

Abstract

Negative allosteric modulators (NAMs) of metabotropic glutamate receptor subtype 5 (mGluR5) have shown promising results in preclinical models for anxiety and drug abuse. Here, we describe a series of aryl-substituted alkynyl analogues of the prototypic mGluR5 NAM 2-methyl-6-(phenylethynyl)pyridine (MPEP, 1). Displacement of [3H]1 binding in rat brain membranes showed that several of these novel compounds displayed high affinity binding (Ki < 10 nM) for mGluR5, with up to a 24-fold increase in affinity over 1. Replacements of the 2-position Me on the pyridyl ring of 1 along with various 3′-CN, 5′-substitutions were generally well tolerated. All of the active analogues in this series had cLog P values in the 2–5 range and displayed inverse agonist characteristics in an ELISA-based assay of Gqα-mediated IP3 production. Compounds 7i and 7j produced in vivo effects in mouse models of anxiety-like behaviors more potently than 1 or 3-3-((2-methyl-4-thiazolyl)ethynyl)pyridine (MTEP, 2), supporting their utility as in vivo tools.
Keywords: glutamate, negative allosteric modulator, inverse agonist, anxiety, light−dark box
The excitatory neurotransmitter glutamate regulates neuronal firing via ionotropic and eight metabotropic glutamate receptor (mGluR) subtypes. The metabotropic glutamate receptor subtype 5 (mGluR5) is a G protein-coupled receptor (GPCR) highly expressed in mesocorticolimbic regions of the brain, primarily localized on postsynaptic glutamatergic synapses of the cortex, amygdala, hippocampus, and basal ganglia (including nucleus accumbens, striatum, and olfactory tubercle). Upon receptor activation, mGluR5 modulates neuronal firing through Gqα-mediated signaling pathways, including the activation of phospholipase C, enhanced production of d-myo-inositol 1,4,5 trisphosphate (IP3), and increased cytosolic calcium.1 Attenuation of mGluR5 signaling has shown promising results in preclinical models for conditions as diverse as Parkinson's disease, anxiety, fragile X syndrome, gastresophageal reflux disease, and drug abuse.2−4 Mice lacking functional mGluR5 show reduced anxiety-like behavioral responses5 and do not self-administer cocaine.6
The orthosteric glutamate binding site on the mGluR5 protein is located within a large bilobed N-terminal domain, a region highly homologous across the mGluRs. Targeting an allosteric binding site located within the transmembrane region provides an opportunity for greater mGluR subtype selectivity, and highly selective agents have been discovered that can either negatively or positively modulate glutamate's actions at mGluR5. The prototypic mGluR5 negative allosteric modulators (NAMs; Figure 1), 2-methyl-6-(phenylethynyl)pyridine (MPEP, 1, Ki = 16 nM) and 3-((2-methyl-4-thiazolyl)ethynyl)pyridine (MTEP, 2, Ki = 42 nM), have served as critical research tools, but notable limitations on receptor selectivity and metabolic stability have limited clinical development.7
Figure 1.

Chemical structures of selective mGluR5 NAMs.
We have previously evaluated structure–activity relationships (SARs) of mGluR5 NAMs with a wide variety of structural motifs, including amides, diaryl amides,8,9 heterobicyclics,10 and quinolines.11 In this report, we have extended previous work on aryl-substituted alkynyl analogues to optimize binding affinity at mGluR5 and expand the pharmacological toolbox for in vivo studies. A 3′-CN substitution has been previously reported to enhance binding affinity at mGluR5 (e.g., compound 3).11,12 More recently, high affinity at mGluR5 resulting from the 3′-CN, 5′-F substitution on the template of either 1 or 2 has been described.13−15 In the present series of compounds, we retained the template of compound 3 and explored substitutions in the 2- and or 5′-positions as the combination of modifications in these two positions has yet to be explored.
The aryltrimethylsilylacetylenes 5 and 6a–c were prepared through the coupling of trimethylsilylacetylene 4 and the respective aryl bromides according to a literature procedure.12 The diaryl alkyne analogues 7a–q were synthesized through the coupling of the aryl bromides with aryltrimethylsilylacetylenes 5 or 6a–c under Sonagashira coupling conditions with good yields (Scheme 1).
Scheme 1. Synthesis of Compounds 7a–q.

Reagents and conditions: (a) Pd(PPh3)2Cl2, CuI, NEt3, RT, overnight, 92–99%. (b) Pd(PPh3)4, CuI, Et3N, DMF, tetrabutylammonium fluoride (TBAF, 1.0 M in THF), 65–70 °C, 28–98%.
Generally, the coupling reaction was not sensitive to higher temperatures. However, if the substituents on the phenyl ring (b ring) were 3′-CN, 5′-F, allowing the reaction temperature to exceed 70 °C resulted in the hydrolysis of the 3′-CN to give the amide as a side product. If the temperature remained >70 °C and the reaction time was long enough, the amide was the only product isolated as shown in Scheme 2. The same result was obtained in the 3′-pyridyl (b ring) series, with the temperature higher than 65 °C.
Scheme 2. Synthesis of Compounds 7r and 7s.

Reagents and conditions: (a) Pd(PPh3)4, CuI, Et3N, DMF, TBAF, 85 °C, 20–57%.
To evaluate the binding affinities of this series of compounds for mGluR5, we developed an assay utilizing [3H]1 as a competitive radioligand for binding site competition in membranes prepared from rat brains. To assess the functional activity of these novel compounds in vitro, we used a competitive immunoassay that measured the production of IP3, a second messenger of Gqα-mediated signaling. The results of these in vitro tests for prototypical ligands, 1 and 2, and the novel alkynyl analogues are listed in Table 1.
Table 1. In Vitro Data for Alkynyl mGluR5 NAMs.
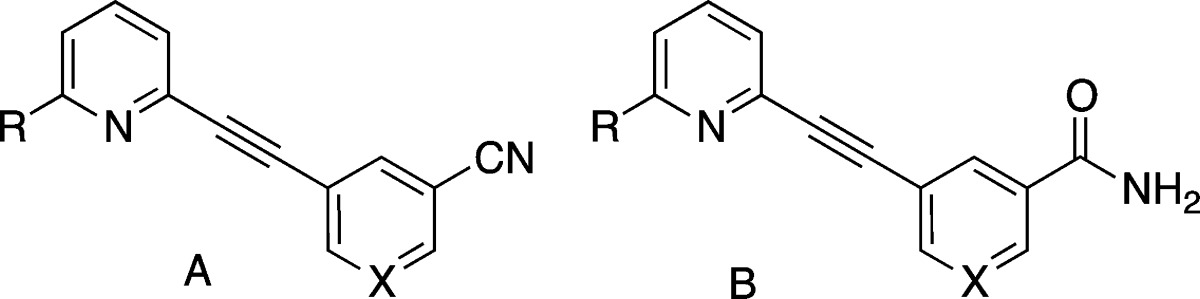
| structure |
nM |
|||||
|---|---|---|---|---|---|---|
| compd ID | template | R | X | cLog Pa | Kib | IC50c |
| 1, MPEP | 3.8 | 16.3 ± 0.5 | 31 ± 8 | |||
| 2, MTEP | 2.1 | 42 ± 1 | 110 ± 50 | |||
| 3d | A | CH3 | CH | 2.9 | 1.04 ± 0.03 | 13 ± 4 |
| 7a | A | CN | CH | 3.7 | 8.2 ± 0.5 | 20 ± 10 |
| 7b | A | HOCH2 | CH | 4.8 | 37 ± 4 | 30 ± 20 |
| 7c | A | CH3O | CH | 2.9 | 200 ± 90 | 300 ± 200 |
| 7d | A | CH3C(O) | CH | 2.5 | 114 ± 3 | 300 ± 100 |
| 7e | A | F | CH | 3.5 | 150 ± 20 | 210 ± 40 |
| 7f | A | Et | CH | 3.5 | 3.4 ± 0.3 | 80 ± 50 |
| 7g | A | n-Bu | CH | 3.0 | 45 ± 3 | 700 ± 400 |
| 7h | A | CH3 | C(OCH3) | 3.4 | 1.29 ± 0.05 | 3.4 ± 0.8 |
| 7ie | A | CH3 | CF | 3.2 | 0.67 ± 0.09 | 2.4 ± 0.3 |
| 7j | A | CN | CF | 2.7 | 2.7 ± 0.2 | 9 ± 4 |
| 7k | A | HOCH2 | CF | 2.3 | 7.5 ± 0.3 | 50 ± 20 |
| 7l | A | CH3O | CF | 3.7 | 40 ± 10 | 400 ± 100 |
| 7m | A | CH3C(O) | CF | 3.1 | 64 ± 2 | 240 ± 90 |
| 7n | A | F | CF | 3.1 | 41 ± 3 | 80 ± 50 |
| 7o | A | CN | N | 1.2 | 52 ± 6 | 500 ± 200 |
| 7p | A | HOCH2 | N | 0.8 | >10000 | |
| 7q | A | CH3O | N | 2.2 | 55 ± 6 | 200 ± 100 |
| 7r | B | CN | CF | 1.9 | >10000 | |
| 7s | B | CN | CH | 1.6 | >10000 | |
Binding studies showed that several of these compounds had affinities in the low nanomolar range for mGluR5: 3, 7a, 7f, 7h, 7i, 7j, and 7k; compound 7i had the highest mGluR5 binding affinity (Ki = 0.67 nM), with an approximately 24-fold improvement over compound 1. Replacement of the 6-Me group with some substituents (e.g., CN, Et, CH2OH) was better tolerated than others, but none led to compounds with higher affinity than the parent pharmacophore. The 5′-F substituents could be replaced with an OMe group (7h; Ki = 1.3 nM) with good results, but modifying the ring to pyridyl—with the nitrogen in this position—significantly reduced or completely abolished activity. Furthermore, when the 5′-CN was reduced to CONH2, inactive compounds resulted. All of the active analogues (Ki < 150 nM) had cLog P values in the 2–5 range.
To evaluate the efficacy of these novel compounds, we utilized an enzyme-linked immunosorbent assay (ELISA)-based immunocompetitive assay in HEK 293A cells stably transfected with rat mGluR519 (Karen O'Malley, Washington University School of Medicine, St. Louis, MO). Agonist stimulation of Gqα GPCRs, such as mGluR5, induces production of the second messenger IP3; the ELISA indirectly measures IP3 production by measuring the accumulation of d-myo-inositol 1 phosphate (IP1), a degradation product of IP3. Activation of mGluR5 by the group I mGluR agonist quisqualic acid (QA) dose dependently increased IP1 levels, as shown in Figure 2. All tested compounds from this series showed inverse agonism, dose dependently reversing IP1 production stimulated by 1 μM QA to levels below vehicle treatment baseline. As shown in Figure 2, compounds 1, 2, 7i, and 7j potently decreased IP1 levels from baseline in the absence of QA. IC50 values, calculated from dose–response curves of the compounds in the absence of QA stimulation, were statistically significantly correlated with Ki values determined via radioligand binding (Pearson r coefficient = 0.4790, two-tailed P value = 0.0326). Compounds 3, 7h, 7i, and 7j were all more potent in this assay than the parent compounds. In further experiments, dose–response curves of each compound were generated in the presence of 1 μM QA, a dose that produces full agonism. In these experiments, the concentration of each compound that induced a 50% reversal of the agonist response could be calculated. The 50% reversal value was highly correlated to the IC50 value of inverse agonism (Pearson r coefficient = 0.6503, two-tailed P value = 0.0019) and very similar to Ki values determined from radioligand binding (Pearson r coefficient = 0.4410, two-tailed P value = 0.0408; best-fit linear regression: 50% reversal (nM) = 0.96 × Ki + 59.8).
Figure 2.
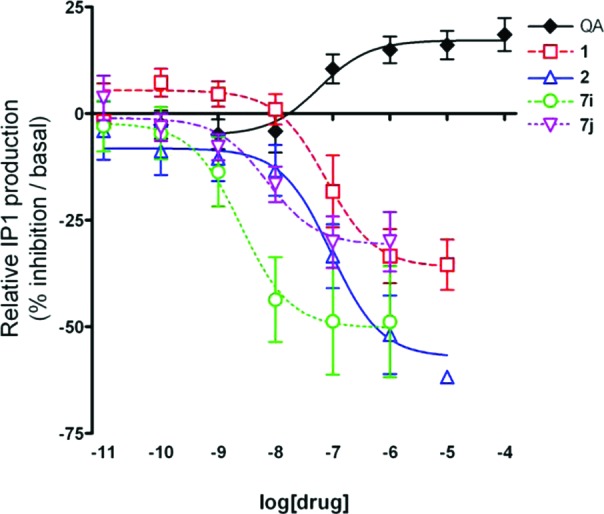
In vitro IP1 accumulation in mGluR5-expressing HEK293 cells is increased by quisqualic acid (QA) and decreased by mGluR5 NAMs. The group I mGluR agonist QA (black ◆) dose dependently enhanced IP3 production, resulting in increased IP1 accumulation. All tested NAMs, including the prototypical mGluR5 NAMs, 1 (red ◻) and 2 (blue △), and the novel NAMs, 7i (green ○) and 7j (purple ▽), showed dose-dependent inverse agonism, reducing IP1 accumulation in the absence of agonist. Values presented are means ± SEMs from at least three experiments.
Compounds 7i (Ki = 0.67 nM) and 7j (Ki = 2.8 nM) were evaluated in mouse models of anxiety-like behaviors and compared to compounds 1 and 2, which exhibit anxiolytic-like activity in a variety of rodent models.20−24 We utilized two behavioral tests, a novel open-field test and the light–dark box test, to evaluate the locomotor effects and the potential anxiolytic-like effects of 7i and 7j; compounds 1 and 2 served as positive controls in these tests. Anxiolysis in the novel open-field test is indicated by increased time spent in the center region of the open field compared to vehicle: the center region is well established to be the most anxiogenic region of the open field.25 Anxiolysis in the light–dark box test is indicated by an increased time spent in the light portion of the test chamber and increased transitions between the light and the dark compartments.26
Overall, all tested compounds produced behavioral effects consistent with anxiolysis. Compounds 1, 2, 7i, and 7j each dose dependently increased locomotor activity (Figure 3A) and the amount of time spent in the center of the novel open-field (Figure 3B). In the light–dark box test, 1, 2, 7i, and 7j each dose dependently increased the proportion of time spent in the light compartment (Figure 3C) and the number of compartment transitions (Figure 3D). Importantly, 7i and 7j were considerably more potent in producing anxiolytic-like effects than 1 or 2. Statistically significant effects consistent with anxiolysis necessitated doses of 3–30 mg/kg for 1 and 10 mg/kg for 2; in comparison, compound 7i was active at 0.3 mg/kg, and 7j was active at 0.1–0.3 mg/kg.
Figure 3.

Anxiolytic-like behavioral effects of mGluR5 NAMs in mice, including the prototypical mGluR5 NAMs, 1 (□) and 2 (Δ), and the novel NAMs, 7i (○) and 7j (▽). For each test, a combined vehicle-only mean (±SEM) is presented as a baseline (⧫), but all statistics were evaluated against the within-group vehicle cohort for each compound. In the novel open field test, mGluR5 NAMs generally increased locomotor activity (A) and also increased the amount of time spent in the center of the open field (B), an indication of anxiolysis. In the light–dark box test, mGluR5 NAMs generally increased proportion of time spent in the light compartment (C) and also increased the number of compartment transitions (D), both indications of anxiolysis. All drugs were suspended in 2% ethanol, 10% Tween 80, and water, except for MTEP, which was dissolved in saline. Drugs were delivered via ip injection 15 min prior to testing. Values presented are means ± SEMs; n = 10–12 for each tested dose. *P < 0.05, **P < 0.01 as compared to vehicle, one-way ANOVA, and Bonferonni's posthoc test.
At the highest tested doses for 1, 7i, and 7j, there appeared to be a loss of anxiolytic effects. For 7j, this is clearly associated with a depression in overall activity at these same doses, but for 1 and 7i, there is no corresponding loss of locomotor activity. The highest tested doses, however, did result in qualitative changes in behavior that suggested sedative effects. Previously, Anderson et al. determined that 1 at 10 mg/kg produced full mGluR5 receptor occupancy in the mouse brain;27 doses above this may produce nonspecific effects. In the absence of pharmacokinetic and metabolic analyses and considering that 7i and 7j have approximately 24- and 6-fold greater binding affinity than 1 at mGluR5, respectively, the behavioral response to doses of 7i and 7j at or above 1 mg/kg may represent nonspecific effects of these compounds.
In summary, although numerous pharmacophores have been mined to optimize mGluR5 binding affinity and in vivo activity, the aryl-substituted alkynyl template of the classic mGluR5 NAMs, compounds 1 and 2, has yielded some of the most promising leads for therapeutic development.15 Herein, we extend SAR in this chemical class and identify two high affinity and potent NAMs (7i and 7j) that demonstrate comparable anxiolytic activity to the parent compounds but at significantly lower doses. We have recently obtained screening data in 63 additional receptors and ion channels (NIDA-ATDP–DPMCDA-Caliper Life Sciences contract) for 7i. At a concentration of 100 nM, there was no significant binding (<13% inhibition) at any of these other targets, highlighting the selectivity of this compound (Supporting Information, Table S1). Further evaluation of 7i in rat models of cocaine self-administration, incubation of cocaine craving, and reinstatement of cocaine-seeking behavior are presently underway.
Acknowledgments
mGluR5-transfected HEK 293A cells were generously provided by Dr. Karen O'Malley of Washington University in St. Louis. We acknowledge the NIDA-DPMCDA ATDP contract N01DA-8-8877 for providing off-target screening data. Thanks to Theresa Kopajtic for help in developing the radioligand binding assay and members of Sergi Ferre's laboratory for help with the ELISA assay. Thanks to Jonathan Katz and Gianluigi Tanda for useful discussions regarding data and figure presentation.
Glossary
Abbreviations
- mGluR
metabotropic glutamate receptor
- GPCR
G protein-coupled receptor
- NAM
negative allosteric modulator
- MPEP
2-methyl-6-(phenylethynyl)pyridine
- MTEP
3-((2-methyl-4-thiazolyl)ethynyl)pyridine
- SAR
structure–activity relationships
- IP3
d-myo-inositol 1,4,5 trisphosphate
- IP1
d-myo-inositol 1 phosphate
- TBAF
tetrabutylammonium formate
Supporting Information Available
Experimental details for the synthesis and purification of the compounds and the in vitro and in vivo pharmacological characterizations of the compounds in this manuscript. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research was funded by the NIDA Intramural Research Program. T.M.K. was supported by an NIH Postdoctoral Intramural Research Training Award (IRTA) Fellowship, and R.P.R. was supported by an NIH Postbaccalaureate IRTA Fellowship.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Niswender C. M.; Conn P. J. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive M. F. Metabotropic glutamate receptor ligands as potential therapeutics for addiction. Curr. Drug Abuse Rev. 2009, 2, 83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. A.; Conn P. J.; Niswender C. M. Glutamate receptors as therapeutic targets for Parkinson’s disease. CNS Neurol. Disord. Drug Targets. 2009, 8, 475–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke G.; Wettstein J. G.; Nordquist R. E.; Spooren W. mGlu5 receptor antagonists and their therapeutic potential. Expert Opin. Ther. Pat. 2008, 18, 123–142. [Google Scholar]
- Brodkin J.; Bradbury M.; Busse C.; Warren N.; Bristow L. J.; Varney M. A. Reduced stress-induced hyperthermia in mGluR5 knockout mice. Eur. J. Neurosci. 2002, 16, 2241–2244. [DOI] [PubMed] [Google Scholar]
- Chiamulera C.; Epping-Jordan M. P.; Zocchi A.; Marcon C.; Cottiny C.; Tacconi S.; Corsi M.; Orzi F.; Conquet F. Reinforcing and locomotor stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nat. Neurosci. 2001, 4, 873–874. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Emmitte K. A. Recent progress in the discovery and development of negative allosteric modulators of mGluR5. Curr. Opin. Drug Discov. Devel. 2009, 12, 446–457. [PubMed] [Google Scholar]
- Kulkarni S. S.; Nightingale B.; Dersch C. M.; Rothman R. B.; Newman A. H. Design and synthesis of noncompetitive metabotropic glutamate receptor subtype 5 antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 3371–3375. [DOI] [PubMed] [Google Scholar]
- Kulkarni S. S.; Newman A. H. Design and synthesis of novel heterobiaryl amides as metabotropic glutamate receptor subtype 5 antagonists. Bioorg. Med. Chem. Lett. 2007, 17, 2074–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni S. S.; Newman A. H. Discovery of heterobicyclic templates for novel metabotropic glutamate receptor subtype 5 antagonists. Bioorg. Med. Chem. Lett. 2007, 17, 2987–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Zou M. F.; Rodriguez A. L.; Conn P. J.; Newman A. H. Structure-activity relationships in a novel series of 7-substituted-aryl quinolines and 5-substituted-aryl benzothiazoles at the metabotropic glutamate receptor subtype 5. Bioorg. Med. Chem. 2010, 18, 3026–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagille D.; Baldwin R. M.; Roth B. L.; Wroblewski J. T.; Grajkowska E.; Tamagnan G. D. Functionalization at position 3 of the phenyl ring of the potent mGluR5 noncompetitive antagonists MPEP. Bioorg. Med. Chem. Lett. 2005, 15, 945–949. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Bates B. S.; Menon U. N.; Jadhav S. B.; Kane A. S.; Jones C. K.; Rodriguez A. L.; Conn P. J.; Olsen C. M.; Winder D. G.; Emmitte K. A. (3-Cyano-5-fluorophenyl)biaryl negative allosteric modulators of mGlu(5): Discovery of a new tool compound with activity in the OSS mouse model of addiction. ACS Chem. Neurosci. 2011, 2, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagille D.; DaCosta H.; Chen Y.; Hemstapat K.; Rodriguez A.; Baldwin R. M.; Conn J. P.; Tamagnan G. D. Potent mGluR5 antagonists: pyridyl and thiazolyl-ethynyl-3,5-disubstituted-phenyl series. Bioorg. Med. Chem. Lett. 2011, 21, 3243–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocher J. P.; Bonnet B.; Bolea C.; Lutjens R.; Le Poul E.; Poli S.; Epping-Jordan M.; Bessis A. S.; Ludwig B.; Mutel V. mGluR5 negative allosteric modulators overview: a medicinal chemistry approach towards a series of novel therapeutic agents. Curr. Top. Med. Chem. 2011, 11, 680–695. [DOI] [PubMed] [Google Scholar]
- Kulkarni S. S.; Zou M. F.; Cao J.; Deschamps J. R.; Rodriguez A. L.; Conn P. J.; Newman A. H. Structure-activity relationships comparing N-(6-methylpyridin-yl)-substituted aryl amides to 2-methyl-6-(substituted-arylethynyl)pyridines or 2-methyl-4-(substituted-arylethynyl)thiazoles as novel metabotropic glutamate receptor subtype 5 antagonists. J. Med. Chem. 2009, 52, 3563–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck T. M.; Rutledge R. P.; Zhang P.; Zou M. F.; Newman A. H.. Identifying Novel mGluR5 Negative Allosteric Modulators As Tools for in Vivo Investigation in Addiction; Program No. 263.13/EE6; Society for Neuroscience: Washington, DC, 2011. [Google Scholar]
- Keck T. M.; Zhang P.; Zou M. F.; Newman A. H.. Novel mGluR5 Negative Allosteric Modulators for in Vivo Investigation; Program No. 272.5/LL5; Society for Neuroscience: San Diego, CA, 2010. [Google Scholar]
- Romano C.; Sesma M. A.; McDonald C. T.; O'Malley K.; Van den Pol A. N.; Olney J. W. Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J. Comp. Neurol. 1995, 355, 455–469. [DOI] [PubMed] [Google Scholar]
- Klodzinska A.; Tatarczynska E.; Chojnacka-Wojcik E.; Nowak G.; Cosford N. D.; Pilc A. Anxiolytic-like effects of MTEP, a potent and selective mGlu5 receptor agonist does not involve GABA(A) signaling. Neuropharmacology 2004, 47, 342–350. [DOI] [PubMed] [Google Scholar]
- Busse C. S.; Brodkin J.; Tattersall D.; Anderson J. J.; Warren N.; Tehrani L.; Bristow L. J.; Varney M. A.; Cosford N. D. The behavioral profile of the potent and selective mGlu5 receptor antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP) in rodent models of anxiety. Neuropsychopharmacology 2004, 29, 1971–1979. [DOI] [PubMed] [Google Scholar]
- Varty G. B.; Grilli M.; Forlani A.; Fredduzzi S.; Grzelak M. E.; Guthrie D. H.; Hodgson R. A.; Lu S. X.; Nicolussi E.; Pond A. J.; Parker E. M.; Hunter J. C.; Higgins G. A.; Reggiani A.; Bertorelli R. The antinociceptive and anxiolytic-like effects of the metabotropic glutamate receptor 5 (mGluR5) antagonists, MPEP and MTEP, and the mGluR1 antagonist, LY456236, in rodents: A comparison of efficacy and side-effect profiles. Psychopharmacology (Berlin, Ger.) 2005, 179, 207–217. [DOI] [PubMed] [Google Scholar]
- Tatarczynska E.; Klodzinska A.; Chojnacka-Wojcik E.; Palucha A.; Gasparini F.; Kuhn R.; Pilc A. Potential anxiolytic- and antidepressant-like effects of MPEP, a potent, selective and systemically active mGlu5 receptor antagonist. Br. J. Pharmacol. 2001, 132, 1423–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spooren W. P.; Vassout A.; Neijt H. C.; Kuhn R.; Gasparini F.; Roux S.; Porsolt R. D.; Gentsch C. Anxiolytic-like effects of the prototypical metabotropic glutamate receptor 5 antagonist 2-methyl-6-(phenylethynyl)pyridine in rodents. J. Pharmacol. Exp. Ther. 2000, 295, 1267–1275. [PubMed] [Google Scholar]
- Simon P.; Dupuis R.; Costentin J. Thigmotaxis as an index of anxiety in mice. Influence of dopaminergic transmissions. Behav. Brain Res. 1994, 61, 59–64. [DOI] [PubMed] [Google Scholar]
- Bourin M.; Hascoet M. The mouse light/dark box test. Eur. J. Pharmacol. 2003, 463, 55–65. [DOI] [PubMed] [Google Scholar]
- Anderson J. J.; Bradbury M. J.; Giracello D. R.; Chapman D. F.; Holtz G.; Roppe J.; King C.; Cosford N. D.; Varney M. A. In vivo receptor occupancy of mGlu5 receptor antagonists using the novel radioligand [3H]3-methoxy-5-(pyridin-2-ylethynyl)pyridine. Eur. J. Pharmacol. 2003, 473, 35–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.