

Abstract
Background
In the honey bee (Apis mellifera), queen and workers have different behavior and reproductive capacity despite possessing the same genome. The primary substance that leads to this differentiation is royal jelly (RJ), which contains a range of proteins, amino acids, vitamins and nucleic acids. MicroRNA (miRNA) has been found to play an important role in regulating the expression of protein-coding genes and cell biology. In this study, we characterized the miRNAs in RJ from two honey bee sister species and determined their possible effect on transcriptome in one species.
Methodology/Principal Findings
We sequenced the miRNAs in RJ either from A. mellifera (RJM) or A. cerana (RJC). We then determined the global transcriptomes of adult A. mellifera developed from larvae fed either with RJM (mRJM) or RJC (mRJC). Finally we analyzed the target genes of those miRNA that are species specific or differentially expressed in the two honey bee species. We show that there were differences in miRNA between RJM and RJC, and that transcriptomes of adult A. mellifera were affected by the two types of RJ. A high proportion (23.3%) of the affected genes were target genes of differential miRNAs.
Conclusion
We show for the first time that there are differences in miRNAs in RJ between A. mellifera and A. cerana. Further, the differences in transcriptomes of bees reared from these two RJs might be related to miRNA differences of the two species. This study provides the first evidence that heterospecific royal jelly can modify gene expression in honey bees through an epigenetic mechanism.
Introduction
The Western honey bee (Apis mellifera) is one of the most important economical insects because of its crucial role in pollination [1]. A honey bee colony is composed of three castes, a fertile queen, hundreds of haploid drones, and thousands of nearly sterile workers [2], [3]. Despite their identical genome, the queen and her workers exhibit vast differences in morphology, behavior, physiology, reproduction and longevity [4]–[6]. The primary substance that leads to this is royal jelly (RJ), which is a yellow milky substance from “nurses” with developed hypopharyngeal and mandibular glands [7]–[9].
RJ contains a range of proteins, carbohydrates, lipids, minerals, vitamins, and a large number of bioactive substances, especially immunological peptides and antibacterial proteins [10]–[13]. Major Royal Jelly Proteins (MRJPs) are the prime RJ ingredient, which are crucial in regulating reproductive maturation [11]. Royalactin is a 57-kDa protein, which can induce larvae developing into queens [14]. Royalactin helps increase body size, promote ovary development and shorten the developmental time. In addition, RJ also contains small amounts of nucleic acids. One study found that RJ contains both DNA and RNA, and there are quantitative differences in nucleic acids in fresh RJ between A. mellifera and A. cerana [13]. The most recent discovery is that RJ contains microRNAs which may play a role in caste differentiation [15].
MicroRNAs (miRNAs) are short (20–22 nucleotides), non-coding, single-stranded RNA molecules that play important roles in post-transcriptional gene regulation and other biological processes in eukaryotes [16]–[18]. These include development, metabolism and regulation of differentiation [19], [20]. miRNAs may specifically bind to partially complementary sites of targeted genes and inhibit mRNA transcription [21], [22]. Animal miRNAs typically bind to targeted mRNAs with sub-optimal complementarity and inhibit or diminish their translation, whereas plant miRNAs bind with high complementarity and mark them for degradation [17]. miRNAs are first characterized in C. elegans, fruit fly (Drosophila), honey bee (A. mellifera), and mosquito [23]–[28]. miRNAs in the brain are found to correlate with age-related behavioral changes in the honey bee [27], [29]. Young workers specialize on feeding larvae (“nurses”) while workers older than 3 weeks old forage for nectar and pollen (“foragers”) [30]. Nurses and foragers have 9 known differentially expressed miRNAs and 67 novel miRNAs [28]. Two recent studies identified 267 novel miRNAs in A. mellifera [31], [32].
While the mechanism by which miRNA modulate gene expression has been well studied [17], [21], [23], it is not clear whether there are differences in RJ originated miRNAs between two honey bee species (A. mellifera and A. cerana), and whether feeding A. mellifera with different RJ might cause differences in transcriptome. A. cerana is considered to the most closely related species to A.mellifera [33]. The two species share common features (such as open nesting) [34] but show differences in behaviors and physiology [35]. It is likely that the two species diverged from a common ancestor around three million years ago [36]. In this study we report the differences in miRNA of RJ either from A. mellifera (RJM) or A. cerana (RJC). We also tested the hypothesis that differentially expressed miRNAs in the RJ of the two species affect A. mellifera transcriptome.
Results
1. Differences in miRNA between Royal Jelly of Two Different Species of Honey Bees
A total of 11,828,863 reads from the RJM library and 10,289,838 reads from the RJC library were obtained after discarding the empty adapters (Table 1). After discarding those sequences that were of low-quality, shorter than 18 nucleotides and single-read sequences, 10,318,386 and 9,493,118 reads for the RJM and RJC remained for analysis respectively. RNAs sequenced by Solexa were in the length of 10–44 nucleotides (nt), and length distributions of small RNAs in the two libraries were significantly different (Contingency Table Analysis, X2>1000, P<0.001, Fig. 1). miRNAs, those with 20–22 nt, in the two types of RJ also showed significantly different length distributions (Contingency Table Analysis, X2>1000, P<0.001).
Table 1. Summary of data cleaning and length distribution of tags produced by small RNA sequencing.
| Total reads | High quality (%) | 3’adapter null (%) | Insert null (%) | 5’adapter contaminants (%) | <18 nt (%) | PolyA (%) | Clean reads (%) | |
| RJM | 11,828,863 | 11,787,392 (100) | 58,528 (0.50) | 2,191 (0.02) | 10,811 (0.09) | 1,397,442 (11.86) | 34 (0) | 10,318,386 (87.54) |
| RJC | 10,289,838 | 10,247,413 (100) | 64,104 (0.63) | 2,469 (0.02) | 5,252 (0.05) | 6,82,430 (6.66) | 40 (0) | 9,493,118 (92.64) |
Figure 1. Length distribution of tags produced by small RNA sequencing in Royal jelly of Apis mellifera (RJM) and Royal jelly of Apis cerana (RJC).

The horizontal axis indicated length nucleic acid (nucleotides, Nt), the ordinate represented distribution frequency (%).
Subsequently, small RNAs were classified into different categories according to their biogenesis and annotations (Table 2). Both RJM and RJC contained several heterogeneous small RNA species which included miRNAs, degraded rRNA fragments and mRNA fragments. In royal jelly, miRNAs were the major fraction of small RNA species. As shown in Table 3, the total reads of miRNAs in RJC (1,872,895 reads) were higher compared to RJM (1,735,052 reads).
Table 2. Different categories of small RNAs in RJM and RJC.
| RJM | RJC | |||
| Unique (%) | Total (%) | Unique (%) | Total (%) | |
| Total sRNAs | 1,176,366 (100.00) | 10,318,386 (100.00) | 1,310,750 (100.00) | 9,493,118 (100.00) |
| miRNA | 503 (0.04) | 8,042 (0.08) | 210 (0.02) | 1,542 (0.02) |
| rRNAetc | 335,464 (28.50) | 3,969,740 (38.45) | 324,232 (24.73) | 3,476,557 (36.62) |
| unann | 841,583 (71.46) | 6,342,898 (61.47) | 986,308 (75.25) | 6,015,019 (63.36) |
Table 3. Summary of common and specific sequences in RJM and RJC.
| Unique sRNAs (%) | Total sRNAs (%) | |
| Total sRNAs | 2,222,699 (100.00) | 19,811,504 (100.00) |
| RJM specific | 1,046,333 (47.07) | 1,735,052 (8.76) |
| RJC specific | 911,949 (41.03) | 1,872,895 (9.45) |
| Shared RJM/RJC | 264,417 (11.90) | 16,203,557 (81.79) |
Solexa sequencing and RNA classification indicated that expression profiles of miRNAs in RJM and RJC are significantly different. By referencing to the mirBase release 13.0 [37], we identified 69 and 48 known miRNAs in RJM and RJC, respectively. There were 23 miRNAs specific to RJM, 2 miRNAs specific to RJC, and 46 shared in both RJ (Table 4). The average expression level of all miRNA in RJM was about 8-fold higher than that of RJC (Table S1). Among these, RJM contained 31 up-expressed, 6 equally expressed, and 2 down-expressed miRNAs compared to RJC (Fig. 2). According to sequence homology, we noticed a high-percentage of miRNA from categories of metabolic process, cell part and catalytic activity (Fig. 3). Cellular process, cell and binding terms were dominant.
Table 4. Summary of known miRNA in RJM and RJC.
| RJM specific (23) |
| ame-mir-1, ame-mir-71, ame-mir-3719, ame-mir-7, ame-mir-1000, ame-mir-210, ame-mir-279, ame-mir-278, ame-mir-iab-4,-ame-mir-3049, ame-ame-mir-263b, ame-mir-3720, ame-mir-316, ame-mir-12, ame-mir-3783, ame-mir-279b, ame-mir-3747b, ame-mir-996, ame-mir-981, ame-mir-282, ame-mir-3732, ame-mir-137, ame-mir-932, ame-mir-305 |
| RJC specific (2) |
| ame-mir-92a, ame-mir-379 |
| Shared RJM/RJC (46) |
| ame-mir-100, ame-mir-3759, ame-mir-184, ame-mir-133, ame-mir-927, ame-mir-275, ame-mir-13b, ame-mir-277, ame-mir-29b, ame-mir-8, ame-mir-92b, ame-mir-283, ame-mir-927b, ame-mir-276, ame-mir-2, ame-mir-3718a, ame-mir-31a, ame-mir-3785, ame-mir-11, ame-mir-190, ame-mir-14, ame-mir-993, ame-mir-315, ame-mir-929, ame-mir-13a, ame-mir-2796, ame-mir-2944, ame-mir-263, ame-mir-375, ame-mir-124, ame-mir-87, ame-mir-34, ame-mir-750, ame-mir-989, ame-mir-3477, ame-mir-281, ame-mir-10, ame-mir-125, ame-mir-9a, ame-let-7, ame-mir-3786, ame-mir-252, ame-bantam, ame-mir-317, ame-mir-306 |
Figure 2. Differential expression analysis of miRNA in RJM and RJC.
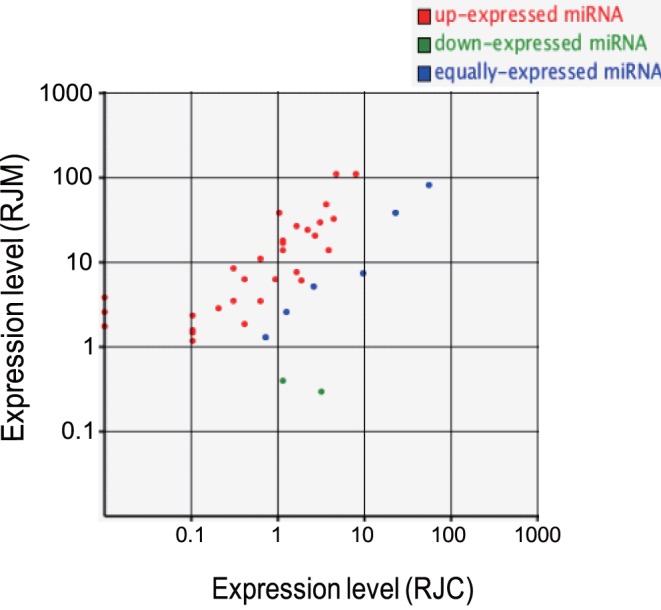
Expression levels were indicated on Y (RJM) or X (RJC). Expression levels were considered different if the threshold of false discovery rate (FDR was 0.001 and the absolute value of log2 Ratio was 1).
Figure 3. Gene Ontology classification of miRNAs in RJM and RJC.

The results are summarized in three main categories: biological process, cellular component and molecular function. The Y-axis indicates the precentage of miRNAs in a category. The X-axis indicates category.
2. Transcriptome Modifications in A. mellifera by Two Different RJs
To test the hypothesis that miRNAs in RJM and RJC affect A. mellifera transcriptome, we determined the global transcriptomes of adult A. mellifera developed from larvae fed either with RJM (mRJM) or RJC (mRJC). The total number of reads for mRJM and mRJC were 48,971,186 and 49,358,642, respectively (Table 5). The distributions of perfectly matched reads to the honey bee genome in mRJM and mRJC were not significantly different (Contingency Table Analysis, X2 = 1.14, P>0.2), however, those uniquely matched to the genome in the two types of bees were significantly different (X2 = 150, P<0.0001). When the mapping was to the honey bee genes (which do not include intron and UTRs), both perfectly matched and uniquely matched reads were significantly differently distributed in mRJM and mRJC (X2>150, P<0.0001 in both cases).
Table 5. Summary of reads in mRJM and mRJC.
| mRJM | mRJC | |||
| reads number | % | reads number | % | |
| Total Reads | 48,971,186 | 100.00 | 49,358,642 | 100.00 |
| Total BasePairs | 4,407,406,740 | 100.00 | 4,442,277,780 | 100.00 |
| Mapped to Genome | ||||
| Total Mapped Reads | 38,461,269 | 78.54 | 38,759,459 | 78.53 |
| Unique Match | 37,970,456 | 77.54 | 38,127,557 | 77.25 |
| Perfect Match | 27,411,616 | 55.97 | 27,638,426 | 56.00 |
| Total Unmapped Reads | 10,509,917 | 21.46 | 10,599,183 | 21.47 |
| Mapped to Gene | ||||
| Total Mapped Reads | 31,019,818 | 63.34 | 31,629,813 | 64.08 |
| Unique Match | 28,160,531 | 57.50 | 28,732,144 | 58.21 |
| Perfect Match | 23,751,463 | 48.50 | 24,053,249 | 48.73 |
| Total Unmapped Reads | 17,951,368 | 36.66 | 17,728,829 | 35.92 |
By use of the Reads Per kb per Million reads (RPKM) method [38], we explored gene expression levels in mRJM and mRJC. This method was adopted to eliminate the influence of variation in gene length and the total reads number. The number of down-regulated genes was more than two times that of up-regulated genes, with 439 down-regulated and 179 up-regulated genes (Fig. 4, Table S2). We systematically examined every differentially expressed gene (DEG) in order to identify genes involved in important pathways. As shown in Table S3, these DEGs were mainly located in endocytosis, focal adhesion, metabolic pathways, regulation of actin cytoskeleton, and RNA transport. Some DEGs were related to pathways on caste differentiation, such as insulin signaling, mTOR, and MAPK. According to sequence homology, we obtained DEGs from categories in metabolic process, cell part and catalytic activity (Fig. 5). Cellular process, cell and binding terms were dominant.
Figure 4. Differential expression analysis of genes in mRJM.
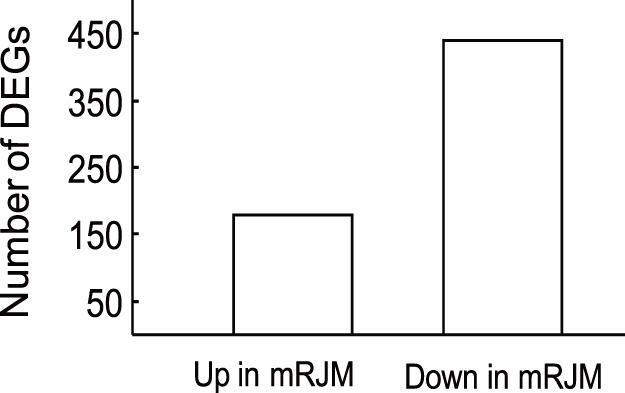
Expression levels were considered different if the threshold of false discovery rate (FDR was 0.001 and the absolute value of log2 Ratio was 1).
Figure 5. Gene Ontology classification of DEGs in mRJM.

The results are summarized in three main categories: biological process, cellular component and molecular function. TheY-axis indicates the precentage of DEGs in a category. The X-axis indicates category.
3. Analysis of miRNA Targeted Genes
We identified the target genes of the following miRNAs: 23 RJM-specific, 2 RJC-specific and 33 differentially expressed miRNA (Table S4). Among the 618 differentially expressed genes between mRJM and mRJC, 144 (23.3%) genes were identified as target genes of miRNAs (Table S4).
Discussion
1. miRNA Differences in RJ of A. mellifera and A. cerana
miRNA in honey bees have been shown to correlate with behavioral plasticity [27]–[29]. In our paper, we were more concerned with miRNAs in royal jelly that may play roles in affecting transcriptome. High-throughput sequencing of small RNAs indicated that there were many small RNAs in the two types of royal jelly (Fig. 1). Small RNAs are of 18–35 nt, which includes three major types: miRNA (20–22 nt), siRNA (24–26 nt), and piRNA (32–34 nt). RJC has a higher percentage of piRNA, especially those with 33 nt (Fig. 1).
We detected 23 unique miRNA in RJM and 2 in RJC. In addition, there were 33 miRNAs differentially expressed in the two types of RJ (Fig. 2). In the up-regulated miRNAs (Table S1), ame-bantam, ame-mir-184, ame-mir-14, ame-mir-252 were the most abundant in RJM. The four miRNAs (ame-let-7, ame-mir-34, ame-mir-100, ame-mir-375) commonly found in other animal bodies or products (such as milk [39] or humans [40] and mouse [41]) were also present in RJ. Only two miRNAs (ame-mir-10, ame-mir-2944) showed higher expression in RJC. Consistent with Guo [15], we also found ame-mir-263, ame-mir-277, and ame-mir-283 in the two types of RJ. However, RJM in our study contained ame-mir-263b, which was absent in their study. This might be due to the fact that our RJ was obtained from 3 day old larvae (largely 4th instar larvae [42]) while they obtained RJ from 4–6th instar larvae.
2. Transcriptome Modification in A. mellifera due to Two Different RJs
Though A. mellifera and A. cerana might diverge from a common ancestor, they show differences in morphology, physiology and disease resistance. After fed with heterospecific royal jelly, A. mellifera showed many DGEs. Kucharski et al. [43] proposed that important elements of glutamatergic synapses are G-protein coupled metabotropic glutamate receptors (GPC mGluRs), which contribute to synaptic plasticity and development. According to their sequence similarity, transduction mechanism and pharmacological profile, mGluRs are divided into three groups: group I (mGluR1 and mGluR5 receptors), group II (AmGluRA), and group III. The mGluR1 receptor links to phospholipase C, which causes phosphoinositide hydrolysis and release of calcium from intracellular stores. In mRJC, the expression level of mGluR1 (GB406151) was lower than that in mRJM, which might affect synaptic plasticity and development in the queen. Myosins [44] are one of three superfamilies of transporting motor proteins, which is involved in organelle formation, vesicle transportation, and cytoskeleton organization. In mRJC, Mhc1 (GB409843, a member of myosins) also decreased. The expression levels of InR-2 (GB725827), NLG-1 (GB724358), and trpgamma (GB410823) also decreased in mRJC. InR-2 is a member of insulin and insulin-like growth factor [45], which is linked to reproductive division of labor and foraging behavior [28]. NLG-1 and trpgamma are closely related to sensory input arising from environmental stimuli [46]–[48]. Four DEGs (GB552209, GB550937, GB410013, and GB409278) were involved in melanogenesis. They showed higher expression in mRJC and could explain prior studies showing darker coloration in mRJC [49], as was also the case in this study. A. cerana has been shown to have a higher sensitivity to odor than A. mellifera [50]. Seven genes (GB552209, GB406100, GB724316, GB551935, GB410013, and GB725569) involved in olfactory transduction were up-regulated in mRJC.
3. Analysis of miRNA Targeted Genes
miRNAs may specifically bind to partially complementary sites of targeted genes and inhibit the mRNA transcription. After feeding with RJM, 179 genes were up-regulated and 439 were down-regulated in Apis mellifera. Out of these 618 DEGs, 144 genes (67 up-regulated and 77 down-regulated) were putative targets of miRNA (Table S4), resulting a rather high 23%. The targeting of miRNAs on DEGs was not specific, with some miRNAs targeting more than 1,000 genes. Some DEGs only affected metabolic pathways (GB726969, GB726367, GB551389, GB100576257, GB724654, GB100577109, GB410748, GB551858, GB724644, GB726961 and GB551379), others were only involved with immune response (GB409078, GB100577433, GB412109, and GB725958), yet others had only limited participation in protein construction (GB100576328, GB410202, GB725868, and GB727133). Still, most DEGs participated in many pathways at the same time, such as GB552530, GB100577495, GB550937, and GB412869. Additionally, we found that some DEGs were involved in caste differentiation related pathways: insulin signaling (GB411959, GB100577495, GB412869, GB725827, GB552209, GB725200, GB724295, GB406096, GB410013, GB677665, and GB413596), Wnt signaling (GB552530, GB552021, GB100576682, GB550937, GB725891, GB724863, GB409278, GB726113, and GB725376), MAPK signaling (GB100577723, GB72624 7, GB725775, GB727172, GB725891, GB725987, GB725025, GB724732, GB410013, and GB100578991), and mTOR signaling pathway (GB412104 and GB100576439).
General Conclusions
We show for the first time that miRNAs in royal jelly are different between A. mellifera and A. cerana. Further, transcriptomes are modified as a result of bees being fed royal jelly of different species. Because a high proportion of the differentially expressed genes were target genes from miRNA, we speculate that the transcriptome modifications are partly caused by miRNA differences of the two species. This study provides the first evidence that miRNA in heterospecific royal jelly can modify gene expression in honey bees. Our results suggested that royal jelly from A. mellifera and A. cerana have different epigenetic effect on gene expression, although these two species are evolutionarily closely related.
Materials and Methods
Honey bee colonies (Apis mellifera and Apis cerana) were raised at the Honeybee Research Institute, Jiangxi Agricultural University, Nanchang, China (28.46°N, 115.49°E) by standard beekeeping techniques.
1. Differences in miRNA between Royal Jelly of Two Different Species of Honey Bees
Harvest of RJM and RJC
RJM and RJC were produced according to standard practices in China [51]. Briefly, the queen was confined inside a queen excluding cage. Queen cups with young larvae (one day old) were introduced into the colony and allowed to be fed by workers for 2 days. We first carefully removed 3 day old larvae by using either a grafting tool or a pair of forceps, then removed the royal jelly by using a spatula.
Measurement of miRNA between RJM and RJC
For miRNA analysis, freshly collected RJM and RJC (N = 4 samples per species, each with 100 mg RJ) were immediately extracted for total RNA. All four samples of each species RJ were pooled to create one sample for RJM and one for RJC. Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s protocol. RNA quality was checked by using an Agilent 2100 Bioanalyzer. RNA fragments of 10–44 bases long were separated from total RNA by using Novex 15% TBE-Urea gel (Invitrogen), followed by 10% TBE-Urea gel. The resulting small RNAs were ligated to 5′ adaptors (Illumina, San Diego, CA, USA) and then combined with 3′adaptors (Illumina). These 5′ and 3′ products were amplified by PCR amplification and excised from 6% TBE-Urea gel (Invitrogen). According to Illumina Genome Analyzer (Beijing Genomics Institute, Shenzhen, China) instructions, the purified DNA segments were directly used for cluster generation and sequence. The sequencer produced image files were then converted to digital-quality data.
To further analyze the RNA secondary structure comprised with the matched Solexa reads, digital-quality sequences with perfect match or one mismatch were maintained. Genomic sequences of 100 nt were taken from these sequences, then the secondary structure was predicted and analyzed by RNAfold and MIREAP [39] under default settings. There are three criteria for candidate miRNA genes: (a) mature miRNAs are present in one arm of the hairpin precursors lacking large internal loops or bulges; (b) the secondary structures of the hairpins are steady, with the free energy of hybridization being lower than −20 kcal/mol; (c) hairpins are located in intergenic regions or introns [39]. Finally, these candidate miRNA reads were analyzed by miRBase database 13.0.
To compare miRNA expression levels between RJM and RJC, the reads of every miRNAs were subjected to the following analysis: a miRNA was considered “altered” only if it had both: (a) 10 copies by Solexa sequencing in both RJM and RJC, and (b) a two-fold difference in copy numbers between RJM and RJC. GO assignments usually has three ontologies: biological process, cellular component and molecular function, which is used to classify the functions of almost all miRNA in our paper.
2. Transcriptome Modification in A. mellifera Fed Two Different RJs
Honey bee (A. mellifera) larvae were reared inside 24-cell tissue culture plates (Costar, NY, USA) inside an incubator (35°C, 75±3% RH). Each cell was primed with 200 µl of freshly collected royal jelly, either from RJM or from RJC before a 1 day old larva was transferred into it. Larvae were transferred every 8 hrs to another plate with new food. For pupation, 6 day old larvae were transferred to 6-cell tissue culture plates lined with a piece of Kimwipe and kept in an incubator (35°C and 78±3% RH) [52]. After adult emergence, we obtained one sample per treatment (mRJM or mRJC), each consisted of 10 adult bees (5 from each of the two colonies) and used their heads for global transcriptome analysis. The two samples were kept at −80°C until use. A total of 1,400 larvae were reared for this experiment.
Measurement of transcriptomes in mRJM and mRJC
Total RNA was extracted with TRIzol regent (Invitrogen, USA) and treated with RNase-free DNase I (Takara Biotechnology, China). Poly(A) mRNA was separated by oligo-dT beads and then treated with the fragmentation buffer. By use of reverse transcriptase and random hexamer primers, the RNA fragments were transcribed into first-strand cDNA. Second strand cDNA synthesis was performed with DNA polymerase I and RnaseH. End-repair was done with T4 DNA polymerase, Klenow fragment, T4 Polynucleotide kinase. Ligation was accomplished with adapter or index adapter using T4 quick DNA ligase. Adaptor ligated fragments were selected according to size. Desired range of cDNA fragments were then excised from the gel. Finally, after validation of Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System, the cDNA library was sequenced by Illumina HiSeq2000.
By use of SOAP [53], a specific transcript with uniquely mapped reads was counted and their sequences assembled. Mapped reads was evaluated according to RPKM (Reads Per kb per Million reads) value of each transcript [38]. The transcript fold change was then calculated by the formula of log2 (mRJM)_RPKM/(mRJC)_RPKM. The formula to calculate the probability of a specific gene being expressed equally between the two samples was defined as
| () |
Where N1 and N2 indicate the total number of clean reads in mRJC and mRJM, respectively, and x and y indicate the mapped clean read counts of the transcript in each sample respectively. Then, the FDR (False Discovery Rate) method was applied to determine the threshold of the p-value in multiple tests. In this study, ‘FDR<0.001’ and the absolute value of log2Ratio >1 were used as the threshold to judge the significance of differentiated gene expression. We used the Blastall program to annotate the pathways of DEGs against the KEGG database.
3. Analysis of miRNA Targeted Genes
To identify possible target sequences of RJ, we used the RNA hybrid software and ftp.ncbi.nih.gov/genomes/Apis_mellifera/RNA/rna.fa.gz, which provided us with a single predicted site of interaction with a minimum free energy.
Supporting Information
The miRNAs expression analysis in RJM and RJC.
(DOC)
Differential expressed genes (DEGs) analysis in mRJM, relative to mRJC.
(DOC)
DEGs in biological process, cellular component, molecular function.
(DOC)
Annotation of DEGs and their related miRNAs as affected by RJM and RJC. Those that were up-methylated by RJM were indicated as bold, and those not bolded were down-regulated.
(DOC)
Acknowledgments
We thank Fei Zhang, Zhen Xiu Zeng, Jun Feng Liu, Liu Qing Tian, Wen Xiang Wang for their help in experiments.
Funding Statement
This work was supported by the Earmarked Fund for China Agriculture Research System (No. CARS-45-KXJ12), the National Natural Science Foundation of China (No. 31060327) and Doctoral Fund of Ministry of Education of China (No.20103603110003). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Winston ML (1987) Biology of the honey bee. Cambridge: Harvard University Press: 296 p.
- 2. Seeley TD (1989) The honey bee colony as a superorganism. American Scientist 77: 546–553. [Google Scholar]
- 3. Smith CR, Toth AL, Suarez AV, Robinson GE (2008) Genetic and genomic analyses of the division of labor in insect societies. Nat Rev Genet 9: 735–748. [DOI] [PubMed] [Google Scholar]
- 4. Jung-Hoffman I (1966) Die determination von Köningin und Arbeiterin der Honigbiene. Zoologie Bienenforsh 8: 296–322. [Google Scholar]
- 5. Weaver N (1966) Physiology of caste determination. Annu Rev Entomol 11: 79–102. [DOI] [PubMed] [Google Scholar]
- 6. Schwander T, Lo N, Beekman M, Oldroyd BP, Keller L (2010) Nature versus nurture in social insect caste differentiation. Trends Ecol Evol 25: 275–282. [DOI] [PubMed] [Google Scholar]
- 7. Chittka A, Chittka L (2010) Epigenetics of royalty. PLoS Biol 11: e1000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tautz J (2008) The buzz about bees. Berlin: Springer. 284 p.
- 9. Maleszka R (2008) Epigenetic integration of environmental and genomic signals in honey bees: the critical interplay of nutritional, brain and reproductive networks. Epigenetics 3: 188–192. [DOI] [PubMed] [Google Scholar]
- 10. Viuda-Martos M, Ruiz-Navajas Y, Fernández-López J, Pérez-Alvarez JA (2008) Functional properties of honey, propolis, and royal jelly. Institute of Food Technologists 73: R117–24. [DOI] [PubMed] [Google Scholar]
- 11. Drapeau MD, Albert S, Kucharski R, Prusko C, Maleszka R (2006) Evolution of the Yellow/Major Royal Jelly Protein family and the emergence of social behavior in honeybees. Genome Res 16: 1385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fang Y, Feng M, Li JK (2010) Royal jelly proteome comparison between A. mellifera ligustica and A. cerana cerana . J Proteome Res 9: 2207–15. [DOI] [PubMed] [Google Scholar]
- 13. Zeng ZJ, Zou Y, Guo DS, Yan WY (2006) Comparative studies of DNA and RNA from the royal jelly of Apis mellifera and Apis cerana. Indian Bee J. 68: 18–21. [Google Scholar]
- 14. Kamakura M (2011) Royalactin induces queen differentiation in honeybees. Nature 473: 478–83. [DOI] [PubMed] [Google Scholar]
- 15.Guo XQ (2010) The development and molecular mechanism of queen-worker differentiation, the miRNAs of royal jelly make a difference to queen-worker differentiation. National Science Library, Chinese Academy of Sciences. 156p.
- 16. Ambros V (2004) The functions of animal microRNAs. Nature 431: 350–355. [DOI] [PubMed] [Google Scholar]
- 17. Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297. [DOI] [PubMed] [Google Scholar]
- 18. He L, Hannon GJ (2004) MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5: 522–531. [DOI] [PubMed] [Google Scholar]
- 19. Filipowicz W, Bhattacharyya SN, Sonenberg N (2008) Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9: 102–114. [DOI] [PubMed] [Google Scholar]
- 20. Wu L, Belasco JG (2008) Let me count the ways: mechanisms of gene regulation by miRNAs and siRNAs. Mol Cell 29: 1–7. [DOI] [PubMed] [Google Scholar]
- 21. Laubinger S, Zeller G, Henz SR, Buechel S, Sachsenberg T, et al. (2010) Global effects of the small RNA biogenesis machinery on the Arabidopsis thaliana transciptome. Proc Natl Acad Sci USA. 107: 17466–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, et al. (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9: 654–659. [DOI] [PubMed] [Google Scholar]
- 23. Behura SK (2007) Insect microRNAs: structure, function and evolution. Insect Biochem Mol Biol 37: 3–9. [DOI] [PubMed] [Google Scholar]
- 24. Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854. [DOI] [PubMed] [Google Scholar]
- 25. Wei Y, Chen S, Yang P, Ma Z, Kang L (2009) Characterization and comparative profiling of the small RNA transcriptomes in two phases of locust. Genome Biology 10: R6 (doi: 10.1186/gb-2009–10–1-r6).. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weaver DB, Anzola JM, Evans JD, Reid JG, Reese JT, et al. (2007) Computational and transcriptional evidence for microRNAs in the honey bee genome. Genome Biol. 8: R97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenberg JK, Xia J, Zhou X, Thatcher SR, Gu X, et al.. (2012) Behavioral plasticity in honey bees is associated with differences in brain microRNA transcriptome. Genes Brain Behav doi: 10.1111/j.1601–183X. 2012.00782-x. [DOI] [PMC free article] [PubMed]
- 28.Liu F, Peng W, Li Z, Li W, Li L, et al. (2012) Next-generation small RNA sequencing for microRNAs profiling in Apis mellifera: comparison between nurses and foragers. Insect Mol Biol. doi: 10.1111/j.1365–2583.2012. 01135.x. [DOI] [PubMed]
- 29. Behura SK, Whitfield CW (2010) Correlated expression patterns of microRNA genes with age-dependent behavioral changes in honeybee. Insect Molecular Biology 19: 431–439. [DOI] [PubMed] [Google Scholar]
- 30. Robinson GE (2002) Genomics and integrative analyses of division of labor in honeybee colonies. Am Nat 160: S160–S172. [DOI] [PubMed] [Google Scholar]
- 31. Hori S, Kaneko K, Saito TH, Takeuchi H (2011) Expression of two microRNAs, ame-mir-276 and-1000, in the adult honeybee (Apis mellifera) brain. Apidologie 42: 89–102. [Google Scholar]
- 32. Chen X, Yu X, Cai Y, Zheng H, Yu D, et al. (2010) Next-generation small RNA sequencing for microRNAs profiling in the honey bee Apis mellifera . Insect Molecular Biology 19: 799–805. [DOI] [PubMed] [Google Scholar]
- 33. Blum MS, Fales HM, Morse RA, Underwood BA (2000) Chemical characters of two related species of giant honeybees (Apis dorsata and A. laboriosa): possible ecological significance. Journal of Chemical Ecology 4: 801–807. [Google Scholar]
- 34.Ruttner F (1988) Biogeography and taxonomy of honeybees. Springer-Verlag Berlin Heidelberg 284 p.
- 35.Hepburn HR, Radloff SE (2011) Honeybees of Asia. Springer-Verlag Berlin Heidelberg 699 p.
- 36.Oldroyd BP, Wongsiri S (2006) Asian honey hees biology, conservation and human interactions. Cambridge: Harvard University Press: 347p.
- 37. Griffiths-Jones S (2004) The microRNA registry. Nucleic Acids Res 32: D109–D111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mortazavi A, Williams BA, McCueK, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- 39. Chen X, Gao C, Li H, Huang L, Sun Q, et al. (2010) Identifcation and characterisation of microRNAs in raw milk during different periods of lactation, commercial fluid, and powdered milk products. Cell Research 20: 1–10. [DOI] [PubMed] [Google Scholar]
- 40. Wang L, Oberg AL, Asmann YW, Sicotte H, McDonnell SK, et al. (2009) Genome-wide transcriptional profiling reveals microRNA-correlated genes and biological processes in human lymphoblastoid cell lines. PLoS One 4: e5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ragan C, Cloonan N, Grimmond SM, Zuker M, Ragan MA (2009) Transcriptome-wide prediction of miRNA targets in human and mouse using FASTH. PLoS One 4: e5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rembold H, Kremer JP (1980) Characterization of postembryonic developmental stages of the female castes of the honey bee, Apis mellifera L. Apidologie. 11: 29–38. [Google Scholar]
- 43. Kucharski R, Mitri C, Maleszka R (2007) Characterization of a metabotropic glutamate receptor in the honeybee (Apis mellifera): implications for memory formation. Invert Neurosci 7: 99–108. [DOI] [PubMed] [Google Scholar]
- 44. Odronitz F, Becker S, Kollmar M (2009) Reconstructing the phylogeny of 21 completely sequenced arthropod species based on their motor proteins. BMC Genomics 10: 173 doi: 10.118 6/1471–2164–10–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. de Azevedo SV, Hartfelder K (2008) The insulin signaling pathway in honey bee (Apis mellifera) caste development-differential expression of insulin-like peptides and insulin receptors in queen and worker larvae. Journal of Insect Physiology 54: 1064–1071. [DOI] [PubMed] [Google Scholar]
- 46.Biswas S, Reinhard J, Oakeshott J, Russell R, Srinivasan MV, et al.. (2010) Sensory regulation of neuroligins and neurexin I in the honeybee brain. PloS One 5: e 9133. doi: 10.1371/journal. pone. 0009133. [DOI] [PMC free article] [PubMed]
- 47. Biswas S, Russell RJ, Jackson CJ, Vidovic M, Ganeshina O, et al. (2008) Bridging the synaptic gap: neuroligins and neurexin I in Apis mellifera . PloS One 3: e3542 doi: 10.1371/journal. pone. 0003542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matsuura H, Sokabe T, Kohno K, Tominaga M, Kadowaki T (2009) Evolutionary conservation and changes in insect TRP channels. BMC Evolutionary Biology 9: 228 doi: 10.1186/1471-2148-9-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tan K, Yang M, Wang Z, Radloff SE, Pirk CW (2012) The pheromones of laying workers in two honeybee sister species: Apis cerana and Apis mellifera . J Comp Physiol A 198: 319–23. [DOI] [PubMed] [Google Scholar]
- 50.Yang GH (1983) Apis cerana. China Agriculture Press. 123 p.
- 51.Zeng ZJ, Jiang YS, Hu FL, Xu BH, Su SK, et al.. (2009) Apiculture. China Agriculture Press. 123 p.
- 52. Shi YY, Huang ZY, Zeng ZJ, Wang ZL, Wu XB, et al. (2011) Diet and cell size both affect queen-worker differentiation through DNA methylation in honey bees (Apis mellifera, Apidae). PLoS One 6: e18808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li R, Yu C, Li Y, Lam TW, Yiu SM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25: 1966–1967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The miRNAs expression analysis in RJM and RJC.
(DOC)
Differential expressed genes (DEGs) analysis in mRJM, relative to mRJC.
(DOC)
DEGs in biological process, cellular component, molecular function.
(DOC)
Annotation of DEGs and their related miRNAs as affected by RJM and RJC. Those that were up-methylated by RJM were indicated as bold, and those not bolded were down-regulated.
(DOC)