

Abstract
Total knee arthroplasty (TKA) is the most common and a cost-effective surgical remediation for older adults with long-standing osteoarthritis. In parallel with the expanding population of older adults, the number of TKAs performed annually is projected to be 3.48 million by 2030. During this surgery, a tourniquet is used to stop blood flow to the operative leg. However, the molecular pathways that are affected by tourniquet use during TKA continue to be elucidated. We hypothesized that components of the catabolic FoxO3a (i.e., MuRF1, MAFbx, and Bnip3) pathway, as well as the cellular stress pathways [i.e., stress-activated protein kinase (SAPK)/JNK and MAPKs], are upregulated during TKA. The purpose of this study was to measure changes in transcripts and proteins involved in muscle cell catabolic and stress-activated pathways. We obtained muscle biopsies from subjects, 70 ± 1.3 yr, during TKA, from the vastus lateralis at baseline (before tourniquet inflation), during maximal ischemia (just before tourniquet release), and during reperfusion. Total tourniquet time was 43 ± 2 min and reperfusion time was 16 ± 1. Significant increases in FoxO3a downstream targets, MAFbx and MuRF1, were present for mRNA levels during ischemia (MAFbx, P = 0.04; MuRF1, P = 0.04), and protein expression during ischemia (MAFbx, P = 0.002; MuRF1, P = 0.001) and reperfusion (MuRF1, P = 0.002). Additionally, stress-activated JNK gene expression (P = 0.01) and protein were elevated during ischemia (P = 0.001). The results of this study support our hypothesis that protein degradation pathways are stimulated during TKA. Muscle protein catabolism is likely to play a role in the rapid loss of muscle volume measured within 2 wk of this surgery.
Keywords: clinical, atrophy, FOXO, JNK, muscle
as a function of our growing population of older adults, it is estimated that by 2030, more than 3.4 million total knee replacement surgeries will be performed annually in the United States (24). Despite the near-universal success of total knee replacement in mitigating chronic knee pain due to osteoarthritis, limitations in physical performance and functional mobility persist. Muscle atrophy of the knee extensors has been shown to be the number one contributor to chronic strength deficits, explaining 77% of the strength loss 1–3 years after surgery and is, therefore, the primary reason for chronic limitations (29).
During total knee arthroplasty (TKA), a tourniquet is routinely placed around the proximal thigh of the operative leg to help ensure rigid bone-implant cementing and to provide the surgeon with a clear surgical field. Previous reports, however, demonstrate that muscle tissue is susceptible to periods of ischemia lasting 15 (1), 30 (1, 39, 42, 43), and 60 min (1, 12, 34, 39, 41), with increasing ischemic times causing more severe tissue injury. It follows then that tourniquet-induced ischemia-reperfusion (I/R) may cause damage to muscle cells during TKA and potentially play a mechanistic role in the rapid, and, more importantly, potentially irrecoverable, muscle loss that results in long-term atrophy-induced functional impairment for these older adults.
We have recently published data demonstrating that proteins regulating cap-dependent translation initiation and elongation were downregulated during and immediately after TKA (36). However, although reductions in muscle protein synthesis can account for the majority of atrophy during disuse, stimulation of muscle protein breakdown, i.e., catabolism, may also occur. Indeed, in our previous report (36), we measured a 12% decrease in midthigh quadriceps volume of the operative (TKA) leg vs. a 6% loss in the nonoperative leg within 2 wk of surgery. We interpreted the differences in the degree of atrophy as potentially being related to the tourniquet-induced I/R injury occurring in the operative (TKA) leg.
Protein synthesis and degradation in skeletal muscle cells are tightly regulated by transcriptional and translational mechanisms (15, 21). These processes define a balance between anabolic and catabolic metabolism and are primarily coordinated through the phosphoinositol 3-kinase (PI3K)/Akt/Forkhead boxO3a (FoxO3a) pathways (16). During anabolic conditions, such as with insulin-like growth factor-1 (IGF1) (7), Akt activation of the mammalian target of rapamycin (mTOR) pathway leads to increases in muscle protein synthesis, while under catabolic conditions, such as with glucocorticoids (3) or disuse (37), downregulation of Akt signaling leads to translocation of FoxO3a to the nucleus and upregulation of proteins controlling muscle atrophy (19, 26). Under catabolic conditions, FoxO3a induces the 26S proteasomal pathway by increasing the expression of, in particular, two muscle-specific E3 ubiquitin ligases, muscle atrophy F-Box (MAFbx; also known as atrogin 1), and muscle RING finger 1 (MuRF1) (6, 22), which are expressed in all muscle atrophy models. In addition to regulating components of the proteasomal pathway, FoxO3a also regulates the autophagic/lysosomal pathway by binding to sites on the promoters of Bcl2/adenovirus EIB 19-kDa-interacting protein 3 (Bnip3) gene. Bnip3 is a proapoptotic member of the Bcl-2 family (28) and has been shown to be upregulated during I/R injury and induction of the autophagic/lysosomal pathway (18). Thus, the Akt/FoxO pathways govern protein catabolism through regulation of the ubiquitin-proteasomal and autophagic/lysosomal pathways.
Our objective, therefore, was to measure transcriptional, translational, and protein changes in the components of the catabolic FoxO3a pathway, i.e., MuRF1, MAFbx, and Bnip3, as well as cell stress pathways, SAPK/JNK and the MAPKs, during TKA. We hypothesized that, in addition to the Akt/4E-BP1 pathway being downregulated (36), the cellular and molecular mechanisms controlling protein catabolism would be upregulated during knee replacement surgery, and as such, help us to further determine how tourniquet-induced I/R injury to muscle cells may contribute to the twofold greater quadriceps atrophy measured in the operative vs. nonoperative leg 2 wk after TKA (36).
METHODS
Ethics approval.
This study was approved by the PeaceHealth Institutional Review Board, Sacred Heart Medical Center, at Riverbend and the Biomedical Institutional Review Board for the University of Oregon and conducted in accordance with the Declaration of Helsinki. All subjects gave informed written consent prior to study participation. This study is registered with ClinicalTrials.gov (identifier: NCT00760383).
Subjects.
We recruited 12 subjects (8 females and 4 males) between 60 and 80 years of age (average age 70 ± 5 yr) from a pool of surgical candidates from the Slocum Center for Orthopedics and Sports Medicine. All subjects were scheduled to undergo a primary total knee arthroplasty (TKA) and had no current untreated endocrine disease, significant heart, kidney, liver, blood or respiratory disease, peripheral vascular disease, active cancer, recent treatment with anabolic steroids, or oral corticosteroids for greater than 1 wk, and no alcohol or drug abuse. Subject characteristics are provided for each subject in Table 1.
Table 1.
Physical characteristics of subjects
| Sex | Age | Ht, cm | Wt, kg | BMI, kg/m2 | Dx | Medications | Tourniquet Time, min | Reperfusion Time, min | Anesthesia |
|---|---|---|---|---|---|---|---|---|---|
| M | 73 | 171.5 | 124 | 42 | OA | Enalapril, Furoseminde, Norvasc, Prozac, Simvastatin, Travatan, Vicodin | 45 | 21 | Gen, FNB |
| F | 76 | 165 | 84 | 31 | OA | Aleve*, nitroglycerine, aspirin, Coreg, Fosinopril, Vytorin, Isosorbide, Furosemide | 40 | 13 | Spinal, FNB |
| F | 70 | 157.5 | 73 | 29 | OA | Crestor, Cephalex, Levothyroxin, Aleve, Triamterene/HCTZ | 33 | 16 | Spinal, FNB |
| M | 72 | 175 | 100 | 33 | OA | Allopurinol, aspirin*, Amlopidene, Crestor, HCTZ, Metoprolol, Norco, Doxazosin, Flomax | 53 | 12 | Gen, FNB |
| F | 79 | 156 | 50 | 21 | OA | TriamtereneHCTZ, Vicodin, aspirin*, Lovastatin, Omeprazole, Sertraline, Naproxen | 44 | 10 | Gen |
| F | 65 | 169 | 66 | 23 | OA | Percocet, Trazodone, Prednisone, Labetalol, Lexapro, Imitrex, Lisinopril, Flexeril, Furosemide, Klonopin | 38 | 15 | Spinal, FNB |
| M | 65 | 170 | 99 | 34 | OA | Pravastatin, Advil*, Clopidogrel, Diltiazem, Lisinopril HCTZ, Metformin, Novolin | 48 | 25 | Gen |
| F | 65 | 157.5 | 76 | 31 | OA | aspirin, Lisinopril, TriamtereneHCTZ, Naproxen, Prilosec | 49 | 11 | Gen, FNB |
| F | 69 | 150 | 91 | 40 | OA | spironolactone, Verapamil, Ropinirole, Simvastatin, Amlopidine, aspirin, Citalopram, Cozaar, Gabapentin, Glipizide, Humulin, Prilosec, oxycodone, Vicodin | 42 | 22 | Gen |
| F | 69 | 155 | 64 | 27 | OA | Lovaza, alpha lipoic acid, Tylenol, Percocet, Ambien, Ranitidine, Plavix, Lyrica | 45 | 15 | Gen, FNB |
| F | 65 | 157.5 | 81 | 33 | OA | Allopurinol, Furosemide, Omeprazole, Simvastatin, Tylenol | 34 | 18 | Gen |
| M | 68 | 180 | 100 | 31 | OA | Enalapril, HCTZ, Lipitor, Norco, Zolpidem | 42 | 11 | Gen, FNB |
Medication withheld week prior to surgery. Dx, diagnosis; OA, osteoarthritis; FNB, femoral nerve block; Gen, general anesthesia; Spinal, spinal anesthesia; Epd, epidural anesthesia. Femoral nerve block: 30 ml of 0.25% to 0.5% bipivicaine or ropivacaine. General anesthesia: intravenous propofol and maintained by inhalation of either desflurane or sevoflurane. Spinal anesthesia: 0.75% bupivicaine +20 μg of fentanyl. Epidural anesthesia: 0.25% bupivicaine. Muscle relaxant: Administered by local injection of rocuronium bromide.
Study design.
Details of the study design have been published previously (36). On the morning of surgery, subjects were admitted to Sacred Heart Medical Center at Riverbend in a fasted state. Anesthesia was administered with either a epidural, spinal, or general anesthetic, along with a preoperative femoral nerve block placed for postoperative analgesia. Intravenous propofol was used to induce general anesthesia and was maintained with inhalational anesthetic (either desflurane or sevoflurane), with or without muscle relaxant (rocuronium bromide) (Table 1). A 10-cm-wide Zimmer tourniquet was positioned around the proximal third of the thigh and not inflated. Just before the surgery was ready to begin, the first of three muscle biopsies was obtained from the vastus lateralis muscle on the operative (TKA) leg using a 5-mm Bergström biopsy needle with applied suction, as previously detailed (36) and Fig. 1. Following the first biopsy, the tourniquet was inflated to 300 mmHg or greater, depending on systolic blood pressure to ensure minimal blood flow to the operative leg. A second muscle biopsy was obtained immediately prior to tourniquet deflation, after completion of the main components of the surgery. The tourniquet was then deflated, allowing for reperfusion of the limb with blood. The final muscle biopsy was obtained in the operating room prior to being moved to postoperative recovery. Total tourniquet time (ischemia) was 43 ± 2 min, and reperfusion time was 16 ± 1. Immediately after each biopsy, muscle samples were blotted, and adipose tissue was removed before being frozen in liquid nitrogen (<1 min). Samples were stored in −80°C until analysis, which was routinely performed within 2 wk.
Fig. 1.
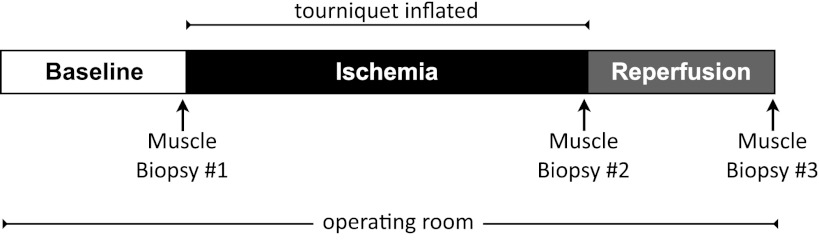
Study design. Arrows indicate skeletal muscle biopsies. A baseline muscle biopsy was acquired in the operating room following anesthesia, immediately prior to tourniquet inflation. The second muscle biopsy was obtained immediately prior to tourniquet deflation. The third muscle biopsy was obtained following deflation of the tourniquet (reperfusion) just prior to the subject leaving the operating room. Please see Table 1 for details of anesthetics used and individual tourniquet (ischemia) and reperfusion times. Average tourniquet time was 43 ± 4 min, and the average reperfusion time was 16 ± 3 min.
Whole-muscle homogenization.
Details for the homogenization procedures have been previously published but have been modified for analysis for this study (10). Frozen muscle samples (25–50 mg) were crushed using Heidolph Brinkmann Silent Crusher M in homogenization (1:9, w/v) buffer containing: 50 mM Tris·HCl, 250 mM mannitol, 50 mM Na pyrophosphate, 1 mM EDTA, 1 mM EGTA, 1.0% Triton X-100, pH 7.4, 1 mM benzamidine, 1 mM DTT, 0.1 mM PMSF, and 5 μP/ml soybean trypsin inhibitor. Samples were centrifuged at 2,817 g at 4°C, and the supernatant was collected. Aliquots of supernatant (30 μl) were subjected to an additional 10 min of boiling at 100°C and further centrifugation at 2,817 g at 4°C for 30 min to isolate 4E-BP1. Protein concentration (mg/ml) was determined in duplicate using a Qubit Protein Assay Kit (no. Q3321; Invitrogen, Carlsbad, CA) on a Qubit 2.0 Fluorometer (Invitrogen).
Nuclear and cytoplasmic homogenization and fractionation.
Isolation of nuclear and cytoplasmic fractions was done using the NE-PER kit (no. 78835; Thermo Fisher Scientific, Waltham, MA). Frozen muscle samples (25–50 mg) were washed with 1× TBS and then homogenized in cytoplasmic extraction reagent I containing 0.5 mM PMSF. Homogenates were then vortexed and incubated on ice for 10 min. Cytoplasmic extraction reagent II was added to the samples, vortexed, and centrifuged at 16,000 g at 4°C for 5 min. The supernatant (cytoplasmic extract) was collected, and nuclear extraction reagent, containing 0.5 mM PMSF, was added to the tube to resuspend the pellet. The samples were vortexed and incubated on ice and then centrifuged at 16,000 g at 4°C for 10 min before the supernatant (nuclear extract) was collected. Protein concentration (mg/ml) was determined in duplicate using a Qubit protein assay kit (no. Q3321; Invitrogen) on a Qubit 2.0 fluorometer (Invitrogen). Adequate separation of cytoplasmic and nuclear fractions was confirmed via Western blot detection of α-tubulin (cytoplasmic protein) and laminin A/C (nuclear protein) (Fig. 2).
Fig. 2.
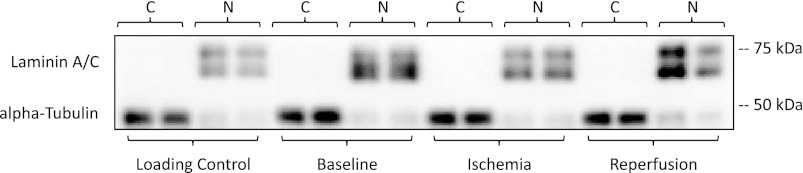
Cytoplasmic and nuclear separation. Representative blots showing adequate separation of cytoplasmic and nuclear fractions were assessed via Western blot analysis detection of α-tubulin, cytoplasmic protein (C), and laminin A/C, nuclear protein (N), in each lane. A single muscle homogenate sample was loaded in duplicate with each gel and used as a between-blot internal loading control. All gels were run in with cytoplasmic and nuclear fractions loaded in adjacent cells, i.e., baseline cytoplasmic next to the baseline nuclear fraction as shown in the representative blot.
SDS-PAGE and immunoblotting.
Details of the immunoblotting procedures have been previously published (36), with specific modifications implemented for this study. Homogenates were loaded in duplicate into TGX all kD precast gels (Bio-Rad) in electrode buffer (0.3% Tris base, 14.4% glycine, 1% SDS in dd-H2O). A single muscle homogenate sample was loaded in duplicate with each gel and used as a between-blot internal loading control (Fig. 2).
Following SDS-PAGE, proteins were transferred to PVDF membranes using Bio-Rad Trans-Blot Turbo Transfer system using the Bio-Rad Midi format, mixed MW presettings (25 V for 7 min). Transfers were verified by staining with Ponceau S.
Protocols for primary and secondary antibody application for all proteins of interest have been previously published (36). Proteins isolated by Western blot analysis were made relative to our loading control, which was run in duplicate for cytoplasmic and nuclear fractions, to account for variance between gels, as well as to an internal loading control within each lane.
Antibodies.
The primary antibodies p-Akt Ser-473 (no. 9271), Akt (no. 9272), p-4E-BP1 Thr-37/46 (no. 9459), 4E-BP1 (no. 9452), p-FoxO3a S253 (no. 9466), FoxO3a (no. 9467), eukaryotic initiation factor 4E phosphorylation (p-eIF-4E) Ser-209 (no. 9741), eIF-4E (no. 9742), p-GSK3α/β S21/9 (no. 9331), GSK3β (no. 9315), Bcl2 (no. 2872), SAPK/JNK (no. 9258), p-38α (no. 2918), p-38β (no. 2339), α-tubulin (no. 2144), laminin A/C (no. 4777) were purchased from Cell Signaling (Beverly, MA, USA). Additional primary antibodies, p-Bcl2 S70 (no. sc-21864), and MAFbx (sc-33782) were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). MuRF1 (no. ab96857) was purchased from Abcam (Cambridge, MA). Bnip3 (no. B7931) was purchased from Sigma-Aldrich (St. Louis, MO). ECL+ Anti-Rabbit IgG, horseradish peroxidase from donkey and mouse were purchased from GE Healthcare and used as our secondary antibodies.
Total RNA isolation and cDNA synthesis.
Details for RNA isolation have been previously published (36) with slight modifications implemented for this study. Skeletal muscle samples (10–20 mg) were homogenized in 1 ml TRI Reagent using Heidolph Brinkmann's Silent Crusher M at 10,000–15,000 rpm in Eppendorph RNase-free tubes. Separation was achieved through the addition of 0.2 ml of chloroform and precipitation with 0.5 ml isopropanol. The RNA pellet was washed twice in 75% ethanol, dried, and then dissolved in 1.5 μl of 0.1 mM EDTA for each 1 mg of starting tissue. RNA concentrations were determined using a Qubit fluorometer (Invitrogen, Carlsbad, CA), and cDNA was reverse transcribed from 1 μg RNA on a CFX96 real-time PCR Detection System (Bio-Rad) using iScript Reaction Mix (Bio-Rad), according to manufacturer's instructions and stored at −80°C for analysis.
Oligonucleotide primers for qPCR.
Oligonucleotide primers were designed using Beacon Design software (ver. 7.91), specific to our Bio-Rad CFX96 real-time PCR Detection System, based on NCBI Entrez Gene ID search results. Primer efficiencies were determined through analysis of four serial dilutions, analyzed in triplicate on a run in triplicate log linear scale: efficiency = 10−1/slope−1. Efficiencies were in accordance with MIQE guidelines (0.95 and 1.05) for all primer pairs (8). Primers were designed for α-tubulin (NM_006082; Fwd: AGATGCTGCCAATAACTATG, Rev: AATTCGGTCCAACACAAG); β-tubulin (NM_030773; Fwd: CAGTGTCCTTCTCAGTTA, Rev: GATTACGCTCTTGTGTATT); β-2-microglobulin (NM_004048; Fwd: TCTCTTATCCAACATCAACATCTT, Rev: GCACGCTTAACTATCTTAACAAG); Forkhead box O3 (FOXO3) (NM_001455; Fwd: GCATTCTTACTGAGGATT, Rev: ACACAAGGCAATATCTAC); atrogin-1 (MAFbx) (NM_058229; Fwd: TGGATTGGAAGAAGATGTAT, Rev: AAAGGATGTGACAGTGTT); muscle ring finger (MuRF) (NM_032588; Fwd: ATGAGGAAGAGGAAGAATT, Rev: CTTACTGGTGTCCTTCTT); activating transcription factor (ATF4) (NM_001675; Fwd: AGATAGGAAGCCAGACTA, Rev: CTCATACAGATGCCACTA); Jun N-terminal kinase (JNK) (NM_002750; Fwd: TAGTGTAGAGATTGGAGATT, Rev: AAGAATGGCATCATAAGC); p-38α (NM_001315; Fwd: GTTCAGTTCCTTATCTACCA, Rev: GCTCACAGTCTTCATTCA); BCL2/adenovirus E1B 19-kDa protein-binding protein 3 (Bnip3) (NM_004052; Fwd: TCATAATCAACAGAAACCAAGA, Rev: ATCACCTAATAATCGGAGACT); B-cell lymphoma 2 (BCL2) (NM_006538; Fwd: ATATCTGCCTGTATCTTG, Rev: TCTGTTGTTATTACTGCTA); and JunB proto-oncogene (JUNB) (NM_002229; Fwd: GAAACGACCTTCTATGAC, Rev: GGTTACTGTAGCCATAAG).
mRNA quantitation by qPCR.
Details on qPCR have been previously published (36), with specific modifications employed for this study. Samples of cDNA were analyzed using SYBR Green fluorescence (iQ SYBR Green Supermix; Bio-Rad, Hercules, CA). Each reaction contained 12.5 μl SYBR Green, 9.5 μl of DEPC-treated nuclease-free water, 0.5 μl forward and 0.5 μl reverse primers, and 2 μl of cDNA template, and was prepared in triplicate. An initial 5-min cycle at 95°C was used to denature the cDNA. This was followed with 50 PCR cycles consisting of denaturation at 95°C for 10 s followed by 30 s of primer annealing at the optimized primer pair-annealing temperature. All PCR cycles were followed by a melt curve analysis. Each gene of interest (GOI) was normalized to the geometric mean of select genes of reference (GOR); α-tubulin, β-tubulin, and β-2-microglobulin. Data for each GOI are expressed as fold change relative to the GOR using the Livak method, also known as the 2-ΔΔCT method (27): ΔCT(test) = CT(GOI, test) − CT (GOR, test) ΔCT(baseline) and ΔΔCT = ΔCT(test) − ΔCT(baseline) 2-ΔΔCT = fold change from baseline.
Statistical analysis.
Statistical evaluation of our data was performed using a repeated-measures ANOVA to compare ischemic and reperfusion samples to baseline. Differences between means were considered significant at P ≤ 0.05. Differences between means were considered a trend at P ≤ 0.10. Analysis for all variables was performed using SAS Institute (2009), Base SAS 9.2. All values are expressed as means ± SE, unless otherwise stated.
RESULTS
Demographics.
Male and female subject characteristics were not different for age, height, weight, and body mass index (P > 0.05); data for all subjects are in Table 1. The average age for our cohort was 70 ± 1.3 yr. The average height, weight, and BMI was 164 ± 2.7 cm, 84 ± 5.8 kg, and 31 ± 1.8 kg/m2, respectively.
Cell signaling.
Adequate tissue samples from all three biopsies were available for only six subjects for the analysis of whole muscle fractions. These samples were used to confirm our previous results (36). Relative to baseline, phosphorylation of Akt at Ser-473 decreased by 43% during ischemia (F = 13.71) (P = 0.013, 80% CI [−72%, −31%]) but was not significant during reperfusion −20% (F = 5.95) (P = 0.08, 80% CI [−51%, −13%]) (Fig. 3A).
Fig. 3.

Protein translation downregulation during total knee arthroplasty (TKA). Data represent Akt at Ser-473 in whole muscle fraction (A) (n = 6); 4E-BP1 Thr-36/47 (B); eukaryotic initiation factor 4E (eIF-4E) at Ser-209 in the cytoplasmic (C) and nuclear fraction (D). Representative phosphorylated, total, and loading control (Ponceau) Western blot are included. Data are expressed as means ± SE (n = 10). *P ≤ 0.05 vs. baseline.
There was adequate tissue to analyze cytoplasmic and nuclear protein fractions for 10 subjects. Relative to baseline eukaryotic initiation factor 4E binding protein (4E-BP1) phosphorylation at Thr-37/46/total 4E-BP1 was significantly decreased by 30% (F = 16.36) (P = 0.003, 80% CI [−26%, −13%]) and 27% (F = 5.57) (P = 0.043, 80% CI [−30%, −8%]) during ischemia and reperfusion, respectively in the cytoplasmic fractions (Fig. 3B). Relative to baseline, eIF-4E at Ser-209 increased significantly in the cytoplasmic fraction during ischemia, by 189% (F = 6.54) [P = 0.031, 80% CI (54%, 182%)] but was not significantly different from baseline during reperfusion (Fig. 3C). Relative to baseline, eIF-4E phosphorylation in the nuclear fraction did not change during ischemia or reperfusion (Fig. 3D).
Relative to baseline, SAPK/JNK protein significantly increased in the cytoplasmic fraction during ischemia by 27% (F = 24.12) [P = 0.001, 80% CI (24%, 43%)] (Fig. 4A), while in the nuclear fraction, there was a trend to decrease during reperfusion, −21% (F = 4.24) [P = 0.073, 80% CI (−24%, −5%)] but was not significant (Fig. 4B). Relative to baseline, p-38α protein did not change in the cytoplasmic fraction during ischemia (Fig. 4C), while in the nuclear fraction, there was a trend for it to increase during ischemia by 34% (F = 3.95) [P = 0.082, 80% CI (5%, 30%)] but was not significant, while during reperfusion, it was significantly decreased by 18% (F = 9.42) (P = 0.015, 80% CI [−17%, −6%]) (Fig. 4D). Relative to baseline, p-38β protein tended to increase in the cytoplasmic fraction during ischemia by 33% (F = 4.12) [P = 0.073, 80% CI (16%, 86%)] but was not significant (Fig. 4E), while in the nuclear fraction, there was a significant increase during ischemia by 25% (F = 9.55) [P = 0.015, 80% CI (5%, 30%)] (Fig. 4F).
Fig. 4.

Stress pathway during TKA. Stress-activated protein kinase (SAPK/JNK) protein in the cytoplasmic (A) and nuclear fraction (B). MAPK p-38α protein in the cytoplasmic (C) and nuclear fraction (D). MAPK p-38β protein in the cytoplasmic (E) and nuclear fraction (F). Representative phosphorylated, total, and loading control (Ponceau) Western blot are included. Data are expressed as means ± SE (n = 10). *P ≤ 0.05 vs. baseline.
Relative to baseline, Bcl2 protein showed a trend to increase in the cytoplasmic fraction during ischemia by 97% (F = 5.10) [P = 0.054, 80% CI (62%, 263%)] but was not significant (Fig. 5A), while in the nuclear fraction, there was no change (Fig. 5B). Bcl2/adenovirus EIB 19-kDa-interacting protein 3 (Bnip3) protein levels did not change in the cytoplasmic fraction during ischemia or reperfusion (Fig. 5C). Bnip3 protein levels in the nuclear fraction significantly decreased during reperfusion by 22% (F = 6.60) [P = 0.033, 80% CI (−139%, −41%)] (Fig. 5D). We did not detect significant changes in phosphorylation status in the cytoplasmic or nuclear fractions for p-GSK3β Ser-9; however, we did detect a significant increase in GSK3β total protein during ischemia 42% (F = 8.26) [P = 0.018, 80% CI (21%, 60%)].
Fig. 5.

Autophagic/lysosomal pathway during TKA. Bcl2 protein in the cytoplasmic (A) and nuclear fraction (B). Bcl2/adenovirus EIB 19-kDa-interacting protein 3 (Bnip3) protein in the cytoplasmic (C) and nuclear fraction (D). Representative phosphorylated, total, and loading control (Ponceau) Western blot images are included. Data are expressed as means ± SE (n = 10). *P ≤ 0.05 vs. baseline.
Relative to baseline, the E3 ubiquitin ligase atrogin-1/MAFbx protein increased in the cytoplasmic fraction during ischemia by 30% (F = 18.30) [P = 0.002, 80% CI (15%, 29%)] (Fig. 6A), while the nuclear fraction significantly decreased during reperfusion by 30% (F = 5.80) (P = 0.043, 80% CI [−97%, −26%]) (Fig. 6B). As well, relative to baseline, MuRF1 protein increased in the cytoplasmic fraction during ischemia by 53% (F = 26.33) [P = 0.0006, 80% CI (36%, 62%)] and remained elevated during reperfusion 31% (F = 18.36) [P = 0.002, 80% CI (18%, 36%)] (Fig. 6C). In the nuclear fraction, MuRF1 protein tended to decrease during reperfusion, by 29% (F = 4.21) [P = 0.074, 80% CI (−106%, −20%)] but was not significant (Fig. 6D).
Fig. 6.

E3 ubiquitin proteasomal pathway during TKA. MAFbx protein in the cytoplasmic (A) and nuclear fraction (B). MuRF1 protein in the cytoplasmic (C) and nuclear fraction (D). Representative phosphorylated, total, and loading control (Ponceau) Western blot images are included. Data are expressed as means ± SE (n = 10). *P ≤ 0.05 vs. baseline.
Gene expression.
Adequate tissue was available to analyze gene expression for seven subjects. Relative to baseline, FoxO3a mRNA increased during ischemia by 157% (F = 18.55) [P = 0.008, 80% CI (66%, 135%)] (Fig. 7A). Relative to baseline, the E3 ubiquitin ligase MAFbx mRNA increased during ischemia by 237% (F = 7.37) [P = 0.042, 80% CI (85%, 288%)] (Fig. 7B). As well, MuRF mRNA increased during ischemia by 288% (F = 7.75) [P = 0.039, 80% CI (161%, 380%)] (Fig. 7C). Relative to baseline, SAPK/JNK mRNA increased during ischemia by 169% (F = 15.16) [P = 0.012, 80% CI (95%, 211%)] (Fig. 7D) and p-38α mRNA increased during ischemia 173% (F = 21.80) [P = 0.006, 80% CI (125%, 242%)] (Fig. 7E). We did not detect significant changes in transcript levels for Bnip3 and Bcl2.
Fig. 7.

mRNA expression during TKA. FoxO3a (A) MAFbx (B), MuRF1 (C), SAPK/JNK (D), and p-38α (E). Data are expressed as means ± SE (n = 7). *P ≤ 0.05 vs. baseline.
DISCUSSION
I/R occurs each time a tourniquet is used to control blood flow during surgeries, such as TKA. However, the cellular and molecular alterations associated with I/R remain to be fully elucidated. We have recently reported that proteins regulating components of the cap-dependent translation initiation and elongation complex are downregulated with I/R during TKA (36). However, at the cellular level, atrophy is defined by the imbalance between proteins controlling both muscle anabolism and catabolism. Our objective, therefore, was to measure changes in the components of the catabolic FoxO3a pathway, as well as cell stress pathways, during TKA. We hypothesized that cellular mechanisms controlling muscle protein catabolism would be upregulated during surgery, and as such, help us to further determine how I/R injury may potentially contribute to the twofold greater quadriceps atrophy measured in the operative vs. nonoperative leg 2 wk after TKA (36).
This study reveals several important and novel findings regarding the acute effects of I/R in stimulating pathways known to play key roles in protein catabolism in muscle cells (Fig. 8). First, we confirm previous findings obtained by our group (36), showing that the Akt/4E-BP1 pathway is downregulated during ischemia and reperfusion and add further to those findings by including the phosphorylation status of the eIF-4E. Second, we found that protein levels for MuRF and MAFbx, two downstream components of FoxO3a pathway, were upregulated during ischemia but only protein levels for MuRF remained elevated through reperfusion. Third, we measured an increased expression of the SAPK/JNK and the p-38 mitogen activated protein kinase (p-38 MAPK α/β). To our knowledge, this study is the first to measure simultaneous changes in key regulatory proteins known to exert control over both the anabolic and catabolic transcriptional and signaling pathways in skeletal muscle cells during TKA.
Fig. 8.

Ischemia-reperfusion stimulates multiple catabolic/autophagic cell signaling pathways during total knee arthroplasty (TKA). A schematic diagram of proteins regulating signaling pathways controlling cap-dependent mRNA translation initiation (36) and autophagy/lysosomal and ubiquitin-proteasomal protein degradation. Ischemia-reperfusion inhibits cap-dependent translation initiation via dephosphorylation of Akt leading to inhibition of the Akt-mTOR pathway and the availability of 4E-BP1 to bind to and inhibit eIF4E association with eIF4G to form an active mRNA cap-binding complex (eIF4F). Dephosphorylation of Akt leads to the dephosphorylation and nuclear translocation of FoxO3a, leading to increased transcription of MAFbx, MuRF1, and Bnip3 and subsequent activation of ubiquitin-proteasomal and autophagic/lysosomal protein degradation pathways. Cell stress signaling increases the expression of SAPK/JNK and p38α/β MAPKs, contributing to the nuclear translocation of FoxO3a and protein degradation.
4E-BP1's binding target eIF-4E showed no changes in the total amount of protein present, but there were measurable changes in phosphorylation in the cytoplasmic fraction during ischemia. eIF-4E phosphorylation at Ser-209 was first thought to enhance its binding to the 5′-cap, but it is now believed that phosphorylation of eIF-4E may have the opposite effect, decreasing its binding affinity (30, 38, 40). Mitogen- or stress- and cytokine-activated signaling regulates eIF-4E phosphorylation to a greater extent than anabolic stimuli (33), and the significance of its phosphorylation is largely undetermined in mammals. It is possible that eIF-4E phosphorylation during TKA is necessary to increase the translation of proapoptotic transcripts that are upregulated during I/R.
Downregulation of Akt leads to dephosphorylation of FoxO3a and translocation into the nucleus, where it promotes the transcription of atrophy-related genes (2). FoxO3a may also be acted upon by the SAPK/JNK pathway. We measured an increase in SAPK/JNK transcript levels and protein concentrations in the cytoplasm during ischemia. Activation of the SAPK/JNK pathway under these conditions is consistent with, and suggests a role for, oxidative stress as a potential stimulating factor initiating the upregulation of the catabolic SAPK/JNK/FoxO pathway (13).
Cytoplasmic protein levels for MuRF1 increased during both ischemia and reperfusion, while MAFbx protein increased only during ischemia. Interestingly, we measured a significant decrease in MAFbx protein in the nuclear fraction during reperfusion which did not appear to mirror reciprocal changes in the cytoplasmic fraction. Previous research has found that MAFbx contains two nuclear localization signals (NLS), which are each sufficient to signal MAFbx translocation into the nucleus (17, 20). We are not sure of the physiological consequences for the decrease in the nuclear fraction but MAFbx cytoplasmic-nuclear shuttling has been shown to occur in myotubes subjected to catabolic conditions (25). It remains that additional work is needed to further define a role for MAFbx colocalization in and out of the nucleus during I/R injury.
The primary function of the Bcl-2 proteins is in the regulation, and stabilization of, the outer mitochondrial membrane (9, 23). While not statistically significant, our data suggest that Bcl-2 protein levels tended to increase in the cytoplasmic fraction during ischemia (P = 0.054), which may be in response to I/R induced oxidative stress as suggested by our p-38β data and hold potential clinical relevance in terms of mitochondrial involvement in respiratory function during tourniquet use. We measured an increase in p-38β in the nuclear fraction during ischemia. It has been shown that p-38 protein colocalizes within the nuclear fraction at rest and translocates to the cytosol upon stimulation (5). Stress-activated p-38 has been shown to bind to and inhibit extracellular signal-regulated kinases 1/2 (ERK1/2), i.e., MAP kinases, which are activated to varying degrees by cytokines, growth factors, and osmotic and other types of cell stress (35). Further research is required in order for us to better understand the contributions of the rapid changes in nuclear and cytoplasmic fractions and how these alterations impact muscle metabolism in the hours and days following TKR.
Our study is limited by the lack of a non-tourniquet control group matched for surgery duration and anesthesia administration. Additionally, we were unable to control for the various preoperative and perioperative medications, analgesics, and muscle relaxants, as well as for levels of physical activity prior to surgery, and we do not have a direct measure of protein synthesis or degradation. As such, we acknowledge the need for further intraoperative and perioperative studies designed to quantify the magnitude of the contribution of I/R injury on muscle protein breakdown.
Perspectives and Significance
Despite the considerable success of TKA in mitigating knee pain due to osteoarthritis, chronic limitations in functional mobility remain and are primarily due to irrecoverable quadriceps muscle loss. Indeed, quadriceps atrophy is the main contributor to long-term strength deficits (29), and by far the most significant long-term clinical barrier following TKA surgery is persistent muscle atrophy (14, 31, 44). Broadly speaking, acute muscle loss for older adults represents an acceleration of sarcopenia, i.e., the gradual ∼1%/year of muscle lost due to aging, per se (11). In other words, a 12% loss in quadriceps volume occurring within 2 wk of surgery (36) would represent ∼12 yr of “normal” aging. This “accelerated sarcopenic muscle loss” (32) is potentially devastating for older women who, compared with older men, demonstrate a blunted capacity to increase muscle volume (4).
In conclusion, we interpret our findings to support our hypothesis that factors resulting from ischemia are sufficient to upregulate components of the FoxO3a catabolic pathway during TKA and that cellular stress pathways may play a role in stimulating this response. These data extend our understanding of the effects of I/R to now include proteins regulating anabolic (36), as well as catabolic and stress-activated, pathways being altered during TKA. Additional studies are necessary to better understand the mechanisms underlying the rapid-onset muscle atrophy that occurs within weeks of this surgery and, in particular, the means by which potentially irrecoverable muscle atrophy may be avoided in older women.
GRANTS
This study was supported by the National Center for Medical Rehabilitation and Research, Eunice Kennedy Shriver National Institute of Child Health & Human Development, National Institutes of Health Grant K01HD57332 (H. C. Dreyer). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.N.B., A.D.H., B.R.V., S.N.S., and B.A.J. performed experiments; A.N.B., A.D.H., B.R.V., and K.S. analyzed data; A.N.B., A.D.H., K.S., and H.C.D. interpreted results of experiments; A.N.B. prepared figures; A.N.B. and H.C.D. drafted manuscript; A.N.B., A.D.H., B.R.V., and H.C.D. edited and revised manuscript; A.N.B., A.D.H., B.R.V., K.S., S.N.S., B.A.J., and H.C.D. approved final version of manuscript; H.C.D. conception and design of research.
ACKNOWLEDGMENTS
We thank Crystal Mills and Alicia Morrison of the Slocum Center for Orthopedics & Sports Medicine for their help with the recruitment and scheduling. We would also like to thank Joni Strub, OR Nurse Manager, and the surgical staff at Sacred Heart Medical Center at RiverBend, as well as the anesthesiologists from Northwest Anesthesia Physicians group for their assistance with the study. We thank Hilary Senesac and Steven Ratchford for assistance with tissue collection.
Patients were recruited at the Slocum Center for Orthopedics and Sports Medicine, Eugene, OR. Surgeries and tissue collection were conducted at the Sacred Heart Medical Center, Riverbend, Springfield, OR. Processing and analysis of all tissue samples were preformed in the Muscle Physiology Laboratory at the University of Oregon, Eugene, OR.
REFERENCES
- 1.Appell HJ, Gloser S, Duarte JA, Zellner A, Soares JM. Skeletal muscle damage during tourniquet-induced ischaemia. The initial step towards atrophy after orthopaedic surgery? Eur J Appl Physiol Occup Physiol 67: 342–347, 1993 [DOI] [PubMed] [Google Scholar]
- 2.Ashrafpour H, Huang N, Neligan PC, Forrest CR, Addison PD, Moses MA, Levine RH, Pang CY. Vasodilator effect and mechanism of action of vascular endothelial growth factor in skin vasculature. Am J Physiol Heart Circ Physiol 286: H946–H954, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Baehr LM, Furlow JD, Bodine SC. Muscle sparing in muscle RING finger 1 null mice: response to synthetic glucocorticoids. J Physiol 589: 4759–4776, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bamman MM, Hill VJ, Adams GR, Haddad F, Wetzstein CJ, Gower BA, Ahmed A, Hunter GR. Gender differences in resistance-training-induced myofiber hypertrophy among older adults. J Gerontol A Biol Sci Med Sci 58: 108–116, 2003 [DOI] [PubMed] [Google Scholar]
- 5.Ben-Levy R, Hooper S, Wilson R, Paterson HF, Marshall CJ. Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr Biol 8: 1049–1057, 1998 [DOI] [PubMed] [Google Scholar]
- 6.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294: 1704–1708, 2001 [DOI] [PubMed] [Google Scholar]
- 7.Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3: 1014–1019, 2001 [DOI] [PubMed] [Google Scholar]
- 8.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55: 611–622, 2009 [DOI] [PubMed] [Google Scholar]
- 9.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL2 family reunion. Mol Cell 37: 299–310, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dreyer HC, Fujita S, Cadenas JG, Chinkes DL, Volpi E, Rasmussen BB. Resistance exercise increases AMPK activity and reduces 4E-BP1 phosphorylation and protein synthesis in human skeletal muscle. J Physiol 576: 613–624, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dreyer HC, Volpi E. Role of protein and amino acids in the pathophysiology and treatment of sarcopenia. J Am Coll Nutr 24: 140S–145S, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duarte JA, Gloser S, Remiao F, Carvalho F, Bastos ML, Soares JM, Appell HJ. Administration of tourniquet. I. Are edema and oxidative stress related to each other and to the duration of ischemia in reperfused skeletal muscle? Arch Orthop Trauma Surg 116: 97–100, 1997 [PubMed] [Google Scholar]
- 13.Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL, Burgering BM. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J 23: 4802–4812, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finch E, Walsh M, Thomas SG, Woodhouse LJ. Functional ability perceived by individuals following total knee arthroplasty compared to age-matched individuals without knee disability. J Orthop Sports Phys Ther 27: 255–263, 1998 [DOI] [PubMed] [Google Scholar]
- 15.Glass DJ. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol 5: 87–90, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol 37: 1974–1984, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA 98: 14440–14445, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ 14: 146–157, 2007 [DOI] [PubMed] [Google Scholar]
- 19.Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci 120: 2479–2487, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Julie LC, Sabrina BP, Marie-Pierre L, Leibovitch SA. Identification of essential sequences for cellular localization in the muscle-specific ubiquitin E3 ligase MAFbx/Atrogin 1. FEBS Lett 586: 362–367, 2012 [DOI] [PubMed] [Google Scholar]
- 21.Kandarian SC, Jackman RW. Intracellular signaling during skeletal muscle atrophy. Muscle Nerve 33: 155–165, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Kettelhut IC, Wing SS, Goldberg AL. Endocrine regulation of protein breakdown in skeletal muscle. Diabetes Metab Rev 4: 751–772, 1988 [DOI] [PubMed] [Google Scholar]
- 23.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275: 1132–1136, 1997 [DOI] [PubMed] [Google Scholar]
- 24.Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am 89: 780–785, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Lagirand-Cantaloube J, Cornille K, Csibi A, Batonnet-Pichon S, Leibovitch MP, Leibovitch SA. Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLos One 4: e4973, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 17: 1807–1819, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 28.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6: 458–471, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Meier WA, Marcus RL, Dibble LE, Foreman KB, Peters CL, Mizner RL, LaStayo PC. The long-term contribution of muscle activation and muscle size to quadriceps weakness following total knee arthroplasty. J Geriatr Phys Ther 32: 35–38, 2009 [PubMed] [Google Scholar]
- 30.Minich WB, Balasta ML, Goss DJ, Rhoads RE. Chromatographic resolution of in vivo phosphorylated and nonphosphorylated eukaryotic translation initiation factor eIF-4E: increased cap affinity of the phosphorylated form. Proc Natl Acad Sci USA 91: 7668–7672, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizner RL, Petterson SC, Snyder-Mackler L. Quadriceps strength and the time course of functional recovery after total knee arthroplasty. J Orthop Sports Phys Ther 35: 424–436, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Phillips SM. Nutrient-rich meat proteins in offsetting age-related muscle loss. Meat Sci 92: 174–178, 2012 [DOI] [PubMed] [Google Scholar]
- 33.Proud CG. Signalling to translation: how signal transduction pathways control the protein synthetic machinery. Biochem J 403: 217–234, 2007 [DOI] [PubMed] [Google Scholar]
- 34.Racz IB, Illyes G, Sarkadi L, Hamar J. The functional and morphological damage of ischemic reperfused skeletal muscle. Eur Surg Res 29: 254–263, 1997 [DOI] [PubMed] [Google Scholar]
- 35.Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene 26: 3100–3112, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Ratchford SM, Bailey AN, Senesac HA, Hocker AD, Smolkowski K, Lantz BA, Jewett BA, Gilbert JS, Dreyer HC. Proteins regulating cap-dependent translation are downregulated during total knee arthroplasty. Am J Physiol Regul Integr Comp Physiol 302: R702–R711, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR, Lecker SH, Goldberg AL. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J 21: 140–155, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Scheper GC, van Kollenburg B, Hu J, Luo Y, Goss DJ, Proud CG. Phosphorylation of eukaryotic initiation factor 4E markedly reduces its affinity for capped mRNA. J Biol Chem 277: 3303–3309, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Sexton WL, Korthuis RJ, Laughlin MH. Microvascular injury after ischemia and reperfusion in skeletal muscle of exercise-trained rats. J Appl Physiol 68: 2329–2336, 1990 [DOI] [PubMed] [Google Scholar]
- 40.Slepenkov SV, Darzynkiewicz E, Rhoads RE. Stopped-flow kinetic analysis of eIF4E and phosphorylated eIF4E binding to cap analogs and capped oligoribonucleotides: evidence for a one-step binding mechanism. J Biol Chem 281: 14927–14938, 2006 [DOI] [PubMed] [Google Scholar]
- 41.Sternbergh WC, 3rd, Adelman B. The temporal relationship between endothelial cell dysfunction and skeletal muscle damage after ischemia and reperfusion. J Vasc Surg 16: 30–39, 1992 [PubMed] [Google Scholar]
- 42.Suval WD, Duran WN, Boric MP, Hobson RW, 3rd, Berendsen PB, Ritter AB. Microvascular transport and endothelial cell alterations preceding skeletal muscle damage in ischemia and reperfusion injury. Am J Surg 154: 211–218, 1987 [DOI] [PubMed] [Google Scholar]
- 43.Suval WD, Hobson RW, 2nd, Boric MP, Ritter AB, Duran WN. Assessment of ischemia reperfusion injury in skeletal muscle by macromolecular clearance. J Surg Res 42: 550–559, 1987 [DOI] [PubMed] [Google Scholar]
- 44.Walsh M, Woodhouse LJ, Thomas SG, Finch E. Physical impairments and functional limitations: a comparison of individuals 1 year after total knee arthroplasty with control subjects. Phys Ther 78: 248–258, 1998 [DOI] [PubMed] [Google Scholar]