

Abstract
Endocrine cancers are a heterogeneous group of diseases that may arise from endocrine cells in any gland of the endocrine system. These malignancies may show an aggressive behavior and resistance to the common anticancer therapies. The etiopathogenesis of these tumors remains mostly unknown. The normal embryological development and differentiation of several endocrine glands are regulated by specific pituitary tropins, which, in adult life, control the function and trophism of the endocrine gland. Pituitary tropins act in concert with peptide growth factors, including the insulin-like growth factors (IGFs), which are considered key regulators of cell growth, proliferation, and apoptosis. While pituitary TSH is regarded as tumor-promoting factor for metastatic thyroid cancer, the role of other pituitary hormones in endocrine cancers is uncertain. However, multiple molecular abnormalities of the IGF system frequently occur in endocrine cancers and may have a role in tumorigenesis as well as in tumor progression and resistance to therapies. Herein, we will review studies indicating a role of IGF system dysregulation in endocrine cancers and will discuss the possible implications of these findings for tumor prevention and treatment, with a major focus on cancers from the thyroid, adrenal, and ovary, which are the most extensively studied.
1. Introduction
Endocrine malignancies, including carcinomas of the thyroid, adrenal, and ovary, are relatively rare tumors deriving from cells present in endocrine glands. Surgery is currently the treatment of choice for these tumors and is often successful in early stages of disease. However, this therapeutic approach for the advanced tumors remains unsatisfactory and is associated with poor prognosis. Thus, a better understanding of the molecular mechanisms and the critical intracellular networks underlying endocrine oncogenesis may help in discovering new targets that could represent promising therapeutic options for these malignancies. As specific pituitary tropins control the trophism and function of specific endocrine glands, it is tempting to speculate about a possible role for these pituitary hormones in endocrine glands tumorigenesis. However, this assumption is controversial as other signaling effectors, including the IGF system, have often a major role in endocrine tumorigenesis.
This paper covers the recent molecular advances in this field focusing on the role of the IGF system in endocrine tumorigenesis with particular attention on the endocrine cancers best characterized until now (i.e., thyroid, adrenal, and ovarian tumors).
2. Regulation of Thyroid, Adrenocortical, and Ovarian Tumor Growth: The Role of Pituitary Hormones
Thyroid cancer growth regulation has been extensively characterized. Several molecular alterations associated with thyroid tumorigenesis have been identified and often converge into the activation of MAPK (mitogen-activated protein kinase) and PI3K (phosphatidylinositol-3-kinase) signaling pathways [1].
Thyroid gland function and trophism is mainly regulated by thyrotropin hormone (TSH). TSH is considered the key player of thyrocyte differentiation and proliferation. Its mitogenic actions are mainly mediated by cAMP, which in turn activates protein kinase A (PKA) dependent and independent pathways. Activating mutations of the TSH receptor (TSHR) or of the gene encoding the Gs α subunit of the heterotrimeric G protein that couples TSHR to adenylyl cyclase (GSP) have been described in 30% of autonomously functioning thyroid adenomas while they are rare in thyroid carcinomas [2, 3]. TSH, however, has a well-known promoting role for thyroid cancer metastases, and TSH suppressive therapy with L-thyroxine is a well-established therapy in the postoperative management of differentiated thyroid cancer [4]. To exert its maximal mitogenic effects, TSH requires concomitant ligand-activated tyrosine kinase receptor (RTK) signaling. Studies carried out in thyroid cell cultures have especially highlighted the importance of the IGF system in regulating thyroid cell growth in response to TSH [5, 6].
TSH makes the cells competent to progress into the G1 phase in response to insulin or IGF-I, which can thus be qualified as the only genuine mitogens [7]. In fact, the protumorigenic effects of TSH are irrelevant in the absence of growth factors, but they are greatly potentiated by the presence of insulin or IGF-I at physiological concentrations [5, 8]. Furthermore, we have recently reported a key role of the IGF system in the biology of follicular thyroid progenitor/stem cells [9]. Insulin/IGF-I signaling pathways are important also in the regulation of thyroid-specific genes transcription, including the TSH receptor [10], thyroglobulin (Tg), and thyroperoxidase (TPO) [11, 12]. Gene expression of both Tg [13] and TPO [14] is mediated predominantly by thyroid transcription factor-2 (TTF-2), a thyroid-specific transcription factor that binds to the promoter of both genes [15, 16] and is stimulated by both the cAMP and the insulin/IGF-I pathways, which may have additive effects [17].
Regarding the adrenocortical cancers, their molecular pathogenesis is still incompletely understood. In contrast to thyroid carcinomas, the cAMP/PKA pathway seems to be less involved in the development of these tumors. Although pituitary adrenocorticotropic hormone (ACTH) stimulates adrenal function by inducing steroidogenic enzymes and increases adrenal gland weight, the proliferative action of ACTH for adrenal tumors has been questioned, and opposite effects, under defined cell culture conditions, have been reported. In vitro inhibition of adrenal cell proliferation by physiological ACTH concentrations has been reported by several groups [18–21]. In support of the growth-inhibiting effect of ACTH, no activating mutations of the ACTH receptor have been found in benign or malignant adrenocortical tumors [22, 23]. Conversely, allelic loss of the ACTH receptor gene has been reported in a subset of sporadic benign and malignant adrenocortical tumors where it was associated with undifferentiated phenotype and worse prognosis [24]. These data tend to exclude a role of ACTH receptor as putative oncogene in adrenal oncogenesis while supporting its role as tumor suppressor. In summary, in the adrenal cortex, the ACTH/PKA signaling is mainly involved in regulating steroid hormone synthesis and cellular differentiation rather than in controlling cellular proliferation and tumor growth. Similarly to thyroid cancer, molecular alterations frequently observed in adrenocortical carcinoma include deregulation of the IGF system as well as mutations in p53 and RAS [25]. In addition to IGF-II overexpression, increased levels of the IGF-IR and IGFBP-2 have been found in advanced human adrenal carcinomas, resulting in increased IGF-dependent cell proliferation and inhibition of the ACTH antiproliferative effect. Although the functional significance of the strong and specific overexpression of IGFs in adrenocortical carcinomas remains still unknown, these factors may regulate both steroidogenic and mitogenic effects and, similarly to what is seen in thyroid cancer, establish autocrine positive loops that promote growth advantage and transformation toward a more malignant phenotype.
In ovarian cancers, the role of the pituitary tropins is still controversial. Pituitary LH and FSH lead to increased sex steroids secretion which may favor ovarian cancer development [26, 27]. A role for gonadotropins in ovarian tumorigenesis is also supported by the observation that ovarian cancer incidence reaches a peak in the postmenopausal period, during which FSH levels are particularly high [28]. Yet, a study on normal rabbit ovarian surface epithelium showed that FSH and LH/hCG stimulate growth in vitro [29]. However, controversial results have been obtained by different research groups. For instance, a more recent study has reported no increase in cell proliferation with LH [30]. Although the mechanisms by which FSH and LH stimulate or inhibit the proliferation of ovarian epithelium remain still unknown, these hormones exert their effects interacting with their specific receptors. Some ovarian cancers, especially those poorly differentiated, lose FSH receptor (FSHR) expression [30]. This observation suggests that FSH may be a growth-promoting factor important at early stages of ovarian epithelial tumorigenesis, with some ovarian tumors losing their requirement for FSH later in tumor development. Like adrenal and thyroid cancers, also in ovarian tumors, IGF system components are often overexpressed. IGF-II appears to increase proliferation and induce differentiation in granulosa cells via the IGF-IR and synergizes with FSH to induce steroidogenesis as well as mitogenesis [31]. This synergism likely involves IGF-induced upregulation of the FSHR [31, 32] and/or the involvement of intracellular mechanisms leading to the activation of intracellular pathways (i.e., protein kinase C, cAMP, MAPK, and PI3K).
A schematic representation of the interplay between pituitary hormones and main signaling pathways of the IGF system in thyroid, adrenal, and ovary cancers is shown in Figure 1.
Figure 1.
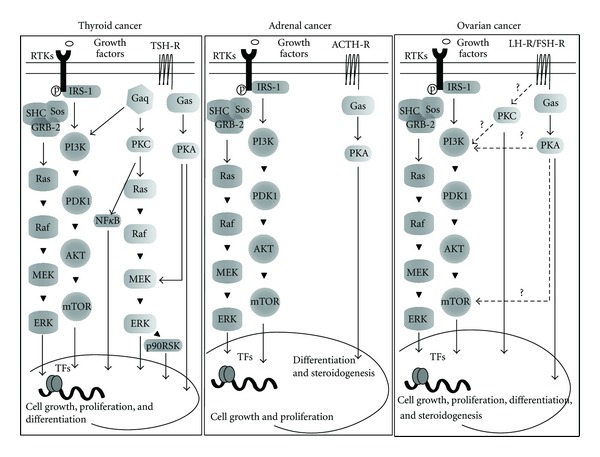
Schematic representation of the interplay between pituitary hormones and main signaling pathways of the IGF system in thyroid, adrenal and ovary cancers. Dot lines: proposed signaling pathways.
Altogether, the lines of evidence reported in these three tumor histotypes suggest that, although the specific pituitary tropin exerts an important role in regulating the growth, differentiation, and function of the target endocrine gland, the interplay of pituitary hormones with other factors, such as the IGF system, is crucial for deregulated cell proliferation and transformation. The IGF system may represent, therefore, a promising therapeutical target for these tumors.
3. The IGF System and Its Involvement in Cancer
In mammals the IGF system includes four receptors (the insulin receptor (IR), the IGF-I receptor (IGF-IR), the insulin-receptor-related receptor (IRR), and the Mannose-6-phosphate/IGF-II receptor (IGF-IIR)), four ligands (proinsulin, insulin, IGF-I, and IGF-II), and six high-affinity binding proteins (IGFP-1 to 6). The human IR exists in two isoforms (IR-A and IR-B) generated by alternative splicing of the IR gene with the exclusion (IR-A) or inclusion (IR-B) of 12 amino acids encoded by exon 11. The IR and the IGF-IR have highly homologous structure, but different functions. Given the high degree of homology, IR and IGF-IR can heterodimerize leading to the formation of insulin/IGF-I hybrid receptors (HRs) [33, 34]. The IGF-IIR is a structurally distinct cell surface receptor whose major function is to induce internalization and degradation of IGF-II, thus modulating its extracellular levels [34].
With regard to the ligands, insulin and IGFs are related peptides, involved in metabolism as well as in growth and reproduction. Insulin largely circulates in free form while more than 90% of IGFs circulates bound to a complex family of IGF-binding proteins (IGFBPs), which regulate both the half-life and the biological effects of IGFs [35]. Insulin and IGFs bind with different affinity IR isoforms and IGF-IR (for more details see [36–38]).
Recently, it has been reported that proinsulin, the insulin prohormone, which is characterized by low metabolic activity compared to mature insulin, is a selective IR-A ligand and may exert a putative role on growth and cell proliferation [39, 40].
After ligand binding, phosphorylated receptors activate two main signaling pathways, the PI3K and the MAPK cascade, involved in the regulation of cell metabolism, proliferation, and survival. Although both the IR and IGF-IR similarly activate these signaling networks, subtle differences exist in the recruitment of certain intracellular mediators and substrates between the two receptors, leading to the specific biological effects of each hormone. Details regarding the IGF system have been previously covered by several reviews to which we refer for more information [34, 36, 41].
Since the IGF system exerts a pivotal role in cell growth and homeostasis, it is not surprising that aberrant expression of receptors belonging to this system might be involved in cancer development, progression, and metastasis. The key role of IGF-IR in oncogenic transformation derives from the studies showing that IGF-IR null cells cannot be transformed by several cellular or viral oncogenes, whereas they become susceptible to the oncogenic mediated transformation after the reintroduction of a functional IGF-IR [42, 43]. However, increased levels of IGF-IR do not result in autonomous receptor signaling in the absence of IGF ligand, while it can induce malignant transformation in presence of its specific ligands [44]. Similarly, in estrogen responsive breast cancer cell lines, growth response to insulin could be specifically inhibited by anti-IR but not anti-IGF-IR blocking antibodies while it can be mimicked by an anti-IR stimulating antibody [45]. These data are in agreement with studies indicating that IR-transfected cells acquire insulin-dependent malignant changes [46, 47] and support the notion that IR may elicit mitogenic and antiapoptotic effects similar to IGF-IR, contributing to cancer development and progression. The first direct evidence that IR may be overexpressed in cancer cells was reported by Papa et al. in breast tumors [37]. Subsequent studies demonstrated that IR is also overexpressed in other human malignancies, including endocrine tumors, such as cancer of the thyroid, ovary, and adrenal glands [38, 48–50]. In most of these tumors, cell growth is dependent on IR activation by insulin, suggesting a mitogenic role of this hormone [51], although IR, isoform A, may also be activated by IGF-II [46, 52, 53]. Both IR isoforms may be overexpressed in cancer, but usually IR-A is predominant, representing 60–100% of total IR. Data showing an increased relative abundance of IR-A are also available for certain endocrine cancers [38, 48, 54]. This observation is particularly interesting, as IR-A is mainly expressed in fetal life, while IR-B predominates in differentiated tissues [38, 55]. Furthermore, at variance with IR-B, which is a highly specific receptor for insulin, IR-A is a high-affinity receptor for insulin; it shows intermediate affinity for IGF-II and low affinity for IGF-I [38]. Although IGF-II is able to bind both to IGF-IR and IR-A with similar affinity, its binding to IR-A has important implications. Indeed, IR-A overexpression amplifies IGF-II effects in cancer cells and serves as a signaling diversification factor, as IR-A and IGF-IR activate different downstream signals.
Because of the high homology between the IR and the IGF-IR [56], in cells coexpressing IRs and IGF-IR [57] hybrid IR/IGF-IR receptors (HRs) may form [58–60]. Functionally, HRs are considered high-affinity IGF-I-binding sites as they bind insulin with much lower affinity [60]. In thyroid cancers large amounts of HRs have been measured both in well-differentiated papillary carcinomas and in poorly differentiated/undifferentiated carcinomas, probably as a consequence of increased IR expression [33]. In these tumors, HRs account for 50–75% of the total IGF-I binding sites and mediate IGF-I mitogenic signaling. No data are available regarding HRs expression in other endocrine malignancies.
3.1. IR and IGF-IR Signaling Pathways: Relevance to Endocrine Malignancies
IR and IGF-IR share many similarities not only in their structures but also in their downstream signaling pathways. Upon ligand binding, the intrinsic tyrosine kinases of both IR and IGF-IR are activated and this results in the phosphorylation of several receptor substrates including the components of the IRS family and Shc. These substrates, in turn, act as multisite “docking” proteins for kinases and adaptors, such as PI3K, Syp, Fyn, Nck, and Grb2, which trigger the activation of downstream kinase cascades [61]. IRS proteins are also involved in the crosstalk with other signaling pathways, including those coming from other growth factors [62], cytokines [63], and integrins [64].
The two main signaling pathways downstream to IR and IGF-IR include the mitogen-activated protein kinases cascade (MAPKs), which involves the sequential activation of a cascade of serine/threonine protein kinases with a key role in the regulation of cellular proliferation and gene expression and the PI3K signaling pathway, which mediates metabolic actions but also stimulates cell growth and survival. Both MAPK and PI3K pathways enhance protein synthesis through mTOR activation and trigger antiapoptotic effects through the phosphorylation and inactivation of Bad [65]. Molecular alterations (mutational and nonmutational) in both PI3K and MAPK have been reported in several malignancies including those from thyroid, ovary, and adrenal glands.
Conditional or constitutive deregulation of MAPK and PI3K cascades is a common event in thyroid cancer and may play a pathogenetic role in this tumor [1]. Indeed, deregulated activation of the MAPK cascade via mutations and/or rearrangements in RET, RAS, and BRAF genes occurs in ~70% of papillary thyroid carcinomas (the most common subtype of thyroid cancers) [66–68]. Thyroid carcinomas also show mutations in PI3K signaling effectors such as PTEN and phosphoinositide-3-kinase, catalytic, alpha polypeptide (PIK3CA). PTEN is downregulated in ~37% of well-differentiated thyroid carcinomas and downregulated or lost in >50% of highly malignant thyroid cancers [69]; point mutations or copy number changes in PIK3CA are found in ~23% of anaplastic thyroid cancers where they can coexist with either RAS or BRAF mutations [70].
In adrenal cancer, pathway analysis for the genomic regions associated with poor prognosis has shown deletions of genes that negatively regulate the activation of ERK1/2 and loss of PTEN gene [71]. Yet, several reports have identified activating RAS mutations [72, 73], while only two papers have analyzed mutations in BRAF gene and found that their prevalence is low [74, 75]. Although functional studies are needed to better characterize the effect of these mutations in adrenocortical tumors, it is possible that deregulation in the MAPK pathway may significantly contribute to aggressive phenotypes.
In ovarian cancer, mutually exclusive mutations of KRAS and BRAF have been described in about 30–50% of low-grade tumors [76–79], while they are rare in high-grade tumors. RAS mutations may promote ovarian tumorigenesis not only through MAPK but also via the interaction with the PI3K/AKT pathway. In ovarian cancers PI3K activation, occurring via either PIK3CA gene amplification/mutations or PTEN protein loss, has been reported by several studies [80–83] with the highest frequency in most malignant histotypes [84].
In the context of the three endocrine tumors mentioned above, the dysregulation of the IGF system may represent one nonmutational mechanism activating MAPK and PI3K signaling cascades. The increased IGF-IR-mediated activation of MAPK/PI3K signaling may, in turn, induce IGF-IR and/or its ligands expression and reduce the expression of IGFBP-3 [85–87]. This close relationship between the IGF system and MAPK/PI3K-mediated signals may contribute to cancer development, progression, invasion, and aggressive behavior.
Indeed, via PI3K/AKT/PTEN and ERK dependent mechanisms [88–90], IGFs control several cycle checkpoints, in particular the G0-G1 transition, increasing cyclin D1 and CDK4 gene expression and down-regulating the cyclin-dependent kinase inhibitor (CDKI) p27. Moreover, through the same pathways, IGFs regulate cell invasion and tumor-dependent angiogenesis modulating the expression of molecular mediators of extracellular matrix remodeling and degradation including type IV collagenases, matrix metalloproteinase-2 (MMP-2) and matrix metalloproteinase-9 (MMP-9), and the membrane type 1 MMP (MMP-14) [91, 92]. These enzymes play an important role in malignant progression and metastatic spread of solid tumors, including endocrine ones. MMPs expression has been found to be elevated in papillary thyroid cancer as compared with normal thyroid tissue [93, 94]. A strong MMP2 expression has been also found in malignant adrenal tumors and considered an unfavorable prognostic factor [95]. In ovarian cancers, MMPs are frequently overexpressed and appear to be an early event of ovarian tumorigenesis suggesting a role of these enzymes in ovarian tumor initiation and not only in tumor progression and invasion [96].
Relevant crosstalks between the IGF system and other signaling pathways also include the involvement of the janus kinase (JAK)-1/2 mediated signaling and the activation of transcription proteins STAT. In particular, STAT-3 may be required for the maintenance of transforming activity of IGF-IR [97]. IGF-I is able to activate STAT-3, but not STAT-5, and this activation is probably mediated by JAK proteins [98]. These mechanisms have been demonstrated in several models, and they may also occur in endocrine cancers, where both STAT and IGF signaling play an important role in tumor invasion and metastasization. Indeed, the STAT-3 pathway is significantly upregulated in metastatic thyroid papillary cancers, suggesting a potential role for activated STAT-3 in lymphatic metastases [91]. Yet, in both ovarian and adrenal cancers a role for STAT signaling in invasion and cancer prognosis has been also identified [99, 100].
Other molecules interacting with the IGF system and involved in the pathogenesis of thyroid, adrenal, and ovarian cancers include the tumor suppressor p53. Inactivating mutations of p53 gene occur in 10%, 47%, and 25% of thyroid, ovary, and adrenal sporadic carcinomas [101, 102], respectively. However, in all these tumors, also when not mutated, p53 activity may be inhibited by other mechanisms among which are an unbalanced expression of isoforms with a dominant negative function, the interaction with Mdm2, and the cooperation with other members of the p53 family such as TAp63α, TAp73α, and their dominant negative variants (ΔNp63 and ΔNp73) [103]. The activity of wild-type p53 reduces IGF axis activity by multiple mechanisms which include inhibition of IGF-IR [104], IR [105], and IGF-II expression [106] with a concomitant increase of IGFBP-3 transcription [107]. Therefore, aberrant p53 (i.e., p53 lacking its suppressor function through point mutations or via other mechanisms) greatly enhances the activity of IGF axis at multiple levels [104]. In the three endocrine tumors mentioned above, the crosstalk between the IGF system and p53 appears an important prerequisite for oncogene-driven tumor cell trasformation, cancer progression, and resistance to anticancer therapies.
3.2. Circulating Levels of Insulin and IGFs and Endocrine Cancers
Epidemiological studies have shown that elevated plasma concentrations of IGFs are associated to increased risk for the development of several human malignancies including cancers of the breast, colon, and prostate as well as sarcomas [61, 108–113]. For instance, several studies have provided strong evidence that premenopausal, but not postmenopausal, women in the highest tertile of serum IGF-I levels had an increased risk of developing breast cancer [114], and that a high IGF-I : IGFBP-3 ratio may be associated with greater breast density and increased breast cancer risk [108, 115].
High circulating IGFs concentrations may exert biological effects in malignant cells not only through IGF-IR but also via IR-A and HRs. As described in more detail below, all these receptors are overexpressed in endocrine cancers [31, 48] as well as in thyroid cancer stem-like cells [9].
A possible role for serum IGF-I in thyroid cancerogenesis has been suggested by the observation that acromegalic patients, who are exposed to sustained high serum IGF-I levels, show an increased frequency of thyroid cancer [116, 117]. In adrenocortical tumors high serum IGFs and low IGFBP3 levels are correlated with cancer risk and are predictive of metastases development [111, 118].
Finally, some haplotypes and SNPs in the IGF components may influence ovarian risk, either directly or by increasing the IGF-I plasma levels. In particular, the following SNPs in the IGFBPs (rs10228265, rs4988515, rs2270628, rs2854746, and rs2854744), in IGF-I (rs11111285, rs1996656 and rs1019731), and in IGF-II (rs4320932, rs4244809, rs680, rs1003483, and rs7924316) have been associated with increased ovarian cancer risk [119, 120].
Not only IGFs but also circulating insulin has been suggested to be involved in the tumorigenesis process. Indeed, a number of population studies have provided substantial and circumstantial lines of evidence that insulin resistance and hyperinsulinemia, common factors underlying obesity and type 2 diabetes mellitus (T2DM), are strong candidates for the increased cancer risk associated with these disorders [121–123]. Although insulin is considered a hormone regulating energy metabolism, it also exerts proliferative, antiapoptotic, and migratory actions, collectively indicated as “mitogenic effects,” via its own receptor (IR). This observation is known from long time and helps in understanding the link between insulin resistance/hyperinsulinemia and cancer.
The involvement of insulin in cell trasformation and cancer development was firstly suggested by in vivo evidence that administration of insulin induced growth of mammary tumor in mice [124] and promoted aberrant crypt foci in the colon of rats [125–127], while insulin deficiency or calorie restriction exerted a protective role [124]. Similarly, in obese mice, insulin levels were positively associated with the proliferation of transplanted lung and colon cancer cells [127].
In light of these experimental lines of evidence, clinical studies have been conducted to investigate the possible role of hyperinsulinemia and insulin resistance in endocrine tumors. At this regard, several case-control and prospective studies have found a strong positive association between overweight/obesity and thyroid cancer risk [128–137], although the data are not entirely consistent [138–145].
The exact nature of the relationship between body mass index (BMI) and thyroid cancer incidence remains still unclear. Besides the high circulating levels of insulin present in overweight/obese patients, other potential mechanisms may include increased levels of inflammatory adipokines [146]. In addition, although obesity is associated with poor prognosis for several malignancies, this relationship has not been reported for thyroid cancer [142].
In partial support with the finding that hyperinsulinemia and insulin resistance are risk factors for thyroid cancer, studies conducted in T2DM patients have shown that higher fasting glucose levels are associated with increased thyroid cancer risk [144, 147]. However, also for this association conflicting results have been obtained [134].
Concerning the association between hyperinsulinemia/insulin resistance and ovarian cancer risk, several lines of evidence suggest that women affected by polycystic ovary syndrome (PCOS), a condition associated with insulin-resistance, are more likely to develop ovarian cancer (OR, 2.5; 95% CI 1.08–5.89) [148]. Furthermore, a meta-analysis of ten cohort studies has shown that overweight and obesity are associated with higher ovarian cancer mortality (OR, 1.6; 95% CI 1.1–2.34) and that, among patients with advanced ovarian cancer, premorbid obesity is associated with worse prognosis (OR, 1.5; 95% CI 1.09–1.93) [149]. However, other studies have not supported these results [150], suggesting that further investigation is needed to firmly establish the association between ovarian cancer and insulin resistance [151].
Finally, it has been suggested that adrenal incidentalomas, usually benign tumors, might be related to hyperinsulinemia and insulin resistance. This hypothesis was postulated for the first time by Reincke et al. [152], who observed a proliferative effect of insulin on adrenal cancer cells without effect on cortisol synthesis [152]. However, a causative role of adrenal incidentalomas for metabolic syndrome cannot be excluded, as some patients show a slight hypercortisolism that may contribute to the insulin resistance. In fact, surgical tumor resection may revert or ameliorate these metabolic alterations [153]. Regarding the link between hyperinsulinemia and malignant adrenal tumors, scanty data are present in the literature so far.
4. IGF System Abnormalities in Specific Endocrine Cancer and Possible Therapeutical Implications
4.1. Thyroid Cancer
Human thyroid carcinomas derived from the thyroid follicular cells (TFCs) include a variety of histotypes ranging from well-differentiated (papillary and follicular) to undifferentiated (anaplastic) cancers. Altogether, they represent approximately 1% (3% in women) of all human cancers [154, 155].
Well-differentiated thyroid carcinomas account for approximately 90% of all thyroid cancers. They retain a variable degree of TSH responsiveness and have a mortality rate of approximately 10%. Poorly differentiated and undifferentiated carcinomas account for only approximately 10% of all thyroid cancers; they have weak or no TSH responsiveness and have a mortality rate ranging from 50% to 100% [48, 156].
As previously mentioned, the IGF-I system plays an important role in regulating normal growth and development in the thyroid [6, 9] and appears also to be involved in thyroid tumorigenesis.
The coexpression of IGF-I and its cognate receptor, IGF-IR, has been documented by various studies in both cultured thyroid cells and tissue specimens. In particular, cultured human and ovine thyrocytes are able to release IGF-I in the culture media [157, 158]. Also, thyroid adenoma cell lines synthesize IGF-I, which stimulates cell growth by autocrine mechanisms [159]. Immunoreactive IGF-I and IGFBPs were also found in the extracts of normal and nodular thyroid tissue specimens obtained at surgery from patients with nontoxic goiter [160–162] (Table 1).
Table 1.
Main molecular alterations involving the IGF system components in thyroid, adrenal, and ovarian cancer.
| Molecular alteration | Thyroid cancer | Adrenal cancer | Ovarian cancer |
|---|---|---|---|
| IGF-IR overexpression | + | + | + |
| IR overexpression | + (IR-A) |
? | + (IR-A) |
| HRs overexpression | + | ? | ? |
| IGFs overexpression | + (IGF-I and IGF-II) |
+ (IGF-II > IGF-I) |
+ (IGF-II > IGF-I) |
| IGFs autocrine production | + (IGF-I and IGF-II) |
+ (IGF-II > IGF-I) |
+ (IGF-I and IGF-II) |
| IGF-II/IR-A loop activation | + | ? | + |
| IGFBPs overexpression | + | + (IGFBP-2, IGFBP-3, IGFBP-6) |
+ (IGFBP-2 > IGFBP-3 > IGFBP-4) |
| Relationship with elevated levels of insulin | + | + | + |
Functional IGF-IR is usually expressed at high levels in thyroid cancer cells. In SW579 thyroid carcinoma cells IGF-I induced angiogenic activity via increased synthesis of HIF-1α transcription factor and consequent stimulation of vascular endothelial growth factor (VEGF) expression [163]. Belfiore et al. measured IGFs and cognate receptors in both thyroid cell lines and tissue specimens. IGF-I content ranged from 104 to 2566 nM/g in cancer tissue and 69 to 680 nM/g in normal thyroid tissue. By using a specific ELISA, they also found that IGF-IR is overexpressed in both thyroid cancer cell lines and specimens as compared to the normal tissue [33] (Table 1).
IGF-IR overexpression in thyroid cancer specimens has been also found by using immunohistochemistry and in situ hybridization, and IGF-I was found to be produced in either paracrine or autocrine manner [8, 164]. IGF-I and IGF-IR immunoreactivity was found to be increased both in adenomas and carcinomas compared with normal thyroid. IGF-I overexpression was more marked in the undifferentiated and poorly differentiated histotypes of thyroid cancer [165].
The above-mentioned study of Vella et al. demonstrated that thyroid cancers overexpress not only IGF-I and IGF-IR, but also IGF-II and IR. In particular, the IGF-II/IR-A autocrine loop is especially activated in poorly differentiated and anaplastic cancers. The relative abundance of IR-A also increases in dedifferentiated cancers. In this context, the IGF-IR seems less important than IR-A in mediating IGF-II mitogenic effects, and blocking antibodies to IR markedly reduced the effects of IGF-II [48].
The concomitant high expression of both IGF-IR and IR-A in thyroid cancer cells causes overexpression of IR/IGF-IR hybrid receptors, which, in most cases, exceed the IGF-IR content. In cells with a high IR/IGF-IR content, blocking antibodies specific to these receptors substantially inhibited IGF-I-induced cell growth. These data indicate that, in addition to IGF-IR and IR-A, also IR/IGF-IR hybrids may be a target in thyroid cancers [33] (Table 1).
Progenitor/stem cells are increasingly considered to be at the origin of most malignancies [166]. Therefore, we recently isolated progenitor/stem cells from both normal and cancer specimens and cultured them as thyrospheres, in order to study the IGF system in this model [9]. We found that IGF-I and IGF-II are produced at high levels by all thyrospheres. However, the IGF-I : IGF-II ratio was approximately 5 : 1 in normal thyrospheres whereas it was 1 : 1 in cancer thyrospheres. IR and IGF-IR in human thyrospheres were markedly overexpressed and with a higher IR : IGF-IR ratio as compared to primary cultures. The IR : IGF-IR ratio was also higher in cancer than in normal thyrospheres. Receptors (IR and IGF-IR) and ligands (IGF-I and IGF-II), all expressed at high levels in thyrospheres, markedly decreased in differentiating cells. IR-A was the predominant isoform in thyrospheres, especially from cancer, while IR-B was predominant in differentiating cells. IR-A relative abundance was associated with characteristics of stemness and with cancer: it ranged from 65 to 86% in cancer thyrospheres, from 50 to 65% in normal thyrospheres, and from 40 to 45% in normal thyroid primary cultures or differentiated sphere-derived thyrocytes [9]. The expression of IR, IGF-IR, and their ligands was evaluated by quantitative real-time PCR. Western blot analysis for IR and IGF-IR confirmed PCR data. Cancer thyrosphere growth was stimulated by insulin and IGFs, while IGF-II was most potent in inducing cell renewal [9] (Table 1).
Considering the involvement of IGF-I system in thyroid cancer [9, 55, 156], Wang et al. studied the potential therapeutic role of anti-IGF-IR humanized monoclonal antibody A12 both in vitro and in vivo. In accordance to other studies, they found that IGF-IR is expressed in various human thyroid cancer cell lines and in normal and neoplastic human thyroid tissues, including surgical specimens of papillary and anaplastic carcinomas. IGF-IR antibody A12 was able to significantly inhibit the proliferation of cultured anaplastic cancer cells by downregulating the IGF-IR signaling pathway. Moreover, administration of A12 also reduced tumor volume in an orthotopic anaplastic cancer nude mouse model and prolonged survival [167] (Table 2).
Table 2.
Preclinical and clinical studies.
| In vitro | In vivo | Trials | References | |
|---|---|---|---|---|
| Thyroid cancer | Ab, IS | Ab, IS | — | Wang et al. 2006 [167]; Aiello et al. 2006 [169]; Chen et al. 2012 [170] |
| Adrenal cancer | TKI, Ab, IS | TKI, Ab | Ab, TKI | Barlaskar et al., 2009 [209]; Shen et al. 2007 [210]; Almeida et al., 2008 [177]; Ferruzzi et al., 2005 [213]; Cantini et al. 2008 [179] |
| Ovarian cancer | Ab, TKI, IS | IS | Ab, IS | Chakrabarty and Kondratick 2006 [223]; Gotlieb et al., 2006 [236]; Liao et al. 2012 [238]; Li et al., 2012 [237]; Romero et al., 2012 [239] |
TKI: tyrosine kinase inhibitor targeting the IGF system; Ab: antibody; IS: insulin sensitizer.
The PPARγ agonists thiazolidinediones and biguanides (metformin) are used as antidiabetic drugs for their insulin-sensitizing effect achieved by different mechanisms [168]. Because of these effects, both these classes of drugs lower circulating insulin levels, and, in principle, they may have favorable effects in patients with IR-overexpressing tumors. Moreover, both thiazolidinediones and metformin have direct and pleiotropic anti-IGF effects in cultured cancer cells. In particular, in anaplastic thyroid cancer (ATC) cells, rosiglitazone antagonized the biological effects of IGF-I by upregulating phosphatase and tensin homolog deleted from chromosome 10 (PTEN) and consequently inhibiting the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway. As a consequence, it reduced anchorage-dependent and -independent growth and migration, increased apoptosis rate, and induced partial redifferentiation in these cancer cells [169]. Rosiglitazone also potentiated the antitumor effect of doxorubicin (Table 2).
Recently, Chen et al. evaluated the effects of metformin, in ATC cell lines and in thyroid cancer stem cells. They found that metformin antagonized the growth-stimulatory effect of insulin in thyroid cancer cell lines. Specifically, metformin inhibited cell cycle progression, inhibited clonal cell growth, and reduced thyroid cancer sphere formation. Moreover, the metformin potentiated the antimitogenic effect of chemotherapeutic agents, such as doxorubicin and cisplatin, in ATC cells [170] (Table 2).
4.2. Adrenal Gland Cancer
Adrenal tumors are classified into benign and malignant groups. Tumor histotypes can be either hormonally silent or hormone secreting. In this case tumors may produce glucocorticoids, androgens, mineralocorticoids, estrogens, and combinations thereof [171]. The vast majority of adrenocortical tumors are benign, while adrenocortical carcinomas (ACCs) are relatively rare; they presents with extremely poor prognosis as a consequence of metastases or local invasion [172]. The frequency of small benign adrenocortical tumors increases with age, ranging between 3 and 7% of all adrenal carcinomas in adults over 50 years. However, ACCs account for only 0.05–0.2% of all cancers [173], with an estimated incidence between 1 and 2 per million and per year in adults in North America and Europe [174, 175]. In children, the incidence is approximately 10-fold lower except in South Brazil where there is a high incidence of pediatric ACC [176].
As previously mentioned, the IGF system has a physiological role in normal adrenal growth and development [177] and is also involved in ACC proliferation and progression [178]. An increased expression of the IGF-IR was demonstrated in SW13 and in H295R human adrenocortical carcinoma cell lines [179]. Both IGF-I and IGF-II are produced by H295R cells [180], and IGF-II increases during proliferation. In the same cell model an IGF-IR blocking antibody (α-IR3) was able to hamper cell growth, demonstrating that autocrine IGF-II production may stimulate cell growth through the IGF-IR [181] (Table 1).
In the reticularis layer of normal adrenal tissues a large number of IGF-I-positive cells with granular cytoplasmic (GC) staining pattern are present [182]. The proportion of these cells increases with the tumorigenesis process; hyperplastic glands show 10–50% of IGF-I-positive cells, while adenomas and carcinomas have over 50% of IGF-I positive cells in 64% and 83% of cases, respectively. Similarly, the IGF-IR is more expressed in adenomatous adrenal tissues than in nontumoral tissues [179]. IGF-II is one of the most expressed genes in adrenocortical carcinomas [183, 184]. The IGF-II gene is located at locus 11p15, which is maternally imprinted and consequently expressed only from the paternal allele. Structural abnormalities, characterized by the loss of maternal allele with the duplication of paternal allele, lead to biallelic expression of IGF-II gene. These alterations are frequently observed in sporadic adult ACCs, but only rarely in adenomas [118, 185, 186]. High IGF-II mRNA levels are associated with a more aggressive phenotype of ACC and a 5-fold increased risk of recurrence [180, 187] (Table 1).
In phosphoenolpyruvate carboxykinase (PEPCK) promoter human IGF-II transgenic mice, postnatal overexpression of IGF-II induced significantly increased adrenal weights, mainly caused by hyperplasia of the zona fasciculate [188]. This is in accordance with elevated serum corticosterone levels in IGF-II transgenic animals [189]. However, the observation that transgenic mice overexpressing IGFs or IGFBP-2 do not develop adrenal tumors indicates that IGF-II alone is not a tumor initiator for adrenal cells but rather a tumor progression factor that requires additional effectors for triggering adrenal tumorigenesis [189]. This notion is also supported by the clinical observation that deregulation of the IGF system is a late event often associated with advanced stage of the disease and poor clinical prognosis [118, 180, 190].
Indeed, in a cohort of pediatric and adult patients with adrenocortical tumors, IGF-II transcripts were mainly overexpressed in adult ACCs compared to adenomas [177]. Yet, a microarray analysis of 24 pediatric adrenocortical tumors (5 adenomas, 18 carcinomas, and 1 undetermined) demonstrated that the median expression of IGF-II in adrenocortical tumors was 18 times higher than in normal adrenal glands [191].
IGF-IR [192] and the IGF-binding protein-2 (IGFBP-2) [180] are also specifically overexpressed in ACCs. These molecular alterations may trigger a cascade of molecular events that can ultimately lead to malignancy in adrenocortical tumor progression [193]. This notion is confirmed by studies in adrenocortical tumor mouse cell line Y1, which have shown that stable transfection with human IGF-IR cDNA results in increased mitogenic response (+140%) to IGF-I as compared with nontransfected Y1 cells. In IGF-IR transfected cells the antiproliferative effect of ACTH was blunted and could be further antagonized by exogenous IGF-I [194] (Table 1).
In order to further clarify the significance of the IGF-IR in tumorigenesis of the human adrenal gland, Weber et al. examined the binding characteristics and concentrations of IGF-IR in normal adult human adrenocortical glands and in adrenocortical tumours of various origin. IGF IR binding in adrenocortical hyperplasias and adenomas was similar than in normal adrenocortical tissue. In contrast, three out of four hormonally active ACCs showed strongly elevated specific IGF-I binding with a 3-4-fold increase in IGF-IR concentration, as compared with normal adrenocortical tissue [194]. H295R cells overexpress also IGFBP-2 [195], which accounts for only 12% of the IGFBP activity in normal adrenocortical cells, but seems to play a specific role in the progression of ACCs by modulating IGF-II activity [196]. In support of the hypothesis of a tumor-growth-promoting effect of IGFBP-2 is the observation that Y-1 mouse adrenocortical tumor cells overexpressing IGFBP-2 show increased tumorigenic potential and cell proliferation [197]. However, the mechanisms of the IGFBP-2-associated increase in adrenal tumorigenesis remain largely unclear (see below).
Although the regulation of IGFBP production by IGFs is highly cell and species specific, a stimulatory effect of IGFs on IGFBP-3 has been reported in a large variety of cell systems [168, 198–204]. Treatment of adult human adrenocortical cells with ACTH predominantly stimulated the abundance of IGFBP-1 and to a lesser extent that of IGFBP-3, while IGF-I and IGF-II selectively induced the accumulation of IGFBP-3 and IGFBP-5 in the medium [205, 206]. Quantification of the specific bands by γ counting revealed that IGFBP-3 accounts for more than half of the detected IGFBP activity, followed by IGFBP-1 with 20% and IGFBP-4 with approximately 10% [196].
The gene expression profiles of IGFs system component may even distinguish malignant and benign tumours [207]. By analyzing the transcriptional profiles in 7 patients with ACCs and 13 with adenomas, Velázquez-Fernández et al. showed that in ACCs several IGF-related genes as IGF-II, IGF-IR, IGFBP3, and IGFBP6 were most significantly upregulated [207].
Recently, miRNAs able to regulate the IGF expression pattern in childhood adrenocortical tumors have been identified. Functional analysis of these miRNAs showed miR-99a and miR-100 regulate expression of IGF-IR, mTOR, and rictor in adrenocortical cancer cells, acting on target sites in their 3′-UTR regions. Downregulation of endogenous miR-100 in H295R and SW-13 cells increased protein expression of mTOR, raptor, and IGF-IR [208].
In order to evaluate the functional consequences of IGF-IR inhibition in adrenal carcinomas, Barlaskar et al. analyzed a large series of benign and malignant human adrenal tumors and a panel of ACC cell lines using a tyrosine kinase inhibitor, NVP-AEW541, and a fully human monoclonal antibody anti-IGF-IR, IMC-A12, both specifically targeting IGF-IR. Treatments with both NVP-AEW541 and IMC-A12 resulted in inhibition of growth of ACC cells in vitro. In xenograft tumors, IGF-IR blockade was more potent than mitotane, the first-line adrenolytic drug used in patients with ACC, and significantly enhanced mitotane response [209]. In vitro (in H295 and in SW-13 cells) efficacy of NVP-AEW541 treatment was confirmed by other studies [177, 210].
Thiazolidinediones (TZDs), a class of antidiabetic drugs, have also been investigated as potential therapeutic agents for ACC. TZDs are ligands for the peroxisome-proliferator-activated receptor (PPAR)-γ, a member of the nuclear receptor superfamily of ligand-dependent transcription factors, that is expressed predominantly in the adipose tissue but also in other tissues, although at much lower levels. PPAR-γ exerts a critical role in several biological processes such as adipogenesis, glucose metabolism, inflammation, cell growth, and differentiation [211]. Nowadays, the molecular basis for the antitumor action of PPAR-γ agonists remains incompletely elucidated. However, numerous studies support the notion that PPAR-γ activation induces apoptosis and thus exerts anticancer effects [212]. Although no differences in the expression of PPAR-γ are seen in normal and tumor tissue, the PPAR-γ agonist rosiglitazone inhibited growth and invasiveness of H295R cells [213]. Indeed, both in SW-13 and H295 ACC cells, rosiglitazone inhibited the signaling pathways downstream IGF-IR, but not the receptor itself [179] (Table 2).
Clinical trials are currently investigating the efficacy of monoclonal antibody IMC-A12, either used as monotherapy or in combination with mitotane (trials NCT00831844 and NCT00778817, resp.). The dual kinase inhibitor of both IGF-IR and IR, OSI-906, is currently being evaluated in ACC patients (trial NCT00924989) (Table 2).
4.3. Ovarian Cancer
Epithelial ovarian cancer (EOC) constitutes 90% of ovarian malignancies [214] and is the most common cause of gynecological cancer-related mortality [215]. It is fairly common in Scandinavia, less common in western Europe and North America, and infrequent in the developing countries and in Japan [216]. A first-degree family history of EOC is associated with approximately 3-fold increased risk [217]. EOCs are subdivided into four major categories: high-grade serous (70%), endometrioid (10%), clear cell (10%), mucinous (3%), and low-grade serous carcinomas (<5%) [214]. The marked clinical differences in ovarian cancer stage at presentation, response to therapy, and survival are manifestations of a complex underlying molecular heterogeneity of ovarian cancers [215]. Relatively little is known about the basic molecular and cellular mechanism that modulates growth of epithelial ovarian cancer and, presently, there are no available treatments capable of curing recurrent ovarian carcinomas due to their rapid evolution into a chemoresistant disease [218].
In physiology, as previously mentioned, regulation of ovarian activity requires a functional IGF axis. Moreover, over the last decade, accumulating data suggest that the insulin/IGF pathway might be a promising therapeutic target in ovarian cancer [219] (Table 1).
In 1991, Yee et al. examined the possibility that the IGF system could be important in regulating the autocrine growth of EOC cells [220]. The expressions of IGF-I-, IGF-IR-, and IGF-binding proteins were studied in ovarian cancer cell lines and tissues. IGF-IR mRNA was found in ovarian cancer cell lines and the primary or metastatic ovarian cancer tissues. In OVCAR-3 cell line, IGF-binding proteins, including IGFBP-2, IGFBP-3 and IGFBP-4, were expressed. In epithelial cells derived from untreated, ovarian cancer specimens, exogenous IGF-I induces cell proliferation. These cells secrete IGFs and IGF-binding proteins and express IGF-IR [221]. Ovary cancer cell lines also express IR; elevated levels of IR and insulin binding capacity were present in six cancer cell lines as compared to normal ovarian epithelium cell lines and were associated with mitogenic signaling in response to low doses of insulin. IR isoform analysis has shown preferential expression of IR-A, suggesting the ability of exogenous IGF-II to stimulate EOC cell proliferation through IR-A [54] (Table 1).
Moreover, studies in NIH-OVCAR3 cells have shown that IGF-I and IGFBP-2 promote ovarian cancer cell growth and invasiveness. IGFBP-2 is dramatically increased in the serum and ovarian cyst fluid of women with epithelial ovarian cancer [222, 223] and is involved in stimulation of cell growth [223]. In agreement with these data, elevated serum levels of IGF-I and IGFBP-2 have been associated with an increased risk of ovarian cancer [224, 225] (Table 1).
Other studies have shown that IGFBP-2 expression level in epithelial ovarian cancers is up to 38-fold higher than in normal ovarian epithelium [226]. Moreover, serum IGFBP-2 levels are elevated in women with early- and advanced-stage ovarian cancer as compared to controls and to patients with benign gynecological conditions, indicating that IGFBP-2 may be useful as a serum biomarker for detection and monitoring of epithelial ovarian cancer. Although the cellular mechanisms through which IGFBP-2 exerts a role in ovarian tumorigenesis are not completely elucidated, experimental data demonstrate that the growth-modulating effects of IGFBP-2 in ovarian cancer cells may be mediated by the activation of three specific cascades controlling cell growth, proliferation, and differentiation, that is, extracellular signal-regulated protein kinases (ERKs), stress-activated protein kinases (SAPKs) or c-Jun N-terminal protein kinases (JNKs) and p38 kinases. Furthermore, it has been seen that IGFBP-2 may regulate the expression of several potential cancer-promoting cytokines including fibroblast growth factors 6 and 7 (FGF-6 and -7), neurotrophin-4 (NT-4), and placental growth factor (PIGF) [223]. Thus, although IGFBPs are potent modulators of the mitogenic effects of IGFs, IGF-independent actions have also been recognized suggesting that IGFBPs are a separate class of growth modulators.
IGF-II is also considered a molecular marker and potential therapeutic target for the most aggressive EOCs. Indeed, when compared with normal ovarian surface epithelium samples, ovarian cancers show approximately 300-fold higher expression of the IGF-II gene. High IGF-II and lower IGFBP-3 expression are associated with high-grade, poorly differentiated, and advanced-stage disease [227, 228].
The association between IGF-II expression and ovarian cancer survival is driven by two specific promoters of IGF-II gene [229]. The IGF-II gene has four promoters, and each initiates a promoter-specific transcript which is expressed in a temporal and spatial-dependent manner. The transcription of three of the four IGF-II promoters, promoters 2, 3, and 4 (P2, P3 and P4), is regulated by DNA methylation [230]. DNA methylation alterations have been identified as being involved in tumorigenesis and disease progression [231, 232].
Using methylation-specific polymerase chain reaction (MSP) assay [233] it has been found that the methylation pattern of P2 and P3 IGF-II promoters as well as the levels of IGF-II mRNA and peptide was significantly different among patients with distinct tumor grade, residual tumor size, and treatment response. Patients with methylated P2 and unmethylated P3 (P2M/P3U) had 5 times higher mRNA expression and nearly 2-fold higher peptide levels compared to those with opposite pattern of methylation (P2U/P3M) [232].
Recently, genetic variations across the IGF components have been correlated with ovarian cancer risk [119, 120]. In primary ovarian cancer tissue, using microarray technology, Spentzos et al. have analyzed the expression patterns of gene families and pathways of IGF axis. Studying sixty-four patients with advanced stages of EOC, they found that expression patterns of IGF axis genes have prognostic significance in this highly lethal disease [234].
However, components of the IGF-I pathway were found overexpressed also in low-grade tumors, which respond to treatment with exogenous IGF-I with increased proliferation and migration [219].
Early data have shown that phosphorothioate antisense oligodeoxynucleotides (S-ODNs) [235] inhibit the function of the IGF-IR in NIH-OVCAR3 ovarian cancer cells and suppress cancer cell growth in vitro, but have small effects in vivo [235]. In the same cell model, a neutralizing antibody to IGFBP-2 also inhibits cell growth and downregulates the expression of a number of potential cancer-promoting cytokines [223].
More recently, it has been shown that NVP-AEW541, an IGF-IR tyrosine kinase inhibitor, is able to inhibit growth in EOC cell lines, OVCAR-3 and OVCAR-4, and to sensitize cells to cisplatin [236] (Table 2).
Today, at least five ongoing clinical trials aim to target the IGF-I axis in EOC patients. A phase II trial is currently investigating the fully human anti-IGF-IR monoclonal antibody AMG 479 in combination with paclitaxel and carboplatin (NCT00718523). Another phase II trial is examining AMG 479 in recurrent platinum-sensitive ovarian cancer (NCT00719212) (Table 2).
The combination between AMG 479 and AMG 655 (an human anti-DR5 monoclonal antibody) is the object of a study (phase I/II study) in patients with EOC and other advanced, refractory solid tumors (NCT00819169). A phase I/II trial is currently evaluating intermittent and continuous OSI-906, in combination with weekly paclitaxel, in patients with recurrent EOCs or other solid tumors (NCT00889382). Another phase I trial (NCT01322802) is testing the safety and immunogenicity of a DNA-plasmid-based vaccine encoding the amino acids 1–163 of IGFBP-2 in patients with advanced EOC.
Recently, an alternative therapeutical approach using insulin sensitizers, such as metformin, has been suggested against ovarian cancer. The rationale for using these drugs comes from evidence that a combination therapy with metformin and LY294002, an inhibitor of PI3K, reduces growth and induces apoptosis in ovarian cancer cells [237] by inhibiting PI3K/AKT and mTOR [238] while activating the AMPK/ACC pathway. In agreement with these preclinical data, an epidemiologic study conducted in 341 patients with EOC has shown that patients with T2DM who used metformin had longer progression-free survival than nonusers, despite receiving similar treatment for ovarian cancer [239]. A close relationship is now established between the use of metformin and progression, survival, and chemosensitivity of EOC [237–240] (Table 2).
Recently, a phase II clinical trial (NCT01579812) started to establish the potential role of metformin as anticancer stem cell agent in EOC patients. The primary objective of this study is to determine if metformin, administered as the time of traditional adjuvant chemotherapy to women with advanced EOC, will improve recurrence-free survival at 18 months compared to controls (Table 2).
5. Conclusions and Perspectives
Advanced endocrine tumors are characterized by poor prognosis and resistance to the common DNA-damaging chemotherapies or radiotherapy. Most extensively characterized endocrine malignancies include thyroid, adrenal, and ovarian cancers. In these tumors, a crosstalk between the IGF system and the pituitary hormones specific for each endocrine gland has been recognized and seems to exert a role in the tumorigenesis process. Recently, the risk of certain malignancies, including endocrine related cancers, has been found 2-3-fold increased in obese and T2DM patients. Insulin resistance and compensatory hyperinsulinemia, typical features of both obesity and diabetes, are the major candidates for cancer risk and are also associated with poor cancer prognosis and resistance to conventional and targeted anticancer therapies. Multiple alterations in the IGF system as well as association with high circulating levels of insulin/IGFs have been reported by several studies for these three endocrine cancer histotypes. This scenario may have important implications for endocrine cancer prevention and treatment. However, the potential role of the IGF system as therapeutical target in these tumors has being only recently evaluated and few clinical trials are currently ongoing.
Today the therapeutical strategies proposed to overcome IGF axis alterations in these malignancies include IGF-IR blocking antibodies, IGF-IR/IR tyrosine-kinase inhibitors, and insulin sensitizers. So far, preclinical results obtained with the first two classes of drugs mentioned above have shown promising hopes although the results are not conclusive and no complete responses have been reported. Furthermore, like in other malignancies, the development of intrinsic and adaptative resistance to IGF axis blockage could occur. Aberrant IR expression, particularly IR-A isoform, as well as HR-A formation and enhanced IGF-II autocrine production are very common alterations in endocrine cancers and could mediate the resistance to IGF-IR blocking drugs. Furthermore, insulin resistance and hyperinsulinemia are side effects of these drugs and may contribute to IR-A overactivation. New approaches aimed at specifically and safely targeting IR-A activation and/or disrupting the autocrine IGF-II/IR-A loop are urgently needed. In light of lines of evidence of association of endocrine cancers risk and hyperinsulinemia, insulin-sensitizers, such as metformin, hold promise as measures useful in cancer prevention.
Author's Contributions
R. Malaguarnera, A. Morcavallo equally contributed to the paper.
Acknowledgments
This workwas supported in part by grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC) to A. Belfiore (Grant no. 10625/12), AIRC Project Calabria 2012 and Fondazione Cassa di Risparmio di Calabria e Lucania to A. Belfiore, from PON (Programma Operativo Nazionale Ricerca e Competitività, Grant 01_01078), and from PRIN-MIUR, Grant 2008BKRFBH_005, to A. Belfiore.
Abbreviations
- RTKs:
Receptors tyrosine kinase
- SHC:
Src homology domain C-terminal
- Sos:
Son of sevenless
- Grb-2:
Growth factor receptor-bound protein 2
- PKA:
Protein kinase A
- PKC:
Protein kinase C
- Gαs:
G-protein (s) subunit alpha
- Gaq:
G-protein (q) subunit alpha
- IRS-1:
Insulin receptor substrate-1
- PI3K:
Phosphoinositide 3-kinase
- PDK1:
Phosphorylated 3-phosphoinoside-dependent protein kinase
- Akt:
Protein kinase B
- mTOR:
Mammalian target of rapamycin
- Ras/Raf/MEK:
Mitogen-activated protein kinase
- ERK kinase:
Extracellular-signal-regulated kinase
- NFκB:
Nuclear factor kappa-B
- p90RSK:
p90 ribosomal S6 kinase
- TFs:
Transcription factors.
References
- 1.Nikiforov YE, Nikiforova MN. Molecular genetics and diagnosis of thyroid cancer. Nature Reviews Endocrinology. 7:569–580. doi: 10.1038/nrendo.2011.142. [DOI] [PubMed] [Google Scholar]
- 2.Russo D, Arturi F, Suarez HG, et al. Thyrotropin receptor gene alterations in thyroid hyperfunctioning adenomas. Journal of Clinical Endocrinology and Metabolism. 1996;81(4):1548–1551. doi: 10.1210/jcem.81.4.8636365. [DOI] [PubMed] [Google Scholar]
- 3.Parma J, Duprez L, van Sande J, et al. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature. 1993;365(6447):649–651. doi: 10.1038/365649a0. [DOI] [PubMed] [Google Scholar]
- 4.Biondi B, Filetti S, Schlumberger M. Thyroid-hormone therapy and thyroid cancer: a reassessment. Nature clinical practice. Endocrinology & metabolism. 2005;1(1):32–40. doi: 10.1038/ncpendmet0020. [DOI] [PubMed] [Google Scholar]
- 5.Milazzo G, La Rosa GL, Catalfamo R, Vigneri R, Belfiore A. Effect of TSH in human thyroid cells: evidence for both mitogenic and antimitogenic effects. Journal of Cellular Biochemistry. 1992;49(3):231–238. doi: 10.1002/jcb.240490305. [DOI] [PubMed] [Google Scholar]
- 6.Dumont JE, Lamy F, Roger P, Maenhaut C. Physiological and pathological regulation of thyroid cell proliferation and differentiation by thyrotropin and other factors. Physiological Reviews. 1992;72(3):667–697. doi: 10.1152/physrev.1992.72.3.667. [DOI] [PubMed] [Google Scholar]
- 7.Ciampolillo A, De Tullio C, Giorgino F. The IGF-I/IGF-I receptor pathway: implications in the pathophysiology of thyroid cancer. Current Medicinal Chemistry. 2005;12(24):2881–2891. doi: 10.2174/092986705774454715. [DOI] [PubMed] [Google Scholar]
- 8.van der Laan BFAM, Freeman JL, Asa SL. Expression of growth factors and growth factor receptors in normal and tumorous human thyroid tissues. Thyroid. 1995;5(1):67–73. doi: 10.1089/thy.1995.5.67. [DOI] [PubMed] [Google Scholar]
- 9.Malaguarnera R, Frasca F, Garozzo A, et al. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. Journal of Clinical Endocrinology and Metabolism. 2011;96(3):766–774. doi: 10.1210/jc.2010-1255. [DOI] [PubMed] [Google Scholar]
- 10.Saji M, Kohn LD. Insulin and insulin-like growth factor-I inhibit thyrotropin-increased iodide transport in serum-depleted FRTL-5 rat thyroid cells: modulation of adenosine 3’,5’-monophosphate signal action. Endocrinology. 1991;128(2):1136–1143. doi: 10.1210/endo-128-2-1136. [DOI] [PubMed] [Google Scholar]
- 11.Santisteban P, Kohn LD, di Lauro R. Thyroglobulin gene expression is regulated by insulin and insulin-like growth factor I, as well as thyrotropin, in FRTL-5 thyroid cells. Journal of Biological Chemistry. 1987;262(9):4048–4052. [PubMed] [Google Scholar]
- 12.Zarrilli R, Formisano S, Di Jeso B. Hormonal regulation of thyroid peroxidase in normal and transformed rat thyroid cells. Molecular Endocrinology. 1990;4(1):39–45. doi: 10.1210/mend-4-1-39. [DOI] [PubMed] [Google Scholar]
- 13.Santisteban P, Acebron A, Polycarpou-Schwarz M, Di Lauro R. Insulin and insulin-like growth factor I regulate a thyroid-specific nuclear protein that binds to the thyroglobulin promoter. Molecular Endocrinology. 1992;6(8):1310–1317. doi: 10.1210/mend.6.8.1406708. [DOI] [PubMed] [Google Scholar]
- 14.Aza-Blanc P, Di Lauro R, Santisteban P. Identification of a cis-regulatory element and a thyroid-specific nuclear factor mediating the hormonal regulation of rat thyroid peroxidase promoter activity. Molecular Endocrinology. 1993;7(10):1297–1306. doi: 10.1210/mend.7.10.8264661. [DOI] [PubMed] [Google Scholar]
- 15.Civitareale D, Lonigro R, Sinclair AJ, Di Lauro R. A thyroid-specific nuclear protein essential for tissue-specific expression of the thyroglobulin promoter. EMBO Journal. 1989;8(9):2537–2542. doi: 10.1002/j.1460-2075.1989.tb08391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Francis-Lang H, Zannini M, de Felice M, Berlingieri MT, Fusco A, Di Lauro R. Multiple mechanisms of interference between transformation and differentiation in thyroid cells. Molecular and Cellular Biology. 1992;12(12):5793–5800. doi: 10.1128/mcb.12.12.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ortiz L, Zannini M, Lauro RD, Santisteban P. Transcriptional control of the forkhead thyroid transcription factor TTF-2 by thyrotropin, insulin, and insulin-like growth factor I. Journal of Biological Chemistry. 1997;272(37):23334–23339. doi: 10.1074/jbc.272.37.23334. [DOI] [PubMed] [Google Scholar]
- 18.Masui H, Garren LD. Inhibition of replication in functional mouse adrenal tumor cells by adrenocorticotropic hormone mediated by adenosine 3’:5’-cyclic monophosphate. Proceedings of the National Academy of Sciences of the United States of America. 1971;68(12):3206–3210. doi: 10.1073/pnas.68.12.3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weidman ER, Gill GN. Differential effects of ACTH or 8 Br cAMP on growth and replication in a functional adrenal tumor cell line. Journal of Cellular Physiology. 1977;90(1):91–104. doi: 10.1002/jcp.1040900112. [DOI] [PubMed] [Google Scholar]
- 20.Morera AM, Saez JM. In vitro mitogenic and steroidogenic effects of ACTH analogues on an adrenal tumor cell line (Y-1) Experimental Cell Research. 1980;127(2):446–451. doi: 10.1016/0014-4827(80)90451-6. [DOI] [PubMed] [Google Scholar]
- 21.Ramachandran J, Suyama AT. Inhibition of replication of normal adrenocortical cells in culture by adrenocorticotropin. Proceedings of the National Academy of Sciences of the United States of America. 1975;72(1):113–117. doi: 10.1073/pnas.72.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Latronico AC, Reincke M, Mendonca BB, et al. No evidence for oncogenic mutations in the adrenocorticotropin receptor gene in human adrenocortical neoplasms. Journal of Clinical Endocrinology and Metabolism. 1995;80(3):875–877. doi: 10.1210/jcem.80.3.7883845. [DOI] [PubMed] [Google Scholar]
- 23.Light K, Jenkins PJ, Weber A, et al. Are activating mutations of the adrenocorticotropin receptor involved in adrenal cortical neoplasia? Life Sciences. 1995;56(18):1523–1527. doi: 10.1016/0024-3205(95)00114-l. [DOI] [PubMed] [Google Scholar]
- 24.Reincke M, Mora P, Beuschlein F, Arlt W, Chrousos GP, Allolio B. Deletion of the adrenocorticotropin receptor gene in human adrenocortical tumors: implications for tumorigenesis. Journal of Clinical Endocrinology and Metabolism. 1997;82(9):3054–3058. doi: 10.1210/jcem.82.9.4211. [DOI] [PubMed] [Google Scholar]
- 25.Kirschner LS. Signaling pathways in adrenocortical cancer. Annals of the New York Academy of Sciences. 2002;968:222–239. doi: 10.1111/j.1749-6632.2002.tb04338.x. [DOI] [PubMed] [Google Scholar]
- 26.Cramer DW, Hutchison GB, Welch WR. Determinants of ovarian cancer risk. I. Reproductive experiences and family history. Journal of the National Cancer Institute. 1983;71(4):711–716. [PubMed] [Google Scholar]
- 27.Cramer DW, Welch WR. Determinants of ovarian cancer risk. II. Inferences regarding pathogenesis. Journal of the National Cancer Institute. 1983;71(4):717–721. [PubMed] [Google Scholar]
- 28.Stadel BV. The etiology and prevention of ovarian cancer. American Journal of Obstetrics and Gynecology. 1975;123(7):772–774. doi: 10.1016/0002-9378(75)90509-8. [DOI] [PubMed] [Google Scholar]
- 29.Nicosia SV, Johnson JH, Streibel EJ. Growth characteristics of rabbit ovarian mesothelial (surface epithelial) cells. International Journal of Gynecological Pathology. 1985;4:58–74. doi: 10.1097/00004347-198501000-00005. [DOI] [PubMed] [Google Scholar]
- 30.Zheng W, Lu JJ, Luo F, et al. Ovarian epithelial tumor growth promotion by follicle-stimulating hormone and inhibition of the effect by luteinizing hormone. Gynecologic Oncology. 2000;76(1):80–88. doi: 10.1006/gyno.1999.5628. [DOI] [PubMed] [Google Scholar]
- 31.Spicer LJ, Aad PY. Insulin-like growth factor (IGF) 2 stimulates steroidogenesis and mitosis of bovine granulosa cells through the IGF1 receptor: role of follicle-stimulating hormone and IGF2 receptor. Biology of Reproduction. 2007;77:18–27. doi: 10.1095/biolreprod.106.058230. [DOI] [PubMed] [Google Scholar]
- 32.Zhou J, Kumar TR, Matzuk MM, Bondy C. Insulin-like growth factor I regulates gonadotropin responsiveness in the murine ovary. Molecular Endocrinology. 1997;11(13):1924–1933. doi: 10.1210/mend.11.13.0032. [DOI] [PubMed] [Google Scholar]
- 33.Belfiore A, Pandini G, Vella V, Squatrito S, Vigneri R. Insulin/IGF-I hybrid receptors play a major role in IGF-I signaling in thyroid cancer. Biochimie. 1999;81(4):403–407. doi: 10.1016/s0300-9084(99)80088-1. [DOI] [PubMed] [Google Scholar]
- 34.Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocrine Reviews. 2009;30(6):586–623. doi: 10.1210/er.2008-0047. [DOI] [PubMed] [Google Scholar]
- 35.Rajaram S, Baylink DJ, Mohan S. Insulin-like growth factor-binding proteins in serum and other biological fluids: regulation and functions. Endocrine Reviews. 1997;18(6):801–831. doi: 10.1210/edrv.18.6.0321. [DOI] [PubMed] [Google Scholar]
- 36.Gallagher EJ, LeRoith D. Minireview: IGF, insulin, and cancer. Endocrinology. 2011;152(7):2546–2551. doi: 10.1210/en.2011-0231. [DOI] [PubMed] [Google Scholar]
- 37.Papa V, Pezzino V, Costantino A, et al. Elevated insulin receptor content in human breast cancer. Journal of Clinical Investigation. 1990;86(5):1503–1510. doi: 10.1172/JCI114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frasca F, Pandini G, Scalia P, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin- like growth factor II receptor in fetal and cancer cells. Molecular and Cellular Biology. 1999;19(5):3278–3288. doi: 10.1128/mcb.19.5.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malaguarnera R, Sacco A, Voci C, Pandini G, Vigneri R, Belfiore A. Proinsulin binds with high affinity the insulin receptor isoform A and predominantly activates the mitogenic pathway. Endocrinology. 2012;153(5):2152–2163. doi: 10.1210/en.2011-1843. [DOI] [PubMed] [Google Scholar]
- 40.Hernández-Sánchez C, Mansilla A, de la Rosa EJ, de Pablo F. Proinsulin in development: new roles for an ancient prohormone. Diabetologia. 2006;49(6):1142–1150. doi: 10.1007/s00125-006-0232-5. [DOI] [PubMed] [Google Scholar]
- 41.LeRoith D, Roberts CT., Jr. The insulin-like growth factor system and cancer. Cancer Letters. 2003;195(2):127–137. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- 42.Morrione A, DeAngelis T, Baserga R. Failure of the bovine papillomavirus to transform mouse embryo fibroblasts with a targeted disruption of the insulin-like growth factor I receptor genes. Journal of Virology. 1995;69(9):5300–5303. doi: 10.1128/jvi.69.9.5300-5303.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sell C, Dumenil G, Deveaud C, et al. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Molecular and Cellular Biology. 1994;14(6):3604–3612. doi: 10.1128/mcb.14.6.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaleko M, Rutter WJ, Miller AD. Overexpression of the human insulinlike growth factor I receptor promotes ligand-dependent neoplastic transformation. Molecular and Cellular Biology. 1990;10(2):464–473. doi: 10.1128/mcb.10.2.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Milazzo G, Giorgino F, Damante G, et al. Insulin receptor expression and function in human breast cancer cell lines. Cancer Research. 1992;52(14):3924–3930. [PubMed] [Google Scholar]
- 46.Giorgino F, Belfiore A, Milazzo G, et al. Overexpression of insulin receptors in fibroblast and ovary cells induces a ligand-mediated transformed phenotype. Molecular Endocrinology. 1991;5(3):452–459. doi: 10.1210/mend-5-3-452. [DOI] [PubMed] [Google Scholar]
- 47.Mastick CC, Kato H, Roberts CT, Jr., LeRoith D, Saltiel AR. Insulin and insulin-like growth factor-I receptors similarly stimulate deoxyribonucleic acid synthesis despite differences in cellular protein tyrosine phosphorylation. Endocrinology. 1994;135(1):214–222. doi: 10.1210/endo.135.1.7516864. [DOI] [PubMed] [Google Scholar]
- 48.Vella V, Pandini G, Sciacca L, et al. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. Journal of Clinical Endocrinology and Metabolism. 2002;87(1):245–254. doi: 10.1210/jcem.87.1.8142. [DOI] [PubMed] [Google Scholar]
- 49.Kamio T, Shigematsu K, Kawai K, Tsuchiyama H. Immunoreactivity and receptor expression of insulinlike growth factor I and insulin in human adrenal tumors. An immunohistochemical study of 94 cases. American Journal of Pathology. 1991;138(1):83–91. [PMC free article] [PubMed] [Google Scholar]
- 50.Beck EP, Russo P, Gliozzo B, et al. Identification of insulin and insulin-like growth factor I, (IGF I) receptors in ovarian cancer tissue. Gynecologic Oncology. 1994;53:196–201. doi: 10.1006/gyno.1994.1115. [DOI] [PubMed] [Google Scholar]
- 51.Pillemer G, Lugasi-Evgi H, Scharovsky G, Naor D. Insulin dependence of murine lymphoid T-cell leukemia. International Journal of Cancer. 1992;50(1):80–85. doi: 10.1002/ijc.2910500117. [DOI] [PubMed] [Google Scholar]
- 52.Mamounas M, Gervin D, Englesberg E. The insulin receptor as a transmitter of a mitogenic signal in Chinese hamster ovary CHO-K1 cells. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(23):9294–9298. doi: 10.1073/pnas.86.23.9294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morrione A, Valentinis B, Xu SQ, et al. Insulin-like growth factor II stimulates cell proliferation through the insulin receptor. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(8):3777–3782. doi: 10.1073/pnas.94.8.3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kalli KR, Falowo OI, Bale LK, Zschunke MA, Roche PC, Conover CA. Functional insulin receptors on human epithelial ovarian carcinoma cells: implications for IGF-II mitogenic signaling. Endocrinology. 2002;143(9):3259–3267. doi: 10.1210/en.2001-211408. [DOI] [PubMed] [Google Scholar]
- 55.Belfiore A. The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Current Pharmaceutical Design. 2007;13(7):671–686. doi: 10.2174/138161207780249173. [DOI] [PubMed] [Google Scholar]
- 56.Ullrich A, Gray A, Tam AW, et al. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO Journal. 1986;5(10):2503–2512. doi: 10.1002/j.1460-2075.1986.tb04528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pandini G, Vigneri R, Costantino A, et al. Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clinical Cancer Research. 1999;5(7):1935–1944. [PubMed] [Google Scholar]
- 58.Moxham CP, Jacobs S. Insulin/IGF-I receptor hybrids: a mechanism for increasing receptor diversity. Journal of Cellular Biochemistry. 1992;48(2):136–140. doi: 10.1002/jcb.240480205. [DOI] [PubMed] [Google Scholar]
- 59.Bailyes EM, Nave BT, Soos MA, Orr SR, Hayward AC, Siddle K. Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochemical Journal. 1997;327, part 1:209–215. doi: 10.1042/bj3270209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soos MA, Nave BT, Siddle K. Immunological studies of type I IGF receptors and insulin receptors: characterisation of hybrid and atypical receptor subtypes. Advances in Experimental Medicine and Biology. 1993;343:145–157. doi: 10.1007/978-1-4615-2988-0_15. [DOI] [PubMed] [Google Scholar]
- 61.Samani AA, Brodt P. The receptor for the type I insulin-like growth factor and its ligands regulate multiple cellular functions that impact on metastasis. Surgical Oncology Clinics of North America. 2001;10(2):289–312. [PubMed] [Google Scholar]
- 62.Argetsinger LS, Hsu GW, Myers MG, Jr., Billestrup N, White MF, Carter- Su C. Growth hormone, interferon-γ, and leukemia inhibitory factor promoted tyrosyl phosphorylation of insulin receptor substrate-1. Journal of Biological Chemistry. 1995;270(24):14685–14692. doi: 10.1074/jbc.270.24.14685. [DOI] [PubMed] [Google Scholar]
- 63.Myers MG, Jr., Grammer TC, Wang LM, et al. Insulin receptor substrate-1 mediates phosphatidylinositol 3’-kinase and p70(S6k) signaling during insulin, insulin-like growth factor-1, and interleukin-4 stimulation. Journal of Biological Chemistry. 1994;269(46):28783–28789. [PubMed] [Google Scholar]
- 64.Vuori K, Ruoslahti E. Association of insulin receptor substrate-1 with integrins. Science. 1994;266(5190):1576–1578. doi: 10.1126/science.7527156. [DOI] [PubMed] [Google Scholar]
- 65.Petley T, Graff K, Jiang W, Yang H, Florini JR. Variation among cell types in the signaling pathways by which IGF-I stimulates specific cellular responses. Hormone and Metabolic Research. 1999;31(2-3):70–76. doi: 10.1055/s-2007-978701. [DOI] [PubMed] [Google Scholar]
- 66.Espinosa AV, Porchia L, Ringel MD. Targeting BRAF in thyroid cancer. British Journal of Cancer. 2007;96(1):16–20. doi: 10.1038/sj.bjc.6603520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fugazzola L, Puxeddu E, Avenia N, et al. Correlation between B-RAFV600E mutation and clinico-pathologic parameters in papillary thyroid carcinoma: data from a multicentric Italian study and review of the literature. Endocrine-Related Cancer. 2006;13(2):455–464. doi: 10.1677/erc.1.01086. [DOI] [PubMed] [Google Scholar]
- 68.Chiloeches A, Marais R. Is BRAF the Achilles’ heel of thyroid cancer? Clinical Cancer Research. 2006;12(6):1661–1664. doi: 10.1158/1078-0432.CCR-06-0051. [DOI] [PubMed] [Google Scholar]
- 69.Halachmi N, Halachmi S, Evron E, et al. Somatic mutations of the PTEN tumor suppressor gene in sporadic follicular thyroid tumors. Genes Chromosomes Cancer. 1998;23:239–243. doi: 10.1002/(sici)1098-2264(199811)23:3<239::aid-gcc5>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 70.Hou P, Liu D, Shan Y, et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clinical Cancer Research. 2007;13(4):1161–1170. doi: 10.1158/1078-0432.CCR-06-1125. [DOI] [PubMed] [Google Scholar]
- 71.Bussey KJ, Demeure MJ. Genomic and expression profiling of adrenocortical carcinoma: application to diagnosis, prognosis and treatment. Future Oncology. 2009;5(5):641–655. doi: 10.2217/fon.09.45. [DOI] [PubMed] [Google Scholar]
- 72.Yashiro T, Hara H, Fulton NC, et al. Point mutations of ras genes in human adrenal cortical tumors: absence in adrenocortical hyperplasia. World Journal of Surgery. 1994;18(4):455–461. doi: 10.1007/BF00353735. [DOI] [PubMed] [Google Scholar]
- 73.Lin SR, Tsai JH, Yang YC, Lee SC. Mutations of K-ras oncogene in human adrenal tumours in Taiwan. British Journal of Cancer. 1998;77(7):1060–1065. doi: 10.1038/bjc.1998.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kotoula V, Sozopoulos E, Litsiou H, et al. Mutational analysis of the BRAF, RAS and EGFR genes in human adrenocortical carcinomas. Endocrine-Related Cancer. 2009;16(2):565–572. doi: 10.1677/ERC-08-0101. [DOI] [PubMed] [Google Scholar]
- 75.Masi G, Lavezzo E, Iacobone M, Favia G, Palù G, Barzon L. Investigation of BRAF and CTNNB1 activating mutations in adrenocortical tumors. Journal of Endocrinological Investigation. 2009;32(7):597–600. doi: 10.1007/BF03346515. [DOI] [PubMed] [Google Scholar]
- 76.Singer G, Kurman RJ, Chang HW, Cho SKR, Shih IM. Diverse tumorigenic pathways in ovarian serous carcinoma. American Journal of Pathology. 2002;160(4):1223–1228. doi: 10.1016/s0002-9440(10)62549-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Singer G, Oldt R, Cohen Y, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. Journal of the National Cancer Institute. 2003;95(6):484–486. doi: 10.1093/jnci/95.6.484. [DOI] [PubMed] [Google Scholar]
- 78.Willner J, Wurz K, Allison KH, et al. Alternate molecular genetic pathways in ovarian carcinomas of common histological types. Human Pathology. 2007;38(4):607–613. doi: 10.1016/j.humpath.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 79.Cho YH, Kim DY, Kim JH, et al. Mutational analysis of KRAS, BRAF, and TP53 genes of ovarian serous carcinomas in Korean women. Yonsei Medical Journal. 2009;50(2):266–272. doi: 10.3349/ymj.2009.50.2.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abubaker J, Bavi P, Al-Haqawi W, et al. PIK3CA alterations in Middle Eastern ovarian cancers. Molecular Cancer. 2009;8, article 51 doi: 10.1186/1476-4598-8-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shayesteh L, Lu Y, Kuo WL, et al. PlK3CA is implicated as an oncogene in ovarian cancer. Nature Genetics. 1999;21(1):99–102. doi: 10.1038/5042. [DOI] [PubMed] [Google Scholar]
- 82.Campbell IG, Russell SE, Choong DYH, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Research. 2004;64(21):7678–7681. doi: 10.1158/0008-5472.CAN-04-2933. [DOI] [PubMed] [Google Scholar]
- 83.Woenckhaus J, Steger K, Sturm K, Münstedt K, Franke FE, Fenic I. Prognostic value of PIK3CA and phosphorylated AKT expression in ovarian cancer. Virchows Archiv. 2007;450(4):387–395. doi: 10.1007/s00428-006-0358-3. [DOI] [PubMed] [Google Scholar]
- 84.Kuo KT, Mao TL, Jones S, et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. American Journal of Pathology. 2009;174(5):1597–1601. doi: 10.2353/ajpath.2009.081000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hwang PH, Kim SY, Lee JC, Kim SJ, Yi HK, Lee DY. PTEN/MMAC1 enhances the growth inhibition by anticancer drugs with downregulation of IGF-II expression in gastric cancer cells. Experimental and Molecular Medicine. 2005;37(5):391–398. doi: 10.1038/emm.2005.49. [DOI] [PubMed] [Google Scholar]
- 86.Yi HK, Kim SY, Hwang PH, et al. Impact of PTEN on the expression of insulin-like growth factors (IGFs) and IGF-binding proteins in human gastric adenocarcinoma cells. Biochemical and Biophysical Research Communications. 2005;330(3):760–767. doi: 10.1016/j.bbrc.2005.03.045. [DOI] [PubMed] [Google Scholar]
- 87.Zhao H, Dupont J, Yakar S, Karas M, LeRoith D. PTEN inhibits cell proliferation and induces apoptosis by downregulating cell surface IGF-IR expression in prostate cancer cells. Oncogene. 2004;23(3):786–794. doi: 10.1038/sj.onc.1207162. [DOI] [PubMed] [Google Scholar]
- 88.Dupont J, Karas M, LeRoith D. The potentiation of estrogen on insulin-like growth factor I action in MCF-7 human breast cancer cells includes cell cycle components. Journal of Biological Chemistry. 2000;275(46):35893–35901. doi: 10.1074/jbc.M006741200. [DOI] [PubMed] [Google Scholar]
- 89.Rosenthal SM, Cheng ZQ. Opposing early and late effects of insulin-like growth factor I on differentiation and the cell cycle regulatory retinoblastoma protein in skeletal myoblasts. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(22):10307–10311. doi: 10.1073/pnas.92.22.10307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science. 1996;272(5263):877–880. doi: 10.1126/science.272.5263.877. [DOI] [PubMed] [Google Scholar]
- 91.Zhang D, Brodt P. Type 1 insulin-like growth factor regulates MT1-MMP synthesis and tumor invasion via PI 3-kinase/Akt signaling. Oncogene. 2003;22(7):974–982. doi: 10.1038/sj.onc.1206197. [DOI] [PubMed] [Google Scholar]
- 92.Mira E, Mañes S, Lacalle RA, Márquez G, Martínez-A C. Insulin-like growth factor I-triggered cell migration and invasion are mediated by matrix metalloproteinase-9. Endocrinology. 1999;140(4):1657–1664. doi: 10.1210/endo.140.4.6623. [DOI] [PubMed] [Google Scholar]
- 93.Maeta H, Ohgi S, Terada T. Protein expression of matrix metalloproteinases 2 and 9 and tissue inhibitors of metalloproteinase 1 and 2 in papillary thyroid carcinomas. Virchows Archiv. 2001;438(2):121–128. doi: 10.1007/s004280000286. [DOI] [PubMed] [Google Scholar]
- 94.Baldini E, Toller M, Graziano FM, et al. Expression of matrix metalloproteinases and their specific inhibitors in normal and different human thyroid tumor cell lines. Thyroid. 2004;14(11):881–888. doi: 10.1089/thy.2004.14.881. [DOI] [PubMed] [Google Scholar]
- 95.Volante M, Sperone P, Bollito E, et al. Matrix metalloproteinase type 2 expression in malignant adrenocortical tumors: diagnostic and prognostic significance in a series of 50 adrenocortical carcinomas. Modern Pathology. 2006;19(12):1563–1569. doi: 10.1038/modpathol.3800683. [DOI] [PubMed] [Google Scholar]
- 96.Cai KQ, Yang WL, Capo-Chichi CD, et al. Prominent expression of metalloproteinases in early stages of ovarian tumorigenesis. Molecular Carcinogenesis. 2007;46(2):130–143. doi: 10.1002/mc.20273. [DOI] [PubMed] [Google Scholar]
- 97.Zong CS, Zeng L, Jiang Y, Sadowski HB, Wang LH. Stat3 plays an important role in oncogenic Ros- and insulin-like growth factor I receptor-induced anchorage-independent growth. Journal of Biological Chemistry. 1998;273(43):28065–28072. doi: 10.1074/jbc.273.43.28065. [DOI] [PubMed] [Google Scholar]
- 98.Zong CS, Chan J, Levy DE, Horvath C, Sadowski HB, Wang LH. Mechanism of STAT3 activation by insulin-like growth factor I receptor. Journal of Biological Chemistry. 2000;275(20):15099–15105. doi: 10.1074/jbc.M000089200. [DOI] [PubMed] [Google Scholar]
- 99.Min H, Wei-Hong Z. Constitutive activation of signal transducer and activator of transcription 3 in epithelial ovarian carcinoma. Journal of Obstetrics and Gynaecology Research. 2009;35(5):918–925. doi: 10.1111/j.1447-0756.2009.01045.x. [DOI] [PubMed] [Google Scholar]
- 100.Doghman M, Karpova T, Rodrigues GA, et al. Increased steroidogenic factor-1 dosage triggers adrenocortical cell proliferation and cancer. Molecular Endocrinology. 2007;21(12):2968–2987. doi: 10.1210/me.2007-0120. [DOI] [PubMed] [Google Scholar]
- 101.Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Human Mutation. 2002;19(6):607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- 102.Reincke M, Beuschlein F, Slawik M, Borm K. Molecular adrenocortical tumourigenesis. European Journal of Clinical Investigation. 2000;30, supplement 3:63–68. doi: 10.1046/j.1365-2362.2000.0300s3063.x. [DOI] [PubMed] [Google Scholar]
- 103.Malaguarnera R, Vella V, Vigneri R, Frasca F. p53 family proteins in thyroid cancer. Endocrine-Related Cancer. 2007;14(1):43–60. doi: 10.1677/erc.1.01223. [DOI] [PubMed] [Google Scholar]
- 104.Werner H, Karnieli E, Rauscher FJ, LeRoith D. Wild-type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(16):8318–8323. doi: 10.1073/pnas.93.16.8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Webster NJG, Resnik JL, Reichart DB, Strauss B, Haas M, Seely BL. Repression of the insulin receptor promoter by the tumor suppressor gene product p53: a possible mechanism for receptor overexpression in breast cancer. Cancer Research. 1996;56(12):2781–2788. [PubMed] [Google Scholar]
- 106.Zhang L, Kashanchi F, Zhan Q, et al. Regulation of insulin-like growth factor II P3 promoter by p53: a potential mechanism for tumorigenesis. Cancer Research. 1996;56(6):1367–1373. [PubMed] [Google Scholar]
- 107.Buckbinder L, Talbott R, Velasco-Miguel S, et al. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature. 1995;377(6550):646–649. doi: 10.1038/377646a0. [DOI] [PubMed] [Google Scholar]
- 108.Tennant MK, Thrasher JB, Twomey PA, Birnbaum RS, Plymate SR. Insulin-like growth factor-binding protein-2 and -3 expression in benign human prostate epithelium, prostate intraepithelial neoplasia, and adenocarcinoma of the prostate. Journal of Clinical Endocrinology and Metabolism. 1996;81(1):411–420. doi: 10.1210/jcem.81.1.8550786. [DOI] [PubMed] [Google Scholar]
- 109.Rasmussen AA, Cullen KJ. Paracrine/autocrine regulation of breast cancer by the insulin-like growth factors. Breast Cancer Research and Treatment. 1998;47(3):219–233. doi: 10.1023/a:1005903000777. [DOI] [PubMed] [Google Scholar]
- 110.Michell NP, Langman MJS, Eggo MC. Insulin-like growth factors and their binding proteins in human colonocytes: preferential degradation of insulin-like growth factor binding protein 2 in colonic cancers. British Journal of Cancer. 1997;76(1):60–66. doi: 10.1038/bjc.1997.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hakam A, Yeatman TJ, Lu L, et al. Expression of insulin-like growth factor-1 receptor in human colorectal cancer. Human Pathology. 1999;30(10):1128–1133. doi: 10.1016/s0046-8177(99)90027-8. [DOI] [PubMed] [Google Scholar]
- 112.DiGiovanni J, Kiguchi K, Frijhoff A, et al. Deregulated expression of insulin-like growth factor 1 in prostate epithelium leads to neoplasia in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(7):3455–3460. doi: 10.1073/pnas.97.7.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Scotlandi K, Manara MC, Serra M, et al. Expression of insulin-like growth factor system components in Ewing’s sarcoma and their association with survival. European Journal of Cancer. 2011;47(8):1258–1266. doi: 10.1016/j.ejca.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 114.Hankinson SE, Willett WC, Colditz GA, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. The Lancet. 1998;351(9113):1393–1396. doi: 10.1016/S0140-6736(97)10384-1. [DOI] [PubMed] [Google Scholar]
- 115.Cohen P, Peehl DM, Baker B, Liu F, Hintz RL, Rosenfeld RG. Insulin-like growth factor axis abnormalities in prostatic stromal cells from patients with benign prostatic hyperplasia. Journal of Clinical Endocrinology and Metabolism. 1994;79(5):1410–1415. doi: 10.1210/jcem.79.5.7525636. [DOI] [PubMed] [Google Scholar]
- 116.Balkany C, Cushing GW. An association between acromegaly and thyroid carcinoma. Thyroid. 1995;5(1):47–50. doi: 10.1089/thy.1995.5.47. [DOI] [PubMed] [Google Scholar]
- 117.Tita P, Ambrosio MR, Scollo C, et al. High prevalence of differentiated thyroid carcinoma in acromegaly. Clinical Endocrinology. 2005;63(2):161–167. doi: 10.1111/j.1365-2265.2005.02316.x. [DOI] [PubMed] [Google Scholar]
- 118.Gicquel C, Bertagna X, Gaston V, et al. Molecular markers and long-term recurrences in a large cohort of patients with sporadic adrenocortical tumors. Cancer Research. 2001;61(18):6762–6767. [PubMed] [Google Scholar]
- 119.Terry KL, Tworoger SS, Gates MA, Cramer DW, Hankinson SE. Common genetic variation in IGF1, IGFBP1 and IGFBP3 and ovarian cancer risk. Carcinogenesis. 2009;30(12):2042–2046. doi: 10.1093/carcin/bgp257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pearce CL, Doherty JA, van den Berg DJ, et al. Genetic variation in insulin-like growth factor 2 may play a role in ovarian cancer risk. Human Molecular Genetics. 2011;20(11):2263–2272. doi: 10.1093/hmg/ddr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fair AM, Dai Q, Shu XO, et al. Energy balance, insulin resistance biomarkers, and breast cancer risk. Cancer Detection and Prevention. 2007;31(3):214–219. doi: 10.1016/j.cdp.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pisani P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Archives of Physiology and Biochemistry. 2008;114(1):63–70. doi: 10.1080/13813450801954451. [DOI] [PubMed] [Google Scholar]
- 123.Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocrine-Related Cancer. 2009;16(4):1103–1123. doi: 10.1677/ERC-09-0087. [DOI] [PubMed] [Google Scholar]
- 124.Heuson JC, Legros N. Influence of insulin deprivation on growth of the 7,12-dimethylbenz(a)anthracene-induced mammary carcinoma in rats subjected to alloxan diabetes and food restriction. Cancer Research. 1972;32(2):226–232. [PubMed] [Google Scholar]
- 125.Steinbach G, Kumar SP, Reddy BS, Lipkin M, Holt PR. Effects of caloric restriction and dietary fat on epithelial cell proliferation in rat colon. Cancer Research. 1993;53(12):2745–2749. [PubMed] [Google Scholar]
- 126.Corpet DE, Jacquinet C, Peiffer G, Taché S. Insulin injections promote the growth of aberrant crypt foci in the colon of rats. Nutrition and Cancer. 1997;27(3):316–320. doi: 10.1080/01635589709514543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tran TT, Medline A, Bruce WR. Insulin promotion of colon tumors in rats. Cancer Epidemiology Biomarkers and Prevention. 1996;5(12):1013–1015. [PubMed] [Google Scholar]
- 128.Rezzónico JN, Rezzónico M, Pusiol E, Pitoia F, Niepomniszcze H. Increased prevalence of insulin resistance in patients with differentiated thyroid carcinoma. Metabolic Syndrome and Related Disorders. 2009;7(4):375–380. doi: 10.1089/met.2008.0062. [DOI] [PubMed] [Google Scholar]
- 129.Rezzonico J, Rezzonico M, Pusiol E, Pitoia F, Niepomniszcze H. Introducing the thyroid gland as another victim of the insulin resistance syndrome. Thyroid. 2008;18(4):461–464. doi: 10.1089/thy.2007.0223. [DOI] [PubMed] [Google Scholar]
- 130.Engeland A, Tretli S, Akslen LA, Bjørge T. Body size and thyroid cancer in two million Norwegian men and women. British Journal of Cancer. 2006;95(3):366–370. doi: 10.1038/sj.bjc.6603249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Oh SW, Yoon YS, Shin SA. Effects of excess weight on cancer incidences depending on cancer sites and histologic findings among men: Korea National Health Insurance Corporation study. Journal of Clinical Oncology. 2005;23(21):4742–4754. doi: 10.1200/JCO.2005.11.726. [DOI] [PubMed] [Google Scholar]
- 132.Suzuki T, Matsuo K, Hasegawa Y, et al. Anthropometric factors at age 20 years and risk of thyroid cancer. Cancer Causes and Control. 2008;19(10):1233–1242. doi: 10.1007/s10552-008-9194-x. [DOI] [PubMed] [Google Scholar]
- 133.Brindel P, Doyon F, Rachédi F, et al. Anthropometric factors in differentiated thyroid cancer in French Polynesia: a case-control study. Cancer Causes and Control. 2009;20(5):581–590. doi: 10.1007/s10552-008-9266-y. [DOI] [PubMed] [Google Scholar]
- 134.Kitahara CM, Platz EA, Freeman LEB, et al. Obesity and thyroid cancer risk among U.S. men and women: a pooled analysis of five prospective studies. Cancer Epidemiology Biomarkers and Prevention. 2011;20(3):464–472. doi: 10.1158/1055-9965.EPI-10-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Clavel-Chapelon F, Guillas G, Tondeur L, Kernaleguen C, Boutron-Ruault MC. Risk of differentiated thyroid cancer in relation to adult weight, height and body shape over life: the French E3N cohort. International Journal of Cancer. 2010;126(12):2984–2990. doi: 10.1002/ijc.25066. [DOI] [PubMed] [Google Scholar]
- 136.Maso LD, Vecchia CL, Franceschi S, et al. A pooled analysis of thyroid cancer studies. V. Anthropometric factors. Cancer Causes and Control. 2000;11(2):137–144. doi: 10.1023/a:1008938520101. [DOI] [PubMed] [Google Scholar]
- 137.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. The Lancet. 2008;371(9612):569–578. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 138.Rapp K, Schroeder J, Klenk J, et al. Obesity and incidence of cancer: a large cohort study of over 145000 adults in Austria. British Journal of Cancer. 2005;93(9):1062–1067. doi: 10.1038/sj.bjc.6602819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Samanic C, Chow WH, Gridley G, Jarvholm B, Fraumeni JF., Jr. Relation of body mass index to cancer risk in 362,552 Swedish men. Cancer Causes and Control. 2006;17(7):901–909. doi: 10.1007/s10552-006-0023-9. [DOI] [PubMed] [Google Scholar]
- 140.Mijović T, How J, Pakdaman M, et al. Body mass index in the evaluation of thyroid cancer risk. Thyroid. 2009;19(5):467–472. doi: 10.1089/thy.2008.0386. [DOI] [PubMed] [Google Scholar]
- 141.Iribarren C, Haselkorn T, Tekawa IS, Friedman GD. Cohort study of thyroid cancer in a San Francisco bay area population. International Journal of Cancer. 2001;93(5):745–750. doi: 10.1002/ijc.1377. [DOI] [PubMed] [Google Scholar]
- 142.Paes JE, Hua K, Nagy R, Kloos RT, Jarjoura D, Ringel MD. The relationship between body mass index and thyroid cancer pathology features and outcomes: a clinicopathological cohort study. Journal of Clinical Endocrinology and Metabolism. 2010;95(9):4244–4250. doi: 10.1210/jc.2010-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Almquist M, Johansen D, Björge T, et al. Metabolic factors and risk of thyroid cancer in the Metabolic syndrome and Cancer project (Me-Can) Cancer Causes and Control. 2011;22(5):743–751. doi: 10.1007/s10552-011-9747-2. [DOI] [PubMed] [Google Scholar]
- 144.Aschebrook-Kilfoy B, Sabra MM, Brenner A, et al. Diabetes and thyroid cancer risk in the National Institutes of Health-AARP Diet and Health Study. Thyroid. 21:957–963. doi: 10.1089/thy.2010.0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gursoy A. Rising thyroid cancer incidence in the world might be related to insulin resistance. Medical Hypotheses. 2010;74(1):35–36. doi: 10.1016/j.mehy.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 146.Vona-Davis L, Rose DP. Adipokines as endocrine, paracrine, and autocrine factors in breast cancer risk and progression. Endocrine-Related Cancer. 2007;14(2):189–206. doi: 10.1677/ERC-06-0068. [DOI] [PubMed] [Google Scholar]
- 147.Rapp K, Schroeder J, Klenk J, et al. Fasting blood glucose and cancer risk in a cohort of more than 140,000 adults in Austria. Diabetologia. 2006;49(5):945–952. doi: 10.1007/s00125-006-0207-6. [DOI] [PubMed] [Google Scholar]
- 148.Chittenden BG, Fullerton G, Maheshwari A, Bhattacharya S. Polycystic ovary syndrome and the risk of gynaecological cancer: a systematic review. Reproductive Biomedicine Online. 2009;19(3):398–405. doi: 10.1016/s1472-6483(10)60175-7. [DOI] [PubMed] [Google Scholar]
- 149.Yang HS, Yoon C, Myung SK, Park SM. Effect of obesity on survival of women with epithelial ovarian cancer: a systematic review and meta-analysis of observational studies. International Journal of Gynecological Cancer. 2011;21(9):1525–1532. doi: 10.1097/IGC.0b013e31822eb5f8. [DOI] [PubMed] [Google Scholar]
- 150.Pierpoint T, McKeigue PM, Isaacs AJ, Wild SH, Jacobs HS. Mortality of women with polycystic ovary syndrome at long-term follow-up. Journal of Clinical Epidemiology. 1998;51(7):581–586. doi: 10.1016/s0895-4356(98)00035-3. [DOI] [PubMed] [Google Scholar]
- 151.Gadducci A, Gargini A, Palla E, Fanucchi A, Genazzani AR. Polycystic ovary syndrome and gynecological cancers: is there a link? Gynecological Endocrinology. 2005;20(4):200–208. doi: 10.1080/09513590400021201. [DOI] [PubMed] [Google Scholar]
- 152.Reincke M, Fassnacht M, Väth S, Mora P, Allolio B. Adrenal incidentalomas: a manifestation of the metabolic syndrome? Endocrine Research. 1996;22(4):757–761. doi: 10.1080/07435809609043773. [DOI] [PubMed] [Google Scholar]
- 153.Midorikawa S, Sanada H, Hashimoto S, Suzuki T, Watanabe T. The improvement of insulin resistance in patients with adrenal incidentaloma by surgical resection. Clinical Endocrinology. 2001;54(6):797–804. doi: 10.1046/j.1365-2265.2001.01274.x. [DOI] [PubMed] [Google Scholar]
- 154.Chen SJ, Yu SN, Tzeng JE, et al. Characterization of the major histopathological components of thyroid nodules using sonographic textural features for clinical diagnosis and management. Ultrasound in Medicine and Biology. 2009;35(2):201–208. doi: 10.1016/j.ultrasmedbio.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 155.Wojciechowska K, Lewinski A. BRAF mutations in papillary thyroid carcinoma. Endocrine Regulations. 2006;40(4):129–138. [PubMed] [Google Scholar]
- 156.Vella V, Sciacca L, Pandini G, et al. The IGF system in thyroid cancer: new concepts. Journal of Clinical Pathology. 2001;54(3):121–124. doi: 10.1136/mp.54.3.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tode B, Serio M, Rotella CM, et al. Insulin-like growth factor-I: autocrine secretion by human thyroid follicular cells in primary culture. Journal of Clinical Endocrinology and Metabolism. 1989;69(3):639–647. doi: 10.1210/jcem-69-3-639. [DOI] [PubMed] [Google Scholar]
- 158.Bachrach LK, Liu FR, Burrow GN, Eggo MC. Characterization of insulin-like growth factor-binding proteins from sheep thyroid cells. Endocrinology. 1989;125(6):2831–2838. doi: 10.1210/endo-125-6-2831. [DOI] [PubMed] [Google Scholar]
- 159.Williams DW, Williams ED, Wynford-Thomas D. Loss of dependence on IGF-1 for proliferation of human thyroid adenoma cells. British Journal of Cancer. 1988;57(6):535–539. doi: 10.1038/bjc.1988.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Onoda N, Ohmura E, Tsushima T, et al. Autocrine role on insulin-like growth factor (IGF)-I in a human thyroid cancer cell line. European Journal of Cancer A. 1992;28(11):1904–1909. doi: 10.1016/0959-8049(92)90033-x. [DOI] [PubMed] [Google Scholar]
- 161.Masood S, Auguste LJ, Westerband A, Belluco C, Valderama E, Attie J. Differential oncogenic expression in thyroid follicular and Hurthle cell carcinomas. American Journal of Surgery. 1993;166(4):366–368. doi: 10.1016/s0002-9610(05)80334-4. [DOI] [PubMed] [Google Scholar]
- 162.Minuto F, Barreca A, del Monte P, Cariola G, Torre GC, Giordano G. Immunoreactive insulin-like growth factor I (IGF-I) and IGF-I-binding protein content in human thyroid tissue. Journal of Clinical Endocrinology and Metabolism. 1989;68(3):621–626. doi: 10.1210/jcem-68-3-621. [DOI] [PubMed] [Google Scholar]
- 163.Poulaki V, Mitsiades CS, McMullan C, et al. Regulation of vascular endothelial growth factor expression by onsulin-like growth factor I in thyroid carcinomas. Journal of Clinical Endocrinology and Metabolism. 2003;88(11):5392–5398. doi: 10.1210/jc.2003-030389. [DOI] [PubMed] [Google Scholar]
- 164.Gydee H, O’Neill JT, Patel A, Bauer AJ, Tuttle RM, Francis GL. Differentiated thyroid crcinomas from children and adolescents express IGF-I and the IGF-I Receptor (IGF-I-R). Cancers with the most intense IGF-I-R expression may be more aggressive. Pediatric Research. 2004;55(4):709–715. doi: 10.1203/01.PDR.0000111282.98401.93. [DOI] [PubMed] [Google Scholar]
- 165.Maiorano E, Ciampolillo A, Viale G, et al. Insulin-like growth factor I expression in thyroid tumors. Applied Immunohistochemistry and Molecular Morphology. 2000;8(2):110–119. doi: 10.1097/00129039-200006000-00005. [DOI] [PubMed] [Google Scholar]
- 166.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea—a paradigm shift. Cancer Research. 2006;66(4):1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 167.Wang Z, Chakravarty G, Kim S, et al. Growth-inhibitory effects of human anti-insulin-like growth factor-1 receptor antibody (A12) in an orthotopic nude mouse model of anaplastic thyroid carcinoma. Clinical Cancer Research. 2006;12(15):4755–4765. doi: 10.1158/1078-0432.CCR-05-2691. [DOI] [PubMed] [Google Scholar]
- 168.Phielix E, Szendroedi J, Roden M. The role of metformin and thiazolidinediones in the regulation of hepatic glucose metabolism and its clinical impact. Trends in Pharmacological Sciences. 2011;32:607–616. doi: 10.1016/j.tips.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 169.Aiello A, Pandini G, Frasca F, et al. Peroxisomal proliferator-activated receptor-γ agonists induce partial reversion of epithelial-mesenchymal transition in anaplastic thyroid cancer cells. Endocrinology. 2006;147(9):4463–4475. doi: 10.1210/en.2005-1610. [DOI] [PubMed] [Google Scholar]
- 170.Chen G, Xu S, Renko K, Derwahl M. Metformin inhibits growth of thyroid carcinoma cells, suppresses self-renewal of derived cancer stem cells, and potentiates the effect of chemotherapeutic agents. Journal of Clinical Endocrinology and Metabolism. 2012;97:E510–E520. doi: 10.1210/jc.2011-1754. [DOI] [PubMed] [Google Scholar]
- 171.Latronico AC, Chrousos GP. Extensive personal, experience: adrenocortical tumors. Journal of Clinical Endocrinology and Metabolism. 1997;82(5):1317–1324. doi: 10.1210/jcem.82.5.3921. [DOI] [PubMed] [Google Scholar]
- 172.Fottner C, Hoeflich A, Wolf E, Weber MM. Role of the insulin-like growth factor system in adrenocortical growth control and carcinogenesis. Hormone and Metabolic Research. 2004;36(6):397–405. doi: 10.1055/s-2004-814563. [DOI] [PubMed] [Google Scholar]
- 173.Shon C, Mladenovski V, Petkov R, et al. Diagnosis and surgical treatment of adrenocortical carcinoma. A review of the literature and report of two cases. Khirurgiia. 2000;56(2):45–49. [PubMed] [Google Scholar]
- 174.Soreide JA, Brabrand K, Thoresen SO. Adrenal cortical carcinoma in Norway, 1970–1984. World Journal of Surgery. 1992;16(4):663–667. doi: 10.1007/BF02067349. [DOI] [PubMed] [Google Scholar]
- 175.Lindholm J, Juul S, Jørgensen JOL, et al. Incidence and late prognosis of Cushing’s syndrome: a population-based study. Journal of Clinical Endocrinology and Metabolism. 2001;86(1):117–123. doi: 10.1210/jcem.86.1.7093. [DOI] [PubMed] [Google Scholar]
- 176.Ribeiro RC, Pinto EM, Zambetti GP, Rodriguez-Galindo C. The international pediatric adrenocortical tumor registry initiative: contributions to clinical, biological, and treatment advances in pediatric adrenocortical tumors. Molecular and Cellular Endocrinology. 2012;351:37–43. doi: 10.1016/j.mce.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 177.Almeida MQ, Fragoso MCBV, Lotfi CFP, et al. Expression of insulin-like growth factor-II and its receptor in pediatric and adult adrenocortical tumors. Journal of Clinical Endocrinology and Metabolism. 2008;93(9):3524–3531. doi: 10.1210/jc.2008-0065. [DOI] [PubMed] [Google Scholar]
- 178.Khandwala HM, McCutcheon IE, Flyvbjerg A, Friend KE. The effects of insulin-like growth factors on tumorigenesis and neoplastic growth. Endocrine Reviews. 2000;21(3):215–244. doi: 10.1210/edrv.21.3.0399. [DOI] [PubMed] [Google Scholar]
- 179.Cantini G, Lombardi A, Piscitelli E, et al. Rosiglitazone inhibits adrenocortical cancer cell proliferation by interfering with the IGF-IR intracellular signaling. PPAR Research. 2008 doi: 10.1155/2008/904041.904041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Boulle N, Logié A, Gicquel C, Perin L, Bouc YL. Increased levels of insulin-like growth factor II (IGF-II) and IGF- binding protein-2 are associated with malignancy in sporadic adrenocortical tumors. Journal of Clinical Endocrinology and Metabolism. 1998;83(5):1713–1720. doi: 10.1210/jcem.83.5.4816. [DOI] [PubMed] [Google Scholar]
- 181.Logié A, Boulle N, Gaston V, et al. Autocrine role of IGF-II in proliferation of human adrenocortical carcinoma NCI H295R cell line. Journal of Molecular Endocrinology. 1999;23(1):23–32. doi: 10.1677/jme.0.0230023. [DOI] [PubMed] [Google Scholar]
- 182.Faiçal S, Maciel RMB, Nosé-Alberti V, Santos MC, Kater CE. Immunodetection of insulin-like growth factor I (IGF-I) in normal and pathological adrenocortical tissue. Endocrine Pathology. 1998;9(1):63–70. doi: 10.1007/BF02739953. [DOI] [PubMed] [Google Scholar]
- 183.Giordano TJ. Gene expression profiling of endocrine tumors using DNA microarrays: progress and Promise. Endocrine Pathology. 2003;14(2):107–116. doi: 10.1385/ep:14:2:107. [DOI] [PubMed] [Google Scholar]
- 184.de Fraipont F, El Atifi M, Cherradi N, et al. Gene expression profiling of human adrenocortical tumors using complementary deoxyribonucleic acid microarrays identifies several candidate genes as markers of malignancy. Journal of Clinical Endocrinology and Metabolism. 2005;90(3):1819–1829. doi: 10.1210/jc.2004-1075. [DOI] [PubMed] [Google Scholar]
- 185.Gicquel C, Bertagna X, Schneid H, et al. Rearrangements at the 11p15 locus and overexpression of insulin-like growth factor-II gene in sporadic adrenocortical tumors. Journal of Clinical Endocrinology and Metabolism. 1994;78(6):1444–1453. doi: 10.1210/jcem.78.6.7911125. [DOI] [PubMed] [Google Scholar]
- 186.Gicquel C, Raffin-Sanson ML, Gaston V, et al. Structural and functional abnormalities at 11p15 are associated with the malignant phenotype in sporadic adrenocortical tumors: study on a series of 82 tumors. Journal of Clinical Endocrinology and Metabolism. 1997;82(8):2559–2565. doi: 10.1210/jcem.82.8.4170. [DOI] [PubMed] [Google Scholar]
- 187.Gicquel C, Boulle N, Logié A, Bourcigaux N, Gaston V, Le Bouc Y. Involvement of the IGF system in the pathogenesis of adrenocortical tumors. Annales d’Endocrinologie. 2001;62(2):189–192. [PubMed] [Google Scholar]
- 188.Wolf E, Hoeflich A, Lahm H. What is the function of IGF-II in postnatal life? Answers from transgenic mouse models. Growth Hormone and IGF Research. 1998;8(3):185–193. doi: 10.1016/s1096-6374(98)80110-x. [DOI] [PubMed] [Google Scholar]
- 189.Weber MM, Fottner C, Schmidt P, et al. Postnatal overexpression of insulin-like growth factor II in transgenic mice is associated with adrenocortical hyperplasia and enhanced steroidogenesis. Endocrinology. 1999;140(4):1537–1543. doi: 10.1210/endo.140.4.6660. [DOI] [PubMed] [Google Scholar]
- 190.Boulle N, Baudin E, Gicquel C, et al. Evaluation of plasma insulin-like growth factor binding protein-2 as a marker for adrenocortical tumors. European Journal of Endocrinology. 2001;144(1):29–36. doi: 10.1530/eje.0.1440029. [DOI] [PubMed] [Google Scholar]
- 191.West AN, Neale GA, Pounds S, et al. Gene expression profiling of childhood adrenocortical tumors. Cancer Research. 2007;67(2):600–608. doi: 10.1158/0008-5472.CAN-06-3767. [DOI] [PubMed] [Google Scholar]
- 192.Weber MM, Auernhammer CJ, Kiess W, Engelhardt D. Insulin-like growth factor receptors in normal and tumorous adult human adrenocortical glands. European Journal of Endocrinology. 1997;136(3):296–303. doi: 10.1530/eje.0.1360296. [DOI] [PubMed] [Google Scholar]
- 193.Blackburn A, Dressendörfer RA, Blum WF, et al. Interactions of insulin-like growth factor (IGF)-II and growth hormone in vivo: circulating levels of IGF-I and IGF-binding proteins in transgenic mice. European Journal of Endocrinology. 1997;137(6):701–708. doi: 10.1530/eje.0.1370701. [DOI] [PubMed] [Google Scholar]
- 194.Weber MM, Fottner C, Wolf E. The role of the insulin-like growth factor system in adrenocortical tumourigenesis. European Journal of Clinical Investigation. 2000;30, supplement 3:69–75. doi: 10.1046/j.1365-2362.2000.0300s3069.x. [DOI] [PubMed] [Google Scholar]
- 195.Reeve JG, Kirby LB, Brinkman A, Hughes SA, Schwander J, Bleechen NM. Insulin-like growth-factor-binding protein gene expression and protein production by human tumour cell lines. International Journal of Cancer. 1992;51(5):818–821. doi: 10.1002/ijc.2910510525. [DOI] [PubMed] [Google Scholar]
- 196.Fottner C, Engelhardt D, Elmlinger MW, Weber MM. Identification and characterization of insulin-like growth factor (IGF)-binding protein expression and secretion by adult human adrenocortical cells: differential regulation by IGFs and adrenocorticotropin. Journal of Endocrinology. 2001;168(3):465–474. doi: 10.1677/joe.0.1680465. [DOI] [PubMed] [Google Scholar]
- 197.Hoeflich A, Fettscher O, Lahm H, et al. Overexpression of insulin-like growth factor-binding protein-2 results in increased tumorigenic potential in Y-1 adrenocortical tumor cells. Cancer Research. 2000;60(4):834–838. [PubMed] [Google Scholar]
- 198.Bale LK, Conover CA. Regulation of insulin-like growth factor binding protein-3 messenger ribonucleic acid expression by insulin-like growth factor I. Endocrinology. 1992;131(2):608–614. doi: 10.1210/endo.131.2.1379162. [DOI] [PubMed] [Google Scholar]
- 199.Camacho-Hubner C, Busby WH, Jr., McCusker RH, Wright G, Clemmons DR. Identification of the forms of insulin-like growth factor-binding proteins produced by human fibroblasts and the mechanisms that regulate their secretion. Journal of Biological Chemistry. 1992;267(17):11949–11956. [PubMed] [Google Scholar]
- 200.Martin JL, Ballesteros M, Baxter RC. Insulin-like growth factor-I (IGF-I) and transforming growth factor-β1 release IGF-binding protein-3 from human fibroblasts by different mechanisms. Endocrinology. 1992;131(4):1703–1710. doi: 10.1210/endo.131.4.1382959. [DOI] [PubMed] [Google Scholar]
- 201.Rechler MM. Insulin-like growthfactor binding proteins. Vitamins and Hormones. 1993;47:1–114. doi: 10.1016/s0083-6729(08)60444-6. [DOI] [PubMed] [Google Scholar]
- 202.Conover CA, de Leon DD. Acid-activated insulin-like growth factor-binding protein-3 proteolysis in normal and transformed cells. Role of cathepsin D. Journal of Biological Chemistry. 1994;269(10):7076–7080. [PubMed] [Google Scholar]
- 203.Conover CA, Bale LK, Durham SK, Powell DR. Insulin-like growth factor (IGF) binding protein-3 potentiation of IGF action is mediated through the phosphatidylinositol-3-kinase pathway and is associated with alteration in protein kinase B/AKT sensitivity. Endocrinology. 2000;141(9):3098–3103. doi: 10.1210/endo.141.9.7660. [DOI] [PubMed] [Google Scholar]
- 204.Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocrine Reviews. 1995;16(1):3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 205.Weber MM, Simmler P, Fottner C, Engelhardt D. Insulin-like growth factor II (IGF-II) is more potent than IGF-I in stimulating cortisol secretion from cultured bovine adrenocortical cells: interaction with the IGF-I receptor and IGF-binding proteins. Endocrinology. 1995;136(9):3714–3720. doi: 10.1210/endo.136.9.7544271. [DOI] [PubMed] [Google Scholar]
- 206.Fottner C, Engelhardt D, Weber MM. Characterization of insulin-like growth factor binding proteins (IGFBPs) secreted by bovine adrenocortical cells in primary culture: regulation by insulin-like growth factors (IGFs) and adrenocorticotropin (ACTH) Hormone and Metabolic Research. 1999;31(2-3):203–208. doi: 10.1055/s-2007-978720. [DOI] [PubMed] [Google Scholar]
- 207.Velázquez-Fernández D, Laurell C, Geli J, et al. Expression profiling of adrenocortical neoplasms suggests a molecular signature of malignancy. Surgery. 2005;138(6):1087–1094. doi: 10.1016/j.surg.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 208.Doghman M, El Wakil A, Cardinaud B, et al. Regulation of insulin-like growth factor-mammalian target of rapamycin signaling by MicroRNA in childhood adrenocortical tumors. Cancer Research. 2010;70(11):4666–4675. doi: 10.1158/0008-5472.CAN-09-3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Barlaskar FM, Spalding AC, Heaton JH, et al. Preclinical targeting of the type I insulin-like growth factor receptor in adrenocortical carcinoma. Journal of Clinical Endocrinology and Metabolism. 2009;94(1):204–212. doi: 10.1210/jc.2008-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Shen W, Xian J, Hu WL, Liu J. Effect of insulin-like growth factor and its receptor-I antibody on growth of human adrenocortical carcinoma SW-13 cell lines in vitro. Nan Fang Yi Ke Da Xue Xue Bao. 2007;27(1):88–91. [PubMed] [Google Scholar]
- 211.Rosen ED, Spiegelman BM. PPARγ: a nuclear regulator of metabolism, differentiation, and cell growth. Journal of Biological Chemistry. 2001;276(41):37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- 212.Clay CE, Namen AM, Atsumi GI, et al. Influence of J series prostaglandins on apoptosis and tumorigenesis of breast cancer cells. Carcinogenesis. 1999;20(10):1905–1911. doi: 10.1093/carcin/20.10.1905. [DOI] [PubMed] [Google Scholar]
- 213.Ferruzzi P, Ceni E, Tarocchi M, et al. Thiazolidinediones inhibit growth and invasiveness of the human adrenocortical cancer cell line H295R. Journal of Clinical Endocrinology and Metabolism. 2005;90(3):1332–1339. doi: 10.1210/jc.2004-0978. [DOI] [PubMed] [Google Scholar]
- 214.Chien JR, Aletti G, Bell DA, Keeney GL, Shridhar V, Hartmann LC. Molecular pathogenesis and therapeutic targets in epithelial ovarian cancer. Journal of Cellular Biochemistry. 2007;102(5):1117–1129. doi: 10.1002/jcb.21552. [DOI] [PubMed] [Google Scholar]
- 215.Kajihara H, Yamada Y, Kanayama S, et al. Clear cell carcinoma of the ovary: potential pathogenic mechanisms (review) Oncology Reports. 2010;23(5):1193–1203. doi: 10.3892/or_00000750. [DOI] [PubMed] [Google Scholar]
- 216.Riman T, Persson I, Nilsson S. Hormonal aspects of epithelial ovarian cancer: review of epidemiological evidence. Clinical Endocrinology. 1998;49(6):695–707. doi: 10.1046/j.1365-2265.1998.00577.x. [DOI] [PubMed] [Google Scholar]
- 217.Auranen A, Grénman S, Mäkinen J, Pukkala E, Sankila R, Salmi T. Borderline ovarian tumors in Finland: epidemiology and familial occurrence. American Journal of Epidemiology. 1996;144(6):548–553. doi: 10.1093/oxfordjournals.aje.a008964. [DOI] [PubMed] [Google Scholar]
- 218.Beauchamp MC, Yasmeen A, Knafo A, Gotlieb WH. Targeting insulin and insulin-like growth factor pathways in epithelial ovarian cancer. Journal of Oncology. 2010;2010:11 pages. doi: 10.1155/2010/257058.257058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.King ER, Zu Z, Tsang YTM, et al. The insulin-like growth factor 1 pathway is a potential therapeutic target for low-grade serous ovarian carcinoma. Gynecologic Oncology. 2011;123(1):13–18. doi: 10.1016/j.ygyno.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Yee D, Morales FR, Hamilton TC, Von Hoff DD. Expression of insulin-like growth factor I, its binding proteins, and its receptor in ovarian cancer. Cancer Research. 1991;51(19):5107–5112. [PubMed] [Google Scholar]
- 221.Conover CA, Hartmann LC, Bradley S, et al. Biological characterization of human epithelial ovarian carcinoma cells in primary culture: the insulin-like growth factor system. Experimental Cell Research. 1998;238(2):439–449. doi: 10.1006/excr.1997.3861. [DOI] [PubMed] [Google Scholar]
- 222.Kalli KR, Conover CA. The insulin-like growth factor/insulin system in epithelial ovarian cancer. Frontiers in Bioscience. 2003;8:d714–d722. doi: 10.2741/1034. [DOI] [PubMed] [Google Scholar]
- 223.Chakrabarty S, Kondratick L. Insulin-like growth factor binding protein-2 stimulates proliferation and activates multiple cascades of the mitogen-activated protein kinase pathways in NIH-OVCAR3 human epithelial ovarian cancer cells. Cancer Biology and Therapy. 2006;5(2):189–197. doi: 10.4161/cbt.5.2.2333. [DOI] [PubMed] [Google Scholar]
- 224.Lukanova A, Lundin E, Toniolo P, et al. Circulating levels of insulin-like growth factor-I and risk of ovarian cancer. International Journal of Cancer. 2002;101(6):549–554. doi: 10.1002/ijc.10613. [DOI] [PubMed] [Google Scholar]
- 225.Tworoger SS, Lee IM, Buring JE, Pollak MN, Hankinson SE. Insulin-like growth factors and ovarian cancer risk: a nested case-control study in three cohorts. Cancer Epidemiology Biomarkers and Prevention. 2007;16(8):1691–1695. doi: 10.1158/1055-9965.EPI-07-0319. [DOI] [PubMed] [Google Scholar]
- 226.Lancaster JM, Sayer RA, Blanchette C, et al. High expression of insulin-like growth factor binding protein-2 messenger RNA in epithelial ovarian cancers produces elevated preoperative serum levels. International Journal of Gynecological Cancer. 2006;16(4):1529–1535. doi: 10.1111/j.1525-1438.2006.00623.x. [DOI] [PubMed] [Google Scholar]
- 227.Sayer RA, Lancaster JM, Pittman J, et al. High insulin-like growth factor-2 (IGF-2) gene expression is an independent predictor of poor survival for patients with advanced stage serous epithelial ovarian cancer. Gynecologic Oncology. 2005;96(2):355–361. doi: 10.1016/j.ygyno.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 228.Lu L, Katsaros D, Wiley A, et al. The relationship of insulin-like growth factor-II, insulin-like growth factor binding protein-3, and estrogen receptor-α expression to disease progression in epithelial ovarian cancer. Clinical Cancer Research. 2006;12(4):1208–1214. doi: 10.1158/1078-0432.CCR-05-1801. [DOI] [PubMed] [Google Scholar]
- 229.Lu L, Katsaros D, Wiley A, et al. Promoter-specific transcription of insulin-like growth factor-II in epithelial ovarian cancer. Gynecologic Oncology. 2006;103(3):990–995. doi: 10.1016/j.ygyno.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 230.Sussenbach JS, Rodenburg RJT, Scheper W, Holthuizen P. Transcriptional and post-transcriptional regulation of the human IGF-II gene expression. Advances in Experimental Medicine and Biology. 1993;343:63–71. doi: 10.1007/978-1-4615-2988-0_7. [DOI] [PubMed] [Google Scholar]
- 231.Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286(5439):481–486. doi: 10.1126/science.286.5439.481. [DOI] [PubMed] [Google Scholar]
- 232.Qian B, Katsaros D, Lu L, et al. IGF-II promoter specific methylation and expression in epithelial ovarian cancer and their associations with disease characteristics. Oncology Reports. 2011;25(1):203–213. [PMC free article] [PubMed] [Google Scholar]
- 233.Beeghly AC, Katsaros D, Wiley AL, et al. IGF-II promoter methylation and ovarian cancer prognosis. Journal of Cancer Research and Clinical Oncology. 2007;133(10):713–723. doi: 10.1007/s00432-007-0211-3. [DOI] [PubMed] [Google Scholar]
- 234.Spentzos D, Cannistra SA, Grall F, et al. IGF axis gene expression patterns are prognostic of survival in epithelial ovarian cancer. Endocrine-Related Cancer. 2007;14(3):781–790. doi: 10.1677/ERC-06-0073. [DOI] [PubMed] [Google Scholar]
- 235.Muller M, Dietel M, Turzynski A, Wiechen K. Antisense phosphorothioate oligodeoxynucleotide down-regulation of the insulin-like growth factor I receptor in ovarian cancer cells. International Journal of Cancer. 1998;77:567–571. doi: 10.1002/(sici)1097-0215(19980812)77:4<567::aid-ijc16>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 236.Gotlieb WH, Bruchim I, Gu J, et al. Insulin-like growth factor receptor I targeting in epithelial ovarian cancer. Gynecologic Oncology. 2006;100(2):389–396. doi: 10.1016/j.ygyno.2005.09.048. [DOI] [PubMed] [Google Scholar]
- 237.Li C, Liu VW, Chan DW, Yao KM, Ngan HY. LY294002 and metformin cooperatively enhance the inhibition of growth and the induction of apoptosis of ovarian cancer cells. International Journal of Gynecological Cancer. 2012;22(1):15–22. doi: 10.1097/IGC.0b013e3182322834. [DOI] [PubMed] [Google Scholar]
- 238.Liao H, Zhou Q, Gu Y, Duan T, Feng Y. Luteinizing hormone facilitates angiogenesis in ovarian epithelial tumor cells and metformin inhibits the effect through the mTOR signaling pathway. Oncology Reports. 2012;27(6):1873–1878. doi: 10.3892/or.2012.1745. [DOI] [PubMed] [Google Scholar]
- 239.Romero IL, McCormick A, McEwen KA, et al. Relationship of type II diabetes and metformin use to ovarian cancer progression, survival, and chemosensitivity. Obstet Gynecol. 2012;119(1):61–67. doi: 10.1097/AOG.0b013e3182393ab3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Bakhru A, Buckanovich RJ, Griggs JJ. The impact of diabetes on survival in women with ovarian cancer. Gynecologic Oncology. 2011;121(1):106–111. doi: 10.1016/j.ygyno.2010.12.329. [DOI] [PubMed] [Google Scholar]