

Abstract
The multistage model of non-melanoma skin carcinogenesis has contributed significantly to our understanding of epithelial cancer in general. We used the Krt1-15CrePR1;R26R transgenic mouse to determine the contribution of keratin 15+ cells from the hair follicle to skin tumor development by following the labeled progeny of the keratin 15 expressing cells into papillomas. We present three novel observations. First, we found that keratin 15 expressing cells contribute to most of the papillomas by 20 weeks of promotion. Second, in contrast to the transient behavior of labeled keratin 15-derived progeny in skin wound healing, keratin 15 progeny persist in papillomas and some malignancies for many months following transient induction of the reporter gene. Third, papillomas have surprising heterogeneity not only in their cellular composition, but also in their expression of the codon 61 signature Ha-ras mutation with approximately 30 percent of keratin 15-derived regions expressing the mutation. Together, these results demonstrate that keratin 15 expressing cells of the hair follicle contribute to cutaneous papillomas with long term persistence and a subset of which express the Ha-ras signature mutation characteristic of initiated cells.
Keywords: skin carcinogenesis, stem cells, hair follicles, papillomas
Introduction
The multistage model of mouse skin carcinogenesis has long been a template for studying cancer mechanisms, and has provided some of the best experimental evidence implicating a role for stem cells in tumor development. In this model, tumors can be induced on the backs of mice first by a single subtumorigenic exposure to a carcinogen such as 7, 12-dimethylbenz[a]anthracene (DMBA; initiation), followed by repetitive chronic epidermal damage (promotion) with 12-O-tetradecanoylphorbol-13-acetate (TPA) [1]. Initiation causes the conversion of some cells into latent neoplastic cells, and promotion elicits expression of a neoplastic change. Effective initiators of skin carcinogenesis such as DMBA have in common the capacity for covalent binding as activated electrophiles to cellular DNA, conferring the ability to form irreversible alterations in the genome [1]. This in turn results in a signature mutation in codon 61 of the c-Ha-ras proto-oncogene, which is considered necessary for papilloma development in the DMBA/TPA model [1]. In this model, it is widely believed that a major target of carcinogens resides in the stem cell compartment within the cutaneous epithelium [2,3,4]. One of the most compelling arguments for the stem cell being the target cell is the permanent nature of the neoplastic lesion, in that tumors can be elicited even with long intervals between initiation and promotion [5]. This argues for a long lived, slowly cycling population of cells that can persist throughout the lifetime of the animal despite the continual renewal of the epidermis and cycling of the hair follicles. Disputed, however, is the stem cell of origin: epidermal, hair follicle, or sebaceous gland [6,7].
Work from our laboratory and from others has supported a stem cell origin for cutaneous tumors from the epidermis as well as hair follicles [4]. However, direct visualization of the tumorigenic process from these stem cells has been thwarted by the lack of specific markers for the stem cell populations. We reported that keratin 15 expressing, slowly cycling cells are contained within the hair follicle, principally in the bulge, but also present in telogen hair germ and in some cells of the outer root sheath during anagen. We previously used a keratin 15 promoter sequence to develop a transgenic mouse that enables us to trace the lineage of cells derived from keratin 15 expressing cells during the hair growth cycle [8] and during wound healing [9]. We now report the use of this mouse to trace the lineage of keratin 15-expressing cells from the hair follicle into skin tumors using the multistage model of skin carcinogenesis. In this communication, we present three major novel observations: 1) progeny of keratin 15 expressing hair follicle cells contribute directly to squamous papilloma development; 2) progeny of keratin 15 expressing cells persist in skin tumors for long intervals with a stable numbers, and 3) the Ha-ras signature mutation is expressed in some but not all of the keratin 15-derived progeny in papillomas.
Materials and Methods
Animals
Female Krt1-15CrePR1;R26R mice reported previously [8] were bred at Columbia University. Female FVB/N mice were obtained from Taconic Farms (Germantown, NY). Animals were maintained in the AAALAC accredited Columbia University Barrier Facility and in the NIEHS animal facility under protocols approved by the Institutional Animal Care and Use Committee. Animals were maintained in accordance with institutional guidelines for the care and use of experimental animals.
Chemicals
7,12-Dimethylbenz[a]anthracene (DMBA), and 12-O-tetradecanoylphorbol-13-acetate (TPA) were purchased from Sigma (St. Louis, MO). Acetone was spectral grade from Fisher Scientific (Norcross, GA). RU486 was purchased from Sigma as a powder, and mixed at a concentration of 1 mg/g with Neutrogena Hand Cream (unscented) for topical application.
Tumor induction
Seven-week old female mice were initiated with a single topical application of 400 nmol DMBA in 200 microliters of acetone. One week later, Cre recombinase was activated following daily topical applications of 1 mg RU486 in Neutrogena Hand Cream for a total of 5 days. One week after the last RU486 treatment, tumors were promoted with 17 nmol of TPA in 200 microliters of acetone applied twice weekly for 20 weeks. Papillomas were harvested from euthanized mice at intervals during promotion, and mice were photographed with a digital camera. Treatment groups and controls are described in Table 1.
Table 1.
Protocols and treatment groups for Krt1-15CrePR1; R26R mice in skin carcinogenesis experiments
| Group | First Treatment* |
Second Treatment† |
Third Treatment‡ |
Number of mice |
|---|---|---|---|---|
| 1 | DMBA | Vehicle | TPA | 63 |
| 2 | DMBA | RU486 | TPA | 76 |
| 3 | RU486 | DMBA | Acetone | 3 |
| 4 | Vehicle | DMBA | Acetone | 2 |
| 5 | DMBA | RU486 | - | 3 |
| 6 | DMBA | Vehicle | - | 2 |
| 7 (Cre-) | DMBA | RU486 | - | 2 |
Seven week old mice were treated topically with 400 nmol of DMBA, or once daily for five days with either 0.1 mg of RU486 in Neutrogena hand cream (vehicle) or with hand cream alone.
One week following the first treatment, mice were treated with Neutrogena hand cream, with RU486, or with DMBA.
One week following the second treatment, the mice were treated twice weekly with TPA or with acetone for intervals up to 20 weeks.
Staining
Each tumor-bearing mouse was photographed following euthanasia. Tumors were then collected, bisected, and lightly fixed in paraformaldehyde. Staining for the reconstituted LacZ activity was accomplished with X-gal reagent (Roche, Indianapolis, IN) following manufacture’s directions. All stained tumors were first photographed as whole mounts with a Zeiss Discovery dissecting microscope. Half of each tumor was then paraffin embedded using isopropanol for processing. Sections were counterstained with Nuclear Fast Red for microscopic examination. Serial sections through papillomas were examined with a Zeiss Axioplan microscope. The epithelial portion of the approximate medial sagittal section was traced with Carl Zeiss Axiovision software, and the blue staining portion was calculated and expressed as percentage.
Laser capture microdissection
Paraffin blocks of beta-galactosidase stained mouse skins were sectioned at seven microns and microdissection was conducted using either the eLeica AS/LMD (Leica Microsystems) or the Zeiss P.A.L.M. (Carl Zeiss, Inc.) platforms.
Sequence analysis of ras mutations
DNA was isolated from captured cells using the PicoPure DNA Extraction kit (Molecular Devices, now part of MDS Analytical Technologies, Mountain View, CA) following manufacture’s directions. Codon 61 of the c-Ha-ras oncogene was amplified by PCR using primers and conditions as described previously [10]. Sequence analysis for the DMBA-induced signature A to T transversion was conducted by the Columbia University DNA Sequencing Facility.
DNA adduct analysis of hair follicle bulge epidermal stem cells
The shaved dorsal skin surface of seven week-old FVB/N female mice (Taconic, Germantown, NY) was initiated with a single dose of DMBA (50 micrograms in 200 microliters of acetone). Twenty-four hours after initiation, skin was collected and keratinocytes harvested as described elsewhere [11] harvesting 5 mice per day on three consecutive days. Keratinocytes were stained with antibodies to CD34 (rat anti-mouse CD34-FITC, eBiosciences; rat anti-human CD49f (alpha-6 integrin), BD Pharmingen) and stem cell (alpha6+CD34+) and basal cell (alpha6+CD34-) populations sorted using Fluorescence Activated Cell Sorting (FACS) as previously described [12]. DNA adduct formation was assessed in genomic DNA isolated from stem and basal keratinoctyes by 32P-postlableing according to protocols outlined in Trempus, et al. [13].
Statistical analysis
In order to analyze the relative numbers of blue, white, and mixed papillomas in RU486-treated and vehicle-treated mice, for each of the three samples (9, 11-15, or 20+ weeks of TPA), we applied a permutation version of the likelihood ratio test, as implemented in the Likelihood Ratio Test Allowing for errors (LTRAE) software. To test whether the proportion of mixed papillomas increases as the number of weeks of TPA increases, and to test whether the proportion of mixed papillomas in the two treatment groups also increases, we applied the linear trend test. Further details of statistical analysis including determination of the minimum sample size, are provided in the Supplemental Information.
Results
We used the Krt1-15CrePR1;R26R bigenic mouse to determine the contribution of keratin 15-expressing cells to skin tumor development. Bigenic mice were exposed to a tumor induction protocol in combination with activation of the Cre recombinase as outlined in Table 1. Groups 1 and 2 were our test groups, with either vehicle (group1) or RU486 (group 2). The remaining groups (3-7) were various controls.
We first discuss the results of the experimental controls. We found that when RU486 was applied prior to DMBA (control groups 3, 4; Table 1), we initially observed a marked epidermal hyperplasia that we felt might alter metabolic activation of DMBA. Therefore, we elected to apply DMBA before RU486 to allow complete metabolism and adduct formation. Epidermal hyperplasia was not observed in the latter sequence. To address the problem of promoter leakiness that could reduce our ability to distinguish true cellular contribution from nonspecific background, we examined mice in groups 5 and 6. We found that skin in group 6 showed blue cells in 35% of the hair follicles compared with 81% of hair follicles in group 5 (Fig. 1A,B, respectively; Text S1). In group 5 (Fig. 1B), we did not observe blue cells in the epidermis, follicular infundibulum, or sebaceous glands. We also examined vehicle treated bigenic skin following 4 weeks of TPA promotion (group 1) to evaluate non-specific blue staining. As shown in Fig. 1C, blue X-gal reaction product could be found in the hair follicles beneath a hyperplastic focus. However, higher magnification revealed that this was not cellular, but instead found to be a crystalline deposit. Although leakiness in other hair follicles was observed in these slides, we concluded that TPA treatment did not induce blue cells. Finally, we examined tumors taken from initiated/promoted R26R reporter mice (group 7; Fig. 1D). None of these tumors were blue, demonstrating lack of recombination in the reporter mouse in the absence of Cre. Taken together, these data demonstrate that although there is background recombination in the bigenic mice, RU486-mediated recombination is significant. Promoter leakiness is discussed further in Table 2, below. The recombination efficiency of the Cre after RU486 application was approximately 30 to 50 percent [9].
Figure 1. Tumor promotion and progression in Krt1-15CrePR1;R26R bigenic mice following initiation with DMBA, induction with RU486, promotion with TPA, and the tissue processed for beta-galactosidase activity.

Figures A-C are skin sections from control mice. The remainder illustrates the spectrum of papillomas from all/all white/ to mixed blue and white. Tumor whole mounts were cut in half, stained for beta-galactosidase activity, and photographed on edge. Serial sections of stained tumors, were inspected microscopically, and approximately median sagittal sections were chosen for analysis based on their continuity with the underlying skin. Scale bar (100μm). A. Section of skin from a mouse initiated with DMBA and treated with Neutrogena hand cream (negative control for RU486) prior to treatment with TPA. Ep, epidermis; D, dermis; HF, hair follicle; SG, sebaceous gland. B. Skin from an initiated Krt1-15PR1;R26R bigenic mouse treated with RU486 prior to treatment with TPA. Note the blue cells in the hair follicle bulge (B). Other arrows as previously noted. C. Section of a squamous hyperplastic focus in an initiated skin treated with Neutrogena hand cream, followed by staining for beta-galactosidase activity. Note the lack of beta-galactosidase activity in this control. D. Section of a small papilloma in an R26R reporter mouse to assess for spontaneous recombination. Note the lack of beta-galactosidase activity in the hair follicles and in the tumor. E. Serial sections through a squamous hyperplastic focus of a bigenic mouse initiated with DMBA followed by induction with RU486 and promotion for four weeks with TPA. Note that this hyperplastic focus and underlying hair follicles are entirely blue. F. Section through the blue papilloma showing the blue epithelium (E) of the papilloma and the white uninvolved epidermis (Ep). G. Section through a papilloma from a bigenic mouse after 11 weeks of tumor promotion. Four blue hair follicles are under the tumor and their bulge stem cell progeny are involved in tumor formation. H. Section through a white papilloma taken after 12 weeks of promotion from vehicle control group. Blue cells were detected in the epithelium of the papilloma due to leakage. I. Section though a papilloma after 21 weeks of promotion in RU486 treated group. J. Bisected squamous cell carcinoma taken at 20 weeks of promotion. Note that this lesion is essentially all blue. K. Bisected squamous cell carcinoma from a bigenic mouse following 20 weeks of TPA treatment. Note that this malignant lesion is essentially all white as is another carcinoma shown in Supplemental Figure1. Panel I after 18 weeks of promotion.
Table 2.
Relative numbers of blue, non-blue, and mixed papillomas during skin tumor promotion of Krt1-15CrePR1;R26R female mice
| Weeks of TPA |
Cre induction | Blue papillomas |
Non-blue papillomas |
Mixed papillomas |
Percentage of blue area in mixed papillomas |
|---|---|---|---|---|---|
| 9 | RU486 | 5 (28%) | 4 (22%) | 9 (50%) | 34.4 |
| Vehicle | 0 (0%) | 6 (50%) | 6 (50%) | 8.5 | |
| 11-15 | RU486 | 2 (7%) | 13 (43%) | 15 (50%) | 31.2 |
| Vehicle | 0 (0%) | 19 (76%) | 6 (24%) | 4.5 | |
| 20+ | RU486 | 0 (0%) | 0 (0%) | 28 (100%) | 32.2 |
| Vehicle | 0 (0%) | 14 (54%) | 12 (46%) | 3.1 |
Krt1-15CrePR1;R26R female mice were treated topically with 400 nmol of DMBA when 7 weeks of age followed one week later with 5 daily topical applications of RU486 in Neutrogena hand cream or with hand cream alone. Papillomas were promoted beginning one week later with twice-weekly treatments with TPA. At intervals thereafter, mice were euthanized and papillomas together with surrounding uninvolved skin were removed, were processed for X-gal staining, and paraffin processing. Approximately median saggital sections through individual papillomas were scored at 25X magnification as entirely blue, non-blue, or mixed containing both blue and non-blue epithelial cells. The percentage of blue area in medial saggital sections from each papilloma was computed with Carl Zeiss Axiovision software. The standard deviations of percentage of blue area of each group are indicated following: 9 weeks (RU486: 24.392, Vehicle: 6.717), 11-15 weeks (RU486: 11.951, Vehicle: 3.390), 20+ weeks (RU486: 20.282, Vehicle: 1.751). The number of samples in each group is indicated following: 9 weeks (RU486: 5, Vehicle: 5), 11-15 weeks (RU486: 12, Vehicle: 17), 20+ weeks (RU486: 12, Vehicle: 17). P values for each interval are: 9 weeks (P=0.0504); 11-15 weeks, (P<0.0001); and 20+weeks (P< 0.0001).
We report here the results of the test groups (groups 1 and 2; Table 1). Serial sections through an early squamous hyperplastic focus in a mouse treated for 4 weeks with TPA reveals blue cells (Fig. 1E, circles), and appear to involve multiple hair follicles. Additionally, some 4-week squamous hyperplastic foci contained few, if any, blue cells (Fig. S1A). We observed that papillomas from RU486-treated, DMBA initiated bigenic mice promoted with TPA could be initially either mostly blue (Fig. 1F; Fig. S1B), mixed blue and non-blue (Fig. 1G; Fig. S1C,D,E), or mostly non-blue (Fig. S1F). In contrast, vehicle controls during this time demonstrated far fewer blue cells (Fig. 1H; Fig. S1G). As in earlier time points following application of RU486, virtually all of these tumors appear to involve multiple hair follicles that are entirely blue possibly due to TPA induced anagen. A rare blue late stage papilloma is shown in Fig. 1I. Shown in Fig. S1H is an advancing keratoacanthoma considered to be a premalignant lesion in mice. The entire epithelial compartment is blue, similar to what was seen in some early papillomas. Additionally, a malignant squamous cell carcinoma (Fig. 1J) also is essentially entirely blue, whereas other SCCs (Fig. 1K; Fig. S1I) are essentially all non-blue. Thus, the persistence of blue cells in the proliferative and differentiative compartments of tumors throughout tumor promotion and progression contrasts with earlier observations in wounding with this model, where keratin 15 progeny initially participated in regeneration but were eventually lost from the epidermis [9].
There was a linear trend in the relative proportions of RU486-treated mice with mixed blue and white papillomas as the number of weeks of TPA exposure increased, as summarized as detailed in Text S2 and Tables S1, S2. That is, as the number of weeks of TPA treatment increased, the proportion of all-blue tumors decreased while the proportion of mixed papillomas in RU486-treated bigenic mice increased. Statistical support for this conclusion is detailed in the Text S3 and Table S3. At all intervals, there were differences between the number of blue papillomas in the RU486-treated and vehicle-treated mice particularly at the 20-week interval (Text S4 and Table S4). However, the proportion of mixed papillomas is the same for each of the “weeks of TPA” categories in the vehicle treated group.
In light of the prevailing concept of tumors being derived from clonal expansion of initiated cells, we wondered whether the keratin 15-derived cells in papillomas increased or decreased over time. We therefore used morphometery to determine the percentage of area containing blue cells in median sagittal sections, and found that the percentage of blue area in mixed tumors remained unexpectedly constant throughout 20 weeks of tumor promotion, at about 30% (Table 2 and Text S4) while the vehicle control group remained at about 5% of blue area. It is interesting that although the number of mixed blue and non-blue papillomas was high in the vehicle controls, the percentage of blue area is significantly smaller than with RU486 treatment. Hence, the blue area due to promoter leakiness is always much smaller than that induced by RU486. The observation that the blue portion of papillomas remained constant was interesting in view of the fact that keratoacanthomas and squamous cell carcinomas were either all blue or all non-blue (Fig. 1J, Fig. S1H (blue) and Fig. 1K, and Fig S1I (non-blue), respectively).
In the two-stage model of skin carcinogenesis, papillomas induced by DMBA often carry a signature A to T transversion in codon 61 of the c-Ha-ras proto-oncogene that is considered to be necessary for tumor development [14,15]. To address the question of whether papillomas derived from the hair follicle bulge progeny express this signature mutation in the ras gene, we conducted laser capture microdissection of multiple blue and non-blue areas of papillomas for DNA isolation and sequence analysis (Fig. S2 A,B; Table 3). We found that 27% of blue areas of papillomas express the mutation (CTA), whereas of the blue areas 73% were wild type (CAA) (Table 3).
Table 3.
Activated Ha-ras expression in skin papillomas of Krt1-15CrePr1;R26R mice following laser capture microdissection
| Captured sample | Number of captured samples |
Wild-type Ras (CAA) | Mutated Ras (CTA) |
|---|---|---|---|
| Blue cells in papilloma | 33 | 24 (72.7%) | 9 (27.3%) |
| Non-blue cells in papilloma |
32 | 11 (34.4%) | 21 (65.6%) |
| Blue cells in hyperplastic epidermis |
3 | 3 (100% | 0 (0%) |
| Non-blue cells in hyperplastic epidermis |
18 | 18 (100%) | 0 (0%) |
Captured cells were collected from X-gal reactive de-paraffinized sections mounted on membrane slides. DNA was isolated, the area around codon 61 in the Ha-ras gene was PCR amplified and sequenced. The total number of tumors was 18, and the total number of captured samples was 86. The positive control was a Tg.AC keratinocyte cell line, and the negative control was a keratinocyte cell line that did not express the mutated codon 61 mutation.
The observation of fewer ras-mutation containing cells among the bulge derived portion of tumors relative to the white cells (Table 3; 27% versus 73%, respectively) suggests that after carcinogen exposure, keratin 15 expressing cells might not be initiated with the same efficiency as the other more proliferative target cells, perhaps because of their quiescent nature. To test this hypothesis, we isolated hair follicle stem cells from initiated FVB/N mice using fluorescent antibodies to CD34 and alpha-6 integrin, which have been shown to be selectable determinates for keratin-15 expressing cells [12]. We analyzed DMBA-induced DNA adduct formation in stem cells compared to CD34 depleted basal cells (Fig. 2 A,B). DMBA is a polycyclic aromatic hydrocarbon that must be metabolized into reactive diol epoxides, which bind to cellular macromolecules, including DNA, forming adducts which eventually result in the signature A to T mutation in the ras oncogenes [16]. Using 32P-postlabeling, we demonstrated that the population of hair follicle stem cells forms adenine adducts as do the non-keratin 15 expressing basal cell population (Fig. 2 A,B). Therefore, these data demonstrate that hair follicle stem cells do form the primary lesion after carcinogen exposure; however, based upon the lower percentage of mutant cells in the blue regions of tumors (Table 3), it is possible that the transition from adduct to mutation may not be as efficient.
Fig. 2. DNA adduct profiles of hair follicle stem cells and basal cells.
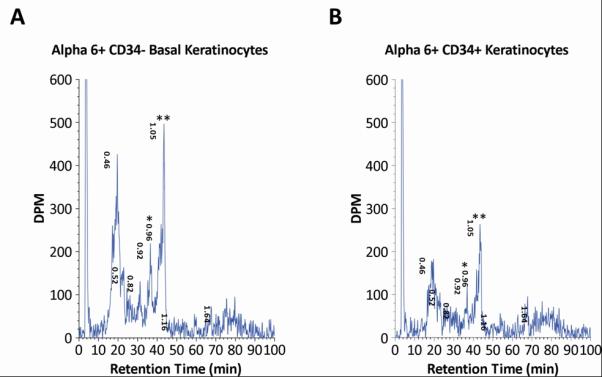
In Fig. A,B, FVB/N mice were treated with 50 μg DMBA, then keratinocytes were harvested 24 h later for DNA isolation and 32P-postlabeling identification of DMBA-3,4-diol-1,2-epoxide DNA adducts in alpha-6 integrin, CD34-sorted keratinocyte basal and hair follicle stem cell populations. Panel A shows adduct profile obtained from the basal (non-hair follicle) population; Panel B shows adduct profile from hair follicle keratinocyte stem cells. syn-dAdenine and anti-dAdenine adducts are identified as * (retention time 0.96 min) and ** (retention time 1.05 min).
Discussion
In this communication, we demonstrate that keratin-15-expressing cells from the hair follicle directly contribute to skin tumor development in mice. Previous work from our laboratory and others has supported the hypothesis that epithelial stem cells, including those from hair follicles from skin are major targets for neoplastic transformation and tumor development. For example, the deletion of Stat3 in the hair follicle using the K15CrePR1 mouse results in decreased tumorigenesis [17], and over-expression of Ha-ras via a truncated keratin 5 promoter biased to the hair follicle increases tumorigenesis suggesting that hair follicle cells are important players in skin tumorigenesis. However, what has been lacking is direct analysis of the cell of origin for papillomas in two-stage skin tumorigenesis. Here, we show that papillomas can be traced to a hair follicle cell origin from early stages of tumor development. This finding is enhancing and valuable to the field.
Our work presented several unexpected observations, including evidence for long term persistence of keratin 15 progeny in skin tumors over several months, tumor heterogeneity from early stages of papilloma development, and heterogeneous expression of the ras signature mutation within papillomas.
The first unexpected finding was the long-term persistence of K15+ progeny in papillomas. We observed that, as in wound healing [9], during tumor promotion keratin 15+ progeny move into the follicular infundibulum and epidermis, but then persist in tumors. Under steady state conditions, bulge stem cells are programmed for maintenance and cycling of the lower hair follicle. In the context of our studies, tumor promotion is causing a regenerative epidermal hyperplasia that recapitulates the wound-healing model where bulge cells are recruited into the epidermis. However, unlike wound-healing in the Krt1-15CrePR1;R26R model where cells of keratin 15 lineage terminally differentiate and are lost from the epidermis, these cells instead persist in the papillomas and perhaps can be further recruited into carcinomas. Other recently reported models have also demonstrated recruitment of hair follicle, including infundibular and suprabulge, progeny into the wounded epidermis 9,18]. It would be interesting to use these other promoters in carcinogenesis studies to determine if other stem cell populations outside of the bulge contribute to skin tumor formation.
The second unanticipated finding was the presence of cellular heterogeneity in papillomas. While we observed contributions of keratin 15 origin in tumors, we also found a surprising number of non-blue cells within the same papillomas. There are several explanations for this, including technical issues such as 1) loss of blue staining during histological preparation of samples, 2) the involvement of other progenitors, or 3) incomplete Cre recombination within the bulge cell population. To address the first issue, we have utilized tissue processing to minimize loss of the X-gal reaction product. Distinguishing between the other two possibilities is difficult using the current system, however, several conclusions can still be reached.
The heterogeneity we observed could be attributed, for example, to contributions from non-keratin 15-expressing stem and progenitor cell populations in the hair follicle, such as those described by Clevers, et al. [19], Nijhof et al. [20], and Jensen et al. [21]. However, because they retain many of the stem cell-like properties described for K15-expressing keratinocytes, a role for these cells in tumorigenesis seems reasonable. Moreover, the cellular heterogeneity we observed could potentially arise from the inclusion of progeny from differentiated progenitors in the suprabasal epidermis. The presence of differentiated cells with the capacity to form tumors has been suggested by the classical work of Brown et al. [7] and Honeycutt et al. [22] who expressed oncogenic ras under the control of promoters involved in differentiation. Their data demonstrated that the target cells for benign papilloma development in their respective models can be derived from the epidermis. Additionally, a recent report from the Ghazizadeh laboratory has shown that in a grafting model of epidermal regeneration, early involucrin-expressing cells give rise to a multi-lineage epithelium [23]. Moreover, Arwert et al. [24] demonstrated that differentiated keratinocytes could initiate skin tumors via inflammatory cells. Nevertheless, while the most important observation is that keratin 15 expressing cells contribute to papillomas, heterogeneity due to incomplete Cre recombination is likely at least in part.
Another point of discussion as regards blue and non-blue cells in papillomas is related to low- and high-risk papillomas differing in susceptibility to malignant conversion as described by Darwiche et al. [25]. Additional evidence for this came from our studies using initiation/promotion in combination with epidermal abrasion, a technique that is used to remove the interfollicular epidermis, thus driving epidermal regeneration from the underlying hair follicles [26]. We demonstrated that mice first initiated with DMBA, then abraded, and finally subjected to TPA promotion developed about 50% fewer papillomas than non-abraded controls. Moreover, these mice developed approximately same number of malignancies, supporting the hypothesis that tumors develop from initiated precursor cells in both the interfollicular epidermis and hair follicles, and that papillomas of hair follicle origin are more likely to progress to malignancy. Unfortunately, in our present study, when we placed our mice in low-risk and high-risk promotion protocols, we failed to find any meaningful association with regards to the number or distribution of blue keratin 15-derived progeny in the papillomas. Clearly, further investigation is needed to illuminate a progenitor-progeny relationship in these two classes of papillomas.
A central theory of the multi-stage experimental skin carcinogenesis model is clonal expansion of initiated cells during promotion into papillomas. In that regard, one would expect to see an increase in the percentage of blue cells in the tumors over time, or all blue or all non-blue tumors depending upon the cell of origin [6]. However, the percentage of blue cells within papillomas remained constant regardless of the length of tumor promotion. Moreover, the numbers of papillomas composed of mixed blue and non-blue cells increased over time, with 100% of papillomas presenting a mixed phenotype by 20 weeks of promotion. In contrast; however, two keratoacanthomas and two squamous cell carcinomas collected from this study were either all blue or all non-blue. It is possible that as papillomas progress to malignancy, the tumors acquire additional genetic and epigenetic “hits” that drive the conversion from benign to malignant These data suggest that as long as the tumor is benign, it is fairly predictable and the relative proportions of non-blue and blue areas are held constant. Our data support the hypothesis that the acquisition of additional genetic hits driving progression to malignancy may target either blue or white progenitors independently. Statistical support of this observation will require further study and a higher multiplicity of carcinomas.
A major goal of this work was to determine not only if follicular keratin 15-expressing cells contribute to tumor development, but whether keratin 15-derived cells in papillomas would contain the signature mutation in the c-Ha-ras proto-oncogene, given that these cells are considered to be a primary carcinogen target population. This lead to another unexpected finding: heterogeneous expression of the ras mutation within individual papillomas. It has long been documented that 90-95% of papillomas arising from DMBA initiation harbor an A to T transversion in the c-Ha-ras proto-oncogene, and that papillomas do not develop in the absence of an initiating event [15]. In our studies, 95% of the papillomas analyzed did in fact have the Ha-ras mutation. In support of our original hypothesis that keratin 15-derived progeny would contain mutant Ha-ras, we found a 30% frequency in the blue regions of all tumors analyzed. The lower percentage of mutant cells from blue regions of papillomas could be due to the relatively lower number of DMBA adducts in the FACS-sorted alpha 6 integrin positive, CD34 positive cells (Figure 2 A,B). Thus, a reduced number of adducts could result in a lower mutation rate due to possible differences in the processing, removal, or mutation of the DNA in stem cells [27,28]. However, to our knowledge, finding areas of wild type Ha-ras distinct from areas containing activated Ha-ras within individual papillomas is the first demonstration of heterogeneity of the mutation in tumors in the multi-stage model of skin tumorigenesis.
Although papillomas could arise from cells of different lineages [25,26], or even from non-initiated cells induced to proliferate as a consequence of the tissue microenvironment, because labeling and tracking of keratin15 positive cells does not completely overlap with the induction of activated Ha-ras, we cannot make clear conclusions on the relative roles of other progenitors or differentiated cells in tumor development.
Of note is recent paper by Lapouge et al. [29] who determined that different epidermal lineages including keratin 15 expressing cells have the ability to initiate papillomas, but that multiple genetic events contribute to malignancy. Also using a transgenic mouse model based on targeted temporal expression of mutant KRas, White et al. [30] determined the inability of hair follicle transit amplifying cells to form tumors, but that benign lesions as well as squamous cell carcinomas are derived from keratin 15 expressing cells or their immediate progeny. Moreover, Schober and Fuchs [31] determined that squamous cell carcinoma initiating and tumor propagating cells express a hair follicle stem cell marker, CD34. Nevertheless, our data support a much more complex tumor evolution than can be readily explained by clonal expansion from a single initiated precursor cell.
Supplementary Material
Acknowledgements
This work was supported by NIH grant CA097957 to RJM and GC. This work was conducted (in part) in the Intramural Research Program of the NIH and National Institute of Environmental Health Sciences (NIEHS). We thank Leon C. King from the National Health and Environmental Effects Research Laboratory, United States Environmental Protection Agency, Research Triangle Park, NC for performing the adduct analysis. We thank Xiao-Jing Wang of the Department of Dermatology, OHSU for providing the CrePr1 construct. We also thank Ann Bode and Ashok Singh of The Hormel Institute for critically reading the manuscript and for their helpful suggestions, and Ashok Singh for assistance with submission of the manuscript.
Footnotes
Competing Financial Interests. The authors declare no competing financial interests.
References
- 1.Abel EL, Angel JM, Kiguichi K, DiGiovanni J. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc. 2009;4:1350–1362. doi: 10.1038/nprot.2009.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perez-Losada J, Balmain A. Stem-cell hierarchy in skin cancer. Nat Rev Cancer. 2003;3:434–443. doi: 10.1038/nrc1095. [DOI] [PubMed] [Google Scholar]
- 3.Gerdes MJ, Yuspa SH. The contribution of epidermal stem cells to skin cancer. Stem Cell Rev. 2005;1:225–231. doi: 10.1385/SCR:1:3:225. [DOI] [PubMed] [Google Scholar]
- 4.Kangsamaksin T, Park H-J, Trempus CS, Morris RJ. A perspective on murine keratinocytes stem cells as targets of chemically induced skin cancer. Mol Carcinog. 2007;46:579–584. doi: 10.1002/mc.20355. [DOI] [PubMed] [Google Scholar]
- 5.Morris RJ, Tacker KC, Fischer SM, Slaga TJ. Quantitation of primary in vitro clonogenic keratinocytes form normal adult murine epidermis, following initiation, and during promotion of epidermal tumors. Cancer Res. 1988;48:6285–6290. [PubMed] [Google Scholar]
- 6.Owens DM, Watt FM. Contribution of stem cells and differentiated cells to epidermal tumors. Nat Rev Cancer. 2003;3:444–451. doi: 10.1038/nrc1096. [DOI] [PubMed] [Google Scholar]
- 7.Brown K, Strathdee D, Bryson S, Lambie W, Balmain A. The malignant capacity of skin tumors induced by expression of a mutant H-ras transgene depends on the cell type targeted. Current Biol. 1998;8:516–524. doi: 10.1016/s0960-9822(98)70203-9. [DOI] [PubMed] [Google Scholar]
- 8.Morris RJ, Liu Y, Marles L, et al. Capturing and profiling adult hair follicle stem cells. Nat Biotech. 2004;22:411–417. doi: 10.1038/nbt950. [DOI] [PubMed] [Google Scholar]
- 9.Ito M, Liu Y, Yang Z, Nguyen J, et al. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat Med. 2005;11:1351–1354. doi: 10.1038/nm1328. [DOI] [PubMed] [Google Scholar]
- 10.Hansen LA, Tennant RW. Follicular origin of epidermal papillomas in v-Ha-ras transgenic TG.AC mouse skin. Proc Natl Acad Sci USA. 1994;91:7822–7826. doi: 10.1073/pnas.91.16.7822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu W-Y, Morris RJ. Method for harvest and assay of in vitro clonogenic keratinocyte stem cells from mice. In: Turksen K, editor. Methods in Molecular Biology. vol 289 Epidermal Cells: Methods and Protocols. Humana Press; Totowa: 2004. pp. 79–86. Totowa. [DOI] [PubMed] [Google Scholar]
- 12.Trempus CS, Morris RJ, Bortner CD, et al. Enrichment for living murine keratinocytes from the hair follicle bulge with the cell surface marker CD34. J Invest Dermatol. 2003;120:501–511. doi: 10.1046/j.1523-1747.2003.12088.x. [DOI] [PubMed] [Google Scholar]
- 13.Trempus CS, Morris RJ, Ehinger M, et al. CD34 expression by hair follicle stem cells is required for skin tumor development in mice. Cancer Res. 2007;67:4173–4181. doi: 10.1158/0008-5472.CAN-06-3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hennings H, Glick AB, Greenhalgh DA, et al. Critical aspects of initiation, promotion, and progression in multistage epidermal carcinogenesis. Proc Soc Exp Biol Med. 1993;202:1–8. doi: 10.3181/00379727-202-43511a. [DOI] [PubMed] [Google Scholar]
- 15.Quintanilla M, Brown K, Ramsden M, Balmain A. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature. 1991;322:78–80. doi: 10.1038/322078a0. [DOI] [PubMed] [Google Scholar]
- 16.Chen JX, Harvey R, Slaga T, Morris R, Tang M-s. Using UvrABC to detect 7, 12-dimethylbenz[a]anthracene-anti-diol-epoxide-DNA binding specificity in the mouse H-ras gene. Chem Res Toxicol. 1996;9:350–354. doi: 10.1021/tx9601115. [DOI] [PubMed] [Google Scholar]
- 17.Kim DJ, Kataoka K, Rao D, Kiguchi K, Cotsarelis G, DiGiovanni J. Targeted disruption of Stat3 reveals a major role for follicular stem cells in skin tumor initiation. Cancer Res. 2009;69:7587–7594. doi: 10.1158/0008-5472.CAN-09-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levy V, Lindon C, Zheng Y, Harfe BD, Morgan BA. Epidermal stem cells arise from the hair follicle after wounding. FASEB J. 2007;21:1358–1366. doi: 10.1096/fj.06-6926com. [DOI] [PubMed] [Google Scholar]
- 19.Snippert HJ, Haegebarth A, Kasper M, et al. Lgr6 marks stem cells in the fair follicle that generate all cell lineages of the skin. Science. 2010;327:1385–1389. doi: 10.1126/science.1184733. [DOI] [PubMed] [Google Scholar]
- 20.Nijhof JG, Braun KM, Giangreco A, et al. The cell surface marker MTS24 identifies a novel population of follicular keratinocytes with characteristics of progenitor cells. Development. 2006;133:3027–37. doi: 10.1242/dev.02443. [DOI] [PubMed] [Google Scholar]
- 21.Jensen UB, Yan X, Triel C, Woo SH, Christensen R, Owens DM. A distinct population of clonogenic and multipotential murine follicular keratinocytes residing in the upper isthmus. J Cell Sci. 2008;121:609–617. doi: 10.1242/jcs.025502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Honeycutt KA, Wailkel RL, Koster MI, Wang XJ, Roop DR. The effect of c-myc on stem cell fate influences skin tumor phenotype. Mol Carcinog. 2010;49:315–319. doi: 10.1002/mc.20617. [DOI] [PubMed] [Google Scholar]
- 23.Mannik J, Alzayady K, Ghazizadeh S. Regeneration of multilineage skin epithelial by differentiated keratinocytes. J Invest Dermatol. 2010;130:388–397. doi: 10.1038/jid.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arwert EN, Lal R, Quist S, Rosewell I, van Rooijen N, Watt FM. Tumor formation initiated by nondividing epidermal cells via an inflammatory infiltrate. Proc Natl Acad Sci USA. 2010;107:19903–19908. doi: 10.1073/pnas.1007404107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darwiche N, Ryscavage A, Perez-Lorenzo R, et al. Expression profile of skin papillomas with high cancer risk displays a unique genetic signature that clusters with squamous cell carcinomas and predicts risk for malignant conversion. Oncogene. 2007;26:6885–6895. doi: 10.1038/sj.onc.1210491. [DOI] [PubMed] [Google Scholar]
- 26.Morris RJ, Tryson KA, Wu KQ. Evidence that epidermal targets of carcinogen action are found in the interfollicular epidermis or infundibulum as well as in the hair follicles. Cancer Res. 2000;60:226–229. [PubMed] [Google Scholar]
- 27.Baer-Dubowska W, Morris RJ, Gill RD, DiGiovanni J. Distribution of covalent DNA adducts in mouse epidermal subpopulations after topical application of benzo(a)pyrene and 7, 12-dimethylbenz(a)anthracene. Cancer Res. 1990;50:3048–3054. [PubMed] [Google Scholar]
- 28.Sotiropoulou PA, Candi A, Mascré G, et al. Bcl-2 and accelerated DNA repair mediates resistance of hair follicle bulge cells to DNA-damage-induced cell death. Nat Cell Biol. 2010;12:572–582. doi: 10.1038/ncb2059. [DOI] [PubMed] [Google Scholar]
- 29.Lapouge G, Youssef KK, Vokaer B, et al. Identifying the cellular origin of squamous tumors. Proc Natl Acad Sci U S A. 2011;108:7431–7436. doi: 10.1073/pnas.1012720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White AC, Tran K, Khuu J, et al. Defining the origins of Ras/p53-mediate squamous cell carcinoma. Proc Natl Acad Sci U S A. 2011;108:7425–7430. doi: 10.1073/pnas.1012670108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schober M, Fuchs E. Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-beta and integrin/focal adhesion kinase (FAK) signaling. Proc Natl Acad Sci U S A. 2011;108:10544–10549. doi: 10.1073/pnas.1107807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.