

Abstract
We have previously reported that mice with a dominant negative transforming growth factor β receptor restricted to T cells (dnTGFβRII mice) develop an inflammatory biliary ductular disease that strongly resembles human primary biliary cirrhosis (PBC). Furthermore, deletion of the gene encoding interleukin (IL)-12p40 resulted in a strain (IL-12p40−/−dnTGFβRII) with dramatically reduced autoimmune cholangitis. To further investigate the role of the IL-12 cytokine family in dnTGFβRII autoimmune biliary disease, we deleted the gene encoding the IL-12p35 subunit from dnTGFβRII mice, resulting in an IL-12p35−/− dnTGFβRII strain which is deficient in two members of the IL-12 family, IL-12 and IL-35. In contrast to IL-12p40−/− mice, the IL-12p35−/− mice developed liver inflammation and bile duct damage with similar severity but delayed onset as the parental dnTGFβRII mice. The p35−/− mice also demonstrated a distinct cytokine profile characterized by a shift from a Th1 to a Th17 response. Strikingly, liver fibrosis was frequently observed in IL-12p35−/− mice. In conclusion, IL-12p35−/− dnTGFβRII mice, histologically and immunologically, reflect key features of PBC, providing a useful generic model to understand the immunopathology of human PBC.
Keywords: Primary biliary cirrhosis, murine models, autoimmunity, cholangitis
Primary biliary cirrhosis (PBC) is an organ-specific autoimmune disease characterized by destruction of intrahepatic small bile duct biliary epithelial cells (1). The serological hallmark of PBC is the presence of anti-mitochondrial auto-antibodies (AMA) directed against the pyruvate dehydrogenase E2 complex (PDC-E2) located within the inner membrane of mitochondria (2–4). We previously reported that mice transgenic for directed expression of a dominant negative form of transforming growth factor beta receptor type II (dnTGFβRII), under the control of the CD4 promoter lacking the CD8 silencer, spontaneously develop an autoimmune biliary ductular disease (5). This disease is associated with the spontaneous production of AMAs directed to the same mitochondrial autoantigens recognized by sera from PBC patients (6) with lymphocytic liver in ltration and periportal in ammation analogous to human PBC. The murine serum cytokine pro le is similar to sera of patients with PBC. These findings indicate that the dnTGFβRII mice are a useful animal model for studying the pathogenic mechanisms of human PBC. We have demonstrated that deleting the p40 chain of IL-12 from dnTGFβRII mice produced a marked diminution in the levels of proinflammatory Th1 cytokines in livers with accompanying reductions in cellular infiltrates in portal tracts and diminished bile duct damage (7).
IL-12, the prototypic member of the heterodimeric family of cytokines, consists of a p40 and a p35 subunit covalently linked by two disulfide linkages. Both p35 and p40 are components of two heterodimeric cytokines in the IL-12 family (8). In order to further examine and differentiate the role of the p35- and p40-containing members of the IL-12 cytokine family in dnTGFβRII disease, we generated an IL-12p35−/− mouse strain on the dnTGFβRII background. Our results indicate that in contrast to the IL-12p40−/− mice that were protected from liver inflammation, the IL-12p35−/− mice developed liver inflammation with similar severity but delayed onset compared to the parental dnTGFβRII mice. The p35−/− mice demonstrate a distinct cytokine profile, with enhanced IL-17, compared to parental dnTGFβRII and p40−/− mice. Strikingly, the deletion of IL-12p35 subunit from dnTGFβRII mice resulted in frequent development of liver fibrosis. This model is unique in that it has resemblance to a number of immunological and histological features of human PBC. Although we do not opine that it recapitulates PBC faithfully, we submit that it is a useful system to dissect the cellular and molecular basis of loss of tolerance and liver damage.
Experimental procedures
Animals
The dnTGFβRII colony on a B6 background (B6.Cg-Tg(Cd4-TGFBR2)16Flv/J ) was maintained at the University of California at Davis animal facility (Davis, CA) and bred as hemizygotes due to the severe inflammatory bowel disease of homozygotes. The dnTGFβRII mice used herein are on a B6 background. Essentially, the transgenic founder mice were backcrossed to B10.BR mice for three generations and thence backcrossed to C57BL/6 mice for 15 generations (9). Further descriptions of the genetics of these mice are found in http://jaxmice.jax.org/strain/005551.html. B6.129S1-Il12btm1Jm/J (IL-12p40−/−) and B6.129S1-Il12atm1Jm/J (IL-12p35−/−) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). IL-12p40−/−dnTGFβRII mice were generated as previously described (5, 7). Similarly, male dnTGFβRII mice were mated with female IL-12p35−/− mice to obtain IL-12p35+/−dnTGFβRII mice, which were subsequently back-crossed with female IL-12p35−/− mice to obtain IL-12p35−/−dnTGFβRII mice. The parental dnTGFβRII and the derived IL-12p35−/−dnTGFβRII mice at 3 to 4 weeks of age were genotyped to confirm the dnTGFβRII gene and IL-12p35−/− in their genomic DNA (5). Male hemizygous dnTGFβRII, hemizygous IL-12p35−/− dnTGFβRII mice and hemizygous IL-12p40−/−dnTGFβRII mice were backcrossed onto female C57BL/6 (B6), IL-35−/− and p40−/− mice, respectively (5). All mice were fed sterile rodent Helicobacter Medicated Dosing System (three-drug combination) diets (Bio-Serv, Frenchtown, NJ) and maintained in individually ventilated cages under specific pathogen-free conditions. Sulfatrim (Hi-tech Pharmacal, Amityville, NY) was delivered through drinking water according to the manufacturer’s instructions. At 12 and 24 weeks of age, animals were sacrificed and their liver and colon tissues were processed as outlined below. In addition, liver mononuclear cells were isolated and analyzed as below. The experimental protocols were approved by the University of California Animal Care and Use Committee.
Serum AMAs
Serum samples collected at different ages were tested for levels of anti-PDC-E2 antibodies using an enzyme-linked immunosorbent assay (ELISA). Briefly, 96-well ELISA plates were coated with 5 μg/ml of purified recombinant PDC-E2 in carbonate buffer (pH 9.6) at 4°C overnight, washed with Tris-Buffered Saline Tween-20 (TBS-T), and blocked with 5% skim milk in TBS for 30 minutes. Serum samples at 1:100 dilution were added to individual wells of the microtiter plate and incubated for 1 hour at room temperature (RT). After washing, horseradish peroxidase (HRP)-conjugated anti-human immunoglobulin (A+M+G) (H+L) (1:2,000) (Zymed, San Francisco, CA) was added. The plates were incubated for 1 hour at RT, then washed. OD450nm was measured after addition of TMB peroxidase substrate (BD Biosciences, San Jose, CA) and incubation at RT for 15 minutes. Previously calibrated positive and negative standards were included with each assay.
Histopathology
The harvested liver and colon tissues were fixed in 4% paraformaldehyde at room temperature for 2 days, embedded in paraffin, and cut into 4-μm sections. The sections were deparaffinized, stained with hematoxylin and eosin (H&E), and evaluated by a “blinded” pathologist for pathological changes. To evaluate the severity of fibrosis, the images of the H&E-stained slides were captured using a microscope at a magnification of 40x. A standard 1063 × 797 pixel region of interest (ROI) was taken from each sample by the “blinded” pathologist and analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA), which provides the percentage of fibrosis area in each ROI. Masson-trichrome and Sirius Red stain was also used for visualizing fibrosis on liver tissue sections.
Flow Cytometry
Liver infiltrating mononuclear cells (MNCs) were isolated as described (10). The cells were re-suspended in staining buffer (0.2% BSA, 0.04% EDTA and 0.05% sodium azide in PBS), divided into 25μl aliquots, and incubated with anti-human FcR blocking reagent (eBioscience) for 15 minutes at 4°C. Cells were then washed and stained for 30 minutes at 4°C with cocktails containing combinations of fluorochrome conjugated monoclonal antibodies for different cell surface markers including CD8a, CD4, NK1.1 (Biolegend, San Diego, CA), CD19 and TCRβ (eBioscience, San Diego, CA). IgG isotype antibodies with matching conjugates were used as negative controls. The cells were then washed once with PBS containing 0.2% BSA. For intracellular cytokine staining, cells were resuspended in 10% FBS RPMI and stimulated with Leukocyte Activation Cocktail in the presence of BD GolgiPlug (BD Pharmingen, San Diego, CA) at 37°C for 4 hours. The cells were stained for surface CD4, NK1.1 and TCRβ, fixed and permeabilized with BD Cytofix/Cytoperm Solution (BD Biosciences, San Diego, CA), then stained for intracellular interferon γ (IFN-γ), IL-4 and IL-17 (BioLegend). Normal IgG isotype controls were used in parallel. A FACScan flow cytometer (BD Immunocytometry Systems, San Jose, CA) upgraded for the detection of 5-colors by Cytek Development (Fremont, CA) was used to acquire data, which were analyzed with Cellquest PRO software (BD Immunocytometry Systems).
Cell Culture and Cytokine Detection
For analysis of secreted cytokines, 2.0 × 105 hepatic MNCs were cultured in 96-well round-bottom plates in 200μl of RPMI supplemented with 10% heat-inactivated fetal bovine serum (FBS) (GIBCO-Invitrogen Corp., Grand Island, NY), 100 μg/mL streptomycin, 100 U/mL penicillin, and 0.5 μg/mL each of anti-CD3 (BioLegend) and anti-CD28 (BioLegend). The cells were incubated for 72 hours at 37°C in a humidified 5% CO2 incubator, then centrifuged. The supernatant was collected and analyzed for concentration of cytokines. IL-22 level was measured using an ELISA kit (BioLegend). The levels of IFN-γ, tumor necrosis factor α (TNF-α), IL-6, and IL-17 were measured simultaneously with a mouse cytometric bead array (CBA) kit (BD Biosciences), using a FACScan flow cytometer with CBA software (BD Biosciences).
Assay of Hepatic Hydroxyproline Content
Hepatic hydroxyproline content were quantified using an hydroxyproline assay kit (BioVision, Mountain View, CA). Briefly, frozen liver tissue from 24 week old mice, in groups of 5–8 mice, were weighed and homogenized in distilled H2O and hydrolyzed. 10 μl of hydrolysate was transferred to a 96-well plate and evaporated to dryness under vacuum; a standard curve was applied following the kit protocol. Total hydroxyproline content was measured and data presented per gram of wet weight liver tissue.
Liver Fibrosis of qPCR Array
A murine fibrosis PCR array (SABiosciences/Qiagen, Frederick, MD) was performed to assess the expression of 84 key genes involved in fibrogenesis or fibronolysis in liver. Briefly, total RNA was extracted from individual liver tissues using the Qiagen RNAeasy Mini Kit (Qiagen, Valencia, CA). For qPCR array analysis, 1 μg of total RNA was reverse transcribed and then quantified on an ABI ViiA™ 7 Real-Time PCR System (Applied Biosystems, Foster City, CA). Amplification was performed for 40 cycles in a total volume of 10 μl and products were detected using SYBR Green (Applied Biosystems, Foster City, CA). The relative level of expression of each target gene was determined by normalizing its mRNA level to internal control genes.
Statistical analysis
Cytokine production, portal inflammation, and bile duct damage in different mouse strains were compared by a two-tailed unpaired Mann-Whitney test. The frequency of fibrosis in the different mouse strains was compared using the Fisher’s exact test. In addition, the Tukey-Kramer Multiple Comparisons Test was performed to compare the quantification of fibrosis in different groups. Hypothesis tests were declared statistically significant for attained significance levels of p < 0.05.
Results
IL-12p35−/−dnTGFβRII mice developed liver inflammation with similar severity but delayed onset
To distinguish the role of IL-12p35 from that of IL-12p40 in dnTGFβRII biliary disease, we generated IL-12p35−/−dnTGFβRII mice and compared the data with the parental dnTGFβRII mice and IL-12p40−/−dnTGFβRII mice (Fig. 1). Histological examination of liver demonstrated that at 12 weeks, both the p40−/− and p35−/− mice had significantly milder portal inflammation compared with the parental dnTGFβRII mice (p<0.05). By 24 weeks, however, the p35−/− mice and the parental dnTGFβRII mice had similar portal tract lymphocyte infiltration; both were significantly more severe than the p40−/− mice (p<0.001) (Fig. 1A and 1B). Biliary duct damage was not observed at 12 weeks in any group except for one out of 9 animals in the dnTGFβRII group (Fig. 1B), but at 24 weeks was readily detectable in the p35−/− and dnTGFβRII mice but not p40−/− mice (Fig. 1A and 1B). In addition, histological analysis of colonic tissues demonstrated the absence of lymphocyte infiltration in both p40−/− and p35−/− mice compared to parental dnTGFβRII mice (p<0.001) (Fig. 1C and 1D), indicating that deletion of p35 differentially affected the liver versus colonic inflammatory response compared to dnTGFβRII mice.
Figure 1.

Liver histopathology in IL-12p40−/−dnTGFβRII (p40−/−), IL-12p35−/−dnTGFβRII (p35−/−) and dnTGFβRII (+/+) mice. Representative H&E staining of liver sections (A) and colon sections (C) at age of 24 weeks. B. Portal inflammation, bile duct damage and colon inflammation (D) were examined in individual animals at 12 weeks and 24 weeks. At each time point 8 to 14 animals from each group were examined. The levels of inflammation and bile duct damage were coded as: 0, none; 1, mild; 2, moderate; and 3, severe. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The liver histological data were largely correlated with the number of MNCs recovered from the liver tissues. At 12 weeks, only dnTGFβRII mice had a significantly enlarged spleen (Fig. 2A). However, a significantly reduced liver weight was observed in p35−/−mice at 12 and 24 weeks (Fig. 2A). Both the p40−/− mice and the p35−/− mice had significantly fewer intrahepatic MNCs than dnTGFβRII mice (p<0.05); by 24 weeks no significant difference was detected between p35−/− mice and dnTGFβRII mice, while the hepatic MNCs in the p40−/− mice remained at significantly lower levels (Fig. 2B). While the numbers of liver-infiltrating CD4 T cells were reduced in both the p40−/− and p35−/−mice compared to the dnTGFβRII mice (p<0.01), the numbers of p35−/− −CD8 T cells increased by 24 weeks and was significantly higher than in p40−/− or dnTGFβRII mice (p<0.05) (Fig. 2B). The numbers of intrahepatic B cells, NK cells and NKT cells were all significantly lower in p40−/− mice than dnTGFβRII mice at both 12 and 24 weeks, while those of the p35−/− mice were in general intermediate between the other two mouse strains (data not shown). In summary, these results indicate that absence of IL-12 p35 resulted in reduced liver inflammation at 12 weeks but not 24 weeks of age compared to dnTGFβRII mice. In contrast, deletion of the IL-12 p40 resulted in complete protection against liver inflammation and bile duct damage at both 12 and 24 weeks.
Figure 2.

Numbers of liver infiltrating MNC, CD4 T cells and CD8 T cells.
A. Weight of spleen and liver at age of 12 and 24 weeks. B. Hepatic MNC were isolated from liver samples of IL-12p40−/−dnTGFβRII (p40−/−), IL-12p35−/−dnTGFβRII (p35−/−) and dnTGFβRII (+/+) mice at 12 and 24 weeks, then analyzed by flow cytometry. At each time point 8 to 14 animals from each group were examined. *, P < 0.05, **, P < 0.01, ***, P < 0.001.
Enhanced liver fibrosis in IL-12p35−/−dnTGFβRII mice
Liver fibrosis is characteristic of human PBC but has not been previously reported in murine models of PBC. To assess the extent of fibrosis, Masson’s trichrome staining (Fig. 3B) and Sirius Red staining (Fig. 3D) were performed to detect collagen distribution. At 24 weeks, fibrosis was detected in 54% (7/13) of the p35−/− mice but none of 14 dnTGFβRII mice (p <0.05, Fisher’s exact test) (Fig. 3C). The results of liver fibrosis quantification with image analysis of Sirius Red staining sections further confirmed the presence of fibrosis in p35−/− mice (Fig. 3E). Mild fibrosis was also observed in one out of eight p40−/− mice. However, the severity of fibrosis in this mouse was substantially lower than the p35−/− mice with fibrosis, based on the area of fibrosis (Fig. 3C and 3E). The hepatic hydroxyproline content was significantly higher in p35−/− mice than p40−/− (p=0.016) and dnTGFβRII mice (p=0.007) at 24 weeks (Fig. 3F).
Figure 3.
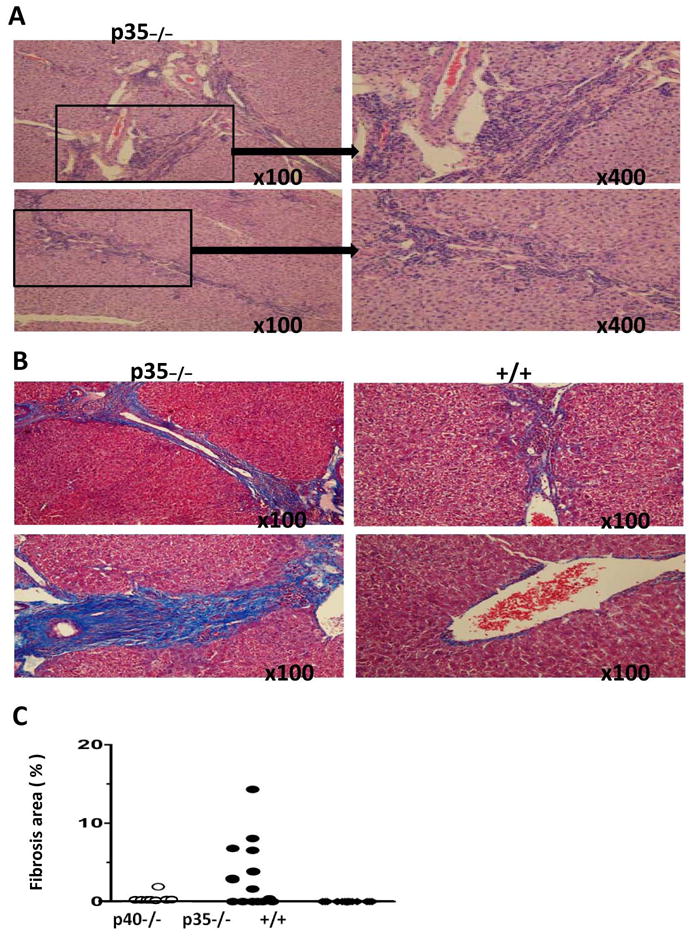

Fibrosis in liver sections from IL-12p40−/−dnTGFβRII (p40−/−), IL-12p35−/−dnTGFβRII (p35−/−) and dnTGFβRII (+/+) mice. A. Representative liver sections from two 24 week-old p35−/−, stained with hematoxylin and eosin (H&E). B. Representative liver sections from two p35−/−) and two dnTGFβRII mice, stained with Masson trichrome. C. Severity of fibrosis, measured as the percentage of fibrosis area, in individual animals of the different mouse strains. D. Representative liver sections from 24 week-old p40−/−, p35−/− and +/+ mice stained with Sirius Red. E. Sirius red positive stained area were quantified using Image J software. Data represent as means ± SD (n=5–6 per group). **P <0.01. F. Hepatic hydroxyproline content was measured from tissues of 24 week-old p40−/− (n=5), p35−/− (n=8) and +/+ mice (n=7). Data represent as μg per gram wet weight. *, P < 0.05, **, P < 0.01.
Enhanced liver fibrosis is associated with decreased expression of Th1 response-related genes
To address the potential mechanism of fibrosis in IL-12p35−/− dnTGFβRII mice, we examined eighty-four genes associated with dysregulated wound healing, tissue repair and remodeling in three groups using a PCR array. A total of 13 genes were significantly downregulated in the IL-12p35−/−dnTGFβRII group (Table 1), including the important negative regulators in liver fibrosis STAT1, IFN-γ, and hepatocyte growth factor (HGF), suggesting that reduced expression levels of IFN-γ/STAT1 signaling and antifibrotic factor-HGF are involved in development of fibrosis in IL-12p35−/−dnTGFβRII mice.
Table 1.
PCR array analysis of fibrosis-related gene expression in liver tissue
| Gene | Protein | IL-12p35−/− dnTGFβRII versus dnTGFβRII mice | IL-12p35−/− dnTGFβRII versus IL-12p40−/−dnTGFβRII mice | ||
|---|---|---|---|---|---|
| p value | Fold Up- or Down- Regulation | p value | Fold Up- or Down- Regulation | ||
| Stat1 | STAT1 | 0.001 | −2.26 | 0.036 | 1.57 |
| Il1a | IL-1α | 0.005 | −1.85 | 0.788 | −1.00 |
| Fasl | Fas Ligand | 0.005 | −2.48 | 0.435 | −1.34 |
| Tgfb1 | TGF-β1 | 0.018 | −1.83 | 0.918 | 1.03 |
| Stat6 | STAT6 | 0.021 | −1.23 | 0.073 | −1.20 |
| Tgfbr2 | TGFβ Receptor2 | 0.022 | −1.40 | 0.504 | −1.08 |
| Serpine1 | Serpine, PAI-1 | 0.022 | −1.95 | 0.615 | 1.27 |
| Ifng | IFN-γ | 0.029 | −2.02 | 0.094 | −1.57 |
| Timp2 | TIMP2 | 0.030 | −1.60 | 0.006 | −1.49 |
| Thbs1 | Thrombospondin 1 | 0.036 | −1.81 | 0.503 | −1.18 |
| Hgf | Hepatocyte growth factor | 0.037 | −1.57 | 0.018 | −1.49 |
| Eng | Endoglin | 0.041 | −1.74 | 0.072 | −1.34 |
| Jun | Jun | 0.042 | −2.38 | 0.425 | −1.20 |
Note: IL-12p35−/−dnTGFβRII mice (n=6), dnTGFβRII mice (n=5), IL-12p40−/−dnTGFβRII (n=5)
Elevated serum AMA level in IL-12p35−/−dnTGFβRII mice
There were no significant differences in the level of serum AMAs among the three mice strains at 12 weeks. By 24 weeks, the serum AMA level was significantly higher in p35−/−mice compared to the other two strains (p<0.05) (Fig. 4).
Figure 4.

Levels of anti PDC-E2 antibody in serum of IL-12p40−/−dnTGFβRII (p40−/−), IL-12p35−/−dnTGFβRII (p35−/−) and dnTGFβRII (+/+) mice. Horizontal bars represent median values. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Increased Th17 response in IL-12p35−/−dnTGFβRII mice
To further confirm decreased IFN-γ/STAT1 signaling and compare Th1, Th2 and Th17 differentiation in the hepatic CD4 T cells of the three strains, we stimulated hepatic MNC and stained for intracellular cytokines IFN-γ, IL-4and IL-17. As shown in Figure 5, the frequency of Th1 (IFN-γ+) cells was significantly lower in both p35−/− and p40−/− mice than the dnTGFβRII mice (p<0.001) at 12 weeks. The relative frequencies of Th2 (IL-4+) cells, in comparison to the Th1 (IFN-γ+) cells, were significantly higher in p40−/− and p35−/− mice (p<0.05) at both 12 weeks and 24 weeks. Most strikingly, the relative frequency of the Th17 (IL-17+) cells, in comparison to the Th1 (IFN-γ+) cells, was significantly higher in the p35−/− mice than either the p40−/− or dnTGFβRII mice at both time points (p<0.05).
Figure 5.

Frequency of Th1, Th2 and Th17 cells in the liver of IL-12p40−/−dnTGFβRII (p40−/−), IL-12p35−/−dnTGFβRII (p35−/−) and dnTGFβRII (+/+) mice. Hepatic MNC were stimulated with Leukocyte Activation Cocktail in the presence of BD GolgiPlug, stained for intracellular IFN-γ, IL-4 and IL-17, and analyzed by flow cytometry to determine the percentage of cytokine producing CD4 T cells. Data are expressed as the mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Consistent with increased frequency of intrahepatic Th17 cells, the p35−/− mice demonstrated increased concentrations of Th17 cytokines secreted from the cultured hepatic MNCs. As shown in Figure 6, while the concentration of secreted IFN-γ was significantly higher in dnTGFβRII mice than the p35−/− and p40−/− mice (p<0.05), the concentrations of IL-17 and IL-22 were both significantly higher in p35−/− mice than p40−/− and dnTGFβRII mice (p<0.05). The level of secreted IL-6 was also significantly higher in p35−/− mice than the other strains at 12 weeks. By 24 weeks, the secreted IL-6 level was significantly lower in p40−/− mice than the other two strains, although in all 3 strains the IL-6 levels are substantially lower than that of 12 weeks. Taken together, these results show an enhanced Th17 response in p35−/− mice.
Figure 6.

Concentration of secreted cytokines from cultured hepatic MNCs. Hepatic MNCs were isolated from liver samples of 12-week and 24-week old IL-12p40−/−dnTGFβRII (p40−/−), IL-12p35−/−dnTGFβRII (p35−/−) and dnTGFβRII (+/+) mice. At each time point 8 to 14 animals from each group were tested. Cells were cultured with anti-CD3 and anti-CD28 for 72 hours before the conditioned media were collected to measure concentrations of various cytokines. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Discussion
The IL-12 family, composed of IL-12, IL-23, IL-27, and IL-35, is an important group of secreted proteins in the cytokine network of the innate and adaptive immune system (8, 11). All four IL-12 family cytokines are heterodimers constructed with an α chain and a β chain, and each cytokine shares at least one chain with another member of the family. Specifically, p40 is shared by IL-12 and IL-23, while p35 is shared by IL-12 and IL-35. We previously reported that p40 deficiency eliminated biliary disease in dnTGFβRII mice (7), suggesting that IL-12 and IL-23 are important in the development of biliary disease. The goal of the current study was to examine the role of the p35-containing cytokines in the pathogenesis of dnTGFβRII mice.
IL-12, IL-23 and IL-27 were initially described as pro-inflammatory/stimulatory cytokines, and have been implicated in various autoimmune diseases including experimental colitis (12), collagen-induced arthritis (13), insulin-dependent diabetes (14), experimental autoimmune encephalomyelitis (EAE) (15), PBC (16) and inflammatory bowel disease (17). In contrast, IL-35, the newest member of the IL-12 family, is distinct from the other three members. Within the CD4 T cell population, IL-35 is expressed by resting and activated Tregs but not effector T cells, hence is considered an inhibitory cytokine that contributes to Treg function (11). The role of IL-35 in infection and autoimmune diseases is a largely uncharted territory. We previously reported that autoimmune cholangitis in dnTGFβRII mice has features strikingly similar to human PBC, and have documented the role of different mononuclear subsets and cytokines in this PBC model (5, 18–21). In particular, dnTGFβRII IL-12p40 knockouts, lacking the pro-inflammatory cytokines IL-12 and IL-23, have no biliary disease (7).
IL-12 is a major cytokine involved in prototype Th1 responses and plays a role in both innate and adaptive immunity (22). A genomewide association analysis of DNA samples from 536 patients with PBC and 1536 controls revealed significant associations between PBC and common genetic variants at IL12A locus (encoding p35 subunit) and IL12RB2 (encoding IL-12 receptor β2) locus, suggesting that the IL-12 signaling pathway is relevant to the pathophysiology of PBC (16). A case report of biliary cirrhosis in an IL-12 deficiency child further suggests that the alteration of IL-12 immunomodulatory signaling is critical to the pathogenesis of PBC (23). We demonstrate herein that similar to IL-12p40−/−dnTGF®RII mice, the IL-12p35−/−dnTGF®RII mice had significantly milder portal inflammation at 12 weeks compared to dnTGF®RII mice. Since the presence of the pro-inflammatory IL-23 alone without IL-12, as in the case of the p35−/− mice, is not sufficient to cause early onset of portal inflammation in dnTGF®RII mice, this suggests that IL-12 plays a dominant role in portal inflammation. However, in the p35−/− mice the IL-12 deficiency in the presence of IL-23 but absence of IL-35 did not prevent dnTGF®RII biliary disease at 24 weeks (Fig. 1), suggesting that the pathological role of IL-23 can be enhanced by the deficiency of IL-35 related Treg functions. Unexpectedly, the absence of both IL-12 and IL-35 resulted in a strikingly high frequency (>50%) of liver fibrosis in the IL-12p35−/−dnTGF®RII mice (Fig. 3), which has not been seen in any other mouse models of PBC. Since IL-35 is expressed only by Tregs, while dnTGFβRII mice clearly lack the ability to regulate the immune response via TGFβ, an important mechanism of Treg mediated tolerance, adding an IL-35 deficiency might cripple the T regulatory mechanisms further; it points to a role for T regulatory activity in control of liver pathogenesis including fibrosis.
In PBC, a predominance of prototype Th1 cytokines and Th1 cells have been reported, and the Th1 response has been highly correlated to the degree of bile duct destruction (24–26). Furthermore, a significant decline of Th2 response has been reported during the late stage of PBC (27). Similar observations have also been reported in other organ-specific autoimmune diseases in which a Th2-type response prevented tissue damage (28, 29). However, the role of the newer IL-12 family cytokines in PBC is not yet clear. We should note that the cell isolation techniques used herein are similar to our previous work; we avoided enzymatic digestion because NK1.1 and the DX5 marker are significantly down-regulated after isolation using enzymatic digestion (30). Our focus was on the correlation between Th1, Th17 and liver fibrosis and thus we used our established technique of mechanical dissection for isolation of mononuclear cells. We do, however, note that cellular infiltration was demonstrated not only by cell counting, but also by histologic examination. Indeed, although a significant decrease was found in hepatic MNC in dnTGFβRII p35−/− mice at 12 weeks of age in comparison with dnTGFβRII mice, no significant difference was found at 24 weeks of age, indicating that the liver infiltration at 24 weeks in the p35−/− mice was as severe as dnTGFβRII mice; such data were supported by histologic examinations.
In the current study we observed a reduced IFN-γ/STAT1 signaling at mRNA level (Table 1) and a lower IFN-γ production in p35−/− mice compared to dnTGFβRII mice (Fig. 5), which is in agreement with previous reports on these mutations on different genetic background (31, 32) and is compatible with the presence of liver inflammation in dnTGFβRII mice but not p35−/− andp40−/− mice at 12 weeks (Fig. 1). The similar levels of liver disease in p35−/− and the parentdnTGFβRII mice at 24 weeks, however, cannot be directly attributed to the Th1/Th2 balance. The most prominent feature in the cytokine profile of p35−/− mice is the significantly enhanced IL-6 and Th17 responses (Fig. 5 and Fig. 6).
IL-17 producing CD4 T cells, or Th17 cells, have attracted attention because of their potent pathogenic role in autoimmune and inflammatory diseases including rheumatoid arthritis, experimental autoimmune encephalomyelitis (EAE), colitis (33–36) and liver disease (37, 38). The pathogenic potential of Th17 cells is conferred by the cytokines they produce, including IL-17A, IL-I7F, IL-21 and IL-22 (39). Recent studies have shown that IL-21 and IL-22 are important in the pathogenesis of inflammatory responses (40). A close relationship between levels of Th17 and PBC has been found in several studies, including a significant increase in the frequency of IL-17+ lymphocytes (41) and the periductal production of IL-17 in association with biliary innate immunity which has been shown to contribute to the pathogenesis of cholangiopathy (42, 43). The elevated level of Th17 cells and Th17-produced cytokines may be one of the contributing factors for the liver disease observed in the 24-week old p35−/− mice. In particular, IL-17 upregulates the expression of CXCL3 on biliary epithelial cells (42), which promotes migration of T cells into the liver, including CD8 T cells that play a pathogenic role in dnTGFβRII biliary damage (44, 45). Consistent with these previous findings, we demonstrate a significantly increased number of intrahepatic CD8 T cells in p35−/− mice by 24 weeks (Fig. 2). In addition, Th17 cells have been shown to participate in the production of autoantibodies (46). This is in agreement with the higher levels of AMA in p35−/− mice compared to the p40−/− and dnTGFβRII mice (Fig. 4).
The association of the distinct cytokine profile and the extensive liver fibrosis in the p35−/− mice is of interest since a similar correlation between Th17 and fibrosis has been documented in patients with autoimmune hepatitis, hepatitis B virus-related liver disease and alcoholic liver disease (37, 47, 48). The expression level of IL-17 strongly correlates with the degree of fibrosis in patients with HBV-related chronic liver diseases (47). IL-17A activated fibroblasts enhances production of IL-6 in one of the mouse models (49). In addition, IL-17A independently induces the activation of collagen-producing cells contributing to BDL- or CCL4-induced liver fibrosis in mice. Thus it is likely that Th17 and IL-6 also play a role in p35−/− liver fibrosis. Of note, IL-22 has recently been shown to be primarily synthesized by the Th17, Th22, γδT, natural killer, and natural killer T cell lineages (39). A weakness of the study herein is the failure to study the mechanism of amplified fibrinogenesis in these mice. Our initial efforts were placed on defining the specific roles of the p35 vs. p40 subunits in modulating the inflammatory disease we have previously reported in dnTGFβRII mice. Hence, the appearance of fibrosis was unexpected and must become a subject of future studies. We note the role of IL-22 in tissue fibrosis remains unclear or controversial (50, 51) and suggest that the fibrosis in p35−/− mice is most likely due to a reduction in IFN-γ and the increase in IL-17. Future directions will focus on the generation of IL-17, p35 double knockout mice as a model to explore these issues including fibrinogenesis and cytokine interactions with hepatic stellate cells.
Interestingly, Th17 cells have been classified as an independent T helper cell subset through the identification of their differentiation factors IL-6 and TGFβ1 (52). Recent studies demonstrate that Th17 cells can also be induced without TGFβ1 but via the effects of IL-6, IL-1β and IL23. These ‘alternative’ Th17 cells are thought to be more pathogenic than the ‘classical’ Th17 cells (53). Since the TGFβ pathway is disrupted in the dnTGFβRII mice, the Th17 cells in the IL-12p35−/−dnTGFβRII are likely to be the ‘alternative’ type with elevated pathogenic character.
In summary, our results reveal distinct phenotypes in terms of fibrosis, liver inflammation and cytokine profile among the dnTGFβRII strain and its two derivative strains, IL-12p35−/− and IL-12p40−/− mice. Importantly, since the deletion of p35 gene disrupts IL-12 and IL-23 while deletion of p40 gene disrupts IL-12 and IL-35, the different phenotypes of 12p35−/− and 12p40−/− reflect the differential effects of a pro-inflammatory cytokine, IL-23, versus a presumably inhibitory cytokine, IL-35, in the absence of IL-12 and a functional TGFβ pathway. In particular, our results suggest a potential role of IL-35 in protection against development of fibrosis. Further investigation of the IL-12 family members, especially the poorly studied IL-35, should shed more light on the immunopathogenic mechanism of the PBC-like liver disease in the dnTGFβRII mice, especially pathogenesis of liver fibrosis.
Acknowledgments
Financial support provided by National Institutes of Health grant DK090019.
Abbreviations
- PBC
Primary biliary cirrhosis
- AMA
anti-mitochondrial auto-antibodies
- PDC-E2
pyruvate dehydrogenase E2 complex
- dnTGFβRII
dominant negative form of transforming growth factor beta receptor type II
- MNCs
mononuclear cells
Contributor Information
Masanobu Tsuda, Email: tsuda.masanobu@gmail.com.
Weici Zhang, Email: ddzhang@ucdavis.edu.
Guo-Xiang Yang, Email: gxyang@ucdavis.edu.
Koichi Tsuneyama, Email: ktsune@med.u-toyama.ac.jp.
Yugo Ando, Email: yugo.ando@gmail.com.
Kazuhito Kawata, Email: kkawata@ucdavis.edu.
Ogyi Park, Email: parkogyi@mail.nih.gov.
Patrick S.C. Leung, Email: psleung@ucdavis.edu.
Ross L. Coppel, Email: ross.coppel@monash.edu.
Aftab A. Ansari, Email: pathaaa@emory.edu.
William M. Ridgway, Email: wridg1@gmail.com.
Bin Gao, Email: bgao@mail.nih.gov.
Zhe-Xiong Lian, Email: zxlian1@ustc.edu.cn.
Richard Flavell, Email: richard.flavell@yale.edu.
Xiao-Song He, Email: xiaosong@stanford.edu.
M. Eric Gershwin, Email: megershwin@ucdavis.edu.
References
- 1.Gershwin ME, Mackay IR. The causes of primary biliary cirrhosis: Convenient and inconvenient truths. Hepatology. 2008;47:737–745. doi: 10.1002/hep.22042. [DOI] [PubMed] [Google Scholar]
- 2.Mao TK, Davis PA, Odin JA, Coppel RL, Gershwin ME. Sidechain biology and the immunogenicity of PDC-E2, the major autoantigen of primary biliary cirrhosis. Hepatology. 2004;40:1241–1248. doi: 10.1002/hep.20491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gershwin ME, Mackay IR, Sturgess A, Coppel RL. Identification and specificity of a cDNA encoding the 70 kd mitochondrial antigen recognized in primary biliary cirrhosis. J Immunol. 1987;138:3525–3531. [PubMed] [Google Scholar]
- 4.Moteki S, Leung PS, Coppel RL, Dickson ER, Kaplan MM, Munoz S, Gershwin ME. Use of a designer triple expression hybrid clone for three different lipoyl domain for the detection of antimitochondrial autoantibodies. Hepatology. 1996;24:97–103. doi: 10.1002/hep.510240117. [DOI] [PubMed] [Google Scholar]
- 5.Oertelt S, Lian ZX, Cheng CM, Chuang YH, Padgett KA, He XS, Ridgway WM, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol. 2006;177:1655–1660. doi: 10.4049/jimmunol.177.3.1655. [DOI] [PubMed] [Google Scholar]
- 6.Miyakawa H, Tanaka A, Kikuchi K, Matsushita M, Kitazawa E, Kawaguchi N, Fujikawa H, et al. Detection of antimitochondrial autoantibodies in immunofluorescent AMA-negative patients with primary biliary cirrhosis using recombinant autoantigens. Hepatology. 2001;34:243–248. doi: 10.1053/jhep.2001.26514. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida K, Yang GX, Zhang W, Tsuda M, Tsuneyama K, Moritoki Y, Ansari AA, et al. Deletion of interleukin-12p40 suppresses autoimmune cholangitis in dominant negative transforming growth factor beta receptor type II mice. Hepatology. 2009;50:1494–1500. doi: 10.1002/hep.23132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collison LW, Vignali DA. Interleukin-35: odd one out or part of the family? Immunol Rev. 2008;226:248–262. doi: 10.1111/j.1600-065X.2008.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 10.Lian ZX, Okada T, He XS, Kita H, Liu YJ, Ansari AA, Kikuchi K, et al. Heterogeneity of dendritic cells in the mouse liver: identification and characterization of four distinct populations. Journal of immunology. 2003;170:2323–2330. doi: 10.4049/jimmunol.170.5.2323. [DOI] [PubMed] [Google Scholar]
- 11.Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 12.Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. The Journal of experimental medicine. 1995;182:1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joosten LA, Lubberts E, Helsen MM, van den Berg WB. Dual role of IL-12 in early and late stages of murine collagen type II arthritis. Journal of immunology. 1997;159:4094–4102. [PubMed] [Google Scholar]
- 14.Trembleau S, Penna G, Bosi E, Mortara A, Gately MK, Adorini L. Interleukin 12 administration induces T helper type 1 cells and accelerates autoimmune diabetes in NOD mice. The Journal of experimental medicine. 1995;181:817–821. doi: 10.1084/jem.181.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falcone M, Bloom BR. A T helper cell 2 (Th2) immune response against non-self antigens modifies the cytokine profile of autoimmune T cells and protects against experimental allergic encephalomyelitis. The Journal of experimental medicine. 1997;185:901–907. doi: 10.1084/jem.185.5.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirschfield GM, Liu X, Xu C, Lu Y, Xie G, Gu X, Walker EJ, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. The New England journal of medicine. 2009;360:2544–2555. doi: 10.1056/NEJMoa0810440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abraham C, Cho JH. IL-23 and autoimmunity: new insights into the pathogenesis of inflammatory bowel disease. Annu Rev Med. 2009;60:97–110. doi: 10.1146/annurev.med.60.051407.123757. [DOI] [PubMed] [Google Scholar]
- 18.Chuang YH, Lian ZX, Yang GX, Shu SA, Moritoki Y, Ridgway WM, Ansari AA, et al. Natural killer T cells exacerbate liver injury in a transforming growth factor beta receptor II dominant-negative mouse model of primary biliary cirrhosis. Hepatology. 2008;47:571–580. doi: 10.1002/hep.22052. [DOI] [PubMed] [Google Scholar]
- 19.Moritoki Y, Zhang W, Tsuneyama K, Yoshida K, Wakabayashi K, Yang GX, Bowlus C, et al. B cells suppress the inflammatory response in a mouse model of primary biliary cirrhosis. Gastroenterology. 2009;136:1037–1047. doi: 10.1053/j.gastro.2008.11.035. [DOI] [PubMed] [Google Scholar]
- 20.Yang GX, Lian ZX, Chuang YH, Moritoki Y, Lan RY, Wakabayashi K, Ansari AA, et al. Adoptive transfer of CD8(+) T cells from transforming growth factor beta receptor type II (dominant negative form) induces autoimmune cholangitis in mice. Hepatology. 2008;47:1974–1982. doi: 10.1002/hep.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Tsuda M, Yang GX, Tsuneyama K, Rong G, Ridgway WM, Ansari AA, et al. Deletion of interleukin-6 in mice with the dominant negative form of transforming growth factor beta receptor II improves colitis but exacerbates autoimmune cholangitis. Hepatology. 2010;52:215–222. doi: 10.1002/hep.23664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 23.Pulickal AS, Hambleton S, Callaghan MJ, Moore CE, Goulding J, Goodsall A, Baretto R, et al. Biliary cirrhosis in a child with inherited interleukin-12 deficiency. J Trop Pediatr. 2008;54:269–271. doi: 10.1093/tropej/fmm119. [DOI] [PubMed] [Google Scholar]
- 24.Martinez OM, Villanueva JC, Gershwin ME, Krams SM. Cytokine patterns and cytotoxic mediators in primary biliary cirrhosis. Hepatology. 1995;21:113–119. [PubMed] [Google Scholar]
- 25.Nagano T, Yamamoto K, Matsumoto S, Okamoto R, Tagashira M, Ibuki N, Matsumura S, et al. Cytokine profile in the liver of primary biliary cirrhosis. J Clin Immunol. 1999;19:422–427. doi: 10.1023/a:1020511002025. [DOI] [PubMed] [Google Scholar]
- 26.Harada K, Van de Water J, Leung PS, Coppel RL, Ansari A, Nakanuma Y, Gershwin ME. In situ nucleic acid hybridization of cytokines in primary biliary cirrhosis: predominance of the Th1 subset. Hepatology. 1997;25:791–796. doi: 10.1002/hep.510250402. [DOI] [PubMed] [Google Scholar]
- 27.Sekiya H, Komatsu T, Isono E, Furukawa M, Matsushima S, Yamaguchi N, Yamauchi K, et al. Decrease in the prevalence of IL-4-producing CD4+ T cells in patients with advanced stage of primary biliary cirrhosis. Am J Gastroenterol. 1999;94:3589–3594. doi: 10.1111/j.1572-0241.1999.01547.x. [DOI] [PubMed] [Google Scholar]
- 28.Cameron MJ, Arreaza GA, Zucker P, Chensue SW, Strieter RM, Chakrabarti S, Delovitch TL. IL-4 prevents insulitis and insulin-dependent diabetes mellitus in nonobese diabetic mice by potentiation of regulatory T helper-2 cell function. J Immunol. 1997;159:4686–4692. [PubMed] [Google Scholar]
- 29.Wilson SB, Kent SC, Patton KT, Orban T, Jackson RA, Exley M, Porcelli S, et al. Extreme Th1 bias of invariant Valpha24JalphaQ T cells in type 1 diabetes. Nature. 1998;391:177–181. doi: 10.1038/34419. [DOI] [PubMed] [Google Scholar]
- 30.Dong ZJ, Wei HM, Sun R, Tian ZG, Gao B. Isolation of murine hepatic lymphocytes using mechanical dissection for phenotypic and functional analysis of NK1. 1+ cells. World J Gastroenterol. 2004;10:1928–1933. doi: 10.3748/wjg.v10.i13.1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, Ferrante J, Stewart C, et al. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity. 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 32.Mattner F, Magram J, Ferrante J, Launois P, Di Padova K, Behin R, Gately MK, et al. Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur J Immunol. 1996;26:1553–1559. doi: 10.1002/eji.1830260722. [DOI] [PubMed] [Google Scholar]
- 33.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. The Journal of experimental medicine. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 35.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annual review of immunology. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 37.Lemmers A, Moreno C, Gustot T, Marechal R, Degre D, Demetter P, de Nadai P, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49:646–657. doi: 10.1002/hep.22680. [DOI] [PubMed] [Google Scholar]
- 38.Lafdil F, Miller AM, Ki SH, Gao B. Th17 cells and their associated cytokines in liver diseases. Cell Mol Immunol. 2010;7:250–254. doi: 10.1038/cmi.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Witte E, Witte K, Warszawska K, Sabat R, Wolk K. Interleukin-22: a cytokine produced by T, NK and NKT cell subsets, with importance in the innate immune defense and tissue protection. Cytokine Growth Factor Rev. 2010;21:365–379. doi: 10.1016/j.cytogfr.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 40.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 41.Lan RY, Salunga TL, Tsuneyama K, Lian ZX, Yang GX, Hsu W, Moritoki Y, et al. Hepatic IL-17 responses in human and murine primary biliary cirrhosis. J Autoimmun. 2009;32:43–51. doi: 10.1016/j.jaut.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harada K, Shimoda S, Sato Y, Isse K, Ikeda H, Nakanuma Y. Periductal interleukin-17 production in association with biliary innate immunity contributes to the pathogenesis of cholangiopathy in primary biliary cirrhosis. Clin Exp Immunol. 2009;157:261–270. doi: 10.1111/j.1365-2249.2009.03947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rong G, Zhou Y, Xiong Y, Zhou L, Geng H, Jiang T, Zhu Y, et al. Imbalance between T helper type 17 and T regulatory cells in patients with primary biliary cirrhosis: the serum cytokine profile and peripheral cell population. Clin Exp Immunol. 2009;156:217–225. doi: 10.1111/j.1365-2249.2009.03898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shields PL, Morland CM, Salmon M, Qin S, Hubscher SG, Adams DH. Chemokine and chemokine receptor interactions provide a mechanism for selective T cell recruitment to specific liver compartments within hepatitis C-infected liver. J Immunol. 1999;163:6236–6243. [PubMed] [Google Scholar]
- 45.Hokeness KL, Deweerd ES, Munks MW, Lewis CA, Gladue RP, Salazar-Mather TP. CXCR3-dependent recruitment of antigen-specific T lymphocytes to the liver during murine cytomegalovirus infection. J Virol. 2007;81:1241–1250. doi: 10.1128/JVI.01937-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hickman-Brecks CL, Racz JL, Meyer DM, LaBranche TP, Allen PM. Th17 cells can provide B cell help in autoantibody induced arthritis. J Autoimmun. 2011;36:65–75. doi: 10.1016/j.jaut.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang L, Chen S, Xu K. IL-17 expression is correlated with hepatitis Brelated liver diseases and fibrosis. Int J Mol Med. 2011;27:385–392. doi: 10.3892/ijmm.2011.594. [DOI] [PubMed] [Google Scholar]
- 48.Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X, Qiu D, et al. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PLoS One. 2011;6:e18909. doi: 10.1371/journal.pone.0018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, Nishihara M, et al. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity. 2008;29:628–636. doi: 10.1016/j.immuni.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 50.Andoh A, Zhang Z, Inatomi O, Fujino S, Deguchi Y, Araki Y, Tsujikawa T, et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology. 2005;129:969–984. doi: 10.1053/j.gastro.2005.06.071. [DOI] [PubMed] [Google Scholar]
- 51.Simonian PL, Wehrmann F, Roark CL, Born WK, O’Brien RL, Fontenot AP. gammadelta T cells protect against lung fibrosis via IL-22. The Journal of experimental medicine. 2010;207:2239–2253. doi: 10.1084/jem.20100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 53.Peters A, Lee Y, Kuchroo VK. The many faces of Th17 cells. Curr Opin Immunol. 2011 doi: 10.1016/j.coi.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]