

Abstract
The recepteur d’origine nantais (RON) receptor tyrosine kinase is overexpressed and stimulates invasive growth in pancreatic cancer cells, yet the mechanisms that underlie RON-mediated phenotypes remain poorly characterized. To better understand RON function in pancreatic cancer cells, we sought to identify novel RON interactants using multidimensional protein identification analysis. These studies revealed plectin, a large protein of the spectrin superfamily, to be a novel RON interactant. Plectin is a multifunctional protein that complexes with integrin-β4 (ITGB4) to form hemidesmosomes, serves as a scaffolding platform crucial to the function of numerous protein signaling pathways and was recently described as an overexpressed protein in pancreatic cancer (Bausch D et al., Clin Cancer Res 2010; Kelly et al., PLoS Med 2008;5:e85). In this study, we demonstrate that on exposure to its ligand, macrophage-stimulating protein, RON binds to plectin and ITGB4, which results in disruption of the plectin-ITGB4 interaction and enhanced cell migration, a phenotype that can be recapitulated by small hairpin ribosomal nucleic acid (shRNA)-mediated suppression of plectin expression. We demonstrate that disruption of plectin-ITGB4 is dependent on RON and phosphoinositide-3 (PI3) kinase, but not mitogen-activated protein kinase (MEK), activity. Thus, in pancreatic cancer cells, plectin and ITGB4 form hemidesmosomes which serve to anchor cells to the extracellular matrix (ECM) and restrain migration. The current study defines a novel interaction between RON and plectin, provides new insight into RON-mediated migration and further supports efforts to target RON kinase activity in pancreatic cancer.
Keywords: RON receptor, plectin, integrin beta-4, pancreatic cancer
Pancreatic cancer is the fourth leading cause of cancer-related mortality in the United States and is projected to account for greater than 40,000 deaths in 2011.1 Despite ongoing efforts to optimize therapy, median survival for pancreatic cancer patients remains <1 year as pancreatic cancer generally metastasizes before clinical detection and is highly chemoresistant.
We have previously shown that recepteur d’origine nantais (RON) is progressively upregulated during murine and human pancreatic carcinogenesis, and that it mediates pancreatic cancer cell migration, invasion and apoptotic resistance.2,3 However, at this time, the mechanisms through which RON regulates oncogenic phenotypes remain unclear. Clarification of the mechanisms through which RON functions may contribute to the development of effective therapies in pancreatic cancer.
To better understand RON function, we utilized mass spectrometry to identify potential RON binding partners. These studies led to the identification of plectin as a RON interactant. Plectin is a large protein (>500 kDa) of the spectrin superfamily that has long been recognized for its ability to bind and link together the three main components of the cytoskeleton: actin, microtubules and intermediate filaments.4 Plectin is known to bind integrin α6β4 as the major structural proteins of the hemidesmosome, a structure that anchors cells to the extracellular matrix.5 While hemidesmosomes are of unquestioned importance in the function of normal cells such as keratinocytes, their functional status and importance in the epithelial cancer cell remains unclear.6 Plectin also regulates protein kinase activation, as well as receptor signaling cascades, and recently plectin was identified as a putative biomarker in pancreatic cancer.7,8
In this study, we sought to characterize the interaction between RON and plectin, and to investigate its relevance to RON-regulated pancreatic cancer cell migration. Our studies demonstrate that absent ligand, RON resides primarily in a cytoplasmic location in pancreatic cancer cells, while plectin is primarily located at the basal cell membrane where it directly interacts with integrin-β4 (ITGB4) to form a hemidesmosome. Exposure to its ligand, macrophage-stimulating protein (MSP), results in cell migration and at the molecular level, translocation of RON to the cell surface to bind plectin resulting in disruption of the plectin-ITGB4 interaction—a process dependent on RON kinase activity. Cell migration is similarly enhanced in the absence of plectin expression. Thus, when associated with ITGB4, plectin serves to restrain pancreatic cancer cell migration, an interaction that is disrupted by RON signaling.
Material and Methods
Cell line and maintenance
The BxPC-3 human pancreatic cancer cell and human embryonic kidney cells (HEK) 293 human embryonic kidney cell lines were obtained from the American Type Culture Collection and were maintained in Roswell Park Memorial Institute culture media (RPMI) and Dulbecco’s modified eagles medium (DMEM) high-glucose medium, respectively. The fast growing (FG) human pancreatic cancer cell line has been previously described and was provided by Dr. David Cheresh (University of California, San Diego) and maintained in DMEM high-glucose medium.9,10 All media were supplemented with 10% fetal bovine serum (FBS) and 2% penicillin–streptomycin unless otherwise noted. All cells were grown in a humidified incubator at 37°C and 5% CO2.
Liquid chromatography—multidimensional protein identification
BxPC3 cells were serum starved overnight, treated with 200 ng/ml of MSP or vehicle for 15 min and protein lysates collected. Fifteen microgram of RON C-20 antibody (Santa Cruz) was conjugated to 75 μl of A/G Ultralink Beads (Pierce) using the Seize X IP Kit (Pierce). Five milligram of each sample was Immunoprecipitated (IP) with the conjugated beads. Precipitated proteins were loaded onto a biphasic capillary column for multidimensional protein identification (MudPIT) then separated and analyzed by 2D-LC separation in combination with tandem MS as described.11
Generation of mass spectrometry data
RAW files were generated from mass spectra using XCalibur version 1.4, and ms2 spectra data extracted using RAW Xtractor (version 1.9.1—available at: http://fields.scripps.edu/?q=content/download). Ms2 spectral data were searched using the SEQUEST algorithm (version 3.0) against the human IPI database (v3.65) database containing 86,382 sequences concatenated to a decoy database in which the sequences for each entry in the original database was reversed. The resulting ms2 spectra matches were assembled and filtered using DTASelect (version 1.9). Peptides with cross-correlation scores greater than 2.0 (+1), 2.5 (+2), 4.0 (+3), delta CN scores greater than 0.09 and percent ion match greater than 49% were included in the final dataset. This resulted in a false positive rate of 1.3% at the peptide level. To eliminate decoy database hits, a minimum peptide length of seven amino acids was imposed and protein identification required the matching of at least two peptides per protein.
Cell transfection
To generate stable plectin knockdown cell lines and corresponding green fluorescent protein (GFP) tagged-missense controls, HEK 293-T cells were plated on a p150 plate to a confluency of ~70% at the time of transfection. On the day of transfection, media was aspirated and replaced with 16 ml of prewarmed Optimem (Invitrogen). Five hundred microliters of prewarmed Optimem without antibiotics was pipetted into two sterile eppendorf tubes. To Tube 1, 80 μl of Lipofectamine 2000 (Invitrogen) was added. To Tube 2, 7 μg of pLKO1 Plec1-SH plasmid, 4.55 μg of rev response element (RRE) plasmid, 1.75 μg of REV plasmid and 2.45 μg of vesicular stomatitis virus glycoprotein (VSVG) plasmid was added. The tubes were inverted five times to mix and then incubated at room temperature for 3 min. The contents in Tube 1 were then added to Tube 2, inverted ten times to mix and then incubated at room temperature for 20 min. This solution was then added dropwise to the HEK 293-T cells. Sixteen hours later, the media was aspirated from the HEK 293-T cells and replaced with 16 ml of fresh complete media. The cells were then incubated at 37° (5% CO2) for 48 hr. Following this period of incubation, the virus-containing media was pipetted into a 50-mL conical vial, centrifuged at 350g and passed through a 0.45 μm filter. The filtered, virus-containing media was then added directly to FG and BxPC-3 cell lines which had been grown to 60% confluency on P100 dishes. After a 6-hr incubation period, the media was changed. FG and BxPC-3 cells which had been successfully transfected were selected in puromycin-containing growth media over 1–2 weeks.
Cell lysates and immunoblot analysis
Cells were lysed in radioimmunoprecipitation assay buffer (RIPA) containing complete protease inhibitors and Phos-STOP phosphatase inhibitors (Roche Applied Science). The lysates were left on ice for 30 min followed by centrifugation at 15,000g for 15 min, and then supernatants were collected. Protein concentration was determined using the Micro bicin-choninic acid assay (BCA) Protein Assay kit (Pierce). Immunoblotting was performed using between 5 and 30 μg of lysate. Samples were analyzed on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by immunoblotting. For immunoprecipitations, 500 μg of cell lysates were incubated with 1 μg of antibody for 30 min on ice followed by the addition of Protein A/G UltraLink Resin (Pierce) for 1 hr at 4°C with rotation. The beads were washed two quick times followed by two 15-min washes in RIPA buffer at 4°C with rotation. After the removal of the final wash, the beads were resuspended in 1× NuPAGE lithium dodecyl sulfate (LDS) sample buffer (Invitrogen) containing 1× NuPAGE sample reducing agent (Invitrogen) and were incubated at 60°C for 30 min to elute the protein from the beads. Samples were analyzed by SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Millipore) for analysis of proteins at 4°C overnight. At this time, the membrane was blocked in blocking buffer (1× tris buffered saline (TBS) + 0.05% Tween + 5% milk) for at least 1 hr. The membrane was then probed with primary antibody. Detection of β-actin (1:10,000; Sigma) served as loading control. Goat anti-mouse–horseradish peroxidase (HRP; Chemicon/Millipore) and goat anti-rabbit-HRP (Santa Cruz Biotechnology) were used as secondary antibodies at 1:5,000 dilution. The reaction was developed with Enhanced Chemiluminescence Plus reagent (GE Healthcare). When appropriate, membranes were stripped with Restore Western blot Stripping Buffer (Pierce) according to the manufacturer’s specifications and reprobed with primary antibody.
Antibodies
For immunoprecipitation, 1 μg of rabbit anti-RON C-20 (Santa Cruz Biotechnology) or 1 μg of mouse monoclonal anti-hemagglutinin (Santa Cruz Biotechnology) antibody was used. For immunoblotting, the following primary antibodies were used: rabbit anti-RON C-20 (1:500; Santa Cruz Biotechnologies), mouse anti-phospho-Akt (1:500), rabbit anti-Akt (1:1,000), rabbit anti-phospho-Erk (1:1,000), rabbit anti-Erk (1:1,000; Cell Signaling) and mouse anti-plectin (1:1000) (Abcam).
Proximity ligation assay
BxPC3 cells were grown to ~50% confluence on eight chamber slides (Nunc Lab Tek). Cells were serum starved overnight, then treated with 100 ng/ml of MSP for 15 min. Cells were fixed with paraformaldehyde for 10 min, then permeablized with phosphate buffered saline (PBS) + 0.1% TritonX-100 (PBS-T). Primary antibody labeling was performed using RON C-20 (Santa Cruz Biotechnology) at 1:1,000 dilution and mouse anti-Plectin (Abcam) at 1 μg/ml in PBS-T overnight at 4°C. The proximity ligation assay (PLA) was then performed as previously described.12 For the RON-Met kinase inhibitor (bristol myers squibb (BMS) 777607, Bristol Myers Squib), serum starved cells were treated with 100 nM of the inhibitor for 1 hr before MSP treatment with BMS being present during MSP stimulation. The phosphoinositide-3 (PI3) kinase inhibitor (LY294002, Cell Signaling) was used on serum starved cells at 50 μM for 1 hr before MSP treatment with the LY294002 compound being present during MSP stimulation. The mitogen-activated protein kinase (MEK) inhibitor U0126 (25 μM) was added to serum starved cells for 1 hr before MSP stimulation (100 ng/ml) with U0126 being present during MSP stimulation.
Immunofluorescence
Cells were plated onto chamber slides (Nunc Lab Tek) to achieve ~80% confluence. The next day cells were serum starved the overnight followed by treatment with 200 ng/ml of MSP for 15 min for BxPC3s and 30 min for FGs. Chambers were washed with PBS and fixed with 3.7% paraformaldehyde for 10 min. Cells were permeablized with PBS + 0.1% TritonX-100 (PBS-T). Primary antibody labeling was performed using rabbit anti RON C-20 (Santa Cruz Biotechnology) at 1:1,000 dilution and mouse anti-plectin (BD Biosciences) at 1:500 dilution in PBS-T overnight at 4°C. Negative control was comprised of normal mouse and rabbit IgG. Secondary antibodies Alexa goat anti Rabbit 594 and goat anti mouse 488 were used at 1:500 dilution for 1 hr at room temperature. The nucleus was labeled with DAPI. Labeling was visualized using a confocal microscope.
Cell proliferation assay
To determine the effect of RON stimulation on cell proliferation, 5 × 103 cells were plated per well in a 96-well plate in 200 μL of appropriate growth medium supplemented with 10% FBS and incubated overnight. The cells were then washed twice with PBS and then appropriate serum free growth media was added. The cells were incubated under these conditions for various time points (0, 24, 48, and 72 hr). Two hours before the designated time point, Alamar Blue (Biosource) was added to a final v/v concentration of 10% and allowed to incubate for 2 hr. An automated fluorescent plate reader at an excitation/emission of 530/590 nm was used to measure the proliferating cell population. For the 0-hr time point, Alamar Blue was added when the complete media was replaced with serum-free media. Cells stimulated with media + 10% FBS served as a positive control for proliferation, whereas media with Alamar Blue alone served as the blank, which was averaged and subtracted from the fluorescent value obtained for each well. Each experiment was done in triplicate and repeated as three independent experiments.
Scratch wound assays
BxPC3 and FG parental or plectin deficient cells were grown in six-well dishes to confluence in complete media. Cells were washed, placed in media containing 0.5% FBS and incubated overnight. The next day, four scratches were made in a plus shape with the end of a p200 barrier pipette tip. Fresh media containing 0.5% FBS was then added to each plate plus either PBS or 100 ng/ml of MSP. Dishes were photographed under ×10 magnification using a Nikon inverted microscope at t = 0 and, 18 hr for BxPC3 and 16 hr for FG cells. The wound area was calculated using the region setting in SPOT imaging software (SPOT Imaging Solutions). Percent wound coverage was calculated as follows: (1 − (area (μm2) at t = final hour/area (μm2) at t = 0)) × 100.
Results
Plectin is a novel RON-binding partner in human pancreatic cancer cells
To better delineate mechanisms of RON function, we first performed mass spectrometry to identify potential RON binding partners. RON protein was IP in the presence and absence of MSP and coprecipitated proteins underwent Mud-PIT analysis. Several interactants were identified; plectin was of particular interest given recent findings that its expression is upregulated in pancreatic cancer, its role in regulating intracellular receptor trafficking and protein interactions, and its importance as part of the hemidesmosome via interaction with ITGB4 and intermediate filaments.12–14 To corroborate MudPIT findings, IP/immunoblot was performed on lysates from two human pancreatic cancer cell lines (BxPC-3 and FG). Under serum-starved conditions, RON and plectin are not associated in BxPC-3 cells and are only modestly associated in FG cells. After exposure to MSP, RON is noted to bind plectin in both cell lines (Fig. 1a). While MudPIT analysis and co-IP verified plectin as a novel RON-binding partner in human pancreatic cancer, the nature of this interaction as either direct or through one or more protein intermediaries, was unclear. To examine this, we utilized a PLA (OLINK Bioscience, Uppsala, Sweden). With this technology, single protein events such as direct protein–protein interaction and protein phosphorylation can be detected. Proteins of interest are first targeted with primary antibodies. PLA probes, which are secondary antibodies with unique oligonucleotide tails, then bind the primary antibodies. In the presence of direct protein interaction (<40 nm proximity), the oligonucleotide tails hybridize to each other to form a closed circle. Fluorescence-labeled oligonucleotides and polymerase are added, and a rolling circle-amplification reaction can proceed, ultimately generating a red fluorescent signal to indicate proximity. Under serum-starved conditions, there is no evidence of RON-plectin interaction. After exposure to MSP, red-fluorescence demonstrates a direct interaction between RON and plectin (Fig. 1b) in FG cells. We observed a similar interaction in BxPc-3 cells (data not shown).
Figure 1.

RON interacts with plectin in the presence of MSP. (a) IP/IB of RON/Plectin in BxPC-3 and FG cells demonstrating RON-plectin interaction after exposure to the RON ligand MSP (100 ng/ml following serum starvation). (b) OLINK proximity ligation assay on FG cells demonstrating that the RON-plectin interaction is direct in the presence of MSP, indicated by the production of red fluorescent dots. (c) Immunofluorescent confocal microscopy on FG cells for RON (red) and plectin (green) showing that before MSP stimulation (100 ng/ml following serum starvation), RON resides primarily in the cytoplasm where there appears to be little direct RON-plectin interaction. After MSP stimulation, the RON receptor relocates to colocalize with plectin at lamellipodia (denoted by yellow color and arrows). (d) Immunofluorescence on BxPC-3 cells demonstrating that the cellular extensions are true lamellipodia. Cells are costained for RON (green) and phalloidin (red), which recognizes the F-actin protein component of the lamellipodia. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
MSP-stimulation drives RON from a peri-nuclear, cytoplasmic location to the cell surface to interact with plectin
To further characterize the RON-plectin interaction, immunofluorescence was performed in both BxPC-3 and FG cells. Cells were serum starved and then treated with either vehicle control or MSP. Fluorescence microscopy was then utilized to study RON-plectin subcellular localization. As seen in Figure 1c, before MSP stimulation, RON resides primarily in the cell cytoplasm near the nucleus, while plectin resides primarily at the cell membrane. There is no evidence of RON-plectin colocalization. After stimulation with MSP, we see some RON protein traffic from its perinuclear, cytoplasmic location to the cell membrane, specifically to extended lamellipodia that appear to be formed, at least in part, by reorganization of plectin. It is at these lamellipodia that RON appears to directly interact with plectin as demonstrated by colocalization (yellow). Note there is an abundance of RON protein in pancreatic cancer cells and even following MSP stimulation, there remains a distinct cellular pool of RON. Finally, to demonstrate that the cell extensions we observed could definitively be classified as lamellipodia, a phalloidin stain for F-actin was performed (Fig. 1d).
Plectin is not required for RON activation or downstream RON signaling
Having demonstrated the direct binding of RON and plectin, we sought to understand the significance of this interaction. Previous authors have shown that plectin serves as a critical scaffold protein for mediation of receptor signaling. Plectin has been demonstrated to function as a cytoskeletal regulator of protein kinase C signaling through sequestration of the protein RACK1.15 Similarly, plectin has been shown to mediate ligand-dependent CXCR4 internalization and activation of downstream extracellular signal regulated kinase 1/2 (ERK 1/2), which is also a RON substrate.16 We, therefore, hypothesized that plectin may act as a scaffold protein important for RON activation. To investigate this hypothesis, plectin-deficient cells were created. Lentiviral constructs expressing small hairpin ribosomal nucleic acid (shRNA) sequences directed against plectin mRNA were transfected into BxPC-3 and FG cells resulting in stable plectin knockdown. Lentiviral construct B7 was the most efficacious and thus was perpetuated for experimental purposes. Likewise, missense controls (designated GFP-missense) were generated via stable transfection of a lentivirus expressing a scrambled shRNA sequence. Immunoblot demonstrated no plectin knockdown in these cells (Supporting Information Fig. 1).
Plectin-deficient cells were serum starved and then exposed to vehicle or MSP. Cell lysates underwent IP for RON and then immunoblot for phosphotyrosine. RON-phosphorylation was unaffected in plectin-deficient cells, indicating that plectin is not critical to RON activation (Fig. 2a). As plectin has been shown to play a significant role in ERK 1/2 activation in the CXCR4 signaling cascade, we then investigated the role of plectin in the activation of downstream effectors of the RON receptor. The mitogen-activated protein kinase (MAPK) and (PI3K)/Akt pathways are critical regulators of oncogenic phenotypes in pancreatic cancer, including cellular proliferation, apoptotic resistance, invasion and migration and have previously been identified as downstream targets of RON signaling.3,17 Plectin-deficient cells were serum starved and then stimulated with vehicle control or MSP. Western blot analysis demonstrated the expected phosphorylation of ERK and AKT, indicating that plectin is not essential to activate these signaling pathways downstream of RON (Fig. 2b).
Figure 2.

Loss of plectin expression does not affect RON signaling. Parental (P), GFP missense and plectin knockdown (KD) of FG and BxPC-3 cells were serum starved overnight followed by treatment of MSP (100 ng/ml) for 30 min in the FG cells and 15 min for the BxPC-3 cells. (a) Immunoprecipitation of RON followed by immunoblotting with phosphor-tyrosine (Millipore #4G10) to determine RON phosphorylation. (b) Phosphorylation of Akt and Erk was determined by immunoblotting using phospho-specific antibodies phospho-Akt (Cell Signaling #9271) and phospho-Erk (Cell Signaling #9109). Note that the loss of plectin expression in plectin knockdown cells does not affect RON phosphorylation or RON-induced activation of either Akt or Erk, thus demonstrating that plectin is not required for RON signaling.
RON signaling disrupts the hemidesmosome in human pancreatic cancer cells
Given our findings that plectin is not critical for RON activation or downstream signaling, we turned our attention to the relationship between RON and the hemidesmosome in pancreatic cancer. Previous reports have demonstrated that multiple tyrosine kinase receptors, including epidermal growth factor receptor (EGFR) and c-Met (the HGF receptor), associate with α6β4 to at least partially induce hemidesmosome disassembly.18,19 MSP-RON signaling in normal human keratinocytes was shown to result in serine phosphorylation of α6β4, binding to RON, and the relocalization of a RON-α6β4 complex from the hemidesmosome to lamellipodia.20 The integrity of the hemidesmosome and its functional significance in carcinoma cells is uncertain, however.6 To study the effect of RON activation on the hemidesmosome in pancreatic cancer cells, we serum starved and then stimulated BxPC-3 and FG cells with MSP. PLA assay demonstrated that under serum starved conditions, plectin and ITGB4 are associated. After MSP stimulation, these proteins dissociate, commensurate with hemidesmosome disassembly (Fig. 3a). These findings were verified by immunoprecipitation for ITGB4 followed by plectin immunoblot (Fig. 3b). We next sought to determine if the MSP-driven RON-α6β4 interaction observed in normal keratinocytes occurs in pancreatic cancer cells. Again, BxPC-3 and FG cells were serum starved and then stimulated with MSP. PLA was then performed and verified the interaction between RON and ITGB4 (Fig. 3c). Immunoprecipitation also verified a RON-ITGB4 association induced by MSP stimulation (Fig. 3d).
Figure 3.
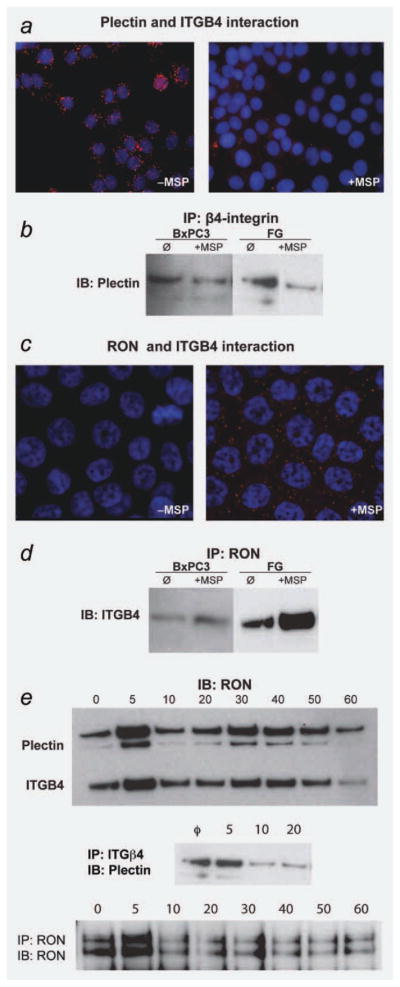
RON signaling regulates integrin beta-4 (ITGB4) interactions. (a) OLINK proximity ligation assay on BxPC-3 cells demonstrating a direct interaction between plectin and ITGB4 in the absence of RON activation, as evidenced by red fluorescent dots. After RON activation, this direct interaction is disrupted as indicated by loss of red signal. (b) IP/IB of ITGB4/Plectin was performed on BxPC-3 and FG cells showing the disruption of the plectin-ITGB4 interaction following exposure to MSP (100 ng/ml following serum starvation). (c) Proximity ligation assay on FG cells demonstrating that MSP (100 ng/ml following serum starvation) prompts the association of RON with ITGB4 as shown by red fluorescent dots. (d) IP/IB of RON/ITGB4 on BxPC-3 and FG cells again demonstrating that MSP (100 ng/ml following serum starvation) prompts the interaction of RON and ITGB4. (e) A time-course experiment was performed in which FG cells were exposed to 100 ng/ml MSP following serum starvation for 5, 10, 20, 30, 40, 50 min and 1 hr. No treatment served as the negative control. Cell lysates were collected and a RON immunoprecipitation was performed, followed by immunoblotting for either plectin (BD Biosciences #611348) or ITGB4 (Santa Cruz #sc-55514). An immunoprecipitation of ITGB4 (Santa Cruz #sc-9090) followed by immunoblotting with Plectin (BD Biosciences #6113348) was also performed on these same lysates looking out 20 min following MSP treatment. To check for equal pull down for the immunoprecipitations, immunoblotting for RON (Santa Cruz #sc-322) was done following the IP. After exposure to MSP, the interaction of RON and plectin/ITGB4 is characterized by repetitive association and disassociation. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Previous reports, coupled with our findings, demonstrate that, once activated, the RON-α6β4 complex relocalizes to extending lamellipodia. Given that lamellipodia extend and withdraw in a continuous cycle to perpetuate cell motility, we sought to characterize the interactions of RON-plectin and RON-ITGB4 following MSP stimulation. To investigate this, we performed a time-course experiment using IP/immunoblotting. FG cells were serum starved (T = 0) or stimulated with MSP for five, 10, 20, 30, 40, 50 or 60 min. Cell lysates were then made, subjected to immunoprecipitation with RON antibody, and immunoblotting first for plectin and then for ITGB4. We also performed a separate IP for ITGB4 followed by immunoblotting for plectin to demonstrate the interaction of these two proteins alone. Figure 3e demonstrates the gradual association and dissociation of RON with plectin and ITGB4 in the presence of MSP.
MSP-induced dissociation of plectin and integrin beta-4 is dependent on RON and PI3-kinase activity
We next sought to use the PLA assay to determine if RON kinase activity was critical for the disruption of the plectin-ITGB4 interaction. Figure 4a demonstrates the expected plectin-ITGB4 interaction under serum-starved conditions (abundant red signal), while MSP-stimulation results in dissociation of the two proteins. Next, before the addition of MSP, we exposed cells to BMS 777607, an ATP-mimetic agent that inhibits RON kinase activity.21 The addition of BMS 777607 blocked dissociation of plectin and ITGB4 in the presence of MSP (Fig. 4a). Thus, it can be concluded that MSP-stimulated breakdown of the hemidesmosome is dependent on RON kinase activation. It has been previously shown that serine phosphorylation (S1356, S1360, and S1364) prevents the interaction of ITGB4 with plectin.22 As the serine-threonine kinase, PI3K, is a RON substrate, we hypothesized that after phosphorylation by RON, PI3K phosphorylates ITGB4 resulting in its dissociation from plectin. To test this idea, we exposed BxPc-3 cells to MSP in the presence and absence of the PI3K inhibitor LY294002. As demonstrated in Figure 4b, the presence of the inhibitor completely blocks plectin-ITGB4 dissociation in cells exposed to MSP thus demonstrating that PI3K acts downstream of RON to mediate dissolution of the hemidesmosome. Finally, we tested the putative role of MEK, which is downstream of the RON substrate ERK 1/2, in dissociation of plectin-ITGB4. As seen in Figure 4c, the MEK inhibitor U0126 has no effect on MSP-induced dissociation of plectin-ITGB4. These experiments suggest that RON signals through PI3K, but not Erk to regulate dissociation of plectin-ITGB4.
Figure 4.
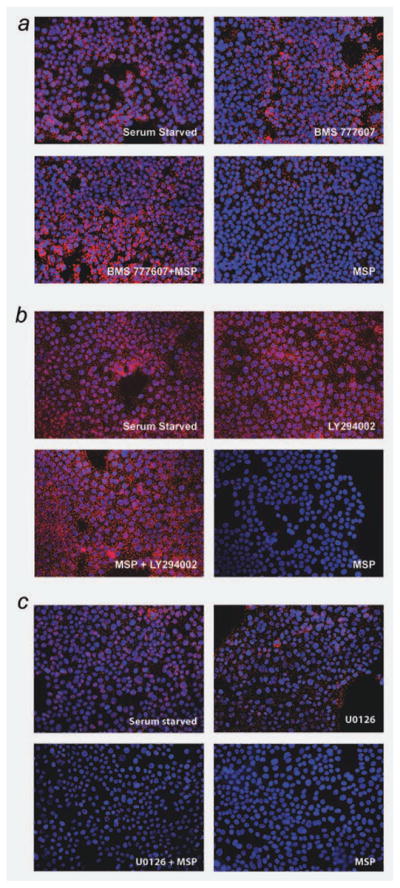
RON and PI3 kinase activity are required for MSP-induced disruption of the plectin-ITGB4 interaction. (a) Proximity ligation assay on BxPC-3 cells showing that RON activity is required for MSP-induced disruption of the plectin-ITGB4 interaction. Serum starved image shows a direct plectin-ITGB4 interaction before 100 ng/ml MSP treatment, as evidenced by red fluorescent dots. For the RON-Met kinase inhibitor (BMS 777607, Bristol Myers Squib), serum starved cells were treated with 100 nM of the inhibitor for 1 hr before MSP treatment with BMS being present during MSP stimulation. As seen in the BMS777607 + MSP image, the presence of the inhibitor prevented MSP to disrupt plectin-ITGB4, demonstrating that RON kinase activity is required. The BMS 777607 image shows that exposure to this inhibitor alone does not affect the plectin-ITGB4 interaction. (b) Proximity ligation assay on BxPC-3 cells showing that PI3 kinase activity is required for MSP-induced disruption of the plectin-ITGB4 interaction. The PI3 kinase inhibitor (LY294002, Cell Signaling) was used on serum starved cells at 50 μM for 1 hr before MSP treatment with the LY294002 compound being present during MSP stimulation. As shown in the MSP + LY294002 image, we see that this inhibitor serves to preserve the plectin-ITGB4 interaction in the presence of MSP. (c) Proximity ligation assay on BxPC-3 cells showing that a MEK inhibitor (U0126, Cell Signaling) does not affect the plectin–ITGB4 interaction in the presence of MSP. The MEK inhibitor U0126 (25 μM) was added to serum starved cells for 1 hr before MSP stimulation (100 ng/ml) with U0126 being present during MSP stimulation. As shown in the MSP + U0126 image, we see that the presence of the inhibitor does not prevent MSP from disrupting the plectin–ITGB4 interaction. This finding suggests that PI3 kinase activity is required for plectin-ITGB4 disruption, and that RON may achieve this disruption via its activation of PI3K. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Loss of plectin increases pancreatic cancer cell migration, recapitulating the MSP-stimulated phenotype
Having demonstrated that MSP-induced RON signaling results in disruption of the plectin-ITGB4 interaction, we sought to investigate its relevance to pancreatic cancer cell migration. To do this, we chose to simulate the RON activation state by examining our plectin-deficient pancreatic cancer cell lines. A scratch wound closure assay was performed in BxPc-3 and FG cells. After 18 hr, the rate of wound closure was compared between parental, plectin-knockdown and missense control cells, both in the absence and presence of MSP. As expected, MSP increased the degree of wound closure from 11.6 to 31.3% in BxPc3 cells and from 44.3 to 56.1% in FG cells (p < 0.001 for both comparisons). Reduced expression of plectin resulted in a significant increase in wound closure at 18 hr for both cells lines (11.6–34.7%-BxPC-3, 44.3–66.5%-FG, p < 0.001 for both comparisons) recapitulating the MSP-stimulated phenotype (Fig. 5). This finding suggests that disruption of the hemidesmosome is a major mechanism by which RON signaling induces pancreatic cancer cell migration. We also observed, however, that loss of plectin does not preclude an additional migratory response to MSP, suggesting that RON induced migration is mediated by additional effects on the cell independent of its effects on the hemidesmosome. We noted no difference in rate of proliferation in plectin-negative cells as compared to parental pancreatic cancer cells, as demonstrated by Alamar Blue proliferation assay (data not shown) demonstrating that scratch wound assay results are not confounded by differing rates of cell proliferation.
Figure 5.

Loss of plectin expression in pancreatic cancer cells enhances migration. (a) Confluent cell monolayers were generated in six-well dishes using BxPC3 and FG parental, GFP missense and plectin knockdown cells. Scratches were made in the monolayer using a p200 tip after which treatment with either PBS or 100 ng/ml of MSP added to its respective well. Images were taken at t = 0 and t = 16 hr for the FG cells and t = 18 hr for the BxPC-3 cells (scratch wound images not shown). Each condition was performed in triplicate, and each experiment was repeated three times. (b) Wound coverage data from scratch assays in FG and BxPC-3 cells is displayed. Scratch would assays were measured by determining the area of the scratch at t = 0 hr and at t = final hour (Final hour for FG cells = 16 hr and for BxPC3 cells = 18 hr) using the region setting in SPOT imaging software. To determine the percent wound coverage, the following equation was used: {1 − (area at tfinal/area at t0)} × 100. The mean value was determined and graphed + SE. Two-tailed Student’s t-test was performed for statistical analysis. Comparisons between PBS vs. treatment were done in parental (grey), GFP missense (white) and in knockdown (black). Significant values were as follows: p < 0.005(**), p < 0.0005(***). In both cell lines, plectin knockdown cells achieved a higher percentage of wound closure in the absence and presence of MSP, ***(p < 0.0005).
Discussion
Pancreatic cancer remains a lethal disease because of its propensity to metastasize before clinical detection as well as its ability to resist chemotherapeutic intervention. The RON receptor tyrosine kinase is upregulated during pancreatic carcinogenesis and mediates cancer cell migration, invasion and apoptotic resistance in pancreatic cancer cells.2,3,23,24 While our previous work has begun to clarify the pathways through which RON mediates apoptotic resistance, the mechanisms through which RON mediates pancreatic cancer cell migration and invasion remain poorly defined.
Our goal in this study was to better understand the means by which RON signaling induces cell migration. We began these studies by performing mass spectrometry to identify potential RON binding partners to derive clues regarding mechanisms of RON function. Of the numerous proteins that were identified by mass spectrometry, plectin was of particular interest not only because of its primary function as a major structural protein that is linked to the actin cytoskeleton—but also because it is upregulated in pancreatic cancer, regulates intracellular receptor trafficking and protein interaction and couples with ITGB4 to comprise the hemidesmosome. In this study, we have confirmed plectin as a novel and direct binding partner of RON and shown that plectin is not required to mediate RON signaling. It is clear that binding of RON and plectin is initiated by exposure to the RON ligand, MSP, which induces trafficking of RON to the cell membrane, specifically at extending lamellipodia. The modest degree of RON-plectin interaction seen in FG cells in the absent of ligand is likely secondary to a basal level of constitutive RON phosphorylation we have observed in FG cells even under serum starved conditions. Co-IP and PLA in unstimulated cells demonstrated that plectin and ITGB4 are directly bound in pancreatic cancer cells, thus demonstrating that hemidesmosomes are intact in these cells.
A recent report by Frijns et al. demonstrated that in keratinocytes, hemidesmosome formation is regulated by the EGFR via its activation of MAPK signaling.25 Our study similarly revealed that in pancreatic cancer cells, MSP induced dissociation of plectin from ITGB4 is dependent on the kinase activity of RON as well as its downstream target PI3K. We have also shown that another RON substrate Erk, has no role in regulating plectin-ITGB4 dissociation. Thus, our results suggest that exposure to MSP, results in the binding of RON to plectin and ITGB4, leading to PI3K phosphorylation of ITGB4 and its dissociation from plectin. The interaction between RON, plectin and ITGB4 is dynamic such that RON is cyclically associating with these two proteins and then dissociating from them. This in turn may allow hemidesmosome assembly and disassembly, suggesting that just as in normal keratinocytes, the migrating pancreatic cancer cell moves along the extracellular matrix (ECM), gripping it via intact hemidesmosomes, releasing it as the hemidesmosome breaks down, extending lamellipodia forward and then re-gripping the ECM as the hemidesmosome reforms. The role of the hemidesmosomal structure in restraining pancreatic cancer cell migration was demonstrated by our experiments in plectin deficient cells which migrate rapidly in the absence of MSP, simulating the MSP-induced phenotype.
Our results leave some questions unanswered and generate new questions that need to be addressed. While we have defined a novel interaction between RON and plectin, the domain of this interaction is yet to be mapped and is the subject of ongoing work. Further, the significance of plectin overexpression in pancreatic cancer remains unclear. The association of plectin with ITGB4 is a crucial step in hemidesmosome stabilization, and our results demonstrate that this interaction in pancreatic cancer cells gives rise to a functional hemidesmosome. Our experiments demonstrate that plectin-deficient cells migrate rapidly recapitulating the MSP-induced phenotype, but their proliferation is unaffected. Taken together, these findings point to plectin as a negative regulator of cell motility. However, plectin is upregulated in pancreatic cancer, a finding that might seem contrary to our results. Thus, it is possible that plectin upregulation is merely an epiphenomenon, that our findings relative to plectin’s effects on migration are cell line dependent, or that plectin overexpression represents an attempt by the cell to check kinase activity and/or cell motility that has become dysregulated.
Although our experiments demonstrate that RON directly binds both plectin and ITGB4, an event dependent on RON kinase activity, we did not find evidence that RON directly phosphorylates either protein. Certainly, it is possible that technical issues prevented our detection of such phosphorylation events. Instead, our data suggest that RON induces dissociation of plectin and ITGB4 via phosphorylation of PI3K. Previously, PI3K has been show to phosphorylate ITGB4 at serine residues 1356, 1360 and/or 1364 and prevent the interaction of ITGB4 with the plectin actin-binding domain.22 It has been shown that EGFR activation results in phosphorylation of ITGB4 via the MAPK pathway contributing to hemidesmosome disassembly in keratinocytes. Again our results show no role for the MAPK pathway in RON-regulated hemidesmosomal disassembly. Thus, it appears likely that RON is facilitating ITGB4 phosphorylation via an alternative downstream protein, PI3K.25 Future studies will be directed at determining the critical residues on ITGB4 required for RON-dependent dissociation from plectin.
Our studies suggest that while RON-dependent breakdown of the hemidesmosome, significantly increases cell migration, RON signaling augments cell migration even in the absence of an intact hemidesmosome. This suggests other RON-mediated effects on cell migration. RON has a variety of other substrates which have been implicated in the migratory response including ERK 1/2, Src kinase and cross-talk with c-met and EGFR.23,26,27 We have shown that MEK, which is downstream of ERK 1/2 is not required for plectin/ITGB4 dissociation. It will be of interest in the future to explore the contribution of other pathways to this RON-induced phenotype. In conclusion, these studies demonstrate an interaction between the RON kinase and plectin, suggest a mechanism by which RON promotes pancreatic cancer cell migration and validate efforts to target RON signaling in pancreatic cancer.
Supplementary Material
Acknowledgments
This work was supported by NIH CA 155620 (A.M.L.).
Footnotes
Additional Supporting Information may be found in the online version of this article
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Logan-Collins J, Thomas RM, Yu P, Jaquish D, Mose E, French R, Stuart W, McClaine R, Aronow B, Hoffman RM, Waltz SE, Lowy AM. Silencing of RON receptor signaling promotes apoptosis and gemcitabine sensitivity in pancreatic cancers. Cancer Res. 2010;70:1130–40. doi: 10.1158/0008-5472.CAN-09-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas RM, Toney K, Fenoglio-Preiser C, Revelo-Penafiel MP, Hingorani SR, Tuveson DA, Waltz SE, Lowy AM. The RON receptor tyrosine kinase mediates oncogenic phenotypes in pancreatic cancer cells and is increasingly expressed during pancreatic cancer progression. Cancer Res. 2007;67:6075–82. doi: 10.1158/0008-5472.CAN-06-4128. [DOI] [PubMed] [Google Scholar]
- 4.Sonnenberg A, Liem RK. Plakins in development and disease. Exp Cell Res. 2007;313:2189–203. doi: 10.1016/j.yexcr.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 5.Leung CL, Green KJ, Liem RK. Plakins: a family of versatile cytolinker proteins. Trends Cell Biol. 2002;12:37–45. doi: 10.1016/s0962-8924(01)02180-8. [DOI] [PubMed] [Google Scholar]
- 6.Litjens SH, de Pereda JM, Sonnenberg A. Current insights into the formation and breakdown of hemidesmosomes. Trends Cell Biol. 2006;16:376–83. doi: 10.1016/j.tcb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 7.Bausch D, Thomas S, Mino-Kenudson M, Fernandez-Del Castillo C, Bauer TW, Williams M, Warshaw AMD, Thayer SP, Kelly KA. Plectin as a novel biomarker for pancreatic cancer. Clin Cancer Res. 2010;17:302–9. doi: 10.1158/1078-0432.CCR-10-0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly KA, Bardeesy N, Anbazhagan R, Gurumurthy S, Berger J, Alencar H, Depinho RA, Mahmood U, Weissleder R. Targeted nanoparticles for imaging incipient pancreatic ductal adenocarcinoma. PLoS Med. 2008;5:e85. doi: 10.1371/journal.pmed.0050085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vezeridis MP, Txanakakis GN, Meitner PA, Doremus CM, Tibbetts LM. In vivo selection of a highly metastatic cell line of a human pancreatic carcinoma in the nude mouse. Cancer. 1992;69:2060–3. doi: 10.1002/1097-0142(19920415)69:8<2060::aid-cncr2820690810>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 10.Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1:50–62. doi: 10.1038/sj.neo.7900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–7. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 12.Gregor M, Zeold A, Oehler S, Marobela KA, Fuchs P, Weigel G, Hardie DG, Wiche G. Plectin scaffolds recruit energy-controlling AMP-activated protein kinase (AMPK) in differentiated myofibres. J Cell Sci. 2006;119:1864–75. doi: 10.1242/jcs.02891. [DOI] [PubMed] [Google Scholar]
- 13.Lunter PC, Wiche G. Direct binding of plectin to Fer kinase and negative regulation of its catalytic activity. Biochem Biophys Res Commun. 2002;296:904–10. doi: 10.1016/s0006-291x(02)02007-7. [DOI] [PubMed] [Google Scholar]
- 14.Koster J, van Wilpe S, Kuikman I, Litjens SH, Sonnenberg A. Role of binding of plectin to the integrin beta4 subunit in the assembly of hemidesmosomes. Mol Biol Cell. 2004;15:1211–23. doi: 10.1091/mbc.E03-09-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Osmanagic-Myers S, Wiche G. Plectin-RACK1 (receptor for activated C kinase 1) scaffolding: a novel mechanism to regulate protein kinase C activity. J Biol Chem. 2004;279:18701–10. doi: 10.1074/jbc.M312382200. [DOI] [PubMed] [Google Scholar]
- 16.Ding Y, Zhang L, Goodwin JS, Wang Z, Liu B, Zhang J, Fan GH. Plectin regulates the signaling and trafficking of the HIV-1 co-receptor CXCR4 and plays a role in HIV-1 infection. Exp Cell Res. 2008;314:590–602. doi: 10.1016/j.yexcr.2007.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Danilkovitch-Miagkova A. Oncogenic signaling pathways activated by RON receptor tyrosine kinase. Curr Cancer Drug Targets. 2003;3:31–40. doi: 10.2174/1568009033333745. [DOI] [PubMed] [Google Scholar]
- 18.Mariotti A, Kedeshian PA, Dans M, Curatola AM, Gagnoux-Palacios L, Giancotti FG. EGF-R signaling through Fyn kinase disrupts the function of integrin alpha6beta4 at hemidesmosomes: role in epithelial cell migration and carcinoma invasion. J Cell Biol. 2001;155:447–58. doi: 10.1083/jcb.200105017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trusolino L, Bertotti A, Comoglio PM. A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell. 2001;107:643–54. doi: 10.1016/s0092-8674(01)00567-0. [DOI] [PubMed] [Google Scholar]
- 20.Santoro MM, Gaudino G, Marchisio PC. The MSP receptor regulates alpha6beta4 and alpha3beta1 integrins via 14–3-3 proteins in keratinocyte migration. Dev Cell. 2003;5:257–71. doi: 10.1016/s1534-5807(03)00201-6. [DOI] [PubMed] [Google Scholar]
- 21.Dai Y, Siemann DW. BMS-777607, a small-molecule met kinase inhibitor, suppresses hepatocyte growth factor-stimulated prostate cancer metastatic phenotype in vitro. Mol Cancer Ther. 2010;9:1554–61. doi: 10.1158/1535-7163.MCT-10-0359. [DOI] [PubMed] [Google Scholar]
- 22.Wilhelmsen K, Litjens SH, Kuikman I, Margadant C, van Rheenen J, Sonnenberg A. Serine phosphorylation of the integrin beta4 subunit is necessary for epidermal growth factor receptor induced hemidesmosome disruption. Mol Biol Cell. 2007;18:3512–22. doi: 10.1091/mbc.E07-04-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Camp ER, Yang A, Gray MJ, Fan F, Hamilton SR, Evans DB, Hooper AT, Pereira DS, Hicklin DJ, Ellis LM. Tyrosine kinase receptor RON in human pancreatic cancer: expression, function, and validation as a target. Cancer. 2007;109:1030–9. doi: 10.1002/cncr.22490. [DOI] [PubMed] [Google Scholar]
- 24.O’Toole JM, Rabenau KE, Burns K, Lu D, Mangalampalli V, Balderes P, Covino N, Bassi R, Prewett M, Gottfredsen KJ, Thobe MN, Cheng Y, et al. Therapeutic implications of a human neutralizing antibody to the macrophage-stimulating protein receptor tyrosine kinase (RON), a c-MET family member. Cancer Res. 2006;66:9162–70. doi: 10.1158/0008-5472.CAN-06-0283. [DOI] [PubMed] [Google Scholar]
- 25.Frijns E, Sachs N, Kreft M, Wilhelmsen K, Sonnenberg A. EGF-induced MAPK signaling inhibits hemidesmosome formation through phosphorylation of the integrin {beta}4. J Biol Chem. 2010;285:37650–62. doi: 10.1074/jbc.M110.138818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benvenuti S, Lazzari L, Arnesano A, Li Chiavi G, Gentile A, Comoglio PM. Ron kinase transphosphorylation sustains MET oncogene addiction. Cancer Res. 2011;71:1945–55. doi: 10.1158/0008-5472.CAN-10-2100. [DOI] [PubMed] [Google Scholar]
- 27.Peace BE, Hill KJ, Degen SJ, Waltz SE. Cross-talk between the receptor tyrosine kinases Ron and epidermal growth factor receptor. Exp Cell Res. 2003;289:317–25. doi: 10.1016/s0014-4827(03)00280-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.