

Abstract
Skeletal muscle atrophy is defined as a decrease in muscle mass and it occurs when protein degradation exceeds protein synthesis. Potential triggers of muscle wasting are long-term immobilization, malnutrition, severe burns, aging as well as various serious and often chronic diseases, such as chronic heart failure, obstructive lung disease, renal failure, AIDS, sepsis, immune disorders, cancer, and dystrophies. Interestingly, a cooperation between several pathophysiological factors, including inappropriately adapted anabolic (e.g., growth hormone, insulin-like growth factor 1) and catabolic proteins (e.g., tumor necrosis factor alpha, myostatin), may tip the balance towards muscle-specific protein degradation through activation of the proteasomal and autophagic systems or the apoptotic pathway. Based on the current literature, we present an overview of the molecular and cellular mechanisms that contribute to muscle wasting. We also focus on the multifacetted therapeutic approach that is currently employed to prevent the development of muscle wasting and to counteract its progression. This approach includes adequate nutritional support, implementation of exercise training, and possible pharmacological compounds.
Keywords: Skeletal muscle atrophy, Cachexia, Proteasome, Autophagy, Apoptosis, Therapy
Introduction
Skeletal muscle is the most abundant tissue in the body of vertebrates and is involved in many different important functions. For instance, skeletal muscle is the primary target of glucose uptake, can regenerate after injury, and is subjected to size remodeling. Overall, skeletal muscle mass represents a determinant of strength, endurance, and physical performance. Reduced muscle bulk or muscle atrophy takes place when protein degradation exceeds protein synthesis, leading to a reduction of the cross-sectional area of myofibers and decreased muscle strength. Though muscle atrophy is a consequence of certain physiological processes, such as aging, it may also be a prominent feature present in several disparate diseases. In this latter case, a general loss of body mass, not exclusively limited to muscle, inherently implies an underlying, usually chronic and severe syndrome with impaired prognosis, known as cachexia. In both physiological and pathological conditions, the clinical implications of muscle wasting translate into poor quality of life, implying exercise intolerance and inability to manage daily activity. Since our knowledge on the mechanisms contributing to muscle wasting has remarkably increased in the past 20 years, the aim of the present review is to provide a comprehensive overview of the different cues, mechanisms, and degradative machineries involved in muscle atrophy. Furthermore, an update is given on current therapeutic approaches.
Muscle atrophy
Muscle atrophy is a physiological consequence of aging (i.e., age-related sarcopenia), defined as the presence of both low muscle mass and low muscle function (strength or performance) [1], but it may also result from prolonged periods of rest or a sedentary lifestyle. Moreover, muscle atrophy represents a clinical feature of cachexia, a multifactorial syndrome implying reduced life expectancy and accompanying many illnesses like chronic heart failure (CHF), chronic obstructive pulmonary disease (COPD), chronic kidney disease (CKD), cancer, HIV, sepsis, immune disorders, and dystrophies [2].
A reduced cross-sectional area of myofibers with subsequent impaired strength is the main characteristic of muscle atrophy [3], mirroring a consistent depletion of contractile proteins. Muscle atrophy is also frequently characterized by a switch of the fiber-type composition. For instance, while the loss of contractile proteins predominantly affects the type II fast fibers during aging or cancer cachexia [3], giving rise to a higher proportion of slow versus fast fibers [4], an increase in type IIX fiber proportions at the expense of either type I or type IIA muscle fibers has been observed in CHF [5–7], suggesting that different pathophysiological cues may differently affect the fiber-type muscular pattern. Besides the loss of muscle mass, inherent deficits in the “quality” of skeletal muscle have been demonstrated. For instance, in symptomatic CHF patients, quadriceps muscle strength is reduced compared to controls, even after correction for reduced circumferential area [8]. Equally important, however, are the prognostic implications of cachexia and muscle loss. Anker et al. [9] provided strong evidence that the identification of a heart failure patient as being cachectic significantly increased the risk of dying, even after correction for age, gender, disease severity, and treatment allocation.
Until recently, the lack of a clear definition for cachexia has hampered structured research as well as the development of treatment targets. Therefore, in order to allow timely clinical recognition, objective criteria to define cachexia have become mandatory. According to the last consensus criteria, the condition sine qua non to define cachexia is weight loss greater than 5 % or weight loss greater than 2 % in individuals already showing depletion according to current body weight and height (BMI <20) or skeletal muscle mass [10]. However, the assessment for classification and clinical management of cachectic patients should include additional domains such as anorexia or reduced food intake, catabolic drive, muscle mass, and strength as well as functional and psychosocial impairment.
Altogether, these observations clearly indicate that different, somewhat apparently unrelated mechanisms may synergistically cooperate to trigger the impairment of body's performance through loss of bulk muscle, thereby inducing critical and often fatal health complications.
Triggers of muscle atrophy
A dysbalance of anabolic versus catabolic factors may alter nitrogen balance leading to consistent protein depletion and muscle atrophy (Fig. 1). During embryonic development as well as in post-natal life, different anabolic factors are required to ensure proper muscle growth. In this context, growth hormone (GH), androgens (testosterone), insulin, and insulin-like growth factor-1 (IGF-1) play a prominent role, with the latter being central due to its unrivaled pleiotropic ability to regulate different muscular mechanisms such as cell proliferation [11] and differentiation [12], myofiber growth [13, 14], and regeneration [13]. Accordingly, low levels of circulating IGF-1 have been associated with sarcopenia [15], CHF [16], cancer [17], and other clinical syndromes [18]. Keeping in mind that IGF-1 exerts pleiotropic effects by stimulating proliferation of resident satellite cells during regeneration as well as growth of preexisting myofibers, it is conceivable that its decline in circulating levels during aging or in consequence of disease may impinge on the muscle architecture. Unlikely, liver-specific IGF-1 knockout mice exhibit normal growth rates [19, 20], suggesting that muscle-produced IGF-1 may compensate for the lack of systemic (hepatic) IGF-1. In support of the pivotal role of IGF-1, it has been reported that muscle-specific transgenic overexpression of IGF-1 promotes muscle hypertrophy and increases physiological muscle strength [12, 13], improves muscle regeneration [13, 21], ameliorates the dystrophic phenotype of mdx mice [22, 23], protects motor neurons in a mouse model of amyotrophic lateral sclerosis (ALS) [24] and also attenuates disease in a mouse model of spinal and bulbar muscular atrophy [25]. In this regard, the beneficial IGF-1 effects mostly rely on the activation of the PI3K/Akt pathway, which promotes protein synthesis and blunts protein degradation [26–28]. Since insulin activates the PI3K/Akt pathway, analogously to IGF-1, insulin resistance plays a consistent role in muscle atrophy of diabetic patients [29]. Indeed, transgenic mice with a dominant-negative IGF-1 receptor specifically targeted to the skeletal muscle-developed insulin resistance due to formation of hybrid receptors between the mutant and the endogenous IGF-I and insulin receptors, thus providing an excellent model to study the molecular mechanisms underlying the development of human type 2 diabetes [30]. While sarcopenia occurring during aging mainly relates to impaired anabolism, because of decreased anabolic factors or reduced anabolic response [31, 32], different conditions leading to muscle wasting show an increase of certain catabolic factors, including tumor necrosis factor alpha (TNFα) [33, 34], transforming growth factor beta (TGFβ) ligands such as myostatin [35–38] and glucocorticoids [39–42]. Several tumor-bearing animal models developing cachexia are characterized by an excess of circulating TNFα [43] as well as by the increased expression of other cachectic cytokines, such as interleukin-1 (IL-1) [44] and IL-6 [45, 46]. Another master regulator of muscle size [38], the TGFβ family member termed myostatin, elicits atrophy when administered to an adult animal [47], as, analogously, glucocorticoids do [48]. Finally, it is worth mentioning that protein degradation in skeletal muscle is stimulated by excessive oxidative stress [49], as discussed in more detail below.
Fig. 1.
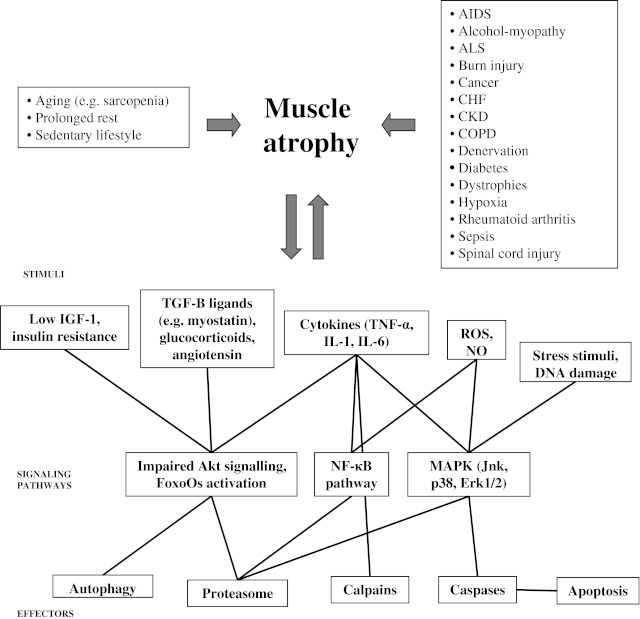
Muscle atrophy may arise as a consequence of many different physiological and pathological conditions. Unraveling the stimuli, signaling pathways and effectors that contribute to muscle depletion is pivotal to develop therapeutic interventions
Molecular pathways underlying muscle atrophy
Unraveling the signaling network that contributes to muscle depletion is pivotal to develop therapeutic interventions (Fig. 1). Among the different pathways controlling muscle size [27, 28, 50, 51], the PI3K/Akt pathway is central and is mainly recruited upon IGF-1/insulin stimuli to promote protein synthesis and to block degradation [52–54]. In the cytoplasm, Akt1 activates S6 kinase 1 (S6K1) via mammalian target of rapamycin (mTOR), which leads to increased protein synthesis [55]. Akt1 phosphorylates also FoxO transcription factors, specifically making FoxO3 unable to enter the nucleus and to drive transcription of certain ubiquitin ligases (e.g., muscle atrophy F-Box (MAFbx/Atrogin-1) and muscle RING finger-1 (MuRF1)) [54] or autophagy-related genes (e.g., LC3 and Bnip3) [56], which account for the protein breakdown mediated by the ubiquitin–proteasomal and autophagic/lysosomal pathways [57], respectively (vide infra). Thus, constitutive Akt1 activation produces rapid muscle hypertrophy in transgenic mice [58, 59], whereas Akt1/Akt2 double-knockout mice exhibit severe skeletal muscle atrophy [60]. Moreover, S6K1 (−/−) myotubes are atrophic, and their hypertrophic response to IGF-1, nutrients, and membrane-targeted Akt1 is blunted [61], definitely corroborating the importance of the IGF-1/Akt axis pathway on the muscle maintenance.
In support of this evidence, muscle atrophy due to reduced Akt signaling is observed in different diseases or pathophysiological conditions, including ALS [62–64], CKD [65, 66], diabetes [29], chronic hypoxia [67], statin-induced myopathy [68], sepsis [69, 70], burn injury [71], and aging [72]. In particular, different molecules behave as proatrophic factors by reducing Akt signaling, including TNFα [73], TNF-related weak inducer of apoptosis [74], glucocorticoids [75, 76], angiotensin [77], and chemotherapy agents such as cisplatin [78]. It is noteworthy that one of the most powerful inducers of muscle atrophy, namely myostatin, can elicit activation of Smad2/3 pathway and increased expression of MAFbx/MuRF1 ubiquitin ligases followed by increased proteasomal activity [79, 80], thus contributing to the breakdown of sarcomeric proteins. Moreover, it has been recently shown that most of the myostatin effects rely on its ability to reduce the Akt signaling [81, 82], as myostatin can inhibit the IGF-1-induced myotube hypertrophy by lowering Akt signaling [83].
Overall, these findings suggest that Akt is a valuable target for improvement of muscle performance in different proatrophic conditions. Of note, sustaining the Akt pathway confers also several advantages to Duchenne's muscular dystrophy (DMD) muscles, as first observed upon IGF-1 administration to mdx mice [23]. Other studies showed that the activation of Akt in DMD muscles promotes hypertrophy [84, 85] prevents the force drop induced by eccentric contractions [86], upregulates the utrophin–glycoprotein complex, promotes sarcolemma stability [87], attenuates muscular degeneration, promotes myofiber regeneration, improves muscle function [88], and mediates the hypertrophic effects promoted by matrix metalloproteinase-9 [89, 90].
Another pathway playing a consistent role in muscle atrophy relies on the activity of transcription factor NF-κB [91, 92], which is frequently activated by several cytokines through a cascade involving the phosphorylation and consequent degradation of NF-κB inhibitor Iκ-Bα [93]. NF-κB pathway was first implicated in a model of skeletal muscle disuse atrophy [94]. Interestingly, NF-κB is able to directly bind the MuRF1 promoter, thus triggering a proteasome-dependent muscle wasting [95]. In addition, NF-κB activation is tightly linked to increased oxidative stress, which in turn controls the rate of protein degradation in skeletal muscle [49]. Indeed, NF-κB induces muscle wasting by stimulating the expression of iNOS, leading to increased nitric oxide (NO) [96–99], as confirmed by the high detection of protein nitration in aged muscle [100–103]. Thus, elevated NO levels are implicated in age-induced skeletal muscle apoptosis [104], in cytokine-induced cachexia [96, 97, 105], and iNOS expression is elevated in COPD [106] and human cancer and AIDS patients suffering from cachexia [107]. In addition, nNOS is commonly dislocated in neuromuscular disorders [108], such as ALS [109], and mediates muscle atrophy via regulation of FoxO transcription factors in a model of unloading-induced muscle atrophy [110]. Thus, the NO pathway may represent an effective target for the therapy of both muscle sarcopenia and cachexia [111].
In physiological conditions, a variety of reactive oxygen species (ROS) are produced in the skeletal muscle to allow the adaptation of the muscle to several stimuli [112]. In contrast, prolonged muscle disuse causes increased radical production and subsequent injury in inactive muscle fibers, as seen upon experimental immobilization of skeletal muscles. However, this effect can be delayed by exogenous antioxidants [113–115]. When ROS levels exceed the healthy threshold, at least three different signaling systems are supposed to play a central role in mediating the proatrophic effects on inactive muscles, namely the cytosolic calcium-dependent calpains; the mitogen-activated protein kinase system that includes Jnk, p38, and Erk1/2 kinases; and the NF-κB pathway [49, 116].
Cellular mechanisms of protein degradation during muscle atrophy
The two major protein degradation pathways in eukaryotic cells are the UPS (ubiquitin–proteasome system) and the autophagy–lysosome pathway (Fig. 1). In normal physiological conditions, the UPS is responsible for the degradation of short-lived proteins, whereas autophagy regulates levels of long-lived proteins and organelles. However, a large body of evidence suggests that both pathways also play an important role in skeletal muscle wasting.
The ubiquitin–proteasome system
Muscle-specific ubiquitin ligases
Ubiquitin is an abundant 8-kDa peptide found in all cell types. To date, free ubiquitin has no biological function but exerts its effects by covalent attachment to other proteins [117]. The substrate protein can be mono-ubiquitinated or additional ubiquitin molecules can be added to produce a poly-ubiquitinated protein that is targeted for degradation by the proteasome [118], while the mono-ubiquitinated substrates are likely degraded in the lysosomes [119]. The ubiquitination of proteins is a multistep reaction [120, 121]. The first step comprises activation of ubiquitin by ubiquitin-activating enzyme (E1), an ATP-hydrolyzing protein that adenylates ubiquitin on its C-terminal glycine and then links the ubiquitin molecule on a cysteine residue at its catalytic site. Subsequently, E1 transfers the activated ubiquitin to the active site of an ubiquitin-conjugating enzyme (E2). Finally, E2 binds to a ligase (E3) that recognizes the substrate and transfers ubiquitin to the target protein. In mammalian cells, there is only one ubiquitin-activating enzyme. The ubiquitin-conjugating enzyme family is composed of several members that specifically interact with a subset of ubiquitin ligases which in turn recognize specific substrates [122, 123].
In 2001, two muscle-specific ubiquitin ligases involved in the ubiquitination process of skeletal muscle proteins have been characterized, namely MAFbx/Atrogin-1 and MuRF1 [124, 125]. MAFbx is member of a multiprotein SCF (Skp1, Cullin1, and F-box) complex, which needs to be assembled first in order to establish E3 activity [125]. MuRF1 contains an N-terminal RING domain, a MuRF family conserved region, a zinc-finger domain (B-box), leucine-rich coiled-coil domains, and an acidic C-terminal tail [126]. Knockout mice for either MAFbx or MuRF1 are phenotypically normal. However, under atrophy conditions, loss of a muscle-specific ubiquitin ligase confers partial protection against muscle wasting [124]. The molecular characterization of these ubiquitin ligases has led to the discovery that MuRF1 binds to the myofibrillar protein titin [126, 127] and more importantly to the myosin heavy chain (MyHC), the most abundant protein in the skeletal muscle [128]. Indeed, MyHC was shown to be degraded by the proteasome [129], and loss of MyHC was blocked by siRNA-mediated inhibition of MuRF1 [128]. Moreover, MuRF1 is able to ubiquitinate MyHC in vitro [128]. More recent work showed that other myofibrillar proteins including myosin light chain and myosin-binding protein C are specifically ubiquitinated by MuRF1 [130]. The relevance of MAFbx in the stimulation of myofibrillar protein degradation is less clear. Hitherto, the only confirmed substrates for MAFbx are MyoD [131] and eIF3f [132]. MyoD is a transcription factor mainly involved in the myogenic program and partially in protein synthesis induction [133], while eIF3f is a translation initiation factor [134]. The role of MAFbx in protein synthesis rather than degradation was then suggested by reports showing no correlation between the rates of protein breakdown and the MAFbx mRNA levels both in vivo [135, 136] and in vitro [137]. Moreover, expression of MAFbx/Atrogin-1 and MuRF1 undergoes different regulation [138, 139]. Because muscle atrophy results from both increased protein degradation and/or reduced synthesis, the two E3 ligases contribute most likely in a distinct manner to the wasting process. These observations should be taken into account when the measurement of mRNA levels of such E3s is taken as an indicator of proteasome-dependent protein degradation.
After characterization of MAFbx and MuRF1, another muscle-specific ubiquitin ligase, termed tripartite motif 32 (TRIM32), was discovered in 2005 [140]. This protein was a TRIM family member containing in its structure a RING finger, a B-box, and a coiled-coil motif. TRIM32 was shown to associate with the thick myofibrillar filament and is able to ubiquitinate actin and dysbindin [140], a protein involved in lysosomal–endosomal trafficking [141]. Mutations in the E3 ubiquitin ligase TRIM32 are involved in skeletal muscle disorders such as limb-girdle muscular dystrophy type 2 H (LGMD2H) and the multisystemic disorder, Bardet–Biedl syndrome type 11 [142]. Skeletal muscles from murine TRIM32 knockout mice (T32KO) reveal mild myopathic changes such as Z-line streaming and a dilated sarcotubular system with vacuoles, thereby replicating phenotypes of LGMD2H and sarcotubular myopathy [143]. Analysis of T32KO neural tissue shows a decreased concentration of neurofilaments and a reduction in myelinated motoraxon diameters, suggesting a shift toward a slower motor unit type, as observed in T32KO soleus muscle. These data suggest that muscular dystrophy due to TRIM32 mutations involves both neurogenic and myogenic characteristics [143]. Finally, in 2007 a novel muscle-specific F-box protein (FBXO40) was found to be upregulated after denervation, though not after starvation [144]. FBXO40 was previously identified in LGMD patients that showed a dramatic decrease in comparison to healthy individuals [145]. Very recently, FBXO40 was found to induce IRS1 ubiquitination in skeletal muscle, thereby providing a negative feedback on the prohypertrophic IGF1R/IRS1/PI3K/Akt pathway [146], as demonstrated by the enhanced body and muscle size of mice null for FBXO40 during the growth phase associated with elevated IGF1 levels. Future studies will reveal new substrates and specific new roles for E3 ubiquitin ligases, while more investigations in humans are needed to validate their potential role as therapeutic targets for the prevention of muscle atrophy.
Proteasome induction/activation in muscle atrophy
Several physiological factors such as classical hormones, insulin, IGF-1 and TNFα can modulate the expression of enzymes controlling ubiquitination and proteasome-dependent protein degradation. For example, insulin reduction as a consequence of fasting or experimental diabetes leads to increased expression of ubiquitin [29, 40], MAFbx [147], as well as UBR1, UBR2, and UBR3 [148, 149]. Similar effects can be found after glucocorticoid administration [39–42], even though most of the glucocorticoid-induced effects are reverted by IGF-1 [41, 42, 150]. Also proinflammatory cytokines such as TNFα are responsible for increased ubiquitin expression and accumulation of ubiquitinated proteins [34, 151]. However, the first observations indicating a role for the ubiquitin system during atrophy came from cancer cachexia models, showing increased ubiquitin and proteasome subunit expression or protein ubiquitination in tumor-bearing animals [152–156]. Later, as MAFbx and MuRF1 were discovered, their levels were reported to be increased in AH-130-bearing rats [17], as well as in MAC16 [157] and colon-26-bearing mice [72, 158]. After the identification of proteasome activation in cancer cachexia, similar observations were reported in atrophying muscles following denervation or starvation [159]. These experimental procedures led to the discovery of the ubiquitin ligases MAFbx and MuRF1 [124, 125]. Worth of note, the atrophy of skeletal muscle following denervation is dependent on the upregulation of specific histone deacetylases (HDAC), namely HDAC4 and HDAC5, which repress Dach2, a negative regulator of myogenin, resulting in the activation of myogenin which in turn drives the increase of MAFbx/Atrogin-1 and MuRF1 expression, leading to proteolytic muscle wasting [160]. Also sepsis-induced muscle atrophy associates with increased expression and enzymatic activity of the proteasome [161]. Subsequently, the raise in the mRNA levels of the ubiquitin ligases was reported [162]. Hitherto, a report showing increased MAFbx or MuRF1 increase exists for almost every experimental condition characterized by muscle wasting, even if evidence for increased proteasome degradation is lacking. These conditions include disuse [124], diabetes [125, 147], CKD [147], glucocorticoid administration [53], rheumatoid arthritis [163], COPD [164], alcohol myopathy [165], AIDS [166], aging [130], and spinal cord injury [167].
Autophagy
Autophagy is an evolutionarily conserved subcellular process for bulk destruction of proteins and entire organelles [168]. The process is classically considered to be a pathway contributing to cellular homeostasis and adaptation to stress and thus may act as a prosurvival mechanism. However, excessive stimulation of the autophagic machinery is deleterious and may lead to caspase-independent cell death [169]. Different autophagic pathways have been described, all sharing the import of cytoplasmic components into the lysosome. For the most prevalent form of autophagy (macroautophagy, further referred to as autophagy), double-membrane vacuoles known as autophagosomes will form, originating from a largely undefined structure known as the phagophore or isolation membrane [168]. The process proceeds in a step-wise fashion, with the elongation of the isolation membrane, maturation of the autophagosome, followed by its fusion with a lysosome, thereby generating an autolysosome. During this final step, the incorporation of the outer autophagosomal membrane with the lysosomal membrane eventually allows the degradation of the remaining inner single membrane and the cytoplasmic content of the autophagosome by lysosomal hydrolases. So far, more than 30 autophagy-related (Atg) genes have been identified that regulate autophagy induction and autophagosome formation [170].
Autophagy is constitutively active at basal levels in skeletal muscle, as shown by the accumulation of autophagosomes in human myopathies caused by genetic deficiency of lysosomal proteins (e.g. Pompe and Danon's disease) [171, 172] or by pharmacological inhibition of lysosomal function, as in chloroquine myopathy [173]. Muscle-specific deletion of the essential autophagy gene Atg7 results in profound muscle atrophy and an age-dependent decrease in muscle force [174, 175], suggesting that basal autophagy plays a beneficial role in controlling muscle mass. Moreover, Atg7 null muscles show accumulation of abnormal mitochondria, sarcoplasmic reticulum distension, disorganization of sarcomere, and formation of aberrant concentric membranous structures [174, 175], indicating that basal autophagy in muscle is responsible for the removal of dysfunctional organelles and required for maintaining myofiber integrity. Because of an abnormal sarcoplasmic reticulum, Atg7 null muscles also reveal activation of ER chaperones, such as BiP and phosphorylation of eIF2a [174, 175], suggesting induction of an unfolded protein response. This condition leads to inhibition of protein synthesis, which further contributes to muscle atrophy.
Even though basal autophagy is constitutively active and sufficient to keep myofibers in a healthy state during normal physiological conditions, several studies demonstrate that autophagy is strongly induced in skeletal muscle following oxidative stress [63, 176] or in response to severe catabolic conditions such as denervation [177] or fasting [175]. The classical pathway that regulates autophagy in mammalian cells acts through mammalian target of rapamycin (mTOR), a protein kinase that plays a key role as an intracellular nutrient sensor and regulator of protein synthesis, cell growth, and metabolism [170]. Surprisingly, mTOR signaling is not the major factor that controls autophagy in adult muscles. Detailed biochemical studies have shown that rapamycin-mediated mTOR inhibition only barely (~10 %) increases protein breakdown in differentiated myotubes [178]. Instead, the transcription factor FoxO3 was found to be necessary (and sufficient) to activate autophagy in myotubes. Indeed, FoxO3 controls the expression of many autophagy-related genes, including microtubule-associated protein-1 light chain 3 (LC3) involved in cargo recruitment and biogenesis of autophagosomes as well as Bnip3 [56, 57]. Because autophagosome formation is not induced in (fed) mice overexpressing GFP-LC3 [175], LC3 induction per se is not sufficient to trigger autophagy. Most likely, LC3 is continuously consumed when autophagy is activated. In this way, upregulation of LC3 is required to replenish the LC3 protein pool and allow progression of autophagy. In contrast, upregulation of Bnip3 has a major role in mediating the effect of FoxO3 on the autophagic process, as shown by the fact that induction of autophagy is markedly decreased by Bnip3 knockdown [56]. Other autophagy-related genes that are induced by FoxO3 during muscle atrophy are the LC3 homologue Gabarapl1, Atg12l, and the lysosomal proteinase cathepsin L [56, 57]. The role of cathepsin L upregulation is still unclear. Possibly, cathepsin L is involved in the degradation of muscle-specific proteins, as shown recently for the sialidase Neu2 [179], a protein involved in myoblast differentiation. Unfortunately, and quite similar to autophagy inhibition, excessive activation of autophagy aggravates muscle wasting by removing portions of cytoplasm, proteins, and organelles [180]. Therefore, tight control of FoxO3-induced gene expression is needed to avoid unbalanced autophagy induction and muscle loss.
Apoptosis and muscle atrophy
Cells undergoing apoptosis are characterized by cytoplasmic and nuclear condensation, oligonucleosomal cleavage of the chromosomal DNA, violent blebbing of the plasma membrane, packaging of the cellular contents into membrane-enclosed vesicles (apoptotic bodies), and elimination of these vesicles by heterophagocytosis [181, 182]. Changes in membrane fluidity and ionic charge, altered behavior of sugar chains or lipids, and conformational changes in membrane proteins are also likely to occur [181, 182]. The morphological features of apoptosis are mainly the result of a subset of proteolytic enzymes, called caspases. These proteases are activated by two principal pathways: an intrinsic and an extrinsic one. The intrinsic pathway begins within the cell and is triggered by stress stimuli such as DNA damage. Hallmark events include mitochondrial outer membrane permeabilization and the release of cytochrome C through the permeability transition pore complex. Once released, cytochrome C binds apoptotic protease-activating factor 1 (Apaf-1) to form a complex known as the apoptosome. This complex also recruits procaspase-9, promoting its self-activation. Active caspase-9, in turn, cleaves downstream effector caspases such as caspase-3 and caspase-7, which rapidly degrade intracellular substrates. The extrinsic pathway overlaps with the intrinsic pathway and is activated by ligand binding to specific death receptors on the cell surface. Examples of ligands to death receptors include TNFα, Fas ligand (FasL), and TNFα-related apoptosis-inducing ligand (TRAIL) that bind to the TNF receptor, Fas, or TRAIL receptor, respectively.
There is overwhelming evidence that apoptosis increases in skeletal muscle during normal aging [183, 184] and thus may contribute to sarcopenia [185] (Fig. 1). First, the mitochondrion-mediated (intrinsic) signaling pathway is activated in aged skeletal muscle. Indeed, the content of several Bcl-2 family members (Bax and Bcl-2) is altered with age, expression of Apaf-1 is enhanced, and the presence of cleaved caspases increases. There is also evidence that caspase-independent mechanisms may contribute to apoptosis through elevated nuclear levels of endonuclease G in aged muscle as well as raised expression levels of apoptosis-inducing factor. Secondly, receptor-mediated apoptotic signaling may be activated in aging muscle, as shown by increased activation of caspase-8, increased expression of FADD, and increased mitochondrial content of tBid. These events most likely occur in response to elevated circulating levels of TNFα. Thirdly, p53 appears to be elevated, although it is unclear whether this may lead to apoptotic cell death, as some studies have shown elevated cytosolic levels while others have seen increases in nuclear p53.
Apart from aging, apoptosis might contribute to muscle atrophy in various other conditions including cancer cachexia [186], CHF [187], muscle denervation [188], dystrophies [189], sepsis [190], hindlimb unweighting [191], muscle disuse [192], and burn injury [193]. However, with regard to interpretation of apoptosis data in muscle, several critical issues should be mentioned. Since skeletal myocytes are multinucleated, apoptosis of a particular nucleus may not equate to apoptosis of the entire muscle fiber. It is not yet known how many nuclei must be lost before the entire fiber undergoes apoptosis or how this process is regulated in skeletal muscle. Therefore, it has been suggested that nuclear apoptosis in skeletal muscle does not necessarily translate into death of the myofiber [184]. On the other hand, the classical terminal deoxynucleotidyl transferase end-labeling (TUNEL) technique is frequently used to detect DNA fragmentation during apoptosis, even though this method lacks specificity as it may also detect single-stranded DNA or RNA breaks. Accordingly, false positive TUNEL reactions can be observed in living muscle cells during active gene transcription [194]. Other difficulties in interpreting cell death data in atrophying muscle stem from the observation that caspase-3 is not only involved in apoptosis but also in myofibrillar protein degradation [195]. As occurs for calpains, caspase-3 might be involved in the release of myofibrillar proteins targeted to proteasome degradation. Also of note, caspase-3 enzyme activity has been reported to be elevated in aging muscle [196], while others have found no change in activity [183]. This discrepancy cannot simply be explained by differences in muscle type, age, or animal strain, although differential expression of some antiapoptotic proteins such as XIAP may help to understand contradictory study results [184].
Therapeutic inhibition of muscle atrophy
It is beyond the scope of this paper to extensively discuss the multitude of therapeutic approaches that have been explored until now to counteract or prevent muscle atrophy, and excellent reviews on this subject have been published [197, 198]. Given that muscle wasting is a complex systemic disorder, it is likely that multiple actions are required to prevent its development and progression. Ideally, a multifacetted approach involves adequate nutritional support, the implementation of exercise training to stimulate muscle hypertrophy and, hopefully, one or more pharmacological compounds that specifically, or because of pleiotropic actions, antagonize “proatrophic” pathways (Table 1).
Table 1.
Overview of different therapeutic approaches employed to counteract muscle wasting
| Therapeutic approach | Class | Molecule | References |
|---|---|---|---|
| Dietary supplementation | BCCAs | Leu, Ile, Val, Hmb | [204, 205] |
| PUFAs | EPA, DHA | [206, 207] | |
| Appetite stimulants | Anti-histaminic drugs, progestational agents, ghrelin | [208] | |
| ACE inhibitors | Enalapril | [211] | |
| Compounds with anabolic activity | Steroidal androgens | Testosterone | [213] |
| SARMs | Ostarine and enobosarm | [214–216] | |
| Growth factors | IGF-1, insulin | [218] | |
| Beta2-agonist | Formoterol | [220] | |
| Anti-inflammatory drugs | TNFα antibodies | Etancercept, infliximab | [221, 222] |
| Cyclooxygenase-2 inhibitors | Celecoxib | [223] | |
| Modulating the energetic crisis of skeletal muscle | Adipocyte-derived cytokine | Adiponectin (diabetes and obesity) | [228] |
| Insulin sensitizer | Thiazolidinediones | [233] | |
| PPARα agonists | Fibrates | [234] | |
| Muscle-wasting inhibitors | Proteasome inhibitors | PSI, LLN, MG132 | [235–239] |
| PS-341, bortezomib, or Velcade | [240–242] | ||
| Exercise training | PRT | PRT, endurance | [232, 243–245] |
| PRT combined with therapeutic strategies | PRT and Eicosapentaenoic acid | [250] |
ACE angiotensin converting enzyme, BCCAs branched-chain amino acids, Hmb β-hydroxy β-methylbutyrate, DHA docosahexaenoic acid, EPA eicosapentaenoic acid, IGF-1 insulin-like growth factor 1, PPARα peroxisome proliferator-activated receptors alpha, PRT physical regular training, PUFAs polyunsaturated fatty acids, SARMs nonsteroidal androgen receptor modulators, TNFα tumor necrosis factor alpha
Malnutrition and weight loss
Patients who suffer from life-threatening diseases are characterized by anorexia. Nevertheless, even bypassing the obstacle of reduced appetite by introducing enteral nutrition, or, if inevitable, total parenteral nutrition, does not reverse the wasting process. Therefore, nutritional optimisation should be considered as a supportive measure in malnourished patients. Deficiencies in micronutrients, vitamins, amino acids, and electrolyte abnormalities need correction. For instance, previous observations in yeast suggested that branched-chain amino acids (BCAAs), leucine, isoleucine, and valine, might be potential candidates in promoting survival [199]. BCAAs increase the activity of mTORC1 (mTORC1; mTOR in complex with raptor), a key regulator of protein synthesis and cell growth [200], thus favoring cell oxidative capacity [201] and PGC-1α-mediated mitochondrial gene expression [202]. In addition, long-term dietary supplementation with a specific BCAA-enriched amino acid mixture reduced oxidative damage, both in cardiac and skeletal muscles of middle aged mice, increasing average lifespan [203]. As such, BCAAs have been reported to have anticachectic effects, presumably resulting from interference with the UPS [204]. Moreover, in vitro experiments indeed demonstrated suppressed expression of MaFbx/atrogin-1 mRNA in a C2C12 cell line [205]. Fish oil derived N-3 polyunsaturated fatty acids (PUFAs) may have anticatabolic effects through anti-inflammatory actions [206, 207]. Despite their long-term application on empirical grounds, appetite stimulants such as antihistaminic drugs, as well as orally active progestational agents (megestrol acetate and medroxyprogesterone acetate) are still evaluated in clinical trials and have been approved for certain indications. The associated tendency for fluid retention limits their use in patients with CHF.
The orexigenic hormone ghrelin as a potential means to target cachexia has recently received considerable attention [208]. The effects of synthetic human ghrelin, as well as ghrelin receptor agonists, are being evaluated in several trials. Ghrelin, which is predominantly produced by the stomach, increases food intake through its actions on the hypothalamus [209]. In addition, serving as a growth hormone secretagogue and mainly mediating its effects via IGF-1, ghrelin also has anabolic effects. Both GH and insulin resistance have indeed been demonstrated in cachexia-related conditions.
Angiotensin II directly induces muscle protein catabolism through the ubiquitin–proteasome proteolytic pathway and may play a role in cancer cachexia, as well as in CHF patients [210]. CHF patients treated with enalapril in the SOLVD study were less likely to develop weight loss [211]. Reduced energy expenditure and weight increase are seen as an undesirable side-effect of beta blockers in general. In CHF patients, similar post-hoc observations in large beta-blocker trials have confirmed that antagonizing high catecholamine levels increases body weight.
Compounds with anabolic activity
In patients with CHF, an overall depletion of circulating anabolic hormones (i.e.; circulating total testosterone and free testosterone, dehydroepiandrosterone sulfate, and IGF-1) is associated with poor outcome [212]. Male hypogonadism is also often found in patients with cancer cachexia. In CHF patients, replacement therapy with testosterone did not seem to impact muscle bulk but elicited a significant improvement in functional status [213]. The positive effects of anabolic steroids, as well as testosterone administration, on protein synthesis, have to be weighed against possible hazards, such as hepatic toxicity, virilising effects, fluid retention, and in the case of testosterone, the development of prostate complications and malignancy. Further development and testing of nonsteroidal androgen receptor modulators (SARMs) might lead to useful alternatives [214], as indeed observed in the case of ostarine [215] and enobosarm [216].
Several cachexia-prone diseases represent a state of GH resistance: high levels of GH and inappropriately low circulating and locally expressed IGF-1, which is considered the main GH mediator [217]. Whereas studies with GH have yielded disappointing results, a more comprehensive approach might be the direct administration of IGF-1. By inhibiting FoxO-1, leading to downregulation of the ubiquitin E3 ligases Murf-1 and MAFbx, sketelal proteosomal protein degradation is reduced; whereas, mainly through the PI3K/Akt pathway, synthesis is stimulated [52, 54]. Recent experimental evidence suggests that IGF-1 supplementation might be a viable option to treat cancer cachexia [218].
Beta-adrenergic agonists inhibit both Ca2+ dependent proteolysis and the FoxO-mediated transcription of E3 ubiquitin ligases. Despite their effective promotion of muscle hypertrophy, their side effects, especially short (tachycardia) as well a long-term (cardiac hypertrophy) cardiovascular risks, preclude current application [219]. In animal models, the newer generation beta-agonist, formoterol, elicited an anabolic response in skeletal muscle at very low doses and might therefore be pursued in future investigations [220].
Anti-inflammatory drugs
Increased levels of proinflammatory cytokines, both in the circulation, as well as locally at the level of the skeletal muscle, have been attributed a key role in the pathophysiology of cachexia and muscle wasting. Hence, the release of potent monoclonal TNFα antibodies opened a new avenue of possible applications. Although not specifically studied as anticachectic drugs, CHF trials with etancercept [221] and infliximab [222] were prematurely stopped because of futility and toxicity, respectively. Nevertheless, these drugs are now successfully used to achieve disease control in patients with rheumatoid arthritis and Crohn's disease. In addition, their potential anticachectic effects are still actively explored in the setting of COPD and cancer. The use of interleukin-6 monoclonal antibodies is still in its early stages. Since a whole cascade of pro- and anti-inflammatory adaptations characterizes cachexia-associated wasting, the “single target approach” of specific cytokine antibodies has been scrutinized. A more general anti-inflammatory approach, as well as the use of so-called “immunomodulators” could be of interest. Cyclooxygenase-2 inhibitors may be beneficial in counteracting muscle wasting. In patients with cancer cachexia, celecoxib indeed led to a significant increase of lean body mass and muscle strength [223]. Thalidomide, pentoxyfilline and statins all exert anti-inflammatory effects but have not been specifically investigated in cachectic patients until now. Despite their potent anti-inflammatory effects, the successful application of 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors in cardiovascular disease was not confirmed in clinical CHF trials [224]. Interestingly, low cholesterol levels, often already found in cachectic patients, may offset the pleiotropic characteristics of statins. Indeed, several in vitro experiments [225] support the neutralizing effects of lipopolysaccharide by cholesterol micelles. The latter may at least partially explain for the unexpectedly improved outcome in CHF patients with elevated plasmalipids [226].
Modulating the energetic crisis of skeletal muscle
Together with GH resistance and downregulation of IGF-1, insulin resistance contributes to the anabolic/catabolic imbalance [227]. The exact mechanisms underlying the initiation of insulin resistance and its relation to muscle wasting remain to be further explored. In CHF patients, increased plasma fatty acids, proinflammatory cytokines, as well as local muscle adiponectin resistance, may culminate into an insulin-resistant state. The adipocyte-derived cytokine adiponectin exerts beneficial insulin-sensitizing and anti-inflammatory effects in situations of insulin resistance, such as obesity and diabetes [228]. Paradoxically, increased adiponectin levels infer poor prognosis in several chronic conditions, such as in the setting of CHF [229] and renal failure [230]. In skeletal biopsies taken from CHF patients, adiponectin expression was significantly upregulated, whereas the reverse was seen for the skeletal muscle adiponectin receptor AdipoR1 [231]. Skeletal muscle metabolic deficiency in CHF was further corroborated by the finding of a deactivated PPARα/AMPK pathway and a concordant downregulation of several target genes, involved in the metabolism of free fatty acids and glucose. Interestingly, these changes were reversible with the introduction of a 4-month exercise training protocol [232]. Targeting insulin resistance by using insulin sensitizer such as thiazolidinediones [233] as well as the application of PPARα agonists have been suggested to beneficially affect protein energy metabolism [234].
Specific proteasome inhibitors
Several compounds designed to act as proteasome inhibitors such as the peptidyl aldehydes carbobenzoxyl-Ile-Glu(O-t-butyl)-Ala-leucinal (PSI), N-acetyl-leucyl-leucyl-norleucinal (LLN), or CBZ-leucyl-leucyl-leucinal (MG132) have helped immensely in understanding the biological role and importance of the ubiquitin–proteasome system during muscle wasting. These compounds reduce the accelerated proteolysis in atrophying muscle and preserve muscle fiber diameter [235–239]. Moreover, inhibition of the proteasome machinery prevents the degradation of IκB, which maintains NF-κB in its inactive state, thus preventing the upregulation of MuRF1 and MaFbx/atrogin-1 as well as the synthesis of TNFα, IL-1, and IL-6 [237, 238]. Other beneficial effects are restoration of Akt phosphorylation, decreased apoptosis, enhanced locomotive activity, and shortening of the rehabilitation period [237, 238]. Besides peptide aldehyde inhibitors, the N-protected dipeptide Pyz-Phe-boroLeu (also known as PS-341, bortezomib, or Velcade) is frequently used to inhibit nonlysosomal protein breakdown. It is a potent proteasome inhibitor and approved by the American Food and Drug Administration for treatment of multiple myeloma. Even though bortezomib can prevent excessive protein degradation and muscle wasting in animal models [240, 241], its use is not yet approved in patients for treatment of muscle atrophy. Indeed, clinical trials with bortezomib for the treatment of cancer cachexia have not shown univocal results [242]. In this light, it is noteworthy that only a partial reduction of denervation-induced atrophy was observed in rat soleus muscle after treatment with bortezomib [240]. This partial effect could be due to other proteolytic mechanisms that are not inhibited by bortezomib, such as lysosomal pathways, which are also involved in muscle wasting (vide supra). Peptide aldehyde inhibitors such as MG132 can repress certain lysosomal cysteine proteases and calpains, which might explain the total rescue of muscle mass in some studies [238].
Exercise training—an all-in-one approach?
The multiple effects of exercise training in reversing atrophy and especially the hypertrophic stimulus of resistance exercises are well known. Regular physical activity, for instance, in patients with CHF, has local and general anti-inflammatory [243, 244] and antioxidative effects [245], decreases insulin and adiponectin resistance [232], has a quiescent influence on the overactivated UPS [246], and reduces skeletal muscle apoptosis [245]. The resulting improvement in terms of physical capacity and quality of life in CHF and COPD patients has stimulated investigators to explore this treatment modality in other conditions, such as cancer, renal failure, and rheumatoid arthritis. Very recent experimental data suggest that in pressure overload-induced heart failure [247], myostatin, produced in the heart itself, translates into remote effects of peripheral muscle wasting. These data are somewhat contradictory to findings from an earlier study, using a different heart failure model (LAD ligation), in which local skeletal muscle myostatin upregulation was shown [248]. Whereas targeted inhibition of myostatin, by using myostatin antibodies or activin receptor IIB blockers, might be a future therapeutic option, it is interesting to mention that exercise training of animals following LAD ligation significantly downregulated skeletal muscle and myocardial myostatin protein overexpression [249]. In that same study, using a C2C12 cell line, TNFα induced the expression of myostatin through a p38 MAPK-dependent pathway involving NF-κB.
Very recently, a combined approach of eicosapentaenoic acid (EPA) and exercise training has been tested on Lewis lung carcinoma-bearing mice [250]. While EPA alone did not prevent the muscle loss induced by tumor growth, the combination with exercise induced a partial rescue of muscle strength and mass in cachectic animals, as corroborated by reduced PAX-7 accumulation and increased PCG-1 protein [250].
Conclusions
Muscle atrophy may arise as a consequence of many different physiological and pathological processes. Regardless of these underlying conditions, counteracting loss of muscle mass is a major issue that should be addressed in order to increase the quality of life in severely ill patients. Based on research conducted in different areas, it is strongly suggested that a plethora of combinations, comprising unbalanced triggers and activated or deactivated pathways, eventually culminate into a hypercatabolic status. These observations clearly lead up to the necessity of elaborating a multifacetted therapeutic approach that involves adequate nutritional support, the implementation of exercise training to stimulate muscle hypertrophy, and one or more pharmacological compounds that specifically antagonize proatrophic pathways. Because of the complexity of each disease and the variation of skeletal muscle wasting among patients, the clinical approach needs to be personalized in order to obtain a targeted therapy and to avoid dangerous side effects.
Acknowledgments
This work was supported by the Fund for Scientific Research (FWO)-Flanders (Belgium), the University of Antwerp, the Associazione “Amici per il Cuore-ONLUS”, Chiari (Brescia)-Italy, and the University of Brescia research fund. FP is an AIRC/Marie Curie fellow in cancer research. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia, and Muscle [251].
Conflict of interest
The authors confirm that there are no conflicts of interest.
Authors' contributions
AF, VC, FP, and WM contributed equally to this paper. All authors read and approved the final manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Cruz-Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, et al. People EWGoSiO. Sarcopenia: European consensus on definition and diagnosis: report of the European Working Group on sarcopenia in older people. Age Ageing. 2010;39:412–23. doi: 10.1093/ageing/afq034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas DR. Loss of skeletal muscle mass in aging: examining the relationship of starvation, sarcopenia and cachexia. Clin Nutr. 2007;26:389–99. doi: 10.1016/j.clnu.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Vandervoort AA. Aging of the human neuromuscular system. Muscle Nerve. 2002;25:17–25. doi: 10.1002/mus.1215. [DOI] [PubMed] [Google Scholar]
- 4.Dedkov EI, Borisov AB, Carlson BM. Dynamics of postdenervation atrophy of young and old skeletal muscles: differential responses of fiber types and muscle types. J Gerontol A Biol Sci Med Sci. 2003;58:984–91. doi: 10.1093/gerona/58.11.b984. [DOI] [PubMed] [Google Scholar]
- 5.Williams AD, Selig S, Hare DL, Hayes A, Krum H, Patterson J, et al. Reduced exercise tolerance in CHF may be related to factors other than impaired skeletal muscle oxidative capacity. J Card Fail. 2004;10:141–8. doi: 10.1016/j.cardfail.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Massie BM, Simonini A, Sahgal P, Wells L, Dudley GA. Relation of systemic and local muscle exercise capacity to skeletal muscle characteristics in men with congestive heart failure. J Am Coll Cardiol. 1996;27:140–5. doi: 10.1016/0735-1097(95)00416-5. [DOI] [PubMed] [Google Scholar]
- 7.Schaufelberger M, Eriksson BO, Lönn L, Rundqvist B, Sunnerhagen KS, Swedberg K. Skeletal muscle characteristics, muscle strength and thigh muscle area in patients before and after cardiac transplantation. Eur J Heart Fail. 2001;3:59–67. doi: 10.1016/s1388-9842(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 8.Harrington D, Anker SD, Chua TP, Webb-Peploe KM, Ponikowski PP, Poole-Wilson PA, et al. Skeletal muscle function and its relation to exercise tolerance in chronic heart failure. J Am Coll Cardiol. 1997;30:1758–64. doi: 10.1016/s0735-1097(97)00381-1. [DOI] [PubMed] [Google Scholar]
- 9.Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, et al. Wasting as independent risk factor for mortality in chronic heart failure. Lancet. 1997;349:1050–3. doi: 10.1016/S0140-6736(96)07015-8. [DOI] [PubMed] [Google Scholar]
- 10.Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 2011;12:489–95. doi: 10.1016/S1470-2045(10)70218-7. [DOI] [PubMed] [Google Scholar]
- 11.Florini JR, Ewton DZ, Magri KA. Hormones, growth factors, and myogenic differentiation. Annu Rev Physiol. 1991;53:201–16. doi: 10.1146/annurev.ph.53.030191.001221. [DOI] [PubMed] [Google Scholar]
- 12.Coleman ME, DeMayo F, Yin KC, Lee HM, Geske R, Montgomery C, et al. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem. 1995;270:12109–16. doi: 10.1074/jbc.270.20.12109. [DOI] [PubMed] [Google Scholar]
- 13.Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, et al. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001;27:195–200. doi: 10.1038/84839. [DOI] [PubMed] [Google Scholar]
- 14.Stewart CE, Rotwein P. Growth, differentiation, and survival: multiple physiological functions for insulin-like growth factors. Physiol Rev. 1996;76:1005–26. doi: 10.1152/physrev.1996.76.4.1005. [DOI] [PubMed] [Google Scholar]
- 15.Harridge SD. Ageing and local growth factors in muscle. Scand J Med Sci Sports. 2003;13:34–9. doi: 10.1034/j.1600-0838.2003.20235.x. [DOI] [PubMed] [Google Scholar]
- 16.Niebauer J, Pflaum CD, Clark AL, Strasburger CJ, Hooper J, Poole-Wilson PA, et al. Deficient insulin-like growth factor I in chronic heart failure predicts altered body composition, anabolic deficiency, cytokine and neurohormonal activation. J Am Coll Cardiol. 1998;32:393–7. doi: 10.1016/s0735-1097(98)00226-5. [DOI] [PubMed] [Google Scholar]
- 17.Costelli P, Muscaritoli M, Bossola M, Penna F, Reffo P, Bonetto A, et al. Rossi Fanelli F. IGF-1 is downregulated in experimental cancer cachexia. Am J Physiol Regul Integr Comp Physiol. 2006;291:R674–83. doi: 10.1152/ajpregu.00104.2006. [DOI] [PubMed] [Google Scholar]
- 18.Jankowska EA, Biel B, Majda J, Szklarska A, Lopuszanska M, Medras M, et al. Anabolic deficiency in men with chronic heart failure: prevalence and detrimental impact on survival. Circulation. 2006;114:1829–37. doi: 10.1161/CIRCULATIONAHA.106.649426. [DOI] [PubMed] [Google Scholar]
- 19.Yakar S, Liu JL, Le Roith D. The growth hormone/insulin-like growth factor-I system: implications for organ growth and development. Pediatr Nephrol. 2000;14:544–9. doi: 10.1007/s004670000363. [DOI] [PubMed] [Google Scholar]
- 20.Yakar S, Liu JL, Fernandez AM, Wu Y, Schally AV, Frystyk J, et al. Liver-specific igf-1 gene deletion leads to muscle insulin insensitivity. Diabetes. 2001;50:1110–8. doi: 10.2337/diabetes.50.5.1110. [DOI] [PubMed] [Google Scholar]
- 21.Pelosi L, Giacinti C, Nardis C, Borsellino G, Rizzuto E, Nicoletti C, et al. Local expression of IGF-1 accelerates muscle regeneration by rapidly modulating inflammatory cytokines and chemokines. FASEB J. 2007;21:1393–402. doi: 10.1096/fj.06-7690com. [DOI] [PubMed] [Google Scholar]
- 22.Denti MA, Rosa A, D'Antona G, Sthandier O, De Angelis FG, Nicoletti C, et al. Body-wide gene therapy of Duchenne muscular dystrophy in the mdx mouse model. Proc Natl Acad Sci U S A. 2006;103:3758–63. doi: 10.1073/pnas.0508917103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barton ER, Morris L, Musaro A, Rosenthal N, Sweeney HL. Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J Cell Biol. 2002;157:137–48. doi: 10.1083/jcb.200108071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L, et al. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol. 2005;168:193–9. doi: 10.1083/jcb.200407021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palazzolo I, Stack C, Kong L, Musaro A, Adachi H, Katsuno M, et al. Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron. 2009;63:316–28. doi: 10.1016/j.neuron.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 2008;23:160–70. doi: 10.1152/physiol.00041.2007. [DOI] [PubMed] [Google Scholar]
- 27.Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol. 2005;37:1974–84. doi: 10.1016/j.biocel.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 28.Glass DJ. Signaling pathways perturbing muscle mass. Curr Opin Clin Nutr Metab Care. 2010;13:225–9. doi: 10.1097/mco.0b013e32833862df. [DOI] [PubMed] [Google Scholar]
- 29.Price SR, Bailey JL, Wang X, Jurkovitz C, England BK, Ding X, et al. Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J Clin Invest. 1996;98:1703–8. doi: 10.1172/JCI118968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernández AM, Kim JK, Yakar S, Dupont J, Hernandez-Sanchez C, Castle AL, et al. Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev. 2001;15:1926–34. doi: 10.1101/gad.908001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balagopal P, Proctor D, Nair KS. Sarcopenia and hormonal changes. Endocrine. 1997;7:57–60. doi: 10.1007/BF02778064. [DOI] [PubMed] [Google Scholar]
- 32.Guillet C, Prod'homme M, Balage M, Gachon P, Giraudet C, Morin L, et al. Impaired anabolic response of muscle protein synthesis is associated with S6K1 dysregulation in elderly humans. FASEB J. 2004;18:1586–7. doi: 10.1096/fj.03-1341fje. [DOI] [PubMed] [Google Scholar]
- 33.García-Martínez C, López-Soriano FJ, Argilés JM. Acute treatment with tumour necrosis factor-alpha induces changes in protein metabolism in rat skeletal muscle. Mol Cell Biochem. 1993;125:11–8. doi: 10.1007/BF00926829. [DOI] [PubMed] [Google Scholar]
- 34.García-Martínez C, Llovera M, Agell N, López-Soriano FJ, Argilés JM. Ubiquitin gene expression in skeletal muscle is increased by tumour necrosis factor-alpha. Biochem Biophys Res Commun. 1994;201:682–6. doi: 10.1006/bbrc.1994.1754. [DOI] [PubMed] [Google Scholar]
- 35.Lee SJ, Lee YS, Zimmers TA, Soleimani A, Matzuk MM, Tsuchida K, et al. Regulation of muscle mass by follistatin and activins. Mol Endocrinol. 2010;24:1998–2008. doi: 10.1210/me.2010-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilson H, Schakman O, Kalista S, Lause P, Tsuchida K, Thissen JP. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am J Physiol Endocrinol Metab. 2009;297:E157–64. doi: 10.1152/ajpendo.00193.2009. [DOI] [PubMed] [Google Scholar]
- 37.McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A. 1997;94:12457–61. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 39.Combaret L, Taillandier D, Dardevet D, Béchet D, Rallière C, Claustre A, et al. Glucocorticoids regulate mRNA levels for subunits of the 19 S regulatory complex of the 26 S proteasome in fast-twitch skeletal muscles. Biochem J. 2004;378:239–46. doi: 10.1042/BJ20031660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wing SS, Goldberg AL. Glucocorticoids activate the ATP-ubiquitin-dependent proteolytic system in skeletal muscle during fasting. Am J Physiol. 1993;264:E668–76. doi: 10.1152/ajpendo.1993.264.4.E668. [DOI] [PubMed] [Google Scholar]
- 41.Chrysis D, Underwood LE. Regulation of components of the ubiquitin system by insulin-like growth factor I and growth hormone in skeletal muscle of rats made catabolic with dexamethasone. Endocrinology. 1999;140:5635–41. doi: 10.1210/endo.140.12.7217. [DOI] [PubMed] [Google Scholar]
- 42.Dehoux M, Van Beneden R, Pasko N, Lause P, Verniers J, Underwood L, et al. Role of the insulin-like growth factor I decline in the induction of atrogin-1/MAFbx during fasting and diabetes. Endocrinology. 2004;145:4806–12. doi: 10.1210/en.2004-0406. [DOI] [PubMed] [Google Scholar]
- 43.Llovera M, García-Martínez C, López-Soriano J, Agell N, López-Soriano FJ, Garcia I, et al. Protein turnover in skeletal muscle of tumour-bearing transgenic mice overexpressing the soluble TNF receptor-1. Cancer Lett. 1998;130:19–27. doi: 10.1016/s0304-3835(98)00137-2. [DOI] [PubMed] [Google Scholar]
- 44.Jagoe RT, Goldberg AL. What do we really know about the ubiquitin-proteasome pathway in muscle atrophy? Curr Opin Clin Nutr Metab Care. 2001;4:183–90. doi: 10.1097/00075197-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 45.Tsujinaka T, Fujita J, Ebisui C, Yano M, Kominami E, Suzuki K, et al. Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J Clin Invest. 1996;97:244–9. doi: 10.1172/JCI118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janssen SP, Gayan-Ramirez G, Van den Bergh A, Herijgers P, Maes K, Verbeken E, et al. Interleukin-6 causes myocardial failure and skeletal muscle atrophy in rats. Circulation. 2005;111:996–1005. doi: 10.1161/01.CIR.0000156469.96135.0D. [DOI] [PubMed] [Google Scholar]
- 47.Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, et al. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296:1486–8. doi: 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]
- 48.Schakman O, Gilson H, Kalista S, Thissen JP. Mechanisms of muscle atrophy induced by glucocorticoids. Horm Res. 2009;72:36–41. doi: 10.1159/000229762. [DOI] [PubMed] [Google Scholar]
- 49.Powers SK, Kavazis AN, McClung JM. Oxidative stress and disuse muscle atrophy. J Appl Physiol. 2007;102:2389–97. doi: 10.1152/japplphysiol.01202.2006. [DOI] [PubMed] [Google Scholar]
- 50.Glass DJ. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol. 2003;5:87–90. doi: 10.1038/ncb0203-87. [DOI] [PubMed] [Google Scholar]
- 51.Lee SJ, Glass DJ. Treating cancer cachexia to treat cancer. Skelet Muscle. 2011;1:2. doi: 10.1186/2044-5040-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, et al. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001;3:1009–13. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- 53.Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, et al. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14:395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- 54.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3:1014–9. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- 56.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–71. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 57.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–83. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 58.Blaauw B, Canato M, Agatea L, Toniolo L, Mammucari C, Masiero E, et al. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. 2009;23:3896–905. doi: 10.1096/fj.09-131870. [DOI] [PubMed] [Google Scholar]
- 59.Lai KM, Gonzalez M, Poueymirou WT, Kline WO, Na E, Zlotchenko E, et al. Conditional activation of akt in adult skeletal muscle induces rapid hypertrophy. Mol Cell Biol. 2004;24:9295–304. doi: 10.1128/MCB.24.21.9295-9304.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, Jacobs J, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17:1352–65. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ohanna M, Sobering AK, Lapointe T, Lorenzo L, Praud C, Petroulakis E, et al. Atrophy of S6K1(-/-) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat Cell Biol. 2005;7:286–94. doi: 10.1038/ncb1231. [DOI] [PubMed] [Google Scholar]
- 62.Léger B, Vergani L, Sorarù G, Hespel P, Derave W, Gobelet C, et al. Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J. 2006;20:583–5. doi: 10.1096/fj.05-5249fje. [DOI] [PubMed] [Google Scholar]
- 63.Dobrowolny G, Aucello M, Rizzuto E, Beccafico S, Mammucari C, Boncompagni S, et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008;8:425–36. doi: 10.1016/j.cmet.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 64.Dobrowolny G, Aucello M, Musarò A. Muscle atrophy induced by SOD1G93A expression does not involve the activation of caspase in the absence of denervation. Skelet Muscle. 2011;1:3. doi: 10.1186/2044-5040-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Price SR, Gooch JL, Donaldson SK, Roberts-Wilson TK. Muscle atrophy in chronic kidney disease results from abnormalities in insulin signaling. J Ren Nutr. 2010;20:S24–8. doi: 10.1053/j.jrn.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang L, Wang XH, Wang H, Du J, Mitch WE. Satellite cell dysfunction and impaired IGF-1 signaling cause CKD-induced muscle atrophy. J Am Soc Nephrol. 2010;21:419–27. doi: 10.1681/ASN.2009060571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Favier FB, Costes F, Defour A, Bonnefoy R, Lefai E, Baugé S, et al. Downregulation of Akt/mammalian target of rapamycin pathway in skeletal muscle is associated with increased REDD1 expression in response to chronic hypoxia. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1659–66. doi: 10.1152/ajpregu.00550.2009. [DOI] [PubMed] [Google Scholar]
- 68.Mallinson JE, Constantin-Teodosiu D, Sidaway J, Westwood FR, Greenhaff PL. Blunted Akt/FOXO signalling and activation of genes controlling atrophy and fuel use in statin myopathy. J Physiol. 2009;587:219–30. doi: 10.1113/jphysiol.2008.164699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crossland H, Constantin-Teodosiu D, Gardiner SM, Constantin D, Greenhaff PL. A potential role for Akt/FOXO signalling in both protein loss and the impairment of muscle carbohydrate oxidation during sepsis in rodent skeletal muscle. J Physiol. 2008;586:5589–600. doi: 10.1113/jphysiol.2008.160150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smith IJ, Lecker SH, Hasselgren PO. Calpain activity and muscle wasting in sepsis. Am J Physiol Endocrinol Metab. 2008;295:E762–71. doi: 10.1152/ajpendo.90226.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sugita H, Kaneki M, Sugita M, Yasukawa T, Yasuhara S, Martyn JA. Burn injury impairs insulin-stimulated Akt/PKB activation in skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E585–91. doi: 10.1152/ajpendo.00321.2004. [DOI] [PubMed] [Google Scholar]
- 72.Penna F, Bonetto A, Muscaritoli M, Costamagna D, Minero VG, Bonelli G, et al. Muscle atrophy in experimental cancer cachexia: is the IGF-1 signaling pathway involved? Int J Cancer. 2010;127:1706–17. doi: 10.1002/ijc.25146. [DOI] [PubMed] [Google Scholar]
- 73.Sishi BJ, Engelbrecht AM. Tumor necrosis factor alpha (TNF-α) inactivates the PI3-kinase/PKB pathway and induces atrophy and apoptosis in L6 myotubes. Cytokine. 2011;54:173–84. doi: 10.1016/j.cyto.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 74.Dogra C, Changotra H, Wedhas N, Qin X, Wergedal JE, Kumar A. TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J. 2007;21:1857–69. doi: 10.1096/fj.06-7537com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zheng B, Ohkawa S, Li H, Roberts-Wilson TK, Price SR. FOXO3a mediates signaling crosstalk that coordinates ubiquitin and atrogin-1/MAFbx expression during glucocorticoid-induced skeletal muscle atrophy. FASEB J. 2010;24:2660–9. doi: 10.1096/fj.09-151480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao W, Qin W, Pan J, Wu Y, Bauman WA, Cardozo C. Dependence of dexamethasone-induced Akt/FOXO1 signaling, upregulation of MAFbx, and protein catabolism upon the glucocorticoid receptor. Biochem Biophys Res Commun. 2009;378:668–72. doi: 10.1016/j.bbrc.2008.11.123. [DOI] [PubMed] [Google Scholar]
- 77.Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, et al. IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J Am Soc Nephrol. 2009;20:604–12. doi: 10.1681/ASN.2008060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fanzani A, Zanola A, Rovetta F, Rossi S, Aleo MF. Cisplatin triggers atrophy of skeletal C2C12 myotubes via impairment of Akt signalling pathway and subsequent increment activity of proteasome and autophagy systems. Toxicol Appl Pharmacol. 2011;250:312–21. doi: 10.1016/j.taap.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 79.Lokireddy S, McFarlane C, Ge X, Zhang H, Sze SK, Sharma M, et al. Myostatin induces degradation of sarcomeric proteins through a Smad3 signaling mechanism during skeletal muscle wasting. Mol Endocrinol. 2011;25:1936–49. doi: 10.1210/me.2011-1124. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 80.Lokireddy S, Mouly V, Butler-Browne G, Gluckman PD, Sharma M, Kambadur R, McFarlane C. Myostatin promotes the wasting of human myoblast cultures through promoting ubiquitin-proteasome pathway-mediated loss of sarcomeric proteins. Am J Physiol Cell Physiol. 2011;301:C1316–24. [DOI] [PubMed]
- 81.Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296:C1258–70. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- 82.Amirouche A, Durieux AC, Banzet S, Koulmann N, Bonnefoy R, Mouret C, et al. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology. 2009;150:286–94. doi: 10.1210/en.2008-0959. [DOI] [PubMed] [Google Scholar]
- 83.Morissette MR, Cook SA, Buranasombati C, Rosenberg MA, Rosenzweig A. Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am J Physiol Cell Physiol. 2009;297:C1124–32. doi: 10.1152/ajpcell.00043.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peter AK, Crosbie RH. Hypertrophic response of Duchenne and limb-girdle muscular dystrophies is associated with activation of Akt pathway. Exp Cell Res. 2006;312:2580–91. doi: 10.1016/j.yexcr.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 85.Gurpur PB, Liu J, Burkin DJ, Kaufman SJ. Valproic acid activates the PI3K/Akt/mTOR pathway in muscle and ameliorates pathology in a mouse model of Duchenne muscular dystrophy. Am J Pathol. 2009;174:999–1008. doi: 10.2353/ajpath.2009.080537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blaauw B, Mammucari C, Toniolo L, Agatea L, Abraham R, Sandri M, et al. Akt activation prevents the force drop induced by eccentric contractions in dystrophin-deficient skeletal muscle. Hum Mol Genet. 2008;17:3686–96. doi: 10.1093/hmg/ddn264. [DOI] [PubMed] [Google Scholar]
- 87.Peter AK, Ko CY, Kim MH, Hsu N, Ouchi N, Rhie S, et al. Myogenic Akt signaling upregulates the utrophin-glycoprotein complex and promotes sarcolemma stability in muscular dystrophy. Hum Mol Genet. 2009;18:318–27. doi: 10.1093/hmg/ddn358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim MH, Kay DI, Rudra RT, Chen BM, Hsu N, Izumiya Y, et al. Myogenic Akt signaling attenuates muscular degeneration, promotes myofiber regeneration and improves muscle function in dystrophin-deficient mdx mice. Hum Mol Genet. 2011;20:1324–38. doi: 10.1093/hmg/ddr015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dahiya S, Bhatnagar S, Hindi SM, Jiang C, Paul PK, Kuang S, et al. Elevated levels of active matrix metalloproteinase-9 cause hypertrophy in skeletal muscle of normal and dystrophin-deficient mdx mice. Hum Mol Genet. 2011;20:4345–59. doi: 10.1093/hmg/ddr362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kumar A, Bhatnagar S. Matrix metalloproteinase inhibitor batimastat alleviates pathology and improves skeletal muscle function in dystrophin-deficient mdx mice. Am J Pathol. 2010;177:248–60. doi: 10.2353/ajpath.2010.091176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kandarian SC, Jackman RW. Intracellular signaling during skeletal muscle atrophy. Muscle Nerve. 2006;33:155–65. doi: 10.1002/mus.20442. [DOI] [PubMed] [Google Scholar]
- 92.Jackman RW, Kandarian SC. The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol. 2004;287:C834–43. doi: 10.1152/ajpcell.00579.2003. [DOI] [PubMed] [Google Scholar]
- 93.Yaron A, Hatzubai A, Davis M, Lavon I, Amit S, Manning AM, et al. Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature. 1998;396:590–4. doi: 10.1038/25159. [DOI] [PubMed] [Google Scholar]
- 94.Hunter RB, Stevenson E, Koncarevic A, Mitchell-Felton H, Essig DA, Kandarian SC. Activation of an alternative NF-kappaB pathway in skeletal muscle during disuse atrophy. FASEB J. 2002;16:529–38. doi: 10.1096/fj.01-0866com. [DOI] [PubMed] [Google Scholar]
- 95.Cai D, Frantz JD, Tawa NE, Melendez PA, Oh BC, Lidov HG, et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119:285–98. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 96.Buck M, Chojkier M. Muscle wasting and dedifferentiation induced by oxidative stress in a murine model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxidants. EMBO J. 1996;15:1753–65. [PMC free article] [PubMed] [Google Scholar]
- 97.Di Marco S, Mazroui R, Dallaire P, Chittur S, Tenenbaum SA, Radzioch D, et al. NF-kappa B-mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR protein, and nitric oxide release. Mol Cell Biol. 2005;25:6533–45. doi: 10.1128/MCB.25.15.6533-6545.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li YP, Reid MB. NF-kappaB mediates the protein loss induced by TNF-alpha in differentiated skeletal muscle myotubes. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1165–70. doi: 10.1152/ajpregu.2000.279.4.R1165. [DOI] [PubMed] [Google Scholar]
- 99.Williams G, Brown T, Becker L, Prager M, Giroir BP. Cytokine-induced expression of nitric oxide synthase in C2C12 skeletal muscle myocytes. Am J Physiol. 1994;267:R1020–5. doi: 10.1152/ajpregu.1994.267.4.R1020. [DOI] [PubMed] [Google Scholar]
- 100.Kanski J, Hong SJ, Schöneich C. Proteomic analysis of protein nitration in aging skeletal muscle and identification of nitrotyrosine-containing sequences in vivo by nanoelectrospray ionization tandem mass spectrometry. J Biol Chem. 2005;280:24261–6. doi: 10.1074/jbc.M501773200. [DOI] [PubMed] [Google Scholar]
- 101.Viner RI, Ferrington DA, Hühmer AF, Bigelow DJ, Schöneich C. Accumulation of nitrotyrosine on the SERCA2a isoform of SR Ca-ATPase of rat skeletal muscle during aging: a peroxynitrite-mediated process? FEBS Lett. 1996;379:286–90. doi: 10.1016/0014-5793(95)01530-2. [DOI] [PubMed] [Google Scholar]
- 102.Haynes V, Traaseth NJ, Elfering S, Fujisawa Y, Giulivi C. Nitration of specific tyrosines in FoF1 ATP synthase and activity loss in aging. Am J Physiol Endocrinol Metab. 2010;298:E978–87. doi: 10.1152/ajpendo.00739.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Beal MF. Oxidatively modified proteins in aging and disease. Free Radic Biol Med. 2002;32:797–803. doi: 10.1016/s0891-5849(02)00780-3. [DOI] [PubMed] [Google Scholar]
- 104.Braga M, Sinha Hikim AP, Datta S, Ferrini MG, Brown D, Kovacheva EL, et al. Involvement of oxidative stress and caspase 2-mediated intrinsic pathway signaling in age-related increase in muscle cell apoptosis in mice. Apoptosis. 2008;13:822–32. doi: 10.1007/s10495-008-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Frost RA, Nystrom GJ, Lang CH. Endotoxin and interferon-gamma inhibit translation in skeletal muscle cells by stimulating nitric oxide synthase activity. Shock. 2009;32:416–26. doi: 10.1097/SHK.0b013e3181a034d2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Agustí A, Morlá M, Sauleda J, Saus C, Busquets X. NF-kappaB activation and iNOS upregulation in skeletal muscle of patients with COPD and low body weight. Thorax. 2004;59:483–7. doi: 10.1136/thx.2003.017640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ramamoorthy S, Donohue M, Buck M. Decreased Jun-D and myogenin expression in muscle wasting of human cachexia. Am J Physiol Endocrinol Metab. 2009;297:E392–401. doi: 10.1152/ajpendo.90529.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Finanger Hedderick EL, Simmers JL, Soleimani A, Andres-Mateos E, Marx R, Files DC, et al. Loss of sarcolemmal nNOS is common in acquired and inherited neuromuscular disorders. Neurology. 2011;76:960–7. doi: 10.1212/WNL.0b013e31821043c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Suzuki N, Mizuno H, Warita H, Takeda S, Itoyama Y, Aoki M. Neuronal NOS is dislocated during muscle atrophy in amyotrophic lateral sclerosis. J Neurol Sci. 2010;294:95–101. doi: 10.1016/j.jns.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 110.Suzuki N, Motohashi N, Uezumi A, Fukada S, Yoshimura T, Itoyama Y, et al. NO production results in suspension-induced muscle atrophy through dislocation of neuronal NOS. J Clin Invest. 2007;117:2468–76. doi: 10.1172/JCI30654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hall DT, Ma JF, Marco SD, Gallouzi IE. Inducible nitric oxide synthase (iNOS) in muscle wasting syndrome, sarcopenia, and cachexia. Aging (Albany NY) 2011;3:702–15. doi: 10.18632/aging.100358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jackson MJ. Free radicals generated by contracting muscle: by-products of metabolism or key regulators of muscle function? Free Radic Biol Med. 2008;44:132–41. doi: 10.1016/j.freeradbiomed.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 113.Kondo H, Miura M, Itokawa Y. Antioxidant enzyme systems in skeletal muscle atrophied by immobilization. Pflugers Arch. 1993;422:404–6. doi: 10.1007/BF00374299. [DOI] [PubMed] [Google Scholar]
- 114.Kondo H, Miura M, Itokawa Y. Oxidative stress in skeletal muscle atrophied by immobilization. Acta Physiol Scand. 1991;142:527–8. doi: 10.1111/j.1748-1716.1991.tb09191.x. [DOI] [PubMed] [Google Scholar]
- 115.Kondo H, Miura M, Nakagaki I, Sasaki S, Itokawa Y. Trace element movement and oxidative stress in skeletal muscle atrophied by immobilization. Am J Physiol. 1992;262:E583–90. doi: 10.1152/ajpendo.1992.262.5.E583. [DOI] [PubMed] [Google Scholar]
- 116.Mastrocola R, Reffo P, Penna F, Tomasinelli CE, Boccuzzi G, Baccino FM, et al. Muscle wasting in diabetic and in tumor-bearing rats: role of oxidative stress. Free Radic Biol Med. 2008;44:584–93. doi: 10.1016/j.freeradbiomed.2007.10.047. [DOI] [PubMed] [Google Scholar]
- 117.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 118.Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–83. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 119.Marmor MD, Yarden Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene. 2004;23:2057–70. doi: 10.1038/sj.onc.1207390. [DOI] [PubMed] [Google Scholar]
- 120.Pickart CM. Ubiquitin enters the new millennium. Mol Cell. 2001;8:499–504. doi: 10.1016/s1097-2765(01)00347-1. [DOI] [PubMed] [Google Scholar]
- 121.Weissman AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol. 2001;2:169–78. doi: 10.1038/35056563. [DOI] [PubMed] [Google Scholar]
- 122.Bartke T, Pohl C, Pyrowolakis G, Jentsch S. Dual role of BRUCE as an antiapoptotic IAP and a chimeric E2/E3 ubiquitin ligase. Mol Cell. 2004;14:801–11. doi: 10.1016/j.molcel.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 123.Berleth ES, Pickart CM. Mechanism of ubiquitin conjugating enzyme E2-230 K: catalysis involving a thiol relay? Biochemistry. 1996;35:1664–71. doi: 10.1021/bi952105y. [DOI] [PubMed] [Google Scholar]
- 124.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–8. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 125.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A. 2001;98:14440–5. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol. 2001;306:717–26. doi: 10.1006/jmbi.2001.4448. [DOI] [PubMed] [Google Scholar]
- 127.McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J Cell Biol. 2002;157:125–36. doi: 10.1083/jcb.200108089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007;6:376–85. doi: 10.1016/j.cmet.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 129.Schulze PC, Fang J, Kassik KA, Gannon J, Cupesi M, MacGillivray C, et al. Transgenic overexpression of locally acting insulin-like growth factor-1 inhibits ubiquitin-mediated muscle atrophy in chronic left-ventricular dysfunction. Circ Res. 2005;97:418–26. doi: 10.1161/01.RES.0000179580.72375.c2. [DOI] [PubMed] [Google Scholar]
- 130.Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, et al. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol. 2009;185:1083–95. doi: 10.1083/jcb.200901052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tintignac LA, Lagirand J, Batonnet S, Sirri V, Leibovitch MP, Leibovitch SA. Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J Biol Chem. 2005;280:2847–56. doi: 10.1074/jbc.M411346200. [DOI] [PubMed] [Google Scholar]
- 132.Lagirand-Cantaloube J, Offner N, Csibi A, Leibovitch MP, Batonnet-Pichon S, Tintignac LA, et al. The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008;27:1266–76. doi: 10.1038/emboj.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ekmark M, Rana ZA, Stewart G, Hardie DG, Gundersen K. De-phosphorylation of MyoD is linking nerve-evoked activity to fast myosin heavy chain expression in rodent adult skeletal muscle. J Physiol. 2007;584:637–50. doi: 10.1113/jphysiol.2007.141457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Goodman CA, Mabrey DM, Frey JW, Miu MH, Schmidt EK, Pierre P, et al. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J. 2011;25:1028–39. doi: 10.1096/fj.10-168799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Krawiec BJ, Frost RA, Vary TC, Jefferson LS, Lang CH. Hindlimb casting decreases muscle mass in part by proteasome-dependent proteolysis but independent of protein synthesis. Am J Physiol Endocrinol Metab. 2005;289:E969–80. doi: 10.1152/ajpendo.00126.2005. [DOI] [PubMed] [Google Scholar]
- 136.Fareed MU, Evenson AR, Wei W, Menconi M, Poylin V, Petkova V, et al. Treatment of rats with calpain inhibitors prevents sepsis-induced muscle proteolysis independent of atrogin-1/MAFbx and MuRF1 expression. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1589–97. doi: 10.1152/ajpregu.00668.2005. [DOI] [PubMed] [Google Scholar]
- 137.Dehoux M, Gobier C, Lause P, Bertrand L, Ketelslegers JM, Thissen JP. IGF-I does not prevent myotube atrophy caused by proinflammatory cytokines despite activation of Akt/Foxo and GSK-3beta pathways and inhibition of atrogin-1 mRNA. Am J Physiol Endocrinol Metab. 2007;292:E145–50. doi: 10.1152/ajpendo.00085.2006. [DOI] [PubMed] [Google Scholar]
- 138.McLoughlin TJ, Smith SM, DeLong AD, Wang H, Unterman TG, Esser KA. FoxO1 induces apoptosis in skeletal myotubes in a DNA-binding-dependent manner. Am J Physiol Cell Physiol. 2009;297:C548–55. doi: 10.1152/ajpcell.00502.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Senf SM, Dodd SL, Judge AR. FOXO signaling is required for disuse muscle atrophy and is directly regulated by Hsp70. Am J Physiol Cell Physiol. 2010;298:C38–45. doi: 10.1152/ajpcell.00315.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kudryashova E, Kudryashov D, Kramerova I, Spencer MJ. Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2 H that binds to skeletal muscle myosin and ubiquitinates actin. J Mol Biol. 2005;354:413–24. doi: 10.1016/j.jmb.2005.09.068. [DOI] [PubMed] [Google Scholar]
- 141.Locke M, Tinsley CL, Benson MA, Blake DJ. TRIM32 is an E3 ubiquitin ligase for dysbindin. Hum Mol Genet. 2009;18:2344–58. doi: 10.1093/hmg/ddp167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Saccone V, Palmieri M, Passamano L, Piluso G, Meroni G, Politano L, et al. Mutations that impair interaction properties of TRIM32 associated with limb-girdle muscular dystrophy 2 H. Hum Mutat. 2008;29:240–7. doi: 10.1002/humu.20633. [DOI] [PubMed] [Google Scholar]
- 143.Kudryashova E, Wu J, Havton LA, Spencer MJ. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum Mol Genet. 2009;18:1353–67. doi: 10.1093/hmg/ddp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ye J, Zhang Y, Xu J, Zhang Q, Zhu D. FBXO40, a gene encoding a novel muscle-specific F-box protein, is upregulated in denervation-related muscle atrophy. Gene. 2007;404:53–60. doi: 10.1016/j.gene.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 145.Zhang Y, Ye J, Chen D, Zhao X, Xiao X, Tai S, et al. Differential expression profiling between the relative normal and dystrophic muscle tissues from the same LGMD patient. J Transl Med. 2006;4:53. doi: 10.1186/1479-5876-4-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shi J, Luo L, Eash J, Ibebunjo C, Glass DJ. The SCF-Fbxo40 Complex Induces IRS1 Ubiquitination in Skeletal Muscle, Limiting IGF1 Signaling. Dev Cell. 2011;21:835–47. [DOI] [PubMed]
- 147.Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 148.Kwon YT, Xia Z, Davydov IV, Lecker SH, Varshavsky A. Construction and analysis of mouse strains lacking the ubiquitin ligase UBR1 (E3alpha) of the N-end rule pathway. Mol Cell Biol. 2001;21:8007–21. doi: 10.1128/MCB.21.23.8007-8021.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lecker SH, Solomon V, Price SR, Kwon YT, Mitch WE, Goldberg AL. Ubiquitin conjugation by the N-end rule pathway and mRNAs for its components increase in muscles of diabetic rats. J Clin Invest. 1999;104:1411–20. doi: 10.1172/JCI7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wing SS, Bedard N. Insulin-like growth factor I stimulates degradation of an mRNA transcript encoding the 14 kDa ubiquitin-conjugating enzyme. Biochem J. 1996;319:455–61. doi: 10.1042/bj3190455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.García-Martínez C, Agell N, Llovera M, López-Soriano FJ, Argilés JM. Tumour necrosis factor-alpha increases the ubiquitinization of rat skeletal muscle proteins. FEBS Lett. 1993;323:211–4. doi: 10.1016/0014-5793(93)81341-v. [DOI] [PubMed] [Google Scholar]
- 152.Llovera M, García-Martínez C, Agell N, Marzábal M, López-Soriano FJ, Argilés JM. Ubiquitin gene expression is increased in skeletal muscle of tumour-bearing rats. FEBS Lett. 1994;338:311–8. doi: 10.1016/0014-5793(94)80290-4. [DOI] [PubMed] [Google Scholar]
- 153.Temparis S, Asensi M, Taillandier D, Aurousseau E, Larbaud D, Obled A, et al. Increased ATP-ubiquitin-dependent proteolysis in skeletal muscles of tumor-bearing rats. Cancer Res. 1994;54:5568–73. [PubMed] [Google Scholar]
- 154.Baracos VE, DeVivo C, Hoyle DH, Goldberg AL. Activation of the ATP-ubiquitin-proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am J Physiol. 1995;268:E996–1006. doi: 10.1152/ajpendo.1995.268.5.E996. [DOI] [PubMed] [Google Scholar]
- 155.Lorite MJ, Thompson MG, Drake JL, Carling G, Tisdale MJ. Mechanism of muscle protein degradation induced by a cancer cachectic factor. Br J Cancer. 1998;78:850–6. doi: 10.1038/bjc.1998.592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Solomon V, Baracos V, Sarraf P, Goldberg AL. Rates of ubiquitin conjugation increase when muscles atrophy, largely through activation of the N-end rule pathway. Proc Natl Acad Sci U S A. 1998;95:12602–7. doi: 10.1073/pnas.95.21.12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Siddiqui RA, Hassan S, Harvey KA, Rasool T, Das T, Mukerji P, et al. Attenuation of proteolysis and muscle wasting by curcumin c3 complex in MAC16 colon tumour-bearing mice. Br J Nutr. 2009;102:967–75. doi: 10.1017/S0007114509345250. [DOI] [PubMed] [Google Scholar]
- 158.Asp ML, Tian M, Wendel AA, Belury MA. Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int J Cancer. 2010;126:756–63. doi: 10.1002/ijc.24784. [DOI] [PubMed] [Google Scholar]
- 159.Wing SS, Haas AL, Goldberg AL. Increase in ubiquitin-protein conjugates concomitant with the increase in proteolysis in rat skeletal muscle during starvation and atrophy denervation. Biochem J. 1995;307:639–45. doi: 10.1042/bj3070639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Moresi V, Williams AH, Meadows E, Flynn JM, Potthoff MJ, McAnally J, et al. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell. 2010;143:35–45. doi: 10.1016/j.cell.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hobler SC, Williams A, Fischer D, Wang JJ, Sun X, Fischer JE, et al. Activity and expression of the 20 S proteasome are increased in skeletal muscle during sepsis. Am J Physiol. 1999;277:R434–40. doi: 10.1152/ajpregu.1999.277.2.R434. [DOI] [PubMed] [Google Scholar]
- 162.Wray CJ, Mammen JM, Hershko DD, Hasselgren PO. Sepsis upregulates the gene expression of multiple ubiquitin ligases in skeletal muscle. Int J Biochem Cell Biol. 2003;35:698–705. doi: 10.1016/s1357-2725(02)00341-2. [DOI] [PubMed] [Google Scholar]
- 163.Granado M, Priego T, Martín AI, Villanúa MA, López-Calderón A. Ghrelin receptor agonist GHRP-2 prevents arthritis-induced increase in E3 ubiquitin-ligating enzymes MuRF1 and MAFbx gene expression in skeletal muscle. Am J Physiol Endocrinol Metab. 2005;289:E1007–14. doi: 10.1152/ajpendo.00109.2005. [DOI] [PubMed] [Google Scholar]
- 164.Doucet M, Russell AP, Léger B, Debigaré R, Joanisse DR, Caron MA, et al. Muscle atrophy and hypertrophy signaling in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;176:261–9. doi: 10.1164/rccm.200605-704OC. [DOI] [PubMed] [Google Scholar]
- 165.Vary TC, Frost RA, Lang CH. Acute alcohol intoxication increases atrogin-1 and MuRF1 mRNA without increasing proteolysis in skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1777–89. doi: 10.1152/ajpregu.00056.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Otis JS, Ashikhmin YI, Brown LA, Guidot DM. Effect of HIV-1-related protein expression on cardiac and skeletal muscles from transgenic rats. AIDS Res Ther. 2008;5:8. doi: 10.1186/1742-6405-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zeman RJ, Zhao J, Zhang Y, Zhao W, Wen X, Wu Y, et al. Differential skeletal muscle gene expression after upper or lower motor neuron transection. Pflugers Arch. 2009;458:525–35. doi: 10.1007/s00424-009-0643-5. [DOI] [PubMed] [Google Scholar]
- 168.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 169.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nishino I. Autophagic vacuolar myopathies. Curr Neurol Neurosci Rep. 2003;3:64–9. doi: 10.1007/s11910-003-0040-y. [DOI] [PubMed] [Google Scholar]
- 172.Raben N, Roberts A, Plotz PH. Role of autophagy in the pathogenesis of Pompe disease. Acta Myol. 2007;26:45–8. [PMC free article] [PubMed] [Google Scholar]
- 173.Suzuki T, Nakagawa M, Yoshikawa A, Sasagawa N, Yoshimori T, Ohsumi Y, et al. The first molecular evidence that autophagy relates rimmed vacuole formation in chloroquine myopathy. J Biochem. 2002;131:647–51. doi: 10.1093/oxfordjournals.jbchem.a003147. [DOI] [PubMed] [Google Scholar]
- 174.Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–15. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 175.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Aucello M, Dobrowolny G, Musarò A. Localized accumulation of oxidative stress causes muscle atrophy through activation of an autophagic pathway. Autophagy. 2009;5:527–9. doi: 10.4161/auto.5.4.7962. [DOI] [PubMed] [Google Scholar]
- 177.Schiaffino S, Hanzlíková V. Studies on the effect of denervation in developing muscle. II. The lysosomal system. J Ultrastruct Res. 1972;39:1–14. doi: 10.1016/s0022-5320(72)80002-9. [DOI] [PubMed] [Google Scholar]
- 178.Sandri M. Autophagy in health and disease. 3. Involvement of autophagy in muscle atrophy. Am J Physiol Cell Physiol. 2010;298:C1291–7. doi: 10.1152/ajpcell.00531.2009. [DOI] [PubMed] [Google Scholar]
- 179.Rossi S, Stoppani E, Martinet W, Bonetto A, Costelli P, Giuliani R, et al. The cytosolic sialidase Neu2 is degraded by autophagy during myoblast atrophy. Biochim Biophys Acta. 2009;1790:817–28. doi: 10.1016/j.bbagen.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 180.Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010;584:1411–6. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]
- 181.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–6. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 182.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–83. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Dirks A, Leeuwenburgh C. Apoptosis in skeletal muscle with aging. Am J Physiol Regul Integr Comp Physiol. 2002;282:R519–27. doi: 10.1152/ajpregu.00458.2001. [DOI] [PubMed] [Google Scholar]
- 184.Dirks-Naylor AJ, Lennon-Edwards S. Cellular and molecular mechanisms of apoptosis in age-related muscle atrophy. Curr Aging Sci. 2011. [PubMed]
- 185.Alway SE, Siu PM. Nuclear apoptosis contributes to sarcopenia. Exerc Sport Sci Rev. 2008;36:51–7. doi: 10.1097/JES.0b013e318168e9dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Belizário JE, Lorite MJ, Tisdale MJ. Cleavage of caspases-1, -3, -6, -8 and -9 substrates by proteases in skeletal muscles from mice undergoing cancer cachexia. Br J Cancer. 2001;84:1135–40. doi: 10.1054/bjoc.2001.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Vescovo G, Dalla Libera L. Skeletal muscle apoptosis in experimental heart failure: the only link between inflammation and skeletal muscle wastage? Curr Opin Clin Nutr Metab Care. 2006;9:416–22. doi: 10.1097/01.mco.0000232902.97286.35. [DOI] [PubMed] [Google Scholar]
- 188.Siu PM, Alway SE. Response and adaptation of skeletal muscle to denervation stress: the role of apoptosis in muscle loss. Front Biosci. 2009;14:432–52. doi: 10.2741/3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sandri M, Carraro U, Podhorska-Okolov M, Rizzi C, Arslan P, Monti D, et al. Apoptosis, DNA damage and ubiquitin expression in normal and mdx muscle fibers after exercise. FEBS Lett. 1995;373:291–5. doi: 10.1016/0014-5793(95)00908-r. [DOI] [PubMed] [Google Scholar]
- 190.Hasselgren PO, Menconi MJ, Fareed MU, Yang H, Wei W, Evenson A. Novel aspects on the regulation of muscle wasting in sepsis. Int J Biochem Cell Biol. 2005;37:2156–68. doi: 10.1016/j.biocel.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 191.Allen DL, Linderman JK, Roy RR, Bigbee AJ, Grindeland RE, Mukku V, et al. Apoptosis: a mechanism contributing to remodeling of skeletal muscle in response to hindlimb unweighting. Am J Physiol. 1997;273:C579–87. doi: 10.1152/ajpcell.1997.273.2.C579. [DOI] [PubMed] [Google Scholar]
- 192.Siu PM. Muscle apoptotic response to denervation, disuse, and aging. Med Sci Sports Exerc. 2009;41:1876–86. doi: 10.1249/MSS.0b013e3181a6470b. [DOI] [PubMed] [Google Scholar]
- 193.Yasuhara S, Perez ME, Kanakubo E, Yasuhara Y, Shin YS, Kaneki M, et al. Skeletal muscle apoptosis after burns is associated with activation of proapoptotic signals. Am J Physiol Endocrinol Metab. 2000;279:E1114–21. doi: 10.1152/ajpendo.2000.279.5.E1114. [DOI] [PubMed] [Google Scholar]
- 194.Conraads VM, Hoymans VY, Vermeulen T, Beckers P, Possemiers N, Maeseneer MD, et al. Exercise capacity in chronic heart failure patients is related to active gene transcription in skeletal muscle and not apoptosis. Eur J Cardiovasc Prev Rehabil. 2009;16:325–32. doi: 10.1097/HJR.0b013e3283244436. [DOI] [PubMed] [Google Scholar]
- 195.Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, et al. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest. 2004;113:115–23. doi: 10.1172/JCI200418330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Pistilli EE, Jackson JR, Alway SE. Death receptor-associated pro-apoptotic signaling in aged skeletal muscle. Apoptosis. 2006;11:2115–26. doi: 10.1007/s10495-006-0194-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.von Haehling S, Lainscak M, Springer J, Anker SD. Cardiac cachexia: a systematic overview. Pharmacol Ther. 2009;121:227–52. doi: 10.1016/j.pharmthera.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 198.Rüegg MA, Glass DJ. Molecular mechanisms and treatment options for muscle wasting diseases. Annu Rev Pharmacol Toxicol. 2011;51:373–95. doi: 10.1146/annurev-pharmtox-010510-100537. [DOI] [PubMed] [Google Scholar]
- 199.Alvers AL, Fishwick LK, Wood MS, Hu D, Chung HS, Dunn WA, et al. Autophagy and amino acid homeostasis are required for chronological longevity in Saccharomyces cerevisiae. Aging Cell. 2009;8:353–69. doi: 10.1111/j.1474-9726.2009.00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Avruch J, Long X, Ortiz-Vega S, Rapley J, Papageorgiou A, Dai N. Amino acid regulation of TOR complex 1. Am J Physiol Endocrinol Metab. 2009;296:E592–602. doi: 10.1152/ajpendo.90645.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Schieke SM, Phillips D, McCoy JP, Aponte AM, Shen RF, Balaban RS, et al. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem. 2006;281:27643–52. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- 202.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–40. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 203.D'Antona G, Ragni M, Cardile A, Tedesco L, Dossena M, Bruttini F, et al. Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metab. 2010;12:362–72. doi: 10.1016/j.cmet.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 204.Eley HL, Russell ST, Tisdale MJ. Effect of branched-chain amino acids on muscle atrophy in cancer cachexia. Biochem J. 2007;407:113–20. doi: 10.1042/BJ20070651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Herningtyas EH, Okimura Y, Handayaningsih AE, Yamamoto D, Maki T, Iida K, et al. Branched-chain amino acids and arginine suppress MaFbx/atrogin-1 mRNA expression via mTOR pathway in C2C12 cell line. Biochim Biophys Acta. 2008;1780:1115–20. doi: 10.1016/j.bbagen.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 206.Colomer R, Moreno-Nogueira JM, García-Luna PP, García-Peris P, García-de-Lorenzo A, Zarazaga A, et al. N-3 fatty acids, cancer and cachexia: a systematic review of the literature. Br J Nutr. 2007;97:823–31. doi: 10.1017/S000711450765795X. [DOI] [PubMed] [Google Scholar]
- 207.van Norren K, Kegler D, Argilés JM, Luiking Y, Gorselink M, Laviano A, et al. Dietary supplementation with a specific combination of high protein, leucine, and fish oil improves muscle function and daily activity in tumour-bearing cachectic mice. Br J Cancer. 2009;100:713–22. doi: 10.1038/sj.bjc.6604905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Palus S, Schur R, Akashi YJ, Bockmeyer B, Datta R, Halem H, et al. Ghrelin and Its Analogues, BIM-28131 and BIM-28125, Improve Body Weight and Regulate the Expression of MuRF-1 and MAFbx in a Rat Heart Failure Model. PLoS One. 2011;6:e26865. doi: 10.1371/journal.pone.0026865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–60. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 210.Sanders PM, Russell ST, Tisdale MJ. Angiotensin II directly induces muscle protein catabolism through the ubiquitin-proteasome proteolytic pathway and may play a role in cancer cachexia. Br J Cancer. 2005;93:425–34. doi: 10.1038/sj.bjc.6602725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Anker SD, Negassa A, Coats AJ, Afzal R, Poole-Wilson PA, Cohn JN, et al. Prognostic importance of weight loss in chronic heart failure and the effect of treatment with angiotensin-converting-enzyme inhibitors: an observational study. Lancet. 2003;361:1077–83. doi: 10.1016/S0140-6736(03)12892-9. [DOI] [PubMed] [Google Scholar]
- 212.Anker SD, Chua TP, Ponikowski P, Harrington D, Swan JW, Kox WJ, et al. Hormonal changes and catabolic/anabolic imbalance in chronic heart failure and their importance for cardiac cachexia. Circulation. 1997;96:526–34. doi: 10.1161/01.cir.96.2.526. [DOI] [PubMed] [Google Scholar]
- 213.Malkin CJ, Pugh PJ, West JN, van Beek EJ, Jones TH, Channer KS. Testosterone therapy in men with moderate severity heart failure: a double-blind randomized placebo controlled trial. Eur Heart J. 2006;27:57–64. doi: 10.1093/eurheartj/ehi443. [DOI] [PubMed] [Google Scholar]
- 214.Bhasin S, Jasuja R. Selective androgen receptor modulators as function promoting therapies. Curr Opin Clin Nutr Metab Care. 2009;12:232–40. doi: 10.1097/MCO.0b013e32832a3d79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zilbermint MF, Dobs AS. Nonsteroidal selective androgen receptor modulator Ostarine in cancer cachexia. Future Oncol. 2009;5:1211–20. doi: 10.2217/fon.09.106. [DOI] [PubMed] [Google Scholar]
- 216.Dalton JT, Barnette KG, Bohl CE, Hancock ML, Rodriguez D, Dodson ST, et al. The selective androgen receptor modulator GTx-024 (enobosarm) improves lean body mass and physical function in healthy elderly men and postmenopausal women: results of a double-blind, placebo-controlled phase II trial. J Cachexia Sarcopenia Muscle. 2011;2:153–61. doi: 10.1007/s13539-011-0034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Anker SD, Volterrani M, Pflaum CD, Strasburger CJ, Osterziel KJ, Doehner W, et al. Acquired growth hormone resistance in patients with chronic heart failure: implications for therapy with growth hormone. J Am Coll Cardiol. 2001;38:443–52. doi: 10.1016/s0735-1097(01)01385-7. [DOI] [PubMed] [Google Scholar]
- 218.Schmidt K, von Haehling S, Doehner W, Palus S, Anker SD, Springer J. IGF-1 treatment reduces weight loss and improves outcome in a rat model of cancer cachexia. J Cachexia Sarcopenia Muscle. 2011;2:105–9. doi: 10.1007/s13539-011-0029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Ryall JG, Lynch GS. The potential and the pitfalls of beta-adrenoceptor agonists for the management of skeletal muscle wasting. Pharmacol Ther. 2008;120:219–32. doi: 10.1016/j.pharmthera.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 220.Kenley RA, Denissenko MF, Mullin RJ, Story J, Ekblom J. Formoterol fumarate and roxithromycin effects on muscle mass in an animal model of cancer cachexia. Oncol Rep. 2008;19:1113–21. [PubMed] [Google Scholar]
- 221.Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL) Circulation. 2004;109:1594–602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- 222.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT, Investigators A-TTACHF. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107:3133–40. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- 223.Mantovani G, Macciò A, Madeddu C, Serpe R, Antoni G, Massa E, et al. Phase II nonrandomized study of the efficacy and safety of COX-2 inhibitor celecoxib on patients with cancer cachexia. J Mol Med (Berl) 2010;88:85–92. doi: 10.1007/s00109-009-0547-z. [DOI] [PubMed] [Google Scholar]
- 224.Tavazzi L, Maggioni AP, Marchioli R, Barlera S, Franzosi MG, Latini R, et al. Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:1223–30. doi: 10.1016/S0140-6736(08)61239-8. [DOI] [PubMed] [Google Scholar]
- 225.Sharma R, von Haehling S, Rauchhaus M, Bolger AP, Genth-Zotz S, Doehner W, et al. Whole blood endotoxin responsiveness in patients with chronic heart failure: the importance of serum lipoproteins. Eur J Heart Fail. 2005;7:479–84. doi: 10.1016/j.ejheart.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 226.Rauchhaus M, Doehner W, Francis DP, Davos C, Kemp M, Liebenthal C, et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation. 2000;102:3060–7. doi: 10.1161/01.cir.102.25.3060. [DOI] [PubMed] [Google Scholar]
- 227.Doehner W, Rauchhaus M, Ponikowski P, Godsland IF, von Haehling S, Okonko DO, et al. Impaired insulin sensitivity as an independent risk factor for mortality in patients with stable chronic heart failure. J Am Coll Cardiol. 2005;46:1019–26. doi: 10.1016/j.jacc.2005.02.093. [DOI] [PubMed] [Google Scholar]
- 228.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–80. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 229.Kistorp C, Faber J, Galatius S, Gustafsson F, Frystyk J, Flyvbjerg A, et al. Plasma adiponectin, body mass index, and mortality in patients with chronic heart failure. Circulation. 2005;112:1756–62. doi: 10.1161/CIRCULATIONAHA.104.530972. [DOI] [PubMed] [Google Scholar]
- 230.Cui J, Panse S, Falkner B. The role of adiponectin in metabolic and vascular disease: a review. Clin Nephrol. 2011;75:26–33. [PubMed] [Google Scholar]
- 231.Van Berendoncks AM, Garnier A, Beckers P, Hoymans VY, Possemiers N, Fortin D, et al. Functional adiponectin resistance at the level of the skeletal muscle in mild to moderate chronic heart failure. Circ Heart Fail. 2010;3:185–94. doi: 10.1161/CIRCHEARTFAILURE.109.885525. [DOI] [PubMed] [Google Scholar]
- 232.Van Berendoncks AM, Garnier A, Beckers P, Hoymans VY, Possemiers N, Fortin D, et al. Exercise training reverses adiponectin resistance in skeletal muscle of patients with chronic heart failure. Heart. 2011;97:1403–9. doi: 10.1136/hrt.2011.226373. [DOI] [PubMed] [Google Scholar]
- 233.Siew ED, Ikizler TA. Insulin resistance and protein energy metabolism in patients with advanced chronic kidney disease. Semin Dial. 2010;23:378–82. doi: 10.1111/j.1525-139X.2010.00763.x. [DOI] [PubMed] [Google Scholar]
- 234.Castillero E, Nieto-Bona MP, Fernández-Galaz C, Martín AI, López-Menduiña M, Granado M, et al. Fenofibrate, a PPAR{alpha} agonist, decreases atrogenes and myostatin expression and improves arthritis-induced skeletal muscle atrophy. Am J Physiol Endocrinol Metab. 2011;300:E790–9. doi: 10.1152/ajpendo.00590.2010. [DOI] [PubMed] [Google Scholar]
- 235.Tawa NE, Odessey R, Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest. 1997;100:197–203. doi: 10.1172/JCI119513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Fischer D, Gang G, Pritts T, Hasselgren PO. Sepsis-induced muscle proteolysis is prevented by a proteasome inhibitor in vivo. Biochem Biophys Res Commun. 2000;270:215–21. doi: 10.1006/bbrc.2000.2398. [DOI] [PubMed] [Google Scholar]
- 237.Jamart C, Raymackers JM, Li An G, Deldicque L, Francaux M. Prevention of muscle disuse atrophy by MG132 proteasome inhibitor. Muscle Nerve. 2011;43:708–16. doi: 10.1002/mus.21949. [DOI] [PubMed] [Google Scholar]
- 238.Caron AZ, Haroun S, Leblanc E, Trensz F, Guindi C, Amrani A, et al. The proteasome inhibitor MG132 reduces immobilization-induced skeletal muscle atrophy in mice. BMC Musculoskelet Disord. 2011;12:185. doi: 10.1186/1471-2474-12-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Carmignac V, Quéré R, Durbeej M. Proteasome inhibition improves the muscle of laminin α2 chain-deficient mice. Hum Mol Genet. 2011;20:541–52. doi: 10.1093/hmg/ddq499. [DOI] [PubMed] [Google Scholar]
- 240.Beehler BC, Sleph PG, Benmassaoud L, Grover GJ. Reduction of skeletal muscle atrophy by a proteasome inhibitor in a rat model of denervation. Exp Biol Med (Maywood) 2006;231:335–41. doi: 10.1177/153537020623100315. [DOI] [PubMed] [Google Scholar]
- 241.Kwon DY, Motley WW, Fischbeck KH, Burnett BG. Increasing expression and decreasing degradation of SMN ameliorate the spinal muscular atrophy phenotype in mice. Hum Mol Genet. 2011;20:3667–77. doi: 10.1093/hmg/ddr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Madeddu C, Mantovani G. An update on promising agents for the treatment of cancer cachexia. Curr Opin Support Palliat Care. 2009;3:258–62. doi: 10.1097/SPC.0b013e3283311c6f. [DOI] [PubMed] [Google Scholar]
- 243.Conraads VM, Beckers P, Bosmans J, De Clerck LS, Stevens WJ, Vrints CJ, et al. Combined endurance/resistance training reduces plasma TNF-alpha receptor levels in patients with chronic heart failure and coronary artery disease. Eur Heart J. 2002;23:1854–60. doi: 10.1053/euhj.2002.3239. [DOI] [PubMed] [Google Scholar]
- 244.Gielen S, Adams V, Möbius-Winkler S, Linke A, Erbs S, Yu J, et al. Anti-inflammatory effects of exercise training in the skeletal muscle of patients with chronic heart failure. J Am Coll Cardiol. 2003;42:861–8. doi: 10.1016/s0735-1097(03)00848-9. [DOI] [PubMed] [Google Scholar]
- 245.Linke A, Adams V, Schulze PC, Erbs S, Gielen S, Fiehn E, et al. Antioxidative effects of exercise training in patients with chronic heart failure: increase in radical scavenger enzyme activity in skeletal muscle. Circulation. 2005;111:1763–70. doi: 10.1161/01.CIR.0000165503.08661.E5. [DOI] [PubMed] [Google Scholar]
- 246.Adams V, Mangner N, Gasch A, Krohne C, Gielen S, Hirner S, et al. Induction of MuRF1 is essential for TNF-alpha-induced loss of muscle function in mice. J Mol Biol. 2008;384:48–59. doi: 10.1016/j.jmb.2008.08.087. [DOI] [PubMed] [Google Scholar]
- 247.Heineke J, Auger-Messier M, Xu J, Sargent M, York A, Welle S, et al. Genetic deletion of myostatin from the heart prevents skeletal muscle atrophy in heart failure. Circulation. 2010;121:419–25. doi: 10.1161/CIRCULATIONAHA.109.882068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Lenk K, Schur R, Linke A, Erbs S, Matsumoto Y, Adams V, et al. Impact of exercise training on myostatin expression in the myocardium and skeletal muscle in a chronic heart failure model. Eur J Heart Fail. 2009;11:342–8. doi: 10.1093/eurjhf/hfp020. [DOI] [PubMed] [Google Scholar]
- 249.Lenk K, Erbs S, Höllriege R, Beck E, Linke A, Gielen S, Möbius Winkler S, Sandri M, Hambrecht R, Schuler G, Adams V. Exercise training leads to a reduction of elevated myostatin levels in patients with chronic heart failure. Eur J Cardiovasc Prev Rehabil. 2011;19:404–11. [DOI] [PubMed]
- 250.Penna F, Busquets S, Pin F, Toledo M, Baccino FM, López-Soriano FJ, et al. Combined approach to counteract experimental cancer cachexia: eicosapentaenoic acid and training exercise. J Cachexia Sarcopenia Muscle. 2011;2:95–104. doi: 10.1007/s13539-011-0028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.von Haehling S, Morley JE, Coats AJ, Anker SD. Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. J Cachexia Sarcopenia Muscle. 2010;1:7–8. doi: 10.1007/s13539-010-0003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]