

Abstract
Novel analogs of the P2 receptor antagonist pyridoxal-5′-phosphate-6-phenylazo-2′,4′-disulfonate (PPADS) were synthesized. Modifications were made through functional group substitution on the sulfophenyl ring and at the phosphate moiety through the inclusion of phosphonates, demonstrating that a phosphate linkage is not required for P2 receptor antagonism. Substituted 6-phenylazo and 6-naphthylazo derivatives were also evaluated. Among the 6-phenylazo derivatives, 5′-methyl, ethyl, propyl, vinyl, and allyl phosphonates were included. The compounds were tested as antagonists at turkey erythrocyte and guinea-pig taenia coli P2Y1 receptors, in guinea-pig vas deferens and bladder P2X1 receptors, and in ion flux experiments by using recombinant rat P2X2 receptors expressed in Xenopus oocytes. Competitive binding assay at human P2X1 receptors in differentiated HL-60 cell membranes was carried out by using [35S]ATP-γ-S. A 2′-chloro-5′-sulfo analog of PPADS (C14H12O9N3ClPSNa), a vinyl phosphonate derivative (C15H12O11N3PS2Na3), and a naphthylazo derivative (C18H14O12N3PS2Na2), were particularly potent in binding to human P2X1 receptors. The potencies of phosphate derivatives at P2Y1 receptors were generally similar to PPADS itself, except for the p-carboxyphenylazo phosphate derivative C15H13O8N3PNa and its m-chloro analog C15H12O8N3ClPNa, which were selective for P2X vs. P2Y1 receptors. C15H12O8N3ClPNa was very potent at rat P2X2 receptors with an IC50 value of 0.82 μM. Among the phosphonate derivatives, [4-formyl-3-hydroxy-2-methyl-6-(2-chloro-5-sulfonylphenylazo)-pyrid-5-yl]methylphosphonic acid (C14H12-O8N3ClPSNa) showed high potency at P2Y1 receptors with an IC50 of 7.23 μM. The corresponding 2,5-disulfonylphenyl derivative was nearly inactive at turkey erythrocyte P2Y1 receptors, whereas at recombinant P2X2 receptors had an IC50 value of 1.1 μM. An ethyl phosphonate derivative (C15H15O11N3PS2Na3), whereas inactive at turkey erythrocyte P2Y1 receptors, was particularly potent at recombinant P2X2 receptors.
Keywords: ATP, nucleotides, ion channels, phospholipase C, smooth muscle, guinea pig, turkey erythrocytes
INTRODUCTION
Extracellular adenine nucleosides and nucleotides, acting through P1 and P2 receptors [North and Barnard, 1997; Burnstock, 1996], are widely accepted as neuro-modulators and neurotransmitters in the central and peripheral nervous systems. Two families of P2 receptors, ligand-gated ion channels (P2X subtype) and G protein-coupled receptors (P2Y subtype), have been recently classified [Abbracchio and Burnstock, 1994]. Numerous subtypes of ATP receptors have been cloned, and it is now clear that uracil nucleotides [Communi and Boeynaems, 1997] are also active or even the preferred ligands at some of the P2Y subtypes. P2X1–7 and P2Y1,2,4,6 designations have been unambiguously assigned to mammalian nucleotide receptors [Burnstock and King, 1996; Fredholm et al., 1997], although there is still uncertainty about the correspondence of these cloned sequences to pharmacologic phenotypes. Other novel mammalian P2Y subtypes, numbering up to P2Y11, have recently been reported [Communi et al., 1997].
The most significant evidence for ATP acting as a cotransmitter was reported for the sympathetic neurotransmission system in which norepinephrine and ATP are coreleased [Burnstock, 1993]. In vas deferens, isolated blood vessels, skeletal muscle, intestine, kidney and skin, norepinephrine and ATP cause synergistic constriction by means of α1-adrenoceptors and P2X receptors, respectively. In rabbit coronary vessels, guinea-pig taenia coli, rat aorta, and rat mesenteric artery, the predominant effect of those transmitters is relaxation by means of β-adrenoceptors and P2Y receptors. Besides sympathetic neurotransmission, P2 receptors are also suggested to function in parasympathetic, sensory-motor NANC inhibitory, and somatic neuromuscular neurotransmission. In platelets, an as yet uncloned P2T receptor [Hourani and Hall, 1996; Greco, 1997] has been shown to be activated mainly by ADP to cause platelet aggregation and is blocked by ATP. More recently, a hypothesis was reported that P2X3 receptors mediate nociception by means of the dorsal root ganglia, whereas activation of P1 receptors seems to result in anti-nociception [Burnstock and Wood, 1996]. The therapeutic potential [Williams, 1996; Williams and Bhagwat, 1996] and physiologic role of P2 receptors also have been reviewed in various biological systems, including brain [Inoue et al., 1996], central nervous [Gibb and Halliday, 1996], sensory nervous [Thorne and Housley, 1996], cardiovascular [Rongen et al., 1997], immune [Di Virgilio et al., 1996], and renal microvascular [Inscho, 1996] systems.
Progress in the field of P2 receptors has been impeded because of the lack of useful ligands, especially P2 receptor selective antagonists and radioligands [Jacobson et al., 1997]. A synthetic polyanionic substance, pyridoxal-5′-phosphate-6-phenylazo-2,4-disulfonate (PPADS), was shown to be a P2 receptor antagonist [Lambrecht et al., 1996]. In smooth muscle assays, it antagonized P2X receptors in rabbit vas deferens [Lambrecht et al., 1992], urinary bladder [Ziganshin et al., 1993], isolated blood vessels [Ziganshin et al., 1994], guinea-pig isolated vas deferens [McLaren et al., 1994], and rat perfused mesenteric arterial bed [Windscheif et al., 1994]. PPADS was initially reported to show moderate selectivity for P2X vs. P2Y receptors [Windscheif et al., 1995a]; however, it was later shown to be more potent than anticipated at P2Y1 receptors [Schachter et al., 1996]. At the P2Y1 subtype, PPADS was a competitive antagonist exhibiting an equilibrium inhibition constant (Ki) value of approximately 1 μM. PPADS had low affinity at the adenylate cyclase-coupled P2Y receptor in rat C6 glioma cells [Boyer et al., 1994], at the P2U receptor in bovine aortic endothelial cells [Brown et al., 1995], and at P2T receptors in human platelets [Windscheif et al., 1995b].
In the present study, the structure-activity relationships (SARs) of a series of pyridoxal-6-arylazo-5-phosphate derivatives and corresponding phosphonate congeners are reported (Fig. 1). Both radioligand binding and pharmacologic assays have been conducted with the aim of developing more potent and selective antagonists for P2 receptor subtypes.
Fig. 1.
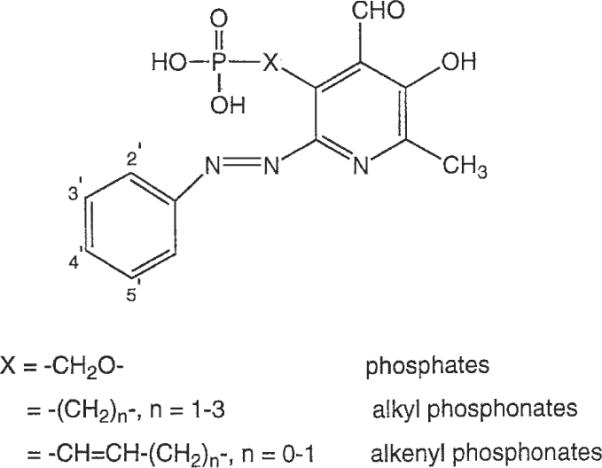
Structure of pyridoxal-6-phenylazo-5′-phosphate or phosphonate derivatives prepared in the present study.
MATERIALS AND METHODS
Pyridoxal-5-phosphate and the reagents for azo coupling reactions were purchased from Aldrich (St. Louis, MO). The aniline derivatives were obtained from Aldrich, Fluka (St. Louis, MO), Lancaster (Windham, NH), Pfaltz & Bauer (Waterbury, CT), and Eastman Kodak (Rochester, NY).
Synthesis
Proton nuclear magnetic resonance spectroscopy was performed on a Varian GEMINI-300 spectrometer and spectra were taken in CDCl3 or D2O. The chemical shifts are expressed as parts per million downfield from tetramethylsilane or as relative parts per million from HOD peaks (4.78 ppm). 31P-NMR spectra were recorded at room temperature by use of Varian XL-300 spectrometer (121.42 MHz); orthophosphoric acid (85%) was used as an external standard. High-resolution fast atom bombardment mass was performed with JEOL SX102 spectrometer using 6-kV Xe atoms. The phosphate and phosphonate derivatives were desorbed from glycerol or magic bullet matrix.
All pyridoxal phosphate and phosphonate derivatives showed more than 95% purity in high pressure liquid chromatography (HPLC) systems. The determinations of purity were performed with a Hewlett-Packard 1090 HPLC system using an OD-5-60 C18 analytical column (250 mm × 4.6 mm, Separation Methods Technologies, Inc., Newark, DE) in two different linear gradient solvent systems. One solvent system (A) was 0.1 M triethylammonium acetate buffer: CH3CN = 95:5 to 40:60 for 20 min with a flow rate of 1 ml/min. The other solvent system (B) was 5 mM tetrabutylammonium phosphate buffer:CH3CN 80:20 to 40:60, in 20 min with a flow rate of 1 ml/min. Peaks were detected by ultraviolet absorption with a diode array detector.
General Procedure of Azo Coupling for the Synthesis of PPADS Analogs
To a stirred solution of each aminoaryl compound (0.4 mmol) in 10 ml of water was added 0.4 ml (0.4 mmol) of 1 N HCl and 28 mg (0.4 mmol) of solid NaNO2 at 0°C. This solution was stirred for 5 min and the pH was adjusted to ~10 with 1 N NaOH (about 0.2 ml). To the mixture was added dropwise a solution of pyridoxal-5-phosphate (100 mg, 0.4 mmol) or a phosphonate (0.4 mmol) previously dissolved in aqueous NaOH (pH ~10). The pH was adjusted to ~9, and the yellow color changed to red. After stirring for 30 min at 0°C, the progress of reaction was monitored by HPLC using solvent system A. The product peaks appeared at retention times of 8–10 min with an ultraviolet absorption maximum at ~410 nm.
The mixture was frozen and lyophilized, leaving a solid. The crude product was dissolved in a minimal volume of water and purified by ion-exchange column chromatography by using Amberlite CG-50 resin (H+ form, weakly acidic) and eluting with water (flow rate 0.5 ml/min). The red fraction showing a single peak in HPLC was collected and lyophilized to give the desired compounds (yield, 50–60%).
Starting with the intermediates 19, 22, 26, 28, and pyridoxal-α5-phosphate (1) and following the general procedure, we have synthesized compounds 2, 4, 7–16, 20, 21, 23–25, 27, 29, 30, and 32–34 (Table 1).
TABLE 1.
MS and HPLC Characterization of PPADS Analogues Synthesizeda
| FAB (M-H+) |
HPLC (rate, min) |
||||
|---|---|---|---|---|---|
| Compoundb | Formula | Calculated | Found | System A | System B |
| 2 | C14H13O6N3PNa2 | 396.0337 | 396.0345 | 10.15 | 13.64 |
| 4 | C14H13O9N3PSNa | 451.9929 | 451.9935 | 8.84 | 14.01 |
| 8 | C15H15O10N3PSNa | 482.0035 | 482.0015 | 8.80 | 14.33 |
| 9 | C15H13O8N3P | 394.0440 | 394.0407 | 9.14 | 14.06 |
| 10 | C15H13O8N3PNa | 416.0260 | 416.0251 | 7.90 | 13.94 |
| 11 | C15H12ON3ClPNa | 449.9870 | 449.9869 | 8.56 | 14.18 |
| 12 | C14H13O6N3ClP | 384.0152 | 384.0155 | 11.93 | 14.93 |
| 13 | C14H12O9N3ClPSNa | 485.9540 | 485.9560 | 9.40 | 15.21 |
| 14 | C15H12O8N3ClPNa | 428.0050 | 428.0049 | 10.09 | 15.33 |
| 15 | C15H12O8N3ClPNa | 450.9948 | 450.9965 | 7.94 | 14.07 |
| 16 | C14H12O8N4ClP | 429.0003 | 428.9997 | 12.07 | 15.11 |
| 19 | C8H10O5NP | 230.0218 | 230.0217 | 7.63 | 8.70 |
| 20 | Cl4Hl1Ol1N3PS2Na3 | 559.9187 | 559.9183 | 8.22 | 15.22 |
| 21 | C14H12O8N3ClPSNa | 469.9591 | 469.9583 | 10.80 | 14.64 |
| 22 | C9H12O5NP | 244.0375 | 244.0380 | 7.54 | 9.76 |
| 23 | C15H15O11N3PS2Na3 | 575.9500 | 575.9501 | 8.13 | 15.62 |
| 24 | C15H14O8ClPSNa | 483.9747 | 483.9756 | 10.32 | 15.00 |
| 25 | C16H15O7N3P | 392.0648 | 392.0645 | 8.49 | 12.41 |
| 26 | C9H10O5NP | 242.0218 | 242.0220 | 8.11 | 9.36 |
| 27 | C15H12O11N3PS2Na3 | 571.9187 | 571.9256 | 7.13 | 13.58 |
| 28 | C10H14O5NP | 258.0531 | 258.0544 | 8.72 | 9.39 |
| 29 | C16H17O11N3PS2Na | 543.9862 | 543.9827 | 9.19 | 15.38 |
| 30 | C16H16O8N3ClPSNa | 497.9904 | 497.9890 | 11.20 | 15.18 |
| 31 | C10H12O5NP | 256.0371 | 256.0375 | 9.25 | 10.05 |
| 32 | C18H14O12N3PS2Na2 | 581.9654 | 581.9670 | 8.37 | 15.82 |
| 33 | C18H14O12N3PS2Na2 | 581.9654 | 581.9647 | 8.29 | 15.78 |
| 34 | C18H14O15N3PS2Na2 | 683.9042 | 683.9044 | 7.50 | 16.74 |
MS, mass spectrum; HPLC, high pressure liquid chromatography; PPADS, pyridoxal-6′-phosphate-b-phenylazo-2,4-disulfonate; FAB, fast atom bombardment. System A was 0.1 M triethylammonium acetate buffer:CH3CN = 95:5 to 40:60 for 20 min wtih flow rate 1 ml/min. System B was 5 mM tetrabutylammonium phosphate buffer:CH3CN 80:20 to 40:60, in 20 min with flow rate 1 ml/min.
6-Phenylazo-pyridoxal-α5-phosphate (sodium salt) (2)
1H NMR (D2O) δ 2.45 (3H, s, CH3), 5.67 (2H, d, J = 5.5 Hz, CH2), 7.49 (3H, m, aromatic H), 7.78 (2H, m, aromatic-H), 10.33 (1H, s, CHO).
Pyridoxal-α5-phosphate-6-phenylazo-3′-sulfonate (sodium salt) (4)
1H-NMR (D2O) δ 2.53 (3H, s, CH3), 5.82 (2H, d, J = 5.9 Hz, CH2), 7.72 (1H, m, Ph), 7.96 (1H, d, J = 7.0 Hz, Ph), 8.12 (1H, d, J = 7.0 Hz, Ph), 8.31 (1H, s, Ph), 10.52 (1H, s, CHO).
Pyridoxal-α5-phosphate-6-phenylazo-2′-methoxy-5′-sulfonate (sodium salt) (8)
1H-NMR (D2O) δ 2.48 (3H, s, CH3), 4.09 (3H, s, OCH3), 5.81 (2H, d, J = 4.9 Hz, CH2), 7.37 (1H, d, J = 8.8 Hz, Ph), 7.94 (1H, d, J = 8.8 Hz, Ph), 8.14 (1H, s, Ph), 10.50 (1H, s, CHO).
Pyridoxal-α5-phosphate-6-phenylazo-3′-carboxylic acid (sodium salt) (9)
1H-NMR (D2O) δ 2.45 (3H, s, CH3), 5.73 (2H, d, J = 5.9 Hz, CH2), 7.63 (1H, t, J = 6.8 Hz, Ph), 8.03 (2H, m, Ph), 8.24 (1H, s, Ph), 10.45 (1H, s, CHO).
Pyridoxal-α5-phosphate-6-phenylazo-4′-carboxylic acid (sodium salt) (10)
1H-NMR (D2O) δ 2.51 (3H, s, CH3), 5.80 (2H, d, J = 5.9 Hz, CH2), 8.00 (4H, bs, Ph), 10.51 (1H, s, CHO).
Pyridoxal-α5-phosphate-6-phenylazo-3′-chloro-2′-carboxylic acid (sodium salt) (11)
1H-NMR (D2O) δ 2.57 (3H, s, CH3), 5.81 (2H, d, J = 6.8 Hz, CH2), 7.52 (1H, t, J = 7.8 Hz, Ph), 7.65 (1H, d, J = 7.8 Hz, Ph), 7.92 (1H, d, J = 7.8 Hz, Ph), 10.58 (1H, s, CHO).
6-(2′-chloro-phenylazo)-pyridoxal-α5-phosphate (sodium salt) (12)
1H-NMR (D2O) δ 2.51 (3H, s, CH3), 5.74 (2H, d, J = 6.0 Hz, CH2), 7.43 (2H, m, Ph), 7.58 (1H, d, J = 7.8 Hz, Ph), 7.74 (1H, d, J = 7.8 Hz, Ph), 10.45 (1H, s, CHO).
Pyridoxal-α5-phosphate-6-phenylazo-2′-chloro-5′-sulfonate (sodium salt) (13)
1H-NMR (D2O) δ 2.48 (3H, s, CH3), 5.81 (2H, d, J = 5.7 Hz, CH2), 7.75 (1H, d, J = 7.8 Hz, Ph), 7.84 (1H, d, J = 7.8 Hz, Ph), 8.13 (1H, s, Ph), 10.49 (1H, s, CHO).
Pyridoxal-α5-phosphate-6-phenylazo-2′-chloro-5′-carboxylic acid (sodium salt) (14)
1H-NMR (D2O) δ 2.53 (3H, s, CH3), 5.83 (2H, d, J = 6.8 Hz, CH2), 7.70 (1H, d, J = 8.7 Hz, Ph), 7.99 (1H, d, J = 8.7 Hz, Ph), 8.18 (1H, s, Ph), 10.55 (1H, s, CHO).
Pyridoxal-α5-phosphate-6-phenylazo-3′-chloro-4′-carboxylic acid (sodium salt) (15)
1H-NMR (D2O) δ 2.49 (3H, s, CH3), 5.78 (2H, d, J = 5.9 Hz, CH2), 7.57 (1H, d, J = 7.8 Hz, Ph), 7.93 (1H, d, J = 7.8 Hz, Ph), 8.03 (1H, s, Ph), 10.50 (1H, s, CHO).
6-(2′-Chloro-5′-nitro-phenylzao)-pyridoxal-α5-phosphate (sodium salt) (16)
1H-NMR (D2O) δ 2.54 (3H, s, CH3), 5.86 (2H, d, J = 6.0 Hz, CH2), 7.86 (1H, d, J = 7.8 Hz, Ph), 8.31 (1H, d, J = 7.8 Hz, Ph), 8.51 (1H, s, Ph), 10.58 (1H, s, CHO).
Dimethyl (3,α4-O-Isopropylidene-3-hydroxy-4-hydroxymethyl-2-methyl-pyrid-5-yl)methylphosphonate (37)
To a solution of 5.11 g (0.0244 mol) of 35 in 100 ml of benzene, was added 1.92 ml (0.0264 mol) of thionyl chloride. The mixture was refluxed for 10 min with stirring and cooled to room temperature. The white solid was collected through filtration, washed with anhydrous acetone, and dried under vacuum to afford 36 (6.1 g, 95%): mass spectrum (MS) (CI, NH3) 228(M++1), 245 (M+NH4+); 1H-NMR (CDCl3) δ 1.64 (6H, s, 2×CH3), 2.79 (3H, s, CH3), 4.65 (2H, s, CH2), 5.12 (2H, s, CH2), 8.31 (1H, s, H6). The solution of 36 in 50 ml of trimethylphosphite was heated at 80°C for 48 h and evaporated under vacuum. The residue was purified by flash silica gel column chromatography with CHCl3:MeOH = 60:1 to afford 37 as a colorless oil (4.76 g, 61%): MS (CI, NH3) 302(M+ + 1), 319(M+NH4+); high resolution mass spectroscopy (HRMS) (EI) calcd. 301.1079, found 301.1064; 1H-NMR (CDCl3) δ 1.55 (6H, s, 2 × CH3), 2.39 (3H, s, CH3), 3.00 (2H, d, J = 21.5 Hz, CH2), 3.68 (3H, s, CH3), 3.72 (3H, s, CH3), 4.93 (2H, s, CH2), 7.92 (1H, s, H6).
Dimethyl (3-hydroxy-4-hydroxymethyl-2-methylpyrid-5-yl)methylphosphonate (38)
A solution of 4.5 g (0.0149 mol) of 37 in 30 ml of 10% formic acid was refluxed for 2 h. After cooling, the mixture was partitioned between CHCl3 and saturated NaHCO3, and the aqueous layer was extracted three times. The combined CHCl3 layer was dried over anhydrous Na2SO4, evaporated, and purified by flash silica gel column chromatography with CHCl3:MeOH = 20:1 to afford 38 as a white solid (2.62 g, 67%): mp = 139–141°C; MS (CI, NH3) 262(M+ + 1); HRMS (EI) calcd. 261.0766, found 261.0752; 1H-NMR (CDCl3) δ 2.48 (3H, s, CH3), 3.14 (2H, d, J = 20.5 Hz, CH2), 3.65 (3H, s, CH3), 3.69 (3H, s, CH3), 4.86 (2H, s, CH2), 7.85 (1H, s, H6).
Dimethyl (4-formyl-3-hydroxy-2-methyl-pyrid-5-yl)methylphosphonate (39)
To a solution of 1.05 g (4.02 mmol) of 38 in 80 ml of anhydrous CHCl3 was added powdered activated MnO2 with vigorous stirring for 2 h at room temperature under N2 atmosphere. The mixture was filtered through a Celite bed, and the filtrate was evaporated under vacuum to afford 39 as a yellow oil (1.01 g, 97%): HRMS (EI) calcd. 259.0609, found 259.0594; 1H-NMR (CDCl3) δ 2.52 (3H, s, CH3), 3.39, 3.46 (2H, 2s, CH2), 3.70 (3H, s, CH3), 3.74 (3H, s, CH3), 8.00 (1H, s, H6), 10.37 (1H, s, CHO), 11.59 (1H, bs, OH).
4-Formyl-3-hydroxy-2-methyl-pyrid-5-yl)methylphosphonic acid (19)
To a stirred solution of 0.8 g (3.09 mmol) of 39 in 30 ml of anhydrous CH3CN was added dropwise 1.68 ml (12.3 mmol) of trimethylsilyl bromide. The mixture was stirred overnight and evaporated under vacuum. The residue was mixed with 10 ml of water and followed by dropwise addition of 6.5 ml of 0.5 M NH4HCO3 solution with stirring until the mixture became a clear solution. The solution was washed with ether and applied to an Amberlite CG-50 (H+ form) ion exchange column followed by elution with water. The pure fractions determined by HPLC were collected and lyophilized to afford 19 as a yellow solid (0.55 g, 77%): mp = 310°C (dec.); 1H-NMR (D2O) δ 2.46 (3H, s, CH3), 3.36 (2H, d, J = 19.5 Hz, CH2), 7.77 (1H, s, H6), 10.23 (1H, s, CHO); 31P-NMR (D2O) δ 17.12 (t, J = 19.4 Hz, phosphonic acid).
5-Formyl-3,α4-O-isopropylidene-3-hydroxy-4-hydroxymethyl-2-methyl-pyridine (40)
To a solution of 5.0 g (0.0239 mol) of 35 in 100 ml of anhydrous CH2Cl2 was added powdered pyridinium dichromate (PDC) with vigorous stirring. After overnight stirring, the mixture was filtered through a Celite bed. The filtrate was evaporated under vacuum and purified by flash silica gel column chromatography with CHCl3:MeOH = 80:1 to afford 40 as a colorless oil (3.81 g, 77%): MS (CI, NH3) 208(M+ + 1), 225 (M+NH4+); 1H-NMR (CDCl3) δ 1.57 (6H, s, 2 × CH3), 2.51 (3H, s, CH3), 5.18 (2H, s, CH2), 8.48 (1H, s, H6), 10.05 (1H, s, CHO).
Ethyl 2-(3,α4-O-isopropylidene-3-hydroxy-4-hydroxymethyl-2-methyl-pyrid-5-yl)propenoate (41)
To a suspension of 1.6 g (0.04 mol) of 60% sodium hydride (prewashed twice with hexanes) in 200 ml of anhydrous tetrahydrofuran (THF) was added dropwise 8.01 ml (0.04 mol) of triethylphosphonoacetate. The mixture became a clear solution with evolution of H2 gas. After 5 min of stirring, 7.56 g (0.0364 mol) of 40 in 2 ml of anhydrous THF was added and the solution stirred for 30 min. The mixture was partitioned between CH2Cl2 and concentrated aqueous NaCl, and the organic layer dried under anhydrous Na2SO4, and evaporated under vacuum. The product was crystallized from hexanes to afford 41 as a white solid (9.65 g, 96%): mp = 81–83°C. Analysis: calcd. C 64.96, H 6.91, N 5.05; found C 65.06, H 6.94, N 5.01; MS (CI, NH3) 278(M+ + 1), 295(M+NH4+); 1H-NMR (CDCl3) δ 1.34 (3H, t, J = 8.8 Hz, CH3), 1.57 (6H, s, 2 × CH3), 2.44 (3H, s, CH3), 4.28 (2H, q, J = 8.8 Hz, CH2), 4.93 (2H, s, CH2), 6.37 (1H, d, J = 16.6 Hz, CH), 7.54 (1H, d, J = 16.6 Hz, CH), 8.27 (1H, s, H-6).
3-(3,α4-O-Isopropylidene-3-hydroxy-4-hydroxymethyl-2-methyl-pyrid-5-yl)-1-propenol (42a)
To a stirred solution of 9.35 g (0.0337 mol) of 41 in 120 ml of anhydrous THF was added 165 ml (0.165 mol) of 1.0 M diisobutyl aluminum hydride (DIBAL-H) solution in THF at 0°C. The mixture was stirred for 2 h at 0°C, an additional 2 h at room temperature, and quenched by 0.1 ml of MeOH. The mixture was poured into ice-cold 0.1 N HCl, the pH was adjusted to 7–8, and the product was extracted with 100 ml of EtOAc three times. The combined EtOAc layer was dried over anhydrous Na2SO4, evaporated under vacuum, and purified by flash silica gel column chromatography with CHCl3:MeOH = 100:1 to afford 42a as a white solid (5.3 g, 67%): mp = 120–122°C; HRMS (EI) calcd. 235.1214, found 235.1208; 1H-NMR (CDCl3) δ 1.54 (6H, s, 2 × CH3), 2.39 (3H, s, CH3), 4.33 (2H, d, J = 4.9 Hz, CH2), 4.81 (2H, s, CH2), 6.27 (1H, dt, J = 4.9, 15.6 Hz, CH), 6.42 (1H, d, J = 15.6 Hz, CH), 8.07 (1H, s, H6).
3-(3,α4-O-Isopropylidene-3-hydroxy-4-hydroxymethyl-2-methyl-pyrid-5-yl)-1-propenylchloride hydro-chloride salt (42b)
5.0 g (0.0212 mol) of 42a reacted by using the same method as for 36 to afford 42b as a white solid (4.1 g, 67%): mp = 187°C (dec.); HRMS (EI) calcd. 253.0870, found 253.0878; MS (CI, NH3) 254(M+ + 1), 271(M+NH4+); 1H-NMR (CDCl3) δ 1.63 (6H, s, 2 × CH3), 2.78 (3H, s, CH3), 4.26 (2H, d, J = 4.9 Hz, CH2), 4.98 (2H, s, CH2), 6.46–6.58 (2H, m, 2 × CH), 8.247 (1H, s, H6).
Dimethyl 3-(3,α4-O-isopropylidene-3-hydroxy-4-hydroxymethyl-2-methyl-pyrid-5-yl)-1-propenylphosphonate (42c)
0.14 g (0.482 mmol) of 42b reacted by using the same method as for 37 except for the reaction temperature (120°C) to afford 42c as a colorless oil (0.07 g, 45%): HRMS (EI) calcd. 327.1235, found 327.1241; 1H NMR (CDCl3) δ 1.55 (6H, s, 2 × CH3), 2.39 (3H, s, CH3), 2.79 (2H, dd, J = 8.8, 22.5 Hz, CH2), 3.77 (3H, s, CH3), 3.81 (3H, s, CH3), 4.83 (2H, s, CH2), 6.02–6.12 (1H, m, CH), 6.36 (1H, dd, J = 4.9, 15.6 Hz, CH), 8.10 (1H, s, H6).
Dimethyl 3-(3-hydroxy-4-hydroxymethyl-2-methyl-pyrid-5-yl)-1-propenylphosphonate (43)
1.5 g (4.58 mmol) of 42b reacted by using the same method as for 38 to afford 43 as a colorless oil (0.97 g, 74%): HRMS (EI) calcd. 287.0922, found 287.0924; 1H-NMR (CDCl3) δ 2.39 (3H, s, CH3), 2.74 (2H, dd, J = 7.8, 22.5 Hz, CH2), 3.73 (3H, s, CH3), 3.77 (3H, s, CH3), 4.95 (2H, s, CH2), 5.79–5.92 (1H, m, CH), 6.46 (1H, dd, J = 4.9, 15.6 Hz, CH), 7.83 (1H, s, H6).
3-(4-Formyl-3-hydroxy-2-methyl-pyrid-5-yl)-1-propenylphosphonic acid (31)
0.2 g (0.696 mmol) of 43 reacted by using the same method as for 38 to 19 to afford 31 as a yellow solid (0.12 g, two steps in 67%): mp = 118°C (dec.); 1H-NMR (D2O) δ 2.39 (3H, s, CH3), 2.62, 2.68 (2H, 2m, CH2), 6.14–6.27 (1H, m, CH), 6.58, 6.88 (1H, 2m, CH), 7.85 (1H, s, H6), 10.26 (1H, s, CHO); 31P-NMR (D2O) δ 20.15 (dq, J = 20.1, 4.9 Hz, phosphonic acid).
Dimethyl 3-(3-hydroxy-4-hydroxymethyl-2-methyl-pyrid-5-yl) propylphosphonate (44)
To a solution of 0.36 g (1.25 mmol) of 43 in CH2Cl2 was added 0.36 g of Pd/C and stirred under H2 atmosphere (1 atm) at room temperature for 5 h. The mixture was filtered through Celite bed. The filtrate was evaporated under vacuum and purified by flash silica gel column chromatography with CHCl3:MeOH = 20:1 to afford 44 as a colorless oil (0.34 g, 93.8%): HRMS (EI) calcd. 289.1079, found 289.1073; 1H-NMR (CDCl3) δ 1.77–1.87 (4H, m, 2 × CH2), 2.56 (3H, s, CH3), 2.71 (2H, t, J = 6.8, CH2), 3.73 (3H, s, OCH3), 3.76 (3H, s, OCH3), 5.03 (2H, s, CH2), 7.75 (1H, s, H6), 9.07 (2H, bs, 2 × OH).
3-(4-Formyl-3-hydroxy-2-methyl-pyrid-5-yl)-propylphosphonic acid (28)
0.23 g (0.8 mmol) of 44 reacted by using the same method as for 38 to 19 to afford 28 as a yellow solid (0.15 g, two steps in 72%): mp = 128°C (dec.); 1H-NMR (D2O + NaOD) δ 1.28–1.37 (2H, m, CH2), 1.58–1.83 (2H, m, CH2), 2.21, (3H, s, CH3), 2.61–2.83 (2H, m, CH2), 7.23(1H, s, H6), 10.28 (1H, s, CHO); 31P-NMR (D2O + NaOD) δ 22.65–23.01 (m, phosphonic acid).
2-(4-Formyl-3-hydroxy-2-methyl-pyrid-5-yl)ethylphosphonic acid (22)
The title compound was synthesized by the reported method [Hullar, 1969] with minor modification of reagents. 1H-NMR (D2O + NaOD) d 1.47–1.58 (2H, m, CH2), 2.22, (3H, s, CH3), 2.75–2.98 (2H, m, CH2), 7.29(1H, s, H6), 10.30 (1H, s, CHO); 31P-NMR (D2O + NaOD) d 21.41–21.71 (m, phosphonic acid).
2-(4-Formyl-3-hydroxy-2-methyl-pyrid-5-yl)-ethenylphosphonic acid (26)
The title compound was synthesized by the reported method [Hullar, 1969] with minor modification of reagents. 1H-NMR (D2O) δ 2.47 (3H, s, CH3), 6.41 (1H, dd, J = 17.6, 20.5 Hz, CH), 7.61 (1H, dd, J = 17.6, 20.5 Hz, CH), 7.89 (1H, s, H6), 10.39 (1H, s, CHO); 31P-NMR (D2O) δ 10.77–11.08 (m, phosphonic acid).
[(4-Formyl-3-hydroxy-2-methyl-6-(2′,5′-disulfonylphenylazo))-pyrid-5-yl]methylphosphonate (sodium salt) (20)
1H-NMR (D2O) δ 2.42 (3H, s, CH3), 3.98, 4.05 (2H, 2s, CH2), 7.82 (1H, d, J = 7.8 Hz, Ph), 8.04 (1H, d, J = 7.8 Hz, Ph), 8.16 (1H, s, Ph), 10.22 (1H, s, CHO).
[(4-Formyl-3-hydroxy-2-methyl-6-(2′-chloro-5′-sulfonylphenylazo))-pyrid-5-yl]methylphosphonate (sodium salt) (21)
1H-NMR (D2O) δ 2.41, (3H, s, CH3), 3.92, 3.99 (2H, s, CH2), 7.63 (1H, d, J = 8.8 Hz, Ph), 7.73 (1H, d, J = 8.8 Hz, Ph), 8.03 (1H, s, Ph), 10.25 (1H, s, CHO).
2-[(4-Formyl-3-hydroxy-2-methyl-6-(2′,5′-disulfonylphenylazo))-pyrid-5-yl]ethylphosphonate (sodium salt) (23)
1H-NMR (D2O) δ 1.88–1.91 (2H, m, CH2), 2.40, (3H, s, CH3), 3.51–3.59 (2H, m, CH2), 7.76(1H, d, J = 7.8 Hz, Ph), 8.01 (1H, d, J = 7.8 Hz, Ph), 8.20 (1H, s, Ph), 10.25 (1H, s, CHO).
2-[(4-Formyl-3-hydroxy-2-methyl-6-(2′-chloro-5′-sulfonylphenylazo))-pyrid-5-yl]ethylphosphonate (sodium salt) (24)
1H-NMR (D2O) δ 1.86–1.97 (2H, m, CH2), 2.40, (3H, s, CH3), 3.59–3.67 (2H, m, CH2), 7.63 (1H, d, J = 8.7 Hz, Ph), 7.74 (1H, d, J = 8.7 Hz, Ph), 8.00 (1H, s, Ph), 10.41 (1H, s, CHO).
2-[(4-Formyl-3-hydroxy-2-methyl-6-(4′-carboxyphenylazo))-pyrid-5-yl]ethylphosphonate (sodium salt) (25)
1H-NMR (D2O) δ 1.83–1.95 (2H, bm, CH2), 2.42, (3H, s, CH3), 3.45–3.62 (2H, bm, CH2), 7.83 (4H, dd, J = 38.1, 6.8 Hz, Ph), 10.35 (1H, s, CHO).
2-[(4-Formyl-3-hydroxy-2-methyl-6-(2′,5′-disulfonylphenylazo))-pyrid-5-yl]ethenylphosphonate (sodium salt) (27)
The title compound was purified by crystallization from EtOH/H2O, due to the instability during the ion-exchange chromatography. 1H-NMR (D2O) δ 2.35 (3H, s, CH3), 6.05 (1H, dd, J = 17.6, 19.5 Hz, CH), 7.78 (1H, dd, J = 17.6, 19.5 Hz, CH), 7.85 (1H, d, J = 8.7 Hz, Ph), 8.08 (1H, d, J = 8.7 Hz, Ph), 8.31 (1H, s, Ph), 10.08 (1H, s, CHO).
3-[(4-Formyl-3-hydroxy-2-methyl-6-(2′,5′-disulfonylphenylazo))-pyrid-5-yl]propylphosphonate (sodium salt) (29)
1H-NMR (D2O) δ 1.77–1.83 (4H, m, 2 × CH2), 2.41, (3H, s, CH3), 3.32–3.39 (2H, m, CH2), 7.75(1H, d, J = 7.8 Hz, Ph), 7.99 (1H, d, J = 7.8 Hz, Ph), 8.16 (1H, s, Ph), 10.21 (1H, s, CHO).
3-[(4-Formyl-3-hydroxy-2-methyl-6-(2′-chloro-5′-disulfonylphenylazo))-pyrid-5-yl]propylphosphonate (sodium salt) (30)
1H-NMR (D2O) δ 1.63–1.86 (4H, m, 2 × CH2), 2.42, (3H, s, CH3), 3.43–3.47 (2H, m, CH2), 7.62 (1H, d, J = 7.8 Hz, Ph), 7.74 (1H, d, J = 7.8 Hz, Ph), 7.94 (1H, s, Ph), 10.37 (1H, s, CHO).
Pyridoxal-α5-phosphate-6-(2′-naphthylazo-5′,7′-disulfonate) (sodium salt) (32)
1H-NMR (D2O) δ 2.48, (3H, s, CH3), 5.74, (2H, bs, CH2), 8.29 (1H, d, J = 7.8 Hz, Naph), 8.41 (1H, s, Naph), 8.57 (2H, bs, Naph), 8.70 (1H, d J = 9.8 Hz, Naph), 10.39 (1H, s, CHO).
Pyridoxal-α5-phosphate-6-(2′-naphthylazo-6′,8′-disulfonate) (sodium salt) (33)
1H-NMR (D2O) δ 2.36, (3H, s, CH3), 5.71, (2H, d, J = 6.6, CH2), 7.97 (2H, pt, Naph), 8.35 (2H, pt, Naph), 9.05 (1H, bs, Naph), 10.39 (1H, s, CHO).
Pyridoxal-α5-phosphate-6-(2′-naphthylazo-3′,6′,8′-trisulfonate) (sodium salt) (34)
1H-NMR (D2O) d 2.58, (3H, s, CH3), 5.87, (2H, d, J = 6.8 Hz, CH2), 8.53 (1H, s, Naph), 8.70 (1H, s, Naph), 8.79 (2H, s, Naph), 10.57 (1H, s, CHO).
Pharmacology
Binding assay in differentiated HL-60 cell membranes
Human HL-60 cells were obtained from American Type Culture Collection (Rockville, MD) and cultured in PRMI 1640 supplemented with 10% fetal bovine serum. To differentiate HL-60 cells, 1 × 106 cells/ml were treated with phorbol myristate acetate (60 ng/ml) for 72 h. The differentiated cells were washed twice with 10 ml of 10 mM Tris, 5 mM ethylenediaminetetraacetic acid buffer, pH 8.25 at 5°C, and then scraped into 5 ml of the same buffer, The cells were homogenized, the suspension was centrifuged at 35,000 × g for 20 min and the resulting pellet was resuspended in 50 mM Tris buffer, pH 8.25 at 5°C, containing 1 mM ethylenediaminetetraacetic acid. Membranes were frozen at −80°C before assay. The binding of 0.2 nM [35S]ATPγS to HL-60 cell membranes was determined as described [Buell et al., 1996] in a final assay volume of 0.25 ml. Incubations for 3 h at 4°C were terminated by vacuum filtration. Non-specific binding was defined by using 10 μM ATPγS.
Antagonism of contractile responses of the guinea-pig urinary bladder and vas deferens and relaxant responses of the guinea-pig taenia coli
Male white guinea-pigs (250–400 g) were killed by a blow to the head and exsanguination. The urinary bladder, vasa deferentia, and ventricular taenia coli were removed and strips of smooth muscle, approximately 2 × 10 mm, were prepared and suspended vertically in 10 ml organ baths for isometric recording of mechanical activity. An initial load of 1 g was applied to the strips which then were allowed to equilibrate for at least 60 min. Electrical field stimulation (EFS) was applied by means of two platinum-wire rings 2.5 mm in diameter, 10 mm apart, through which the strip was threaded. The modified Krebs solution used in these experiments had the following composition (mM): NaCl 133, KCl 4.7, NaHCO3 16.4, MgSO4 0.6, NaH2PO4 0.8, CaCl2 2.5 and glucose 7.7, gassed with 95% O2/5% CO2 (pH 7.3–7.4) and maintained at 37 ± 1°C. Contractions were recorded with a Linton FSG-01 force-displacement transducer, acquired by Biopac MP100WSW Data Acquisition System and displayed on a PC screen. EFS was provided by a Grass S9 stimulator and applied at a given frequency with a pulse width of 0.5 ms and supramaximal voltage. Adenosine-5′-triphosphate disodium salt (ATP), phentolamine, atropine sulphate and carbamylcholine chloride (carbachol) were obtained from Sigma (St. Louis, MO).
Atropine (0.3 μM) and phentolamine (1 μM) were present in a Krebs solution throughout experiments on urinary bladder and vas deferens. Tissue was stimulated with EFS (0.5–64 Hz) until a maximal contraction was reached and the tone declined by approximately 30%. Intervals of 1 min were maintained between stimulations. All contractile responses were calculated as a percentage of the response evoked by KCl at a concentration of 240 mM, which was added at the end of the experiment.
In experiments on taenia coli, relaxant responses to ATP (1, 10, and 100 μM) and to EFS (0.5–8 Hz) were measured by using carbachol (25 nM) -precontracted tissue. Intervals of 10 min were maintained between two subsequent precontractions by carbachol for washout. Relaxant responses were calculated as a percentage of maximal relaxation possible.
Responses to EFS and ATP were examined in preparations before and after incubation with a given compound at a concentration of 10 and 30 μM for at least 30 min. Time-control preparations were maintained in parallel and established that there was no significant change in response to EFS or ATP throughout experiments.
Each concentration of compound was tested three times on tissues from three different animals (n = 3). Means were compared by the paired Student's t-test. A probability of =0.05 was considered as significant. Data are presented as mean (M) ± s.e.m.
Antagonist activity at recombinant P2X2 receptors
Xenopus oocytes were harvested and prepared as previously described [King et al., 1997]. Defolliculated oocytes were injected cytosolically with rat P2X2 cRNA (30 nL, 1 μg/ml) [Brake et al., 1994] incubated for 24 h at 18°C in Barth's solution and kept for up to 12 days at 4°C until used in electrophysiologic experiments.
ATP-activated membrane currents (Vh = −90 mV) were recorded from cRNA-injected oocytes by using the twin-electrode voltage-clamp technique (Axoclamp 2B amplifier). Voltage recording (1–2 MΩ tip resistance) and current-recording microelectrodes (5 MΩ tip resistance) were filled with 0.6 M K2SO4 and 3.0 M KCl, respectively. Oocytes were held in an electrophysiologic chamber and superfused with Ringer's solution (5 mM/min, at 18°C) containing (mM) NaCl, 110; KCl, 2.5; HEPES, 5; CaCl2, 1.8, adjusted to pH 7.5.
ATP (10 μM, approximately the EC70 value) was superfused over the oocytes for 120 s then washed out for a period of 5 min. For inhibition curves, data were normalized to the current evoked by ATP at pH 7.5. Test substances were added for 5 min before ATP exposure; all compounds were tested for reversibility of their effects. The concentration required to inhibit the ATP response by 50% (IC50) was taken from Hill plots constructed by using the formula log(I/Imax - 1), where I is the current evoked by ATP in the presence of an antagonist. Data are presented as mean ± s.e.m. (n = 4) for data from different batches of oocytes.
Phospholipase C assay
P2Y1 receptor-promoted stimulation of inositol phosphate formation by adenine nucleotide analogs was measured in turkey erythrocyte membranes as previously described [Harden et al., 1988; Boyer et al., 1989]. The K0.5 values were averaged from three to eight independently determined concentration-effect curves for each compound. Briefly, 1 ml of washed turkey erythrocytes was incubated in inositol-free medium (DMEM; Gibco, Gaithersburg, MD) with 0.5 mCi of 2-[3H]myo-inositol (20 Ci/mmol; American Radiolabeled Chemicals, Inc., St. Louis, MO) for 18–24 h in a humidified atmosphere of 95% air 5% CO2 at 37°C. Erythrocyte ghosts were prepared by rapid lysis in hypotonic buffer (5 mM sodium phosphate, pH 7.4, 5 mM MgCl2, 1 mM EGTA) as previously described [Boyer et al., 1989]. Phospholipase C activity was measured in 25 μl of [3H]inositol-labeled ghosts (~175 μg of protein, 200–500,000 cpm/assay) in a medium containing 424 μM CaCl2, 0.91 mM MgSO4, 2 mM EGTA, 115 mM KCl, 5 mM KH2PO4, and 10 mM HEPES, pH 7.0. Assays (200-μl final volume) contained 1 μM GTPγS and the indicated concentrations of nucleotide analogs. Ghosts were incubated at 30°C for 5 min, and total [3H]inositol phosphates were quantitated by anion exchange chromatography as previously described [Harden et al., 1988; Boyer et al., 1989].
Agonist potencies were calculated by using a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). Antagonist IC50 values (mean ± standard error) represent the concentration needed to inhibit by 50% the effect elicited by 10 nM 2-methylthioadenosine-5′-diphosphate. All concentration-effect curves were repeated in at least three separate experiments carried out with different membrane preparations by using duplicate or triplicate assays.
RESULTS
Synthesis
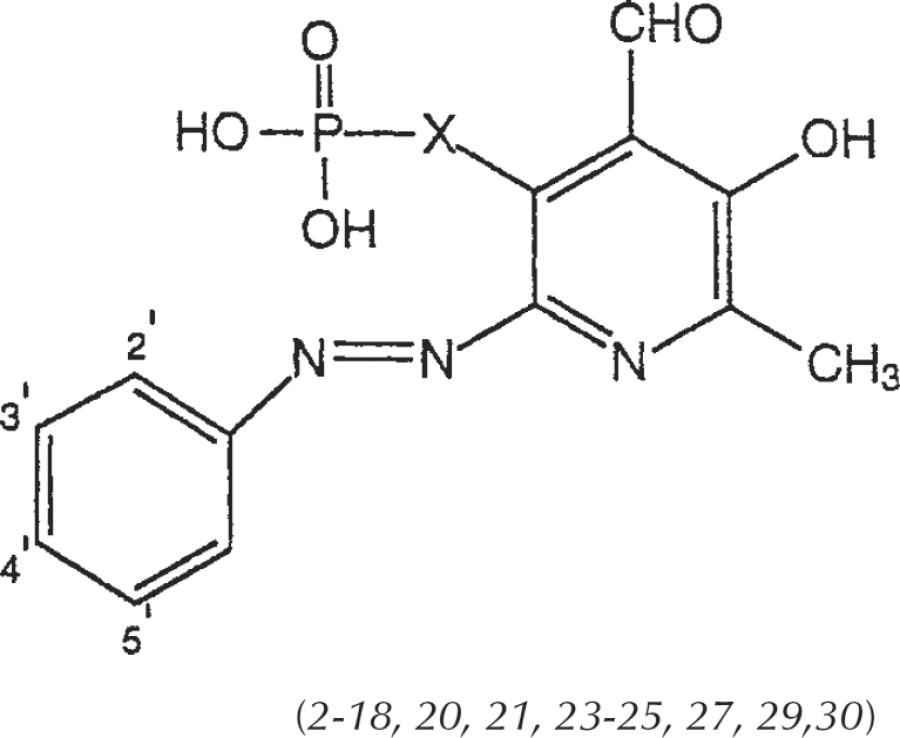
The 6-arylazopyridoxal-5-phosphate analogs that were prepared and characterized (Table 1) could be divided into three categories. Among substituted-phenylazo derivatives and their precursors (Table 2) there are both 5-phosphate (compounds 1–18) and 5-phosphonate (compounds 19–31) analogs, and several naphthylazo pyridoxal-5-phosphate derivatives (compounds 32–34) have been included. The phosphate analogs were modified through substitution of the phenyl ring with sulfo, chloro, nitro, and carboxylate groups. The phosphonate analogs all contain 5′-sulfo and either chloro- or sulfo-groups at the 2′-position. Several compounds (compounds 1–3, 5, 6, 17, and 18) previously reported by Lambrecht [1996] are included in this study for comparison.
TABLE 2.
Structures of Pyridoxal-6-azoaryl-5-phosphate Derivatives Examined as Antagonists at P2 Receptors
| |||||
|---|---|---|---|---|---|
|
| |||||
| Compounda | 2′ | 3′ | 4′ | 5′ | X |
| 2 | H | H | H | H | CH2O |
| 3 | SO3H | H | H | H | CH2O |
| 4 | H | SO3H | H | H | CH2O |
| 5 | H | H | SO3H | H | CH2O |
| 6 (PPADS) | SO3H | H | SO3H | H | CH2O |
| 7 (IsoPPADS) | SO3H | H | H | SO3H | CH2O |
| 8 | OCH3 | H | H | SO3H | CH2O |
| 9 | H | COOH | H | H | CH2O |
| 10 | H | H | COOH | H | CH2O |
| 11 | COOH | Cl | H | H | CH2O |
| 12 | Cl | H | H | H | CH2O |
| 13 | Cl | H | H | SO3H | CH2O |
| 14 | Cl | H | H | COOH | CH2O |
| 15 | H | Cl | COOH | H | CH2O |
| 16 | Cl | H | H | NO2 | CH2O |
| 17 | NO2 | H | H | NO2 | CH2O |
| 18 | H | H | NO2 | H | CH2O |
| 20 | SO3H | H | H | SO3H | CH2 |
| 21 | Cl | H | H | SO3H | CH2 |
| 23 | SO3H | H | H | SO3H | CH2CH2 |
| 24 | Cl | H | H | SO3H | CH2CH2 |
| 25 | H | H | COOH | H | CH2CH2 |
| 27 | SO3H | H | H | SO3H | CH=CH |
| 29 | SO3H | H | H | SO3H | CH2CH2CH2 |
| 30 | Cl | H | H | SO3H | CH2CH2CH2 |
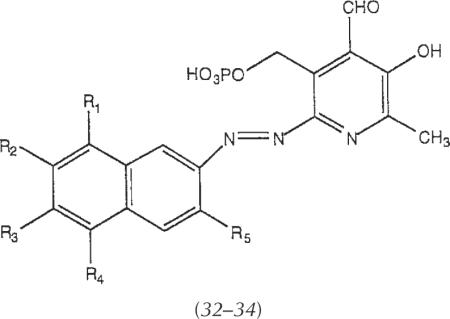
| |||||
|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | |
| 32 | H | SO3H | H | SO3H | H |
| 33 | SO3H | H | SO3H | H | H |
| 34 | H | SO3H | H | SO3H | SO3H |
Compounds 1 (X=CH2O), 19 (X=CH2), 22 (X=CH2CH2), 26 (X=CH=CH), and 28 (X=CH2CH2CH2), and 31 (X=CH=CHCH2) are non–azo-linked precursors (see Schemes 1 and 2). Compounds 1, 2, 3, 5, 6, 17, and 18 were reported by Lambrecht etal., 1996.
The synthetic procedures for the various 3-hydroxy-2-methyl-pyridine-5-alkyl or 5-olefinic phosphonic acid derivatives are shown in Scheme 1. The starting isopropylidene derivative, 35, was synthesized from pyridoxine according to the previous report [Yang et al., 1991]. The transformation of hydroxymethyl group of 35 to the corresponding chloromethyl group of 36 [Tomita et al., 1966], followed by an Arbuzov reaction using trimethylphosphite, afforded the phosphonate compound, 37 with an overall yield of 61%. After the hydrolytic cleavage of isopropylidene group of 37 with 10% formic acid, oxidation of 4-hydroxymethyl group of 38 with activated manganese (IV) oxide provided the 4-formyl compound 39 in high yield. A mild deprotection of the dimethyl ester of the phosphonate was accomplished by an ester exchange reaction by using trimethylsilyl bromide and followed by hydrolysis of the silyl esters. After purification, by using ion exchange chromatography (Amberlite CG-50, H+ form), 19 was isolated in moderate yield.
Scheme 1.

a: SOCl2 in benzene, reflux, 10 min. b: P(OCH3)3, reflux, 48 h. c: 10% formic acid reflux, 2 h. d: Activated MnO2 in CHCl3, 2 h. e: TMS-Br in CH3CN, 24 h. f: PDC in CH2Cl2, 24 h. g: EtOCOCH2P(O)(OEt)2, NaH in THF, 30 min. h: DIBAL-H in THF at 0°C, 2 h. i: 1 atmosphere H2, Pd/C in CH2Cl2, 5 h.
The precursor for chain extension between the pyridine ring and the phosphonic acid group was compound 40, which was obtained by oxidation of 35 by using PDC in CH2Cl2. 5-trans-Vinyl and 5-ethyl phosphonate derivatives, 26 and 22, were synthesized by the reported procedures [Hullar, 1969] with minor modification of reagents. 5-Allyl and 5-propyl phosphonic acid derivatives were prepared through a Wittig reaction of the aldehyde group [Tomita et al., 1966] in 40 leading to the trans-vinyl carboxylic acid ester, 41. Selective reduction of the ester in 41 with DIBAL-H afforded the alcohol 42a, which was converted to phosphonate 31 by using the same procedures as for the preparation of 19 from 35. A saturated propyl phosphonate, 28, was prepared through catalytic reduction of the dimethyl phosphonate intermediate, 43, followed by oxidation and deprotection.
The phosphonic acid derivatives, 19, 22, 26, and 28 were subjected to the azo coupling reaction (Scheme 2) with two different aromatic amine moieties, aniline-2,5-disulfonic acid and aniline-2-chloro-5-sulfonic acid. The azo-linked products obtained were compounds 20, 21, 23–25, 27, 29, and 30 (Table 2). The chemical characterization and HPLC retention times in two solvent systems of these compounds are reported in Table 1.
Scheme 2.
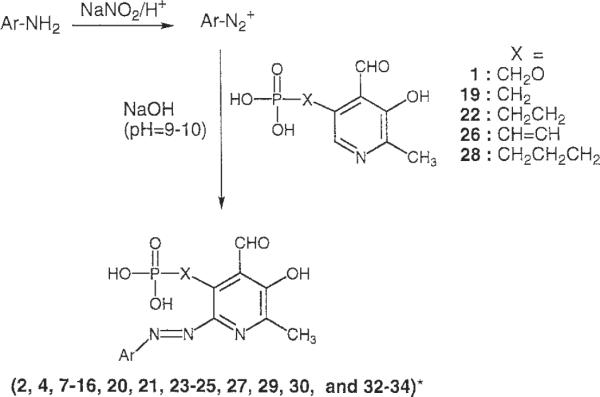
Ar, substituted phenyl or naphthyl. The precise structures are presented in Table 2.
Radioligand Binding Studies at Human P2X1 Receptors
Assays at P2X receptors consisted of both radioligand binding and biological assays. The binding assay involved inhibition of specific binding of [35S]ATP-γ-S to membranes of HL-60 cells. The P2X1 receptor is the principal subtype of P2 receptors expressed in differentiated HL-60 cells, and agonist binding selectivities determined with HL-60 membranes were shown by Buell et al. [1996] to correspond to the affinity of agonists at the human P2X1 receptor. We verified that the order of potency in inhibition of radioligand binding by known receptor agonists and antagonists was approximately as expected from the published potencies at P2X1 receptors. For example, the following IC50 values (all nM) were measured: ATP, 5.51 ± 0.37; ATP-γ-S, 3.88 ± 1.14; 2-Cl-ATP, 6.55 ± 1.60; L-β,γ-me-ATP; 24,800 ± 5400; suramin, 730 ± 171.
The IC50 values for most of the PPADS derivatives in the P2X1 receptor binding assay were greater than 100 μM (Table 3). Even PPADS, 6, displayed only 18% inhibition at 100 μM, which was somewhat weaker than expected. IsoPPADS, 7, was somewhat more potent than PPADS in inhibition of binding. Several derivatives displayed relatively favorable affinity in the binding assay. For example, a phenylazo phosphonate derivative, 27, and a naphthylazo phosphate derivative, 32, exhibited IC50 values of 10 and 15 μM, respectively. Both of these compounds contained two sulfonate groups on the aryl rings. Compound 2, an unsubstituted phenylazo phosphate derivative, was of intermediate affinity with an IC50 value of 45 μM.
TABLE 3.
Pharmacologic Activities of Pyridoxal-6-azoaryl-5-phosphate and Phosphonate Derivatives at P2 Receptors (Structures in Table 2 and Schemes 1 and 2)
| P2X1 |
P2X2 |
P2Y1 |
|||||
|---|---|---|---|---|---|---|---|
| Compound | Human HL-60 cells Inhibition of binding % at 100 μMa | Guinea pig urinary bladder Inhibition of contraction % of 30 μMb | Guinea pig vas deferens Inhibition of contraction % of 30 μMb | Recombinant rat, in oocytes Inhibition of ion flux IC50 (μM)c | Guinea Pig taenia coli Inhibition of relaxation by 2 Hz stim., % at 30 μMd | Guinea pig taenia coli Inhibition of relaxation by KB (μM), or % at 30 μMd | Turkey erythrocyte Inhibition of PLC IC50 (μM)e |
| 2 | 55 ± 1 | 31 | 1.2 ± 0.2 | 6.3 | 22.5 | ||
| 4 | 25 ± 1 | 43 ± 20 | 91 ± 2 | 63 ± 11 | n.a. | 20.2 ± 0.5 | |
| 6 | 18 ± 1 | 1.6 ± 0.1 | 16.6 ± 2.5 | ||||
| 7 | 31 ± 1 | 0.398 ± 0.129 | 1.1–3.7h | 21.4 ± 9.0 | |||
| 8 | 14 ± 6 | 57 ± 10 | 95 ± 1 | 58 ± 19 | 38 ± 19% | 11.8 ± 3.9 | |
| 9 | 10 ± 7 | 36 ± 8 | 74 ± 13 | 2.6 ± 0.0 | 70 ± 3 | 43 ± 27% | 25.1 ± 6.3 |
| 10 | 25 ± 6 | 61 ± 19 | 87 ± 2 | 11.9 ± 1.4 | n.a. | n.a. | >100 |
| 11 | 16 ± 5 | 30 ± 11 | 56 ± 8 | 41 ± 17 | 49 ± 12% | 32.5 ± 7.5 | |
| 12 | 52 ± 1 | n.a.f | 86 ± 6 | 46 ± 8 | n.a. | 6.94 ± 1.75 | |
| 13 | 53 ± 5 | 71 ± 7 | 91 ± 3 | 35 ± 6 | 44 ± 19% | 12.7 ± 5.6 | |
| 14 | 42 ± 2 | 51 ± 7 | 76 ± 4 | 63 ± 3 | n.a. | 8.23 ± 1.30 | |
| 15 | 11 ± 2 | 59 ± 14 | n.a.g | 0.819 ± 0.279 | n.a. | n.a. | >100 |
| 16 | 64 ± 5 | n.a.f | 85 ± 5 | 47 ± 9 | n.a. | 13.8 ± 2.2 | |
| 19 | 22 ± 1 | 34 ± 6 | 60 ± 12 | 61 ± 12 | 64 ± 8% | >100 | |
| 20 | 43 ± 1 | 67 ± 3 | 85 ± 6 | 1.1 ± 0.0 | n.a. | n.a. | >100 |
| 21 | 28 ± 1 | n.a. | 88 ± 5 | n.a. | n.a. | 7.23 ± 2.10 | |
| 22 | 7 ± 2 | 22 ± 15 | 78 ± 11 | 33.0 ± 11.0 | 63 | n.a. | |
| 23 | 25 ± 3 | 40 ± 16 | 84 ± 10 | 1.5 ± 0.1 | 12.5 | n.a. | |
| 24 | 17 ± 0 | 13.1 ± 1.2 | |||||
| 25 | 53 ± 18 | 97 ± 1 | 2.4 ± 0.3 | 30 ± 10 | n.a. | >100 | |
| 26 | 15 ± 1 | >100 | |||||
| 27 | 81 ± 1 | >100 | |||||
| 28 | 28 ± 1 | n.a.f | 46 ± 15 | n.a. | n.a. | n.a. | |
| 29 | 22 ± 3 | n.a. | 83 ± 5 | 10.7 ± 1.0 | 54 ± 11 | 56 ± 8% | >100 |
| 30 | 18 ± 1 | n.a. | 89 ± 3 | n.a. | Enhancementi | 20.2 ± 3.4 | |
| 31 | 11 ± 2 | n.a. | n.a. | n.a. | n.a. | >100 | |
| 32 | 71 ± 2 | 41 ± 20 | 86 ± 1 | 31 ± 20 | n.a. | 15.6 ± 3.4 | |
| 33 | 30 ± 10 | 34 ± 9 | 75 ± 13 | 25 ± 20 | Enhancementi | 14.7 ± 3.2 | |
| 34 | 26 ± 1 | 55 ± 16 | 80 ± 3 | 36 ± 13 | 75 ± 4% | 9.31 ± 3.64 | |
Inhibition of specific binding of [35S]ATPγS (mean ± s.e.m., n = 3). The IC50 values determined for compounds 2, 7, 27, and 32 were 45, 90,10, and 15 μM, respectively.
Inhibition of contractile response (mean ± s.e.m., n = 3) in isolated smooth muscle to electrical field stimulations 8 Hz, unless noted). For compounds reported by Lambrecht et al. [1996], IC50 values (μM) of twitch contractions at P2X receptors in rabbit isolated vas deferens with 0.5 Hz electrical field stimulation were 1 (34.6), 2 (3.1), 3 (2.6), 5 (3.1), 6 (1.4), 17 (3.97), and 18 (5.14). n.a., inactive.
Inhibition of ion current (mean ± s.e.m., n = 3) induced by ATP (10 μM) at recombinant P2X2 receptors expressed in Xenopus oocytes.
P2Y receptors in carbachol-precontracted guinea-pig taenia coli, inhibition of relaxation (mean ± s.e.m., n = 3) induced by electrical field stimulations (2 Hz) or by application of ATP (1 μM), unless noted, n.a., inactive.
Inhibition of 10 nM 2-MeSADP-stimulated phospholipase C in turkey erythrocyte membranes (mean ± s.e.m. for n ≥ 3), labeled using [3H|inositol. n.a., inactive.
~40% inhibition at ≥16 Hz.
~90% inhibition at ≤ 16 Hz.
Apparent Kd value (μM) in guinea-pig taenia coli against ADPβS [Bültmann et al., 1996].
Enhanced relaxation observed upon applicaiton of compounds 30 (93.0 ± 4.2% vs. 44.3 ± 11.0% for 10 μM ATP control) and 33 (52.7 ± 9.2% vs. 32.7 ± 13.3% for 1 μM ATP control).
Smooth Muscle Assays at Guinea-pig P2X1 Receptors
Functional assays consisted of classic smooth muscle systems, specifically antagonism of electric field-stimulated contraction (P2X system) of vas deferens and guinea-pig urinary bladder. Most of the phosphate and a few phosphonate derivatives displayed antagonism to some degree in the smooth muscle assays of guinea-pig P2X1 receptors in the urinary bladder [Ziganshin et al., 1993] and vas deferens [McLaren et al., 1994]. The three most potent analogs in the human P2X1 receptor binding assay, i.e., 2, 27, and 32, showed considerable potency of varying degrees in these smooth muscle assays. The compounds that consistently showed the greatest degree of antagonism in both bladder and vas deferens were: 8, 10, 13, 20, and 34. Curiously, compounds 12, 16, 21, and 28–30 antagonized electric field-induced contraction in the vas deferens but not in the bladder. Compound 10, a phosphate derivative that contained a carboxyl group at the 4-position of the phenylazo ring, displayed a particularly favorable degree of antagonism in both bladder (Fig. 2A) and vas deferens (Fig. 2B). The m-chloro-p-carboxyphenylazo derivative 15 inhibited contraction in the bladder, whereas in the vas deferens inhibition occurred at = 16 Hz but not at 8 Hz stimulation.
Fig. 2.

P2X1 receptor-mediated contractile responses of control (white bars) and compound 10 (10 μM, slash bars; 30 μM, black bars) calculated as percentage of the response evoked by KCl at a concentration of 240 mM, which was added at the end of experiments. (A) Guinea-pig urinary bladder and (B) guinea-pig vas deferens were stimulated with electrical field stimulation (0.5–64 Hz) in a Krebs solution containing atropine (0.3 μM) and phentolamine (1 μM) until a maximal contraction was reached and the tone declined by approximately a third. Intervals of 1 min were always kept between two subsequent stimulations. *, P < 0.05 from control; ^, P < 0.05 from 10 μM (n = 3).
Smooth Muscle Assay at Guinea-pig P2Y1 Receptors and Biochemical Assay at Turkey Erythrocyte P2Y1 Receptors
Antagonism at P2Y1 receptors was established by using a smooth muscle system, i.e. guinea-pig taenia coli, in which antagonism of either agonist-induced or electric field–induced relaxation was measured [Windscheif et al., 1995a]. The phenylazo derivative phosphate derivative 2 and the ethylphosphonate analog of isoPPADS 23 had measurable KB values in antagonizing ATP-induced relaxation of 6.3 and 12.5 μM, respectively. Other compounds that consistently showed the greatest degree of antagonism in the guinea-pig taenia coli were 8, 9, 11, 19, 29, and 34. The ethyl phosphonate derivative 25 was essentially inactive at guinea-pig P2Y1 receptors and was also inactive as an antagonist at P2Y receptors of C6 glioma cells (data not shown) [Boyer et al., 1994]. The compounds were further evaluated at the phospholipase C-coupled P2Y1 receptor of turkey erythrocytes. At both guinea-pig and turkey erythrocyte P2Y1 receptors, most of the phosphate derivatives, except for compounds 10 and 15, showed activity similar to PPADS, 6. Compounds 10 and 15 were either inactive or very weak at both taenia coli (Fig. 3) and turkey erythrocyte P2Y1 receptors. Potency of antagonism at turkey erythrocyte P2Y1 receptors among compounds with IC50 values of <10 μM decreased in the order 12, 21, 14, 34>8, 13, 24, 16, 33, 32, 6. Phosphonate derivatives, except for compounds 21 and 24, were inactive or very weak at turkey erythrocyte P2Y1 receptors. Compound 21, although relatively potent at turkey erythrocyte P2Y1 receptors (IC50 value of 7.23 μM), was inactive in the guinea-pig taenia coli. Among the phosphonate congeners, the homologous 2-chloro-5-sulfonyl-phenylazo phosphonate derivatives 21, 24, and 30 displayed a trend of decreasing potency at turkey erythrocyte P2Y1 receptors, depending on the chain length between the pyridine ring and phosphorous atom (Table 3). Nevertheless, a measurable potency (IC50 value of 20.2 μM) was observed even for the propyl phosphonate 30.
Fig. 3.
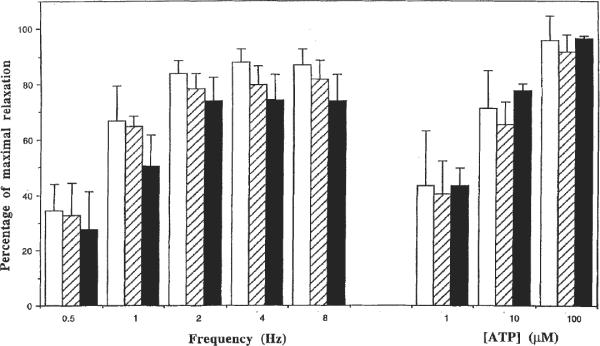
P2Y1 receptor-mediated relaxant responses of control (white bars) and compound 10 (10 μM, slash bars; 30 μM, black bars) calculated as a percentage of maximal relaxation possible: guinea-pig taenia coli relaxant responses to ATP (1, 10, and 100 μM) and to electrical field stimulation (0.5–8 Hz) were tested on carbachol-precontracted tissue (25 nM). Intervals of 10 min were kept between two subsequent precontractions by carbachol for washout.
Curiously, the propyl phosphonate derivative 30 and the naphthylazo derivative 33 seemed to enhance the relaxation of the taenia coli in response to ATP but not to electrical stimulation. This phenomenon may conceivably occur through potential inhibition of enzymes which degrade ATP, such as nucleotidases, by the analogs. Numerous derivatives that act as ATP antagonists are also known to inhibit ecto-nucleotidases [Jacobson et al., 1997].
Although pyridoxal-5-phosphate 1 itself has P2-antagonist properties [Trezise et al., 1994], the presence of the 6-arylazo moiety (with or without substitution) clearly enhanced potency at P2 receptors. The other non–azo-linked pyridoxal-5-phosphate/phosphonate precursors 19, 22, 26, and 28 were all essentially inactive at turkey erythrocyte P2Y1 receptors.
Recombinant Rat P2X2 Receptor Ion Channel Assay
Compounds were tested in a functional assay of recombinant rat P2X2 receptors expressed in Xenopus oocytes, in which ATP-induced currents were inhibited (Fig. 4) [King et al., 1997]. Of the novel compounds synthesized in our study, compound 15 was the most potent at rat P2X2 receptors, with an IC50 value of 0.82 μM. Thus, 15 was intermediate in potency between isoPPADS, 7, and PPADS, 6, which had IC50 values of 0.40 and 1.6 μM, respectively. Other compounds that displayed roughly the same potency as PPADS at rat P2X2 receptors were the unsubstituted phenylazo derivative 2, the m-carboxyphenylazo derivative 9, the methylphosphonate analog of isoPPADS 20, and the corresponding ethylphosphonate 23, and the p-carboxyphenylazo ethylphosphonate derivative 25. The order of potency at rat P2X2 receptors evident in Figure 4 was 7 > 15 > 20, 2 > 23, 6 > 25, 9. Thus, the particular combination of phosphonate and substituted phenylazo units has indications of displaying relatively high potency at the P2X2 receptor subtype.
Fig. 4.
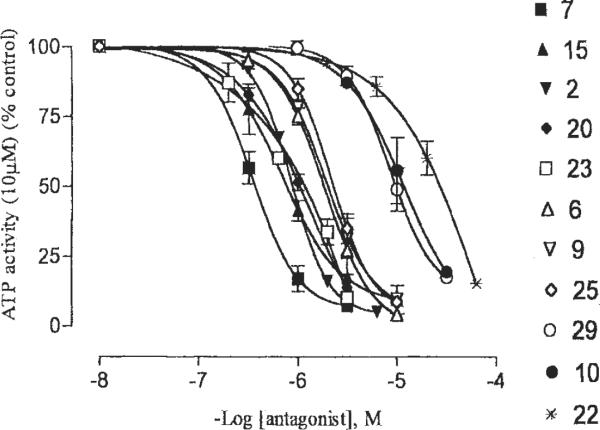
Effects of various analogs as indicated on current induced by activation by 10 μM ATP (approximate EC70 value) of recombinant rat P2X2 receptors, expressed in Xenopus oocytes, by using the twin-electrode voltage-clamping technique. The medium consisted of Ca2+ Ringer's buffer at pH 7.50.
PPADS has been shown to inhibit P2X receptors with slow reversibility [Windscheif et al., 1995a; Humphrey et al., 1995]. The effects of some derivatives (2, 7, 15, 20, 23, and 25) at the rat P2X2 receptor were readily reversible, whereas others (6, 9, 10, 22, and 29) were only slowly reversible (data not shown).
DISCUSSION
In contrast to the initial reports, it seems that PPADS is not selective for P2X receptors. As reconfirmed in this study, PPADS also has considerable affinity for P2Y receptors [Schachter et al., 1996; Boyer et al., 1994; Brown et al., 1995]. In the present study, PPADS and isoPPADS and numerous analogs thereof, were compared in a variety of P2X and P2Y receptor assays. We have shown that at rat P2X2 and turkey erythrocyte P2Y1 receptors, isoPPADS is more potent as an antagonist than PPADS. Recently we have shown that isoPPADS is more potent than PPADS also at recombinant rat P2X1 receptors [Jacobson et al., 1998]. Thus, the 2′,5′-disulfonate substitution of the phenylazo ring is a more favorable lead structure for ligand development than the 2′,4′-isomer.
In the present study, we have greatly expanded the range of PPADS and isoPPADS analogs for the study of SARs and selectivity at various P2 receptor subtypes. Based on mutagenesis results, the 4-aldehyde moiety of PPADS is proposed to aid in binding of the antagonist through the formation of a Schiff base with the ε-amino group of a specific lysine residue on the large extracellular domain of P2X1 and P2X2 receptors [Humphrey et al., 1995]. All of the present analogs have retained the aldehyde group. The potency of PPADS as a P2X antagonist has been reported to be not highly dependent on substituents in the 6-phenylazo ring, except for electron-withdrawing substituents, such as a nitro group, which decreases the potency [Lambrecht et al., 1996]. We have confirmed that the unsubstituted phenylazo derivative 2 acts as a P2 receptor antagonist and found further substitution patterns on the arylazo ring that serve to modulate the potency and selectivity of the derivatives.
A major finding of the present study is that the phosphate group of PPADS may be replaced with a phosphonate resulting in preservation of P2 receptor antagonist properties. The 5-methylphosphate group of PPADS was previously shown to be required for its activity as a P2 receptor antagonist [Trezise et al., 1994], and the present study reveals that a phosphonate substitution is also acceptable for P2 receptor antagonism. The phosphonate group may be linked to the pyridine ring at the 5-position through a hydrocarbon chain consisting of methyl, ethyl, propyl, vinyl or allyl groups. Among the these analogs, the saturated chain-linked phosphonate compound 23 showed significant selectivities for P2X2 receptors vs. P2X1 or P2Y1 receptors, whereas in the case of vinyl phosphonate analogs the potency at P2X1 receptors was restored. These results indicate that altering the electron density or electrostatic potential surrounding the phosphate group in PPADS may be a means of resolving selectivity among the subtypes.
Compounds 10 and 15, both of which contained a carboxyl group at the 4-position of the phenylazo ring, displayed particularly good selectivity for P2X compared with P2Y1 receptors (Figs. 2, 3). Of the novel compounds synthesized in our study, compound 15, a 3′-chloro-4′-carboxylic acid derivative, was the most potent at P2X2 receptors. Compound 15 was essentially inactive at guinea-pig and turkey P2Y1 receptors and at 30 μM only moderately antagonized guinea-pig bladder P2X1 receptors (see below). Thus, compound 15 is of further interest as a selective pharmacologic probe of P2X2 receptors. Compound 10, a 4′-carboxylic acid derivative, and compound 20, a methylphosphonate derivative, were not very selective within P2X subtypes examined but were highly selective when compared with P2Y1 receptors. Also, compounds 23 and 25 were relatively potent at rat P2X2 receptors, whereas 23 was relatively weak at both P2X1 and P2Y1 receptors (Table 3). Considerable differences between the taenia coli and turkey P2Y receptors in antagonist potency were evident (Table 3), suggesting the presence of different subtypes. Similarly among agonists, α,β-me-ATP is active in the taenia coli [Burnstock et al, 1994] but not at recombinant P2Y1 receptors.
The development of potent and selective antagonists of P2 receptors remains a challenge. Among nucleotide antagonists, a P2Y1 selective antagonist N6-methyl-2′-deoxyadenosine-3′,5′-bisphosphate (MRS 2179) having a Ki value of approximately 100 nM was recently reported [Camaioni et al., 1998; Boyer et al., 1998]. Non-nucleotide heterocyclic P2 receptor antagonists (PPADS, Reactive Blue 2, suramin, etc.) have not shown subtype selectivity before the present study [Jacobson et al., 1997]. In the present study, we have identified several novel derivatives of PPADS, that display encouraging selectivity, including the p-carboxyphenylazo phosphate derivative 10 (MRS 2159) and its m-chloro analog 15 (MRS 2160), which were selective for P2X vs. P2Y1 receptors. Among the phosphonate derivatives, [4-formyl-3-hydroxy-2-methyl-6-(2-chloro-5-sulfonylphenylazo)-pyrid-5-yl]methylphosphonic acid, 21 (MRS 2192), showed high potency at P2Y1 receptors with an IC50 of 7.23 μM. The corresponding 2,5-disulfonylphenyl derivative 20 (MRS 2191) was nearly inactive at turkey erythrocyte P2Y1 receptors, whereas at recombinant P2X2 receptors had an IC50 value of 1.1 μM. Thus a single ring substitution, sulfo instead of chloro, has a major effect on the selectivity of these methylphosphonates as P2Y receptor antagonists. An ethyl phosphonate derivative 23 (MRS 2142) was selective for recombinant P2X2 vs. either P2X1 or turkey erythrocyte P2Y1 receptors.
The present study demonstrates that it is possible to achieve selectivity for P2 receptor subtypes by using a relatively nonselective lead structure, PPADS. It will be necessary to study the present set of PPADS analogs at other P2 receptor subtypes and compare potencies within the same species to establish the complete pharmacologic profiles as antagonists. PPADS itself does not potently antagonize P2Y2, P2Y4, or P2Y6 receptors. Also, at P2X receptors, PPADS is much more potent at P2X1–P2X3 and P2X5 receptors than at P2X4, P2X6, and P2X7 receptors. Thus, by optimizing the pharmacologic properties of PPADS analogs, it may be possible to achieve high affinity and selectivity for subtypes within P2X and P2Y receptor families.
ACKNOWLEDGMENTS
We thank Gilead Sciences (Foster City, CA) and the Cystic Fibrosis Foundation (Silver Spring, MD) for financial support to E.C. We thank Dr. Lewis Pannell and Wesley White for determination of HRMS and NMR, and Mary Furr for technical assistance.
This work was supported by USPHS grants GM328213 and HL54889.
Grant sponsor: United States Public Health Service; Grant number: GM38213; Grant number: HL54889.
Abbreviations
- ATP
adenosine 5′-triphosphate
- DIBAL-H
diisobutyl aluminum hydride
- EFS
electrical field stimulation
- HPLC
high pressure liquid chromatography
- HRMS
high resolution mass spectroscopy
- Ki
equilibrium inhibition constant
- me-ATP
adenosine-5′-methylenetriphosphate, (α,β) or (β,γ) isomers
- 2-MeSATP
2-methylthioadenosine-5′-triphosphate
- MRS 2179
N6-methyl-2′-deoxyadenosine 3′,5-bisphosphate
- MS
mass spectrum
- PDC
pyridinium dichromate
- PPADS
pyridoxal-5′-phosphate-6-phenylazo-2,4-disulfonate
- SAR
structure-activity relationship
- THF
tetrahydrofuran
Footnotes
This is a US Government work and, as such, is in the public domain in the United States of America.
REFERENCES
- Abbracchio MP, Burnstock G. Purinoceptors: are there families of P2x and P2y purinoceptors? Pharmacol Ther. 1994;64:445–475. doi: 10.1016/0163-7258(94)00048-4. [DOI] [PubMed] [Google Scholar]
- Boyer JL, Downes CP, Harden TK. Kinetics of activation of phospholipase C by P2Y purinergic receptor agonists and guanine nucleotides. J Biol Chem. 1989;264:884–890. [PubMed] [Google Scholar]
- Boyer JL, Zohn IE, Jacobson KA, Harden TK. Differential-effects of P2-purinoceptor antagonists on phospholipase C-coupled and adenylyl cyclase-coupled P2Y-purinoceptors. Br J Pharmacol. 1994;113:614–620. doi: 10.1111/j.1476-5381.1994.tb17034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JL, Mohanram A, Camaioni E, Jacobson KA, Harden TK. Competitive and selective antagonism of P2Y1 receptors by N6-methyl 2′-deoxyadenosine 3′,5′-biphosphate. Brit J Pharmacol. 1998;124:1–3. doi: 10.1038/sj.bjp.0701837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brake AJ, Wagenbach MJ, Julius D. New structural motif for ligand-gated ion channels defined by an ionotropic ATP receptor. Nature. 1994;371:519–523. doi: 10.1038/371519a0. [DOI] [PubMed] [Google Scholar]
- Brown C, Tanna B, Boarder MR. PPADS: an antagonist at endothelial P2Y-purinoceptors but not P2U- purinoceptors. Br J Pharmacol. 1995;116:2413–2416. doi: 10.1111/j.1476-5381.1995.tb15088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buell G, Michel AD, Lewis C, Collo G, Humphrey PPA, Surprenant A. P2X1 receptor activation in HL-60 cells. Blood. 1996;87:2659–2664. [PubMed] [Google Scholar]
- Bültmann R, Dudeck O, Starke K. Evaluation of P2-purinoceptor antagonists at two relaxation-mediating P2-purinoceptors in guinea-pig taenia coli. Naunyn Schmiedebergs Arch Pharmacol. 1996;353:445–451. doi: 10.1007/BF00261442. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Physiological and pathological roles of purines: an update. Drug Dev Res. 1993;28:195–206. [Google Scholar]
- Burnstock G, Fischer B, Maillard M, Ziganshin A, Ralevic V, Knight G, Brizzolara A, von Isakovics A, Boyer JL, Harden TK, Jacobson KA. Structure activity relationships for derivatives of adeno-sine-5′-triphosphate as agonists at P2 purinoceptors: heterogeneity within P2X- and P2Y- subtypes. Drug Devel Res. 1994;31:206–219. doi: 10.1002/ddr.430310308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. P2 purinoceptors: historical-perspective and classification. Ciba Found Symp. 1996;198:1–34. doi: 10.1002/9780470514900.ch1. [DOI] [PubMed] [Google Scholar]
- Burnstock G, King BF. Numbering of cloned P2 purinoceptors. Drug Dev Res. 1996;38:67–71. [Google Scholar]
- Burnstock G, Wood JN. Purinergic receptors: their role in nociception and primary afferent neurotransmission. Curr Opin Neurobiol. 1996;6:526–532. doi: 10.1016/s0959-4388(96)80060-2. [DOI] [PubMed] [Google Scholar]
- Camaioni E, Boyer JL, Mohanram A, Harden TK, Jacobson KA. Deoxyadenosine bisphosphate derivatives as potent antagonists at P2Y1 receptors. J Med Chem. 1998;41:183–190. doi: 10.1021/jm970433l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Communi D, Boeynaems JM. Receptors responsive to extracellular pyrimidine nucleotides. Trends Pharmacol Sci. 1997;18:83–86. doi: 10.1016/s0165-6147(96)01035-8. [DOI] [PubMed] [Google Scholar]
- Communi D, Govaerts C, Parmentier M, Boeynaems JM. Cloning of a human purinergic P2Y receptor-coupled to phospholipase C and adenylyl cyclase. J Biol Chem. 1997;272:31969–31973. doi: 10.1074/jbc.272.51.31969. [DOI] [PubMed] [Google Scholar]
- Di Virgilio F, Ferrari D, Falzoni S, Chiozzi P, Munerati M, Steinberg TH, Baricordi OR. P2 purinoceptors in the immune-system. Ciba Found Symp. 1996;198:290–305. doi: 10.1002/9780470514900.ch17. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Abbracchio MP, Burnstock G, Dubyak GR, Harden TK, Jacobson KA, Schwabe U, Williams M. Toward a revised nomenclature for P1 and P2 receptors. Trends Pharmacol Sci. 1997;18:79–82. doi: 10.1016/s0165-6147(96)01038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb AJ, Halliday FC. Fast purinergic transmission in the central-nervous-system. Semin Neurosci. 1996;8:225–232. [Google Scholar]
- Greco NJ. Functional expression of a P2T ADP receptor in Xenopus oocytes injected with megakaryocyte (CMK-11-5) RNA. Arterioscler Thromb Vasc Biol. 1997;17:769–777. doi: 10.1161/01.atv.17.4.769. [DOI] [PubMed] [Google Scholar]
- Harden TK, Hawkins PT, Stephens L, Boyer JL, Downes P. Phosphoinositide hydrolysis by guanosine 5′-(gamma-thio)-triphosphate-activated phospholipase C of turkey erythrocyte membranes. Biochem J. 1988;252:583–593. doi: 10.1042/bj2520583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourani SMO, Hall DA. P2T purinoceptors: ADP receptors on platelets. Ciba Found Symp. 1996;198:53–70. doi: 10.1002/9780470514900.ch3. [DOI] [PubMed] [Google Scholar]
- Hullar TL. Pyridoxal phosphate: I. Phosphonic acid analogs of pyridoxal phosphate. Synthesis via Wittig reactions and enzyme evaluation. J Med Chem. 1969;12:58–63. doi: 10.1021/jm00301a016. [DOI] [PubMed] [Google Scholar]
- Humphrey PPA, Buell G, Kennedy I, Khakh BS, Michel AD, Surprenant A, Trezise DJ. New insights on P2X purinoceptors. Naunyn Schmiedebergs Arch Pharmacol. 1995;352:585–596. doi: 10.1007/BF00171316. [DOI] [PubMed] [Google Scholar]
- Inoue K, Koizumi S, Ueno S. Implication of ATP receptors in brain functions. Prog Neurobiol. 1996;50:483–492. doi: 10.1016/s0301-0082(96)00037-8. [DOI] [PubMed] [Google Scholar]
- Inscho EW. Purinoceptor-mediated regulation of the renal microvasculature. J Auton Pharmacol. 1996;16:385–388. doi: 10.1111/j.1474-8673.1996.tb00059.x. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Kim YC, Camaioni E, vanRhee AM. Structure activity relationships of P2 receptor agonists and antagonists. In: Turner JT, Gary Weisman G, Fedan J, editors. The receptors: The P2 nucleotide receptors. Humana Press; Clifton, NJ: 1997. pp. 81–107. [Google Scholar]
- Jacobson KA, Kim YC, Wildman SS, Mohanram A, Harden TK, Boyer JL, King BF, Burnstock G. A pyridoxine cyclic phosphate and its 6-azoaryl derivative selectively potentiate and antagonize activation of P2X1 receptors. J Med Chem. 1998;41:2201–2206. doi: 10.1021/jm980183o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BF, Wildman SS, Ziganshina LE, Pintor J, Burnstock G. Effects of extracellular pH on agonism and antagonism at a recombinant P2X2 receptor. Br J Pharmacol. 1997;121:1445–1453. doi: 10.1038/sj.bjp.0701286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht G, Friebe T, Grimm U, Windscheif U, Bungardt E, Hildebrandt C, Baumert HG, Spatzkumbel G, Mutschler E. PPADS, a novel functionally selective antagonist of P2 purinoceptor-mediated responses. Eur J Pharmacol. 1992;217:217–219. doi: 10.1016/0014-2999(92)90877-7. [DOI] [PubMed] [Google Scholar]
- Lambrecht G, Ardanuy U, Baumert HG, Bo X, Hoyle CHV, Nickel P, Pfaff O, Ralevic V, Windschief U, Zinashin AU, Ziyal R, Mutscheler E, Burnstock G. Persp Recept Res. 1996. Design and pharmacological characterization of selective P2-purinoceptor antagonists; pp. 337–350. [Google Scholar]
- McLaren GJ, Lambrecht G, Mutschler E, Baumert HG, Sneddon P, Kennedy C. Investigation of the actions of PPADS, a novel of P2X-purinoceptor antagonist, in the guinea-pig isolated vas deferens. Br J Pharmacol. 1994;111:913–917. doi: 10.1111/j.1476-5381.1994.tb14825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA, Barnard EA. Nucleotide receptors. Curr Opin Neurobiol. 1997;7:346–357. doi: 10.1016/s0959-4388(97)80062-1. [DOI] [PubMed] [Google Scholar]
- Schachter JB, Li Q, Boyer JL, Nicholas RA, Harden TK. Second messenger cascade specificity and pharmacological selectivity of the human P2Y1-purinoceptor. Br J Pharmacol. 1996;118:167–173. doi: 10.1111/j.1476-5381.1996.tb15381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongen GA, Floras JS, Lenders J, Thien T, Smits P. Cardiovascular pharmacology of purines. Clin Sci. 1997;92:13–24. doi: 10.1042/cs0920013. [DOI] [PubMed] [Google Scholar]
- Thorne PR, Housley GD. Purinergic signaling in sensory systems. Semin Neurosci. 1996;8:233–246. [Google Scholar]
- Tomita I, Brooks HG, Metzler DE. Synthesis of vitamin B6 derivatives. II. 3-hydroxy-4-hydeoxymethyl-2-methyl-5-pyridine acetic acid and related substances (1) J Heterocycl Chem. 1966;3:178–183. [Google Scholar]
- Trezise DJ, Bell NJ, Khakh BS, Michel AD, Humphrey PPA. P2 purinoceptor antagonist properties of pyridoxal-5-phosphate. Eur J Pharmacol. 1994;259:295–300. doi: 10.1016/0014-2999(94)90656-4. [DOI] [PubMed] [Google Scholar]
- Williams M. Challenges in developing P2 purinoceptor-based therapeutics. Ciba Found Symp. 1996;198:309–321. doi: 10.1002/9780470514900.ch18. [DOI] [PubMed] [Google Scholar]
- Williams M, Bhagwat SS. P2 purinoceptors: a family of novel therapeutic targets. Annu Rep Med Chem. 1996;31:21–30. [Google Scholar]
- Windscheif U, Ralevic V, Bäumert HG, Mutschler E, Lambrecht G, Burnstock G. Vasoconstrictor and vasodilator responses to various agonists in the rat perfused mesenteric arterial bed: selective-inhibition by PPADS of contractions mediated via P2X-purinoceptors. Br J Pharmacol. 1994;113:1015–1021. doi: 10.1111/j.1476-5381.1994.tb17094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windscheif U, Pfaff O, Ziganshin AU, Hoyle CHV, Bäumert HG, Mutschler E, Burnstock G, Lambrecht G. Inhibitory-action of PPADS on relaxant responses to adenine- nucleotides or electrical-field stimulation in guinea-pig taenia-coli and rat duodenum. Br J Pharmacol. 1995a;115:1509–1517. doi: 10.1111/j.1476-5381.1995.tb16644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windscheif U, Radziwon P, Breddin HK, Baumert HG, Lambrecht G, Mutschler E. Two different inhibitory effects of pyridoxal-phosphate-6-azophenyl- 2′,4′-disulfonic acid on adenosine diphosphate-induced human platelet aggregation. Arzneim Forsch. 1995b;2:994–997. [PubMed] [Google Scholar]
- Yang D, Shih Y, Liu H. Chemical synthesis of stereospecifically labeled pyridoxamine 5′-phosphate. J Org Chem. 1991;56:2940–2946. [Google Scholar]
- Ziganshin AU, Hoyle C, Bo XN, Lambrecht G, Mutschler E, Bäumert HG, Burnstock G. PPADS selectively antagonizes P2X-purinoceptor-mediated responses in the rabbit urinary-bladder. Br J Pharmacol. 1993;110:1491–1495. doi: 10.1111/j.1476-5381.1993.tb13990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziganshin AU, Hoyle CHV, Lambrecht G, Mutschler E, Baumert HG, Burnstock G. Selective antagonism by PPADS at P2X-purinoceptors in rabbit isolated blood vessels. Br J Pharmacol. 1994;111:923–929. doi: 10.1111/j.1476-5381.1994.tb14827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]