor protein that binds to specific TGF-b proteins.149 Mutations in ENG are more often found in individuals with hereditary hemorrhagic telangiectasia (HHT). The combined syndrome of JPS and HHT (termed JPS/HHT) may be present in 15% to 22% of individuals with a SMAD4 mutation and has also been associated with ENG. The prevalence of ENG mutations in patients with JP without HHT has yet to be adequately described.150
Hereditary Mixed Polyposis Syndrome
The term “hereditary mixed polyposis syndrome” (HMPS) unites a collection of polyposis syndromes showing a mixture of various types of polyp. In some patients, mutations of the PTEN or the BMPR1A gene are demonstrated. These cases should be treated as variants of Cowden syndrome and JPS and treated accordingly.151
PTEN Mutations (Cowden Disease and Bannayan-Ruvalcaba-Riley Syndrome)
PTEN mutations lead to the Cowden syndrome, Bannayan–Riley–Ruvalcaba syndrome, Proteus syndrome, and Proteus-like syndrome. These are grouped into PTEN hamartoma tumor syndrome (PHTS).152 Cowden disease (also known as Cowden syndrome or multiple hamartoma syndrome) is an autosomal dominant disease characterized by facial trichilemmomas, oral papillomas, multinodular goiter, fibrocystic breast disease, esophageal glycogenic acanthosis, and intestinal hamartomas.152,153 Breast and thyroid cancer risk are most pronounced in Cowden disease, with colon cancer developing in up to 10% of patients. Autosomal dominant germline mutations of the PTEN gene have been identified in the majority of patients with Cowden disease and predispose to Bannayan-Ruvalcaba-Riley syndrome, which shares characteristics with Cowden disease and additionally includes slowed psychomotor development and pigmentary spotting of the penis.154,155
In comparison to the gatekeeper function of the APC gene and the caretaker roles of the mismatch repair and MYH genes, the genes predisposing to hamartomatous polyposis syndromes have been dubbed ''landscaper'' genes.156 In sporadic circumstances, nonneoplastic hamartomatous polyps are not believed to confer a significant cancer risk. In comparison, germline mutations and somatic inactivation of the STK11, SMAD4, BMPR1A, and PTEN genes in hamartomatous polyposis syndromes are believed to create an epithelial milieu (or landscape) at risk for neoplastic development.157
Cronkhite-Canada Syndrome
Cronkhite-Canada syndrome is a rare systemic disease first reported in 1955 by Cronkhite and Canada.158 Patients of European or Asian descent are most frequently affected. The estimated incidence of CCS is one per million based on the largest study performed to date.159,160 The mean age of onset is estimated to be in the fifth to sixth decade, with a slight male predominance in 3:2 ratio.161 The etiology of CCS is currently unknown. So far, there is no strong evidence to suggest a familial predisposition (Table 5). However, clinicians will often be asked to differentiate this syndrome from other polyposis syndromes.
Table 5. Lower Gastrointestinal Tract Cancer Predisposition Syndromes.
| Syndromes | Gene | Inheritance | Gastrointestinal Polyp Histology | Extracolonic Cancers | Other Associations | Estimated Cumulative Colorectal Cancer Risk |
|---|---|---|---|---|---|---|
| Peutz-Jeghers syndrome | STK11 | AD | Hamartoma*, adenoma† | Cervical, uterus, ovarian, breast, Sertoli cell tumors, entire GI tract, pancreato-biliary | Hyperestrogenism, mucosal pigmentation + , Polyps in gallbladder, ureter, nasal and bronchial passages | 39% by 70 years |
| Juvenile syndrome | BMPR1A SMAD4ENG | AD | Hamartoma*, adenoma† | Gastric, small intestine, pancreas | HHT | 17% 68% by 60 years |
| Familial adenomatous polyposis | APC | AD | Adenoma*, cystic fundic gland polyp | Duodenal, hepatoblastoma medulloblastoma, papillary thyroid | Desmoid tumors, osteomas, CHRPE, dental anomalies, gastric polyps | 90% by 45 years (69% by 80 years attenuated FAP) |
| Lynch syndrome | MLH1, MSH2, PMS2, MSH6 | AD | Adenoma† | Endometrial, ovarian, gastric, small intestine, pancreato-biliary, renal pelvis and ureter, sebaceous carcinoma, keratoacanthoma, glioblastoma | Sebaceous adenoma | 80% by 75 years |
| MYH-associated polyposis | MYH | AR | Adenoma*, hyperplastic†, gastric fundic gland polyp | Duodenal | Duodenal adenoma, gastric polyps CHRPE, osteomas, dental anomalies, desmoid tumors | 80% by 70 years |
AD, autosomal dominant; AR, autosomal recessive; CHRPE, congenital hypertrophy of retinal pigment epithelium; GI, gastrointestinal; HHT, hereditary hemorrhagic telangiectasis.
Very common in this condition.
Seen in this condition.
From Shah NB, Lindor NM. Lower gastrointestinal tract cancer predisposition syndromes. Hematol Oncol Clin N Am 2010;24:1229–1252. Reprinted with kind permission of Elsevier.
Several Cronkhite-Canada cases have been associated with elevated antinuclear antibody (ANA) and IgG4 levels.159,162,163 There is also an association between CCS to hypothyroidism164 and various autoimmune diseases such as systemic lupus erythematous, rheumatoid arthritis, and scleroderma.165 Other authors have reported mental stress and physical fatigue may also contribute to the etiology.160 The symptoms of CCS can vary, but classically the syndrome is characterized by the presence of diffuse gastrointestinal polyposis, dystrophic changes in nails, alopecia, cutaneous hyperpigmentation, diarrhea, and weight loss. Other symptoms such as hypogeusia, altered or blunted taste sensation, and xerostomia, dry mouth, have also been described in the literature.166
Endoscopic appearance of CCS varies according to current literature. Gastric mucosa has been described as being thickened hypertrophic gastric folds mimicking Menetrier disease to atrophic appearing with polypoid lesions.167 Colonic polyps have been characterized as sessile and “strawberry like” in one study.168
The optimal treatment of CCS is currently unknown due in part to its rarity. Nutritional support aimed at improving electrolytes, vitamin, and mineral deficiencies can rarely induce complete remission. In fact, current literature favors a combination therapy based on nutritional support and corticosteroids.169 TPN therapy in combination with partial bowel rest may provide crucial nutritional support while allowing the disease process to enter remission. Other therapies such as antihistamine receptor antagonist agents and cromolyn sodium have also been used as supplement therapy in cases where degranulating eosinophils and mast cells are seen on biopsy.170 One case report has also suggested improvement of CCS after eradication of Helicobacter pylori infection.171 The total treatment period is not well defined, varying from 6 to 12 months of combination therapy.
Numerous complications can rise from CCS, with the most notable being the development of malignancy. This can be as high as 15%.172 Both gastric and colorectal cancers have been reported, with sigmoid colon and the rectum being the most common initial site of cancer.173
Unfortunately, due to the rarity of this disease, optimal screening protocols have not been developed, although annual endoscopic surveillance has been widely practiced. The long-term prognosis is quite poor according to early studies. One study is reporting a 55% mortality rate in a cohort of 55 patients.165 However, due to improved in medical therapy and increased recognition of the syndrome, the prognosis is now thought to be better compared with earlier case reports.174
Conclusion
In the 21st century, genetic information will play an increasing role in the management of common medical disorders. This will be especially important in the management of colorectal cancer where the role of heredity in the phenotypic expression of disease is likely to affect the diagnosis, treatment, and prevention of colorectal cancer in patients who are at risk. All providers who care for patients with gastrointestinal cancers should be aware of genetically inherited colorectal cancer syndromes. This knowledge allows us to appropriately institute screening measures that can reduce mortality from CRC. Families with Lynch syndrome will likely derive the most benefit because it is by far the most common hereditary CRC. Families with FAP are easy to identify and potentially cure. Most of the other eponymous syndromes are rare and are seen more often in pediatric patients.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun M J. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Mecklin J P. Frequency of hereditary colorectal carcinoma. Gastroenterology. 1987;93(5):1021–1025. doi: 10.1016/0016-5085(87)90565-8. [DOI] [PubMed] [Google Scholar]
- 3.Ponz de Leon M, Sassatelli R, Sacchetti C, Zanghieri G, Scalmati A, Roncucci L. Familial aggregation of tumors in the three-year experience of a population-based colorectal cancer registry. Cancer Res. 1989;49(15):4344–4348. [PubMed] [Google Scholar]
- 4.Jasperson K W, Tuohy T M, Neklason D W, Burt R W. Hereditary and familial colon cancer. Gastroenterology. 2010;138(6):2044–2058. doi: 10.1053/j.gastro.2010.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samowitz W S, Curtin K, Lin H H. et al. The colon cancer burden of genetically defined hereditary nonpolyposis colon cancer. Gastroenterology. 2001;121(4):830–838. doi: 10.1053/gast.2001.27996. [DOI] [PubMed] [Google Scholar]
- 6.Papaemmanuil E, Carvajal-Carmona L, Sellick G S. et al. CORGI Consortium. Deciphering the genetics of hereditary non-syndromic colorectal cancer. Eur J Hum Genet. 2008;16(12):1477–1486. doi: 10.1038/ejhg.2008.129. [DOI] [PubMed] [Google Scholar]
- 7.Gallagher D J, Smith J D, Offit K, Stadler Z K. Diagnosing hereditary colorectal cancer. Clin Colorectal Cancer. 2010;9(4):205–211. doi: 10.3816/CCC.2010.n.030. [DOI] [PubMed] [Google Scholar]
- 8.Järvinen H J, Aarnio M, Mustonen H. et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118(5):829–834. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- 9.de Jong A E, Hendriks Y M, Kleibeuker J H. et al. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology. 2006;130(3):665–671. doi: 10.1053/j.gastro.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 10.Regula J, Rupinski M, Kraszewska E. et al. Colonoscopy in colorectal-cancer screening for detection of advanced neoplasia. N Engl J Med. 2006;355(18):1863–1872. doi: 10.1056/NEJMoa054967. [DOI] [PubMed] [Google Scholar]
- 11.Dijk D A van, Oostindiër M J, Kloosterman-Boele W M, Krijnen P, Vasen H F. ; Hereditary Tumor Study Group of the Comprehensive Cancer Centre West. Family history is neglected in the work-up of patients with colorectal cancer: a quality assessment using cancer registry data. Fam Cancer. 2007;6(1):131–134. doi: 10.1007/s10689-006-9114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nielsen M, Joerink-van de Beld M C, Jones N. et al. Analysis of MUTYH genotypes and colorectal phenotypes in patients with MUTYH-associated polyposis. Gastroenterology. 2009;136(2):471–476. doi: 10.1053/j.gastro.2008.10.056. [DOI] [PubMed] [Google Scholar]
- 13.Jones N Vogt S Nielsen M et al. Increased colorectal cancer incidence in obligate carriers of heterozygous mutations in MUTYH Gastroenterology 20091372489–494., 494, e1, quiz 725–726 [DOI] [PubMed] [Google Scholar]
- 14.Gruber S B, Kohlmann W. The genetics of hereditary non-polyposis colorectal cancer. J Natl Compr Canc Netw. 2003;1(1):137–144. doi: 10.6004/jnccn.2003.0014. [DOI] [PubMed] [Google Scholar]
- 15.Rhyu M S. Molecular mechanisms underlying hereditary nonpolyposis colorectal carcinoma. J Natl Cancer Inst. 1996;88(5):240–251. doi: 10.1093/jnci/88.5.240. [DOI] [PubMed] [Google Scholar]
- 16.Chung D C, Rustgi A K. DNA mismatch repair and cancer. Gastroenterology. 1995;109(5):1685–1699. doi: 10.1016/0016-5085(95)90660-6. [DOI] [PubMed] [Google Scholar]
- 17.Lazar V, Grandjouan S, Bognel C. et al. Accumulation of multiple mutations in tumour suppressor genes during colorectal tumorigenesis in HNPCC patients. Hum Mol Genet. 1994;3(12):2257–2260. doi: 10.1093/hmg/3.12.2257. [DOI] [PubMed] [Google Scholar]
- 18.Hendriks Y M, Wagner A, Morreau H. et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology. 2004;127(1):17–25. doi: 10.1053/j.gastro.2004.03.068. [DOI] [PubMed] [Google Scholar]
- 19.Piñol V, Castells A, Andreu M. et al. Gastrointestinal Oncology Group of the Spanish Gastroenterological Association. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA. 2005;293(16):1986–1994. doi: 10.1001/jama.293.16.1986. [DOI] [PubMed] [Google Scholar]
- 20.Choi Y H, Cotterchio M, McKeown-Eyssen G. et al. Penetrance of colorectal cancer among MLH1/MSH2 carriers participating in the colorectal cancer familial registry in Ontario. Hered Cancer Clin Pract. 2009;7(1):14. doi: 10.1186/1897-4287-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lagerstedt Robinson K, Liu T, Vandrovcova J. et al. Lynch syndrome (hereditary nonpolyposis colorectal cancer) diagnostics. J Natl Cancer Inst. 2007;99(4):291–299. doi: 10.1093/jnci/djk051. [DOI] [PubMed] [Google Scholar]
- 22.Hampel H, Frankel W L, Martin E. et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352(18):1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 23.Wijnen J T, Vasen H F, Khan P M. et al. Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. N Engl J Med. 1998;339(8):511–518. doi: 10.1056/NEJM199808203390804. [DOI] [PubMed] [Google Scholar]
- 24.Hampel H, Frankel W, Panescu J. et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66(15):7810–7817. doi: 10.1158/0008-5472.CAN-06-1114. [DOI] [PubMed] [Google Scholar]
- 25.Lindor N M, Rabe K, Petersen G M. et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA. 2005;293(16):1979–1985. doi: 10.1001/jama.293.16.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mueller-Koch Y, Vogelsang H, Kopp R. et al. Hereditary non-polyposis colorectal cancer: clinical and molecular evidence for a new entity of hereditary colorectal cancer. Gut. 2005;54(12):1733–1740. doi: 10.1136/gut.2004.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Llor X, Pons E, Xicola R M. et al. Gastrointestinal Oncology Group of the Spanish Gastroenterological Association. Differential features of colorectal cancers fulfilling Amsterdam criteria without involvement of the mutator pathway. Clin Cancer Res. 2005;11(20):7304–7310. doi: 10.1158/1078-0432.CCR-05-0965. [DOI] [PubMed] [Google Scholar]
- 28.Valle L, Perea J, Carbonell P. et al. Clinicopathologic and pedigree differences in Amsterdam I-positive hereditary nonpolyposis colorectal cancer families according to tumor microsatellite instability status. J Clin Oncol. 2007;25(7):781–786. doi: 10.1200/JCO.2006.06.9781. [DOI] [PubMed] [Google Scholar]
- 29.Jass J R. Hereditary non-polyposis colorectal cancer: the rise and fall of a confusing term. World J Gastroenterol. 2006;12(31):4943–4950. doi: 10.3748/wjg.v12.i31.4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdel-Rahman W M, Ollikainen M, Kariola R. et al. Comprehensive characterization of HNPCC-related colorectal cancers reveals striking molecular features in families with no germline mismatch repair gene mutations. Oncogene. 2005;24(9):1542–1551. doi: 10.1038/sj.onc.1208387. [DOI] [PubMed] [Google Scholar]
- 31.Shah N B, Lindor N M. Lower gastrointestinal tract cancer predisposition syndromes. Hematol Oncol Clin N Am. 2010;24:1229–1252. doi: 10.1016/j.hoc.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watson P, Lynch H T. Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer. 1993;71(3):677–685. doi: 10.1002/1097-0142(19930201)71:3<677::aid-cncr2820710305>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 33.Watson P, Vasen H F, Mecklin J P, Järvinen H, Lynch H T. The risk of endometrial cancer in hereditary nonpolyposis colorectal cancer. Am J Med. 1994;96(6):516–520. doi: 10.1016/0002-9343(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 34.Aarnio M, Mecklin J P, Aaltonen L A, Nyström-Lahti M, Järvinen H J. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer. 1995;64(6):430–433. doi: 10.1002/ijc.2910640613. [DOI] [PubMed] [Google Scholar]
- 35.Kastrinos F, Mukherjee B, Tayob N. et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302(16):1790–1795. doi: 10.1001/jama.2009.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vasen H F, Möslein G, Alonso A. et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer) J Med Genet. 2007;44(6):353–362. doi: 10.1136/jmg.2007.048991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hendriks Y M, Wagner A, Morreau H. et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology. 2004;127(1):17–25. doi: 10.1053/j.gastro.2004.03.068. [DOI] [PubMed] [Google Scholar]
- 38.Broaddus R R, Lynch H T, Chen L M. et al. Pathologic features of endometrial carcinoma associated with HNPCC: a comparison with sporadic endometrial carcinoma. Cancer. 2006;106(1):87–94. doi: 10.1002/cncr.21560. [DOI] [PubMed] [Google Scholar]
- 39.Vasen H F, Mecklin J P, Khan P M, Lynch H T. The International Collaborative Group on hereditary nonpolyposis colorectal cancer (ICG-HNPCC) Dis Colon Rectum. 1991;34(5):424–425. doi: 10.1007/BF02053699. [DOI] [PubMed] [Google Scholar]
- 40.Vasen H F, Watson P, Mecklin J P, Lynch H T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116(6):1453–1456. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez-Bigas M A, Boland C R, Hamilton S R. et al. A National Cancer Institute workshop on hereditary nonpolyposis colorectal cancer syndrome: meeting highlights and Bethesda Guidelines. J Natl Cancer Inst. 1997;89(23):1758–1762. doi: 10.1093/jnci/89.23.1758. [DOI] [PubMed] [Google Scholar]
- 42.Syngal S, Fox E A, Eng C, Kolodner R D, Garber J E. Sensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J Med Genet. 2000;37(9):641–645. doi: 10.1136/jmg.37.9.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Umar A, Boland C R, Terdiman J P. et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hampel H, Frankel W L, Martin E. et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352(18):1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 45.Grover S, Syngal S. Risk assessment, genetic testing, and management of Lynch syndrome. J Natl Compr Canc Netw. 2010;8(1):98–105. doi: 10.6004/jnccn.2010.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hampel H, Frankel W, Panescu J. et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66(15):7810–7817. doi: 10.1158/0008-5472.CAN-06-1114. [DOI] [PubMed] [Google Scholar]
- 47.Barnetson R A, Tenesa A, Farrington S M. et al. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med. 2006;354(26):2751–2763. doi: 10.1056/NEJMoa053493. [DOI] [PubMed] [Google Scholar]
- 48.Tenesa A, Farrington S M, Prendergast J G. et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet. 2008;40(5):631–637. doi: 10.1038/ng.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tomlinson I P, Webb E, Carvajal-Carmona L. et al. CORGI Consortium; EPICOLON Consortium. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet. 2008;40(5):623–630. doi: 10.1038/ng.111. [DOI] [PubMed] [Google Scholar]
- 50.Balmaña J, Stockwell D H, Steyerberg E W. et al. Prediction of MLH1 and MSH2 mutations in Lynch syndrome. JAMA. 2006;296(12):1469–1478. doi: 10.1001/jama.296.12.1469. [DOI] [PubMed] [Google Scholar]
- 51.Chen S, Wang W, Lee S. et al. Colon Cancer Family Registry. Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA. 2006;296(12):1479–1487. doi: 10.1001/jama.296.12.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balaguer F, Balmaña J, Castellví-Bel S. et al. Gastrointestinal Oncology Group of the Spanish Gastroenterological Association. Validation and extension of the PREMM1,2 model in a population-based cohort of colorectal cancer patients. Gastroenterology. 2008;134(1):39–46. doi: 10.1053/j.gastro.2007.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grover S, Stoffel E M, Mercado R C. et al. Colorectal cancer risk perception on the basis of genetic test results in individuals at risk for Lynch syndrome. J Clin Oncol. 2009;27(24):3981–3986. doi: 10.1200/JCO.2008.18.6940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suspiro A, Fidalgo P, Cravo M. et al. The Muir-Torre syndrome: a rare variant of hereditary nonpolyposis colorectal cancer associated with hMSH2 mutation. Am J Gastroenterol. 1998;93(9):1572–1574. doi: 10.1111/j.1572-0241.1998.00487.x. [DOI] [PubMed] [Google Scholar]
- 55.Kruse R, Rütten A, Lamberti C. et al. Muir-Torre phenotype has a frequency of DNA mismatch-repair-gene mutations similar to that in hereditary nonpolyposis colorectal cancer families defined by the Amsterdam criteria. Am J Hum Genet. 1998;63(1):63–70. doi: 10.1086/301926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.South C D, Hampel H, Comeras I, Westman J A, Frankel W L, de la Chapelle A. The frequency of Muir-Torre syndrome among Lynch syndrome families. J Natl Cancer Inst. 2008;100(4):277–281. doi: 10.1093/jnci/djm291. [DOI] [PubMed] [Google Scholar]
- 57.Galiatsatos P, Foulkes W D. Familial adenomatous polyposis. Am J Gastroenterol. 2006;101(2):385–398. doi: 10.1111/j.1572-0241.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 58.Rozen P, Macrae F. Familial adenomatous polyposis: the practical applications of clinical and molecular screening. Fam Cancer. 2006;5(3):227–235. doi: 10.1007/s10689-005-5674-2. [DOI] [PubMed] [Google Scholar]
- 59.Lipton L, Tomlinson I. The genetics of FAP and FAP-like syndromes. Fam Cancer. 2006;5(3):221–226. doi: 10.1007/s10689-005-5673-3. [DOI] [PubMed] [Google Scholar]
- 60.Houlston R, Crabtree M, Phillips R, Crabtree M, Tomlinson I. Explaining differences in the severity of familial adenomatous polyposis and the search for modifier genes. Gut. 2001;48(1):1–5. doi: 10.1136/gut.48.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pérez Segura P, Guillén Ponce C, Ramón y Cajal T, Serrano Blanch R, Aranda E. TTD consensus document on the diagnosis and management of hereditary colorectal cancer. Clin Transl Oncol. 2010;12(5):356–366. doi: 10.1007/s12094-010-0517-5. [DOI] [PubMed] [Google Scholar]
- 62.Lal G, Gallinger S. Familial adenomatous polyposis. Semin Surg Oncol. 2000;18(4):314–323. doi: 10.1002/(sici)1098-2388(200006)18:4<314::aid-ssu6>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 63.Berk T Cohen Z Bapat B Gallinger S Negative genetic test result in familial adenomatous polyposis: clinical screening implications Dis Colon Rectum 1999423307–310., discussion 310–312 [DOI] [PubMed] [Google Scholar]
- 64.Petersen G M, Slack J, Nakamura Y. Screening guidelines and premorbid diagnosis of familial adenomatous polyposis using linkage. Gastroenterology. 1991;100(6):1658–1664. doi: 10.1016/0016-5085(91)90666-9. [DOI] [PubMed] [Google Scholar]
- 65.Rustin R B, Jagelman D G, McGannon E, Fazio V W, Lavery I C, Weakley F L. Spontaneous mutation in familial adenomatous polyposis. Dis Colon Rectum. 1990;33(1):52–55. doi: 10.1007/BF02053203. [DOI] [PubMed] [Google Scholar]
- 66.Zippel D B, Temple W J. When is a neoplasm not a neoplasm? When it is a desmoid. J Surg Oncol. 2007;95(3):190–191. doi: 10.1002/jso.20637. [DOI] [PubMed] [Google Scholar]
- 67.Sturt N J Clark S K Current ideas in desmoid tumours Fam Cancer 200653275–285., discussion 287–288 [DOI] [PubMed] [Google Scholar]
- 68.Bertario L, Russo A, Sala P. et al. Hereditary Colorectal Tumours Registry. Genotype and phenotype factors as determinants of desmoid tumors in patients with familial adenomatous polyposis. Int J Cancer. 2001;95(2):102–107. doi: 10.1002/1097-0215(20010320)95:2<102::aid-ijc1018>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 69.Lynch H T, Fitzgibbons R. Surgery, desmoid tumors, and familial adenomatous polyposis: case report and literature review. Am J Gastroenterol. 1996;91(12):2598–2601. [PubMed] [Google Scholar]
- 70.Eccles D M, der Luijt R van, Breukel C. et al. Hereditary desmoid disease due to a frameshift mutation at codon 1924 of the APC gene. Am J Hum Genet. 1996;59(6):1193–1201. [PMC free article] [PubMed] [Google Scholar]
- 71.Chen C S, Phillips K D, Grist S. et al. Congenital hypertrophy of the retinal pigment epithelium (CHRPE) in familial colorectal cancer. Fam Cancer. 2006;5(4):397–404. doi: 10.1007/s10689-006-0011-y. [DOI] [PubMed] [Google Scholar]
- 72.Arvanitis M L, Jagelman D G, Fazio V W, Lavery I C, McGannon E. Mortality in patients with familial adenomatous polyposis. Dis Colon Rectum. 1990;33(8):639–642. doi: 10.1007/BF02150736. [DOI] [PubMed] [Google Scholar]
- 73.Church J M, McGannon E, Hull-Boiner S. et al. Gastroduodenal polyps in patients with familial adenomatous polyposis. Dis Colon Rectum. 1992;35(12):1170–1173. doi: 10.1007/BF02251971. [DOI] [PubMed] [Google Scholar]
- 74.Kadmon M, Tandara A, Herfarth C. Duodenal adenomatosis in familial adenomatous polyposis coli. A review of the literature and results from the Heidelberg Polyposis Register. Int J Colorectal Dis. 2001;16(2):63–75. doi: 10.1007/s003840100290. [DOI] [PubMed] [Google Scholar]
- 75.Groves C J, Saunders B P, Spigelman A D, Phillips R K. Duodenal cancer in patients with familial adenomatous polyposis (FAP): results of a 10 year prospective study. Gut. 2002;50(5):636–641. doi: 10.1136/gut.50.5.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park J G, Park K J, Ahn Y O. et al. Risk of gastric cancer among Korean familial adenomatous polyposis patients. Report of three cases. Dis Colon Rectum. 1992;35(10):996–998. doi: 10.1007/BF02253505. [DOI] [PubMed] [Google Scholar]
- 77.Brosens L A, Keller J J, Offerhaus G J, Goggins M, Giardiello F M. Prevention and management of duodenal polyps in familial adenomatous polyposis. Gut. 2005;54(7):1034–1043. doi: 10.1136/gut.2004.053843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cetta F, Mazzarella L, Bon G, Zuckermann M, Casorelli A, Nounga H. Genetic alterations in hepatoblastoma and hepatocellular carcinoma associated with familial adenomatous polyposis. Med Pediatr Oncol. 2003;41(5):496–497. doi: 10.1002/mpo.10362. [DOI] [PubMed] [Google Scholar]
- 79.Herraiz M, Barbesino G, Faquin W. et al. Prevalence of thyroid cancer in familial adenomatous polyposis syndrome and the role of screening ultrasound examinations. Clin Gastroenterol Hepatol. 2007;5(3):367–373. doi: 10.1016/j.cgh.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 80.Hamilton S R, Liu B, Parsons R E. et al. The molecular basis of Turcot's syndrome. N Engl J Med. 1995;332(13):839–847. doi: 10.1056/NEJM199503303321302. [DOI] [PubMed] [Google Scholar]
- 81.Knudsen A L, Bisgaard M L, Bülow S. Attenuated familial adenomatous polyposis (AFAP). A review of the literature. Fam Cancer. 2003;2(1):43–55. doi: 10.1023/a:1023286520725. [DOI] [PubMed] [Google Scholar]
- 82.Fearnhead N S, Britton M P, Bodmer W F. The ABC of APC. Hum Mol Genet. 2001;10(7):721–733. doi: 10.1093/hmg/10.7.721. [DOI] [PubMed] [Google Scholar]
- 83.Näthke I S. The adenomatous polyposis coli protein: the Achilles heel of the gut epithelium. Annu Rev Cell Dev Biol. 2004;20:337–366. doi: 10.1146/annurev.cellbio.20.012103.094541. [DOI] [PubMed] [Google Scholar]
- 84.Sieber O, Segditsas S, Knudsen A. et al. Disease severity and genetic pathways in attenuated FAP vary greatly but depend on the site of the germline mutation. Gut. 2006;55:1440–1448. doi: 10.1136/gut.2005.087106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bertario L, Russo A, Sala P. et al. Hereditary Colorectal Tumor Registry. Multiple approach to the exploration of genotype-phenotype correlations in familial adenomatous polyposis. J Clin Oncol. 2003;21(9):1698–1707. doi: 10.1200/JCO.2003.09.118. [DOI] [PubMed] [Google Scholar]
- 86.Caspari R, Olschwang S, Friedl W. et al. Familial adenomatous polyposis: desmoid tumours and lack of ophthalmic lesions (CHRPE) associated with APC mutations beyond codon 1444. Hum Mol Genet. 1995;4(3):337–340. doi: 10.1093/hmg/4.3.337. [DOI] [PubMed] [Google Scholar]
- 87.Enomoto M, Konishi M, Iwama T, Utsunomiya J, Sugihara K I, Miyaki M. The relationship between frequencies of extracolonic manifestations and the position of APC germline mutation in patients with familial adenomatous polyposis. Jpn J Clin Oncol. 2000;30(2):82–88. doi: 10.1093/jjco/hyd017. [DOI] [PubMed] [Google Scholar]
- 88.Ficari F, Cama A, Valanzano R. et al. APC gene mutations and colorectal adenomatosis in familial adenomatous polyposis. Br J Cancer. 2000;82(2):348–353. doi: 10.1054/bjoc.1999.0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brensinger J D, Laken S J, Luce M C. et al. Variable phenotype of familial adenomatous polyposis in pedigrees with 3′ mutation in the APC gene. Gut. 1998;43(4):548–552. doi: 10.1136/gut.43.4.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nieuwenhuis M H, Vasen H F. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol. 2007;61(2):153–161. doi: 10.1016/j.critrevonc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 91.Lefevre J H, Parc Y, Svrcek M. et al. APC, MYH, and the correlation genotype-phenotype in colorectal polyposis. Ann Surg Oncol. 2009;16(4):871–877. doi: 10.1245/s10434-008-0297-0. [DOI] [PubMed] [Google Scholar]
- 92.Al-Tassan N, Chmiel N H, Maynard J. et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat Genet. 2002;30(2):227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 93.Lipton L, Tomlinson I. The multiple colorectal adenoma phenotype and MYH, a base excision repair gene. Clin Gastroenterol Hepatol. 2004;2(8):633–638. doi: 10.1016/s1542-3565(04)00286-1. [DOI] [PubMed] [Google Scholar]
- 94.Holter S, Gallinger S. New York, NY: Springer Science Business Media; 2009. MUTYH-associated polyposis; pp. 173–181. [Google Scholar]
- 95.Cheadle J P, Sampson J R. MUTYH-associated polyposis—from defect in base excision repair to clinical genetic testing. DNA Repair (Amst) 2007;6(3):274–279. doi: 10.1016/j.dnarep.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 96.Croitoru M E, Cleary S P, Di Nicola N. et al. Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst. 2004;96(21):1631–1634. doi: 10.1093/jnci/djh288. [DOI] [PubMed] [Google Scholar]
- 97.Nielsen M, Franken P F, Reinards T H. et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP) J Med Genet. 2005;42(9):e54. doi: 10.1136/jmg.2005.033217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Halford S E, Rowan A J, Lipton L. et al. Germline mutations but not somatic changes at the MYH locus contribute to the pathogenesis of unselected colorectal cancers. Am J Pathol. 2003;162(5):1545–1548. doi: 10.1016/S0002-9440(10)64288-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Okamoto K, Toyokuni S, Uchida K. et al. Formation of 8-hydroxy-20-deoxyguanosine and 4-hydroxy-2-nonenal-modified proteins in human renal-cell carcinoma. Int J Cancer. 1994;58:825–829. doi: 10.1002/ijc.2910580613. [DOI] [PubMed] [Google Scholar]
- 100.Malins D C, Haimanot R. Major alterations in the nucleotide structure of DNA in cancer of the female breast. Cancer Res. 1991;51(19):5430–5432. [PubMed] [Google Scholar]
- 101.Jaruga P, Zastawny T H, Skokowski J, Dizdaroglu M, Olinski R. Oxidative DNA base damage and antioxidant enzyme activities in human lung cancer. FEBS Lett. 1994;341(1):59–64. doi: 10.1016/0014-5793(94)80240-8. [DOI] [PubMed] [Google Scholar]
- 102.DeMarzo A M, Nelson W G, Isaacs W B, Epstein J I. Pathological and molecular aspects of prostate cancer. Lancet. 2003;361(9361):955–964. doi: 10.1016/S0140-6736(03)12779-1. [DOI] [PubMed] [Google Scholar]
- 103.Charbonnier F, Olschwang S, Wang Q. et al. MSH2 in contrast to MLH1 and MSH6 is frequently inactivated by exonic and promoter rearrangements in hereditary nonpolyposis colorectal cancer. Cancer Res. 2002;62(3):848–853. [PubMed] [Google Scholar]
- 104.Sampson J R, Dolwani S, Jones S. et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362(9377):39–41. doi: 10.1016/S0140-6736(03)13805-6. [DOI] [PubMed] [Google Scholar]
- 105.Sieber O M, Lipton L, Crabtree M. et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348(9):791–799. doi: 10.1056/NEJMoa025283. [DOI] [PubMed] [Google Scholar]
- 106.Jenkins M A, Croitoru M E, Monga N. et al. Risk of colorectal cancer in monoallelic and biallelic carriers of MYH mutations: a population-based case-family study. Cancer Epidemiol Biomarkers Prev. 2006;15(2):312–314. doi: 10.1158/1055-9965.EPI-05-0793. [DOI] [PubMed] [Google Scholar]
- 107.Farrington S M, Tenesa A, Barnetson R. et al. Germline susceptibility to colorectal cancer due to base-excision repair gene defects. Am J Hum Genet. 2005;77(1):112–119. doi: 10.1086/431213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Webb E L Rudd M F Houlston R S Colorectal cancer risk in monoallelic carriers of MYH variants Am J Hum Genet 2006794768–771., author reply 771–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lubbe S J, Di Bernardo M C, Chandler I P, Houlston R S. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol. 2009;27(24):3975–3980. doi: 10.1200/JCO.2008.21.6853. [DOI] [PubMed] [Google Scholar]
- 110.Avezzù A, Agostini M, Pucciarelli S. et al. The role of MYH gene in genetic predisposition to colorectal cancer: another piece of the puzzle. Cancer Lett. 2008;268(2):308–313. doi: 10.1016/j.canlet.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 111.Nielsen M, Poley J W, Verhoef S. et al. Duodenal carcinoma in MUTYH-associated polyposis. J Clin Pathol. 2006;59(11):1212–1215. doi: 10.1136/jcp.2005.031757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Boardman L A. Heritable colorectal cancer syndromes: recognition and preventive management. Gastroenterol Clin North Am. 2002;31(4):1107–1131. doi: 10.1016/s0889-8553(02)00049-3. [DOI] [PubMed] [Google Scholar]
- 113.Anyanwu S N. Sporadic Peutz-Jeghers syndrome in a Nigerian. Cent Afr J Med. 1999;45(7):182–184. doi: 10.4314/cajm.v45i7.8481. [DOI] [PubMed] [Google Scholar]
- 114.Yoon K A, Ku J L, Choi H S. et al. Germline mutations of the STK11 gene in Korean Peutz-Jeghers syndrome patients. Br J Cancer. 2000;82(8):1403–1406. doi: 10.1054/bjoc.1999.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hearle N, Schumacher V, Menko F H. et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12(10):3209–3215. doi: 10.1158/1078-0432.CCR-06-0083. [DOI] [PubMed] [Google Scholar]
- 116.Mehenni H, Resta N, Park J G, Miyaki M, Guanti G, Costanza M C. Cancer risks in LKB1 germline mutation carriers. Gut. 2006;55(7):984–990. doi: 10.1136/gut.2005.082990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Olschwang S, Boisson C, Thomas G. Peutz-Jeghers families unlinked to STK11/LKB1 gene mutations are highly predisposed to primitive biliary adenocarcinoma. J Med Genet. 2001;38(6):356–360. doi: 10.1136/jmg.38.6.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Aaltonen L. New York, NY: McGraw-Hill; 2002. Peutz-Jeghers syndrome; pp. 337–41. [Google Scholar]
- 119.Riegert-Johnson D, Boardman L. New York, NY: Springer; 2009. Peutz-Jeghers syndrome; pp. 193–198. [Google Scholar]
- 120.Amos C I, Keitheri-Cheteri M B, Sabripour M. et al. Genotype-phenotype correlations in Peutz-Jeghers syndrome. J Med Genet. 2004;41(5):327–333. doi: 10.1136/jmg.2003.010900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rowan A, Churchman M, Jefferey R, Hanby A, Poulsom R, Tomlinson I. In situ analysis of LKB1/STK11 mRNA expression in human normal tissues and tumours. J Pathol. 2000;192(2):203–206. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH686>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 122.Boudeau J, Baas A F, Deak M. et al. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003;22(19):5102–5114. doi: 10.1093/emboj/cdg490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ylikorkala A, Avizienyte E, Tomlinson I P. et al. Mutations and impaired function of LKB1 in familial and non-familial Peutz-Jeghers syndrome and a sporadic testicular cancer. Hum Mol Genet. 1999;8(1):45–51. doi: 10.1093/hmg/8.1.45. [DOI] [PubMed] [Google Scholar]
- 124.Karuman P, Gozani O, Odze R D. et al. The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol Cell. 2001;7(6):1307–1319. doi: 10.1016/s1097-2765(01)00258-1. [DOI] [PubMed] [Google Scholar]
- 125.Gruber S B, Entius M M, Petersen G M. et al. Pathogenesis of adenocarcinoma in Peutz-Jeghers syndrome. Cancer Res. 1998;58(23):5267–5270. [PubMed] [Google Scholar]
- 126.Flageole H, Raptis S, Trudel J L, Lough J O. Progression toward malignancy of hamartomas in a patient with Peutz-Jeghers syndrome: case report and literature review. Can J Surg. 1994;37(3):231–236. [PubMed] [Google Scholar]
- 127.Nakau M, Miyoshi H, Seldin M F, Imamura M, Oshima M, Taketo M M. Hepatocellular carcinoma caused by loss of heterozygosity in Lkb1 gene knockout mice. Cancer Res. 2002;62(16):4549–4553. [PubMed] [Google Scholar]
- 128.Jishage K, Nezu J, Kawase Y. et al. Role of Lkb1, the causative gene of Peutz-Jegher's syndrome, in embryogenesis and polyposis. Proc Natl Acad Sci U S A. 2002;99(13):8903–8908. doi: 10.1073/pnas.122254599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hemminki A, Markie D, Tomlinson I. et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391(6663):184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 130.Jenne D E, Reimann H, Nezu J. et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18(1):38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- 131.Giardiello F M, Hamilton S R, Kern S E. et al. Colorectal neoplasia in juvenile polyposis or juvenile polyps. Arch Dis Child. 1991;66(8):971–975. doi: 10.1136/adc.66.8.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Aaltonen L, Jass J, Howe J. Lyon, France: IARC Press; 2000. Juvenile polyposis; pp. 130–132. [Google Scholar]
- 133.Burt R W, Bishop D T, Lynch H T, Rozen P, Winawer S J. ; WHO Collaborating Centre for the Prevention of Colorectal Cancer. Risk and surveillance of individuals with heritable factors for colorectal cancer. Bull World Health Organ. 1990;68(5):655–665. [PMC free article] [PubMed] [Google Scholar]
- 134.Nugent K P, Talbot I C, Hodgson S V, Phillips R K. Solitary juvenile polyps: not a marker for subsequent malignancy. Gastroenterology. 1993;105(3):698–700. doi: 10.1016/0016-5085(93)90885-g. [DOI] [PubMed] [Google Scholar]
- 135.Brosens L A, Hattem A van, Hylind L M. et al. Risk of colorectal cancer in juvenile polyposis. Gut. 2007;56(7):965–967. doi: 10.1136/gut.2006.116913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Merg A, Lynch H T, Lynch J F, Howe J R. Hereditary colorectal cancer-part II. Curr Probl Surg. 2005;42(5):267–333. doi: 10.1067/j.cpsurg.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 137.Roth S, Sistonen P, Salovaara R. et al. SMAD genes in juvenile polyposis. Genes Chromosomes Cancer. 1999;26(1):54–61. doi: 10.1002/(sici)1098-2264(199909)26:1<54::aid-gcc8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 138.Brosens L A, Hattem W A van, Jansen M, de Leng W W, Giardiello F M, Offerhaus G J. Gastrointestinal polyposis syndromes. Curr Mol Med. 2007;7(1):29–46. doi: 10.2174/156652407779940404. [DOI] [PubMed] [Google Scholar]
- 139.Howe J R, Sayed M G, Ahmed A F. et al. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. J Med Genet. 2004;41(7):484–491. doi: 10.1136/jmg.2004.018598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Höfting I Pott G Stolte M [The syndrome of juvenile polyposis] Leber Magen Darm 1993233107–108., 111–112 [PubMed] [Google Scholar]
- 141.Chow E, Macrae F. A review of juvenile polyposis syndrome. J Gastroenterol Hepatol. 2005;20(11):1634–1640. doi: 10.1111/j.1440-1746.2005.03865.x. [DOI] [PubMed] [Google Scholar]
- 142.Sayed M G, Ahmed A F, Ringold J R. et al. Germline SMAD4 or BMPR1A mutations and phenotype of juvenile polyposis. Ann Surg Oncol. 2002;9(9):901–906. doi: 10.1007/BF02557528. [DOI] [PubMed] [Google Scholar]
- 143.Pyatt R E, Pilarski R, Prior T W. Mutation screening in juvenile polyposis syndrome. J Mol Diagn. 2006;8(1):84–88. doi: 10.2353/jmoldx.2006.050072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kim B G, Li C, Qiao W. et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature. 2006;441(7096):1015–1019. doi: 10.1038/nature04846. [DOI] [PubMed] [Google Scholar]
- 145.Kinzler K W, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280(5366):1036–1037. doi: 10.1126/science.280.5366.1036. [DOI] [PubMed] [Google Scholar]
- 146.Friedl W, Uhlhaas S, Schulmann K. et al. Juvenile polyposis: massive gastric polyposis is more common in MADH4 mutation carriers than in BMPR1A mutation carriers. Hum Genet. 2002;111(1):108–111. doi: 10.1007/s00439-002-0748-9. [DOI] [PubMed] [Google Scholar]
- 147.Handra-Luca A, Condroyer C, de Moncuit C. et al. Vessels' morphology in SMAD4 and BMPR1A-related juvenile polyposis. Am J Med Genet A. 2005;138A(2):113–117. doi: 10.1002/ajmg.a.30897. [DOI] [PubMed] [Google Scholar]
- 148.Sweet K, Willis J, Zhou X P. et al. Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis. JAMA. 2005;294(19):2465–2473. doi: 10.1001/jama.294.19.2465. [DOI] [PubMed] [Google Scholar]
- 149.Blanco F J, Santibanez J F, Guerrero-Esteo M, Langa C, Vary C P, Bernabeu C. Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-beta receptor complex. J Cell Physiol. 2005;204(2):574–584. doi: 10.1002/jcp.20311. [DOI] [PubMed] [Google Scholar]
- 150.Howe J R, Haidle J L, Lal G. et al. ENG mutations in MADH4/BMPR1A mutation negative patients with juvenile polyposis. Clin Genet. 2007;71(1):91–92. doi: 10.1111/j.1399-0004.2007.00734.x. [DOI] [PubMed] [Google Scholar]
- 151.Zbuk K M, Eng C. Hamartomatous polyposis syndromes. Nat Clin Pract Gastroenterol Hepatol. 2007;4(9):492–502. doi: 10.1038/ncpgasthep0902. [DOI] [PubMed] [Google Scholar]
- 152.Dahia P L. PTEN, a unique tumor suppressor gene. Endocr Relat Cancer. 2000;7(2):115–129. doi: 10.1677/erc.0.0070115. [DOI] [PubMed] [Google Scholar]
- 153.Burt R, Neklason D W. Genetic testing for inherited colon cancer. Gastroenterology. 2005;128(6):1696–1716. doi: 10.1053/j.gastro.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 154.Marsh D J, Dahia P L, Zheng Z. et al. Germline mutations in PTEN are present in Bannayan-Zonana syndrome. Nat Genet. 1997;16(4):333–334. doi: 10.1038/ng0897-333. [DOI] [PubMed] [Google Scholar]
- 155.Liaw D, Marsh D J, Li J. et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16(1):64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 156.Kinzler K W, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280(5366):1036–1037. doi: 10.1126/science.280.5366.1036. [DOI] [PubMed] [Google Scholar]
- 157.Gryfe R. Inherited colorectal cancer syndromes. Clin Colon Rectal Surg. 2009;22(4):198–208. doi: 10.1055/s-0029-1242459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cronkhite L W, Canada W J. Generalized gastrointestinal polyposis; an unusual syndrome of polyposis, pigmentation, alopecia and onychotrophia. N Engl J Med. 1955;252(24):1011–1015. doi: 10.1056/NEJM195506162522401. [DOI] [PubMed] [Google Scholar]
- 159.Riegert-Johnson D L, Osborn N, Smyrk T, Boardman L A. Cronkhite-Canada syndrome hamartomatous polyps are infiltrated with IgG4 plasma cells. Digestion. 2007;75(2-3):96–97. doi: 10.1159/000102963. [DOI] [PubMed] [Google Scholar]
- 160.Goto A. Cronkhite-Canada syndrome: epidemiological study of 110 cases reported in Japan. Nihon Geka Hokan. 1995;64(1):3–14. [PubMed] [Google Scholar]
- 161.Ward E M, Wolfsen H C. Review article: the non-inherited gastrointestinal polyposis syndromes. Aliment Pharmacol Ther. 2002;16(3):333–342. doi: 10.1046/j.1365-2036.2002.01172.x. [DOI] [PubMed] [Google Scholar]
- 162.Sampson J E, Harmon M L, Cushman M, Krawitt E L. Corticosteroid-responsive Cronkhite-Canada syndrome complicated by thrombosis. Dig Dis Sci. 2007;52(4):1137–1140. doi: 10.1007/s10620-006-9375-y. [DOI] [PubMed] [Google Scholar]
- 163.Takeuchi Y, Yoshikawa M, Tsukamoto N. et al. Cronkhite-Canada syndrome with colon cancer, portal thrombosis, high titer of antinuclear antibodies, and membranous glomerulonephritis. J Gastroenterol. 2003;38(8):791–795. doi: 10.1007/s00535-002-1148-6. [DOI] [PubMed] [Google Scholar]
- 164.Qiao M, Lei Z, Nai-Zhong H, Jian-Ming X. Cronkhite-Canada syndrome with hypothyroidism. South Med J. 2005;98(5):575–576. doi: 10.1097/01.SMJ.0000157528.71614.C4. [DOI] [PubMed] [Google Scholar]
- 165.Daniel E S, Ludwig S L, Lewin K J, Ruprecht R M, Rajacich G M, Schwabe A D. The Cronkhite-Canada Syndrome. An analysis of clinical and pathologic features and therapy in 55 patients. Medicine (Baltimore) 1982;61(5):293–309. [PubMed] [Google Scholar]
- 166.Blonski W C, Furth E E, Kinosian B P, Compher C, Metz D C. A case of Cronkhite-Canada syndrome with taste disturbance as a leading complaint. Digestion. 2005;71(4):201–205. doi: 10.1159/000086135. [DOI] [PubMed] [Google Scholar]
- 167.Samoha S, Arber N. Cronkhite-Canada syndrome. Digestion. 2005;71(4):199–200. doi: 10.1159/000086134. [DOI] [PubMed] [Google Scholar]
- 168.Yashiro M, Kobayashi H, Kubo N, Nishiguchi Y, Wakasa K, Hirakawa K. Cronkhite-Canada syndrome containing colon cancer and serrated adenoma lesions. Digestion. 2004;69(1):57–62. doi: 10.1159/000076560. [DOI] [PubMed] [Google Scholar]
- 169.Anderson R D Patel R Hamilton J K Boland C R Cronkhite-Canada syndrome presenting as eosinophilic gastroenteritis Proc (Bayl Univ Med Cent) 2006193209–212. (Baylor University Medical Center) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mönkemüller K, Neumann H, Evert M. Cronkhite-Canada syndrome: panendoscopic characterization with esophagogastroduodenoscopy, endoscopic ultrasound, colonoscopy, and double balloon enteroscopy. Clin Gastroenterol Hepatol. 2008;6(10):A26. doi: 10.1016/j.cgh.2008.03.027. [DOI] [PubMed] [Google Scholar]
- 171.Chadalavada R, Brown D K, Walker A N, Sedghi S. Cronkhite-Canada syndrome: sustained remission after corticosteroid treatment. Am J Gastroenterol. 2003;98(6):1444–1446. doi: 10.1111/j.1572-0241.2003.07509.x. [DOI] [PubMed] [Google Scholar]
- 172.Ward E, Wolfsen H C, Ng C. Medical management of Cronkhite-Canada syndrome. South Med J. 2002;95(2):272–274. [PubMed] [Google Scholar]
- 173.Okamoto K, Isomoto H, Shikuwa S, Nishiyama H, Ito M, Kohno S. A case of Cronkhite-Canada syndrome: remission after treatment with anti-Helicobacter pylori regimen. Digestion. 2008;78(2-3):82–87. doi: 10.1159/000165354. [DOI] [PubMed] [Google Scholar]
- 174.Kao K T, Patel J K, Pampati V. Cronkhite-Canada syndrome: a case report and review of literature. Gastroenterol Res Pract. 2009;(2009):619378. doi: 10.1155/2009/619378. [DOI] [PMC free article] [PubMed] [Google Scholar]