

Abstract
Renal cell carcinoma (RCC) accounts for ~4% of all human malignancies and is the 9th leading cause of male cancer death in the United States. The purpose of this study was to determine the effect of variation within microRNA (miRNA) binding sites of genes in the VHL-HIF1α pathway on RCC risk. We identified 429 miRNA binding site single nucleotide polymorphisms (SNPs) in 102 pathway genes and assessed 53 tagging-SNPs for 31 of these genes for risk in a case-control study consisting of 894 RCC cases and 1,516 controls. Results showed that five SNPs were significantly associated with RCC risk. The most significant finding was rs743409 in MAPK1. Under the additive model, the variant was associated with a 10% risk reduction (OR: 0.90, 95% CI, 0.77-0.98). Other significant findings were for SNPs in CDCP1, TFRC, and DEC1. Cumulative effects analysis showed that subjects carrying four or five unfavorable genotypes had a 2.14-fold increase in risk (95% CI, 1.03-4.43, P = 0.04) than those with no unfavorable genotypes. Potential higher-order gene-gene interactions were identified and categorized subjects into different risk groups. The OR of the high-risk group defined by two SNPs: CDCP1:rs6773576 (GG) and DEC1:rs10982724 (GG) was 4.46-times higher than that of low-risk reference group (95% CI, 1.31-15.08). Overall, our study provides the first evidence supporting a connection between miRNA binding site SNPs within the VHL-HIF1α pathway and RCC risk. These novel genetic risk factors might help identify individuals at high risk to enable detection of tumors at an early, curable stage.
Keywords: VHL-HIF1α pathway, microRNA, renal cell carcinoma
Introduction
Renal cell carcinoma (RCC) accounts for ~4% of all human malignancies and is the 9th leading cause of male cancer death in the United States [1]. In 2012, it is estimated that approximately 64,770 cases of kidney and renal pelvis cancers will be diagnosed, resulting in 13,570 deaths in the United States [1]. The incidence of RCC has been increasing by an estimated 2-3% annually for the past two decades [2]. The 5-year cumulative survival rate of patients with RCC was 70% and has remained unchanged for 10 years [3]. For patients with tumors that have spread to regional lymph nodes, the 5-year cancer-specific survival rate is 63.3%, and once RCC has metastasized, the 5-year survival rate is 11.1% [4]. Because RCC is a “quiet” disease and typically diagnosed at an advanced stage when systemic chemotherapy has limited or no effectiveness [5,6], it is vital to develop new approaches for risk prediction and early detection to improve overall survival in RCC patients.
The VHL-HIF1α pathway has been well established as playing a major role in the pathogenesis of RCC. Mutations in this pathway, particularly in the tumor suppressor Von Hippel-Lindau (VHL), are associated with familial and sporadic renal cancer with VHL mutations detected in 61% of sporadic clear cell RCCs [7]. VHL promoter hypermethylation was also detected in more than 20% of sporadic clear cell RCC cases [8]. The main function of VHL is its E3-ubiquitin ligase activity which results in degradation of target proteins. Hypoxia inducible factor (HIF) is a critical downstream target of VHL. HIF is a basic helix-loop-helix transcription factor that consists of an oxygen-sensitive α-subunit HIF-α, and a constitutively expressed beta-subunit, HIF-β (encoded for by ARNT). HIF is normally bound to VHL and then degraded by an oxygen dependent ubiquitin-proteasome-mediated pathway [9]. Under normal conditions, the HIF-α subunit is hydroxylated by specific prolyl-hydroxylases and targeted for rapid degradation by VHL [10,11]. In a hypoxic environment or in the absence of functional VHL, HIF-1α is not degraded and translocates to the nucleus, where it dimerizes with HIF-β to form active HIF [10]. Besides VHL, cytoplasm HIF-1α is also regulated by the p53-MDM2 complex, MAPKs, AKT, and HSP90 [12,13]. HIF is involved in the regulation of many biological processes that facilitate both oxygen delivery and oxygen deprivation adaptation, as well as cell proliferation and angiogensis through vascular endothelial growth factor (VEGF), platelet-derived growth factor β (PDGF-β) and transforming growth factor-α (TGF-α) and metastasis through the repression of E-cadherin [14-16]. Research has also demonstrated that HIF inhibition is sufficient to decrease RCC tumor formation [17]. Inhibition of VEGF, PDGF, and TGF-α by sorafenib, axitinib, sunitinib, and bevacizumab are the basis for current treatment of metastatic RCC [18,19]. In addition, inhibition of HIF by PI3K/Akt/mTOR pathway inhibitors is another strategy under development [18,20].
MicroRNAs (miRNA) are a class of small noncoding RNA molecules 19-22 nucleotides in length. MiRNAs regulate gene expression by binding to target gene 3′ untranslated regions (3′UTR). As of November 2011, 1,527 human miRNA genes had been identified and deposited in the miRNA database (http://www.mirbase.org). Each miRNA may regulate hundreds of different protein-coding genes and depending on context it can function as either an oncogene or tumor suppressor. Previous studies have demonstrated that mutations, deletions, amplifications, over expression, and epigenetic silencing of miRNAs are involved in cancer [21]. MiRNAs regulate gene expression through complementarity to targeted 3′ UTR [22]. Recently, several genes known to play a role in RCC pathogenesis (mTOR, VHL, HIF-1α, PDGFβ) were detected as potential targets of miRNAs that were dysregulated in RCC [23]. Recently, our group identified genetic variations within miRNAs and miRNA-processing genes that modify RCC risk, survival, and recurrence [24,25]. However, there has been no report regarding the effect of genetic variation within miRNA binding sites of important cellular pathways involved in RCC pathogenesis on RCC risk.
Polymorphisms in miRNA binding sites have attracted attention because they can create a new binding site, disrupt an existing site, or change miRNA binding affinity. These alterations can thus affect normal miRNA function, potentially leading to cancer development. The Polymorphism in miRNA Target Site (PolymiRTS) database scans for SNPs located in the 3′UTR of each gene and predicts the effect on putative miRNA target sites [26]. We utilized the wealth of information in this database to scan more than one hundred genes that function in the VHL-HIF1α pathway for variants that potentially alter miRNA binding sites. The tagging SNPs for these variants were then assessed for RCC risk in a large case-control population. To our knowledge, these variants have not been previously assessed for association with cancer development.
Patients and Methods
Study Population and Epidemiologic Data
The proposed study was carried out within the framework of the ongoing case-control study of RCC which has been recruiting incident cases from The University of Texas MD Anderson Cancer Center in Houston, Texas since 2002. All patients were residents of Texas, have histologically confirmed RCC and were previously untreated by chemotherapy or radiotherapy. There are no age, sex, ethnicity, and cancer stage restrictions for participation in the study. Population-based controls are recruited using an established modified random digit dialing protocol. The control participants were frequency matched to the cases in terms of age, sex, ethnicity, and county of residence. We also included controls from an ongoing bladder cancer case-control study who were involved in a previously published GWAS of bladder cancer [27]. For the current analysis, we included 894 cases and 1,156 controls who had previously been included in our genome-wide scan analysis [28]. This population was restricted to non-Hispanic whites to limit the confounding effect from population stratification. The study has been approved by the Institutional Review Board of MD Anderson Cancer Center. Written informed consent has been obtained from all study participants.
Epidemiologic data were collected through in-person interview using a pre-tested risk factor questionnaire conducted by trained MD Anderson staff personnel. Following the interview, a 40-mL peripheral blood sample was obtained for DNA extraction and storage.
Selection of miRNA binding site tagging SNPs in VHL-HIFα pathway genes
We utilized Ingenuity systems pathway analysis (Ingenuity Systems, Inc. Redwood City, CA) and PubMed to identify an extensive list of VHL-HIFα pathway-related genes. A total of 102 candidate genes were selected for analysis with RCC risk. Next, we scanned for miRNA binding site SNPs within the 3′UTR of these genes using the PolymiRTS database (http://compbio.uthsc.edu/miRSNP/). We further selected SNPs based on a minor allele frequency (MAF) greater than 0.01 as reported in dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/). The complete set of 429 SNPs were screened with software package SNAP (http://www.broadinstitute.org/mpg/snap/) to identify tagging SNPs with r2 threshold larger than 0.8. In the case of multiple binding site SNPs within the same haplotype block (defined by the linkage disequilibrium coefficient r2 > 0.8), only one tagging SNP was selected. After SNAP screening of this set of SNPs, 53 tagging SNPs in 31 genes were present on the Illumina 610 array (Illumina, San Diego, CA). We then extracted the genotyping information from our existing data [28] generated using this chip to perform statistical analysis.
Statistical analysis
Lab and epidemiological data was merged and cleaned using system files created with the SAS software package (Cary, NC). Most analyses were performed using STATA 10.0 (Stata Corporation, College Station, TX) and HelixTree (Golden Helix, Bozeman, MT). Distributions of characteristics between cases and controls were evaluated by the χ2 test (for categorical variables) or student’s t-test (for continuous variables with normal distribution). Multivariate unconditional logistic regression model was applied to calculate odds ratios (ORs) and 95% confidence intervals (95% CIs) for analyzing the main effects of selected SNPs on RCC risk under three different genetic models of inheritance: dominant, recessive, and additive. The best-fitting model was the one with the smallest P value. Bootstrap resampling was performed 1000 times to internally validate the results from our analyses [29]. The combined effects of unfavorable genotypes analysis included those SNPs showing statistical significance in the main analysis (P < 0.05). Higher-order gene–gene interactions were evaluated using Classification and Regression Tree (CART) analysis implemented in the HelixTree software. We also performed 10,000 bootstrap runs to internally validate our CART analysis results. All statistical analyses were two-sided.
Results
Characteristics of the study population
This study included 894 cases and 1,516 controls, who were all non-Hispanic whites (Table 1). The mean age was 63.23 ± 10.91 years for controls and 59.69 ± 10.69 for the cases, which was a significantly different between the two groups (P < 0.001). A significant difference in sex was also observed (P < 0.001) with the controls having a higher number of male participants. There was no significant difference in terms of smoking status between these two groups (P > 0.05).
Table 1.
Characteristics of study population (N= 2410, 894/1516)
| Variables | Control (n %) | Case (n %) | P value |
|---|---|---|---|
| Age, mean(SD) | 63.23 (10.91) | 59.69 (10.69) | <0.001 |
| Sex | |||
| Male | 1174 (77.44) | 609 (68.12) | |
| Female | 342 (22.56) | 285 (31.88) | <0.001 |
| Smoking status | |||
| Ever | 853 (56.27) | 515 (57.61) | |
| Never | 663 (43.73) | 379 (42.39) | 0.52 |
Main effects of miRNA binding site genetic variation in VHL-HIFα pathway genes
Fifty-three tagging SNPs for miRNA binding site variants were identified from 31 selected genes (Table 2). Among these tagging SNPs, five were significantly associated with RCC risk (P < 0.05, Table 3). The most significant associations were observed for two SNPs in MAPK1 (mitogen-activated protein kinase 1). Rs743409 was associated with a 10% reduction in risk (HR, 0.90, 95% CI 0.77-0.98; P = 0.02) under the additive model. MAPK1:rs9607241 resulted in a 14% increase in risk (HR, 1.14, 95% CI, 1.02-1.29, P = 0.03) under the additive model. In addition, rs6773576 in CDCP1 (CUB domain-containing protein 1) resulted in an 18% increase in risk (HR, 1.18, 95% CI, 1.01-1.37, P = 0.03) under the additive model, TFRC (transferrin receptor):rs406271 resulted in an 88% increase in risk (HR, 1.88, 95% CI, 0.77-1.00, P = 0.04) under the additive model, and rs10982724 in DEC1 (deleted in esophageal cancer 1) resulted in an greater than 2-fold increase in risk (HR, 2.18, 95% CI, 1.01-4.74, P = 0.05) under the recessive model. Three of these five top SNPs (rs13943, rs1063311, and rs12947) had highly consistent results in bootstrap analysis for internal validation, with bootstrap P values < 0.05 for more than 800 of 1000 samplings (Table 3).
Table 2.
Tagging SNPs of genes carrying miRNA binding site SNP (MAF > 0.01)
| Gene name | Tagging SNP |
|---|---|
| APC | rs11950612 |
| CASR | rs2134225 |
| CASR | rs9740 |
| CCDN1 | rs649392 |
| CCDN1 | rs7178 |
| CCND1 | rs1789172 |
| CCND1 | rs9344 |
| CDCP1 | rs17077237 |
| CDCP1 | rs9863819 |
| CITED2 | rs1131431 |
| CRKL | rs2266953 |
| CRKL | rs2539918 |
| CRKL | rs4822700 |
| DEC1 | rs10982724 |
| EGLN3 | rs1680699 |
| EPAS1 | rs10495933 |
| ESR1 | rs2459111 |
| ESR1 | rs3798577 |
| ETS1 | rs4937333 |
| ETS1 | rs6590330 |
| ETS1 | rs8705 |
| HIF1A | rs12435848 |
| HK2 | rs10496197 |
| HK2 | rs943 |
| HSP90AA1 | rs7155973 |
| HSPA4 | rs4705990 |
| IGF2 | rs3802971 |
| LDHA | rs10766473 |
| MAPK1 | rs2876980 |
| MAPK1 | rs743409 |
| MAPK1 | rs9607241 |
| MMP2 | rs1861320 |
| NOCA1 | rs9309308 |
| NOTCH3 | rs1044009 |
| PGF | rs2268613 |
| PI3K | rs7614305 |
| POU5F1 | rs6905862 |
| RBX1 | rs138354 |
| TFRC | rs406271 |
| TFRC | rs570 |
| TFRC | rs6773576 |
| TGFA | rs10172814 |
| TGFA | rs10496180 |
| TGFA | rs11466297 |
| TGFA | rs3771515 |
| TGFA | rs488065 |
| TGFA | rs549386 |
| TSC1 | rs10491534 |
| TSC1 | rs10491535 |
| TSC1 | rs2809244 |
| VEGFA | rs3025033 |
| VHL | rs1642742 |
| IGF2 | rs7115054 |
Table 3.
List of significant genes carrying miRNA binding site polymorphisms
| Case/Control |
No. Times in bootstrap sample |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| miRNA Binding Site SNP* |
Tagging SNP |
Gene | Genotype | Best Model‡ |
ww | wv | vv | OR (95%CI)† |
P value |
P<0.05 |
| rs13943 | rs743409 | MAPK1 | G/A | ADD | 261/396 | 436/734 | 197/386 | 0.90 (0.77-0.98) |
0.02 | 952 |
| rs1063311 | rs9607241 | MAPK1 | T/C | ADD | 282/541 | 434/711 | 178/264 | 1.14 (1.02-1.29) |
0.03 | 903 |
| rs12947 | rs6773576 | CDCP1 | G/A | ADD | 573/1037 | 281/427 | 40/52 | 1.18 (1.01-1.37) |
0.03 | 848 |
| rs406271 | rs406271 | TFRC | A/G | ADD | 425/665 | 389/675 | 80/176 | 0.88 (0.77-1.00) |
0.04 | 662 |
| rs3750505 | rs10982724 | DEC1 | A/G | REC | 706/1233 | 173/271 | 15/12 | 2.18 (1.01-4.74) |
0.05 | 514 |
miRNA binding site SNPs and tagging SNPs are within the same haplotype block (r2 > 0.8)
Adjusted by age, sex, smoking status.
Best model: the model with smallest P value; ADD: additive model, REC: recessive model.
ww, homozygous wild-type genotype; wv, heterozygous variant genotype; vv, homozygous variant genotype
Cumulative effects of SNPs on RCC risk
To further assess the effects of miRNA binding site variants on RCC risk, we performed a cumulative analysis of these five SNPs identified as significant in the main effects analysis. The risk differed significantly among these five groups (Pfor trend <0.001; Table 4) with an increase in risk with an increase in the number of unfavorable genotypes. Compared with the reference group, subjects carrying four or five unfavorable genotypes had a 2.14-fold increase in risk (95% CI, 1.03-4.43, P = 0.04).
Table 4.
Number of unfavorable genotypes and RCC risk
| Number | Control (n %) | Case (n %) | OR (95% CI)* | P* value |
|---|---|---|---|---|
| 0 | 29(1.91) | 11(1.23) | 1(reference) | |
| 1 | 261(17.22) | 128(14.32) | 1.35(0.65-2.83) | 0.42 |
| 2 | 275(18.14) | 149(16.67) | 1.52(0.73-3.17) | 0.26 |
| 3 | 681(44.92) | 396(44.07) | 1.62(0.79-3.31) | 0.19 |
| 4-5 | 270(17.81) | 210(23.44) | 2.14(1.03-4.43) | 0.04 |
| P for trend | <0.001 |
Adjusted for age, sex, smoking status.
Higher-order gene-gene interactions
We next explored higher-order gene-gene interactions to determine whether or not complex interactions among these significant SNPs could further modulate RCC risk. The final tree structure identified several potential interactions among the top five SNPs (Figure 1). CDCP1:rs6773576, which tags rs12947 located in a binding site for hsa-miR-379, hsa-miR-380, hsa-miR-3924, hsa-miR-411, and hsa-miR-4495 was identified as the initial split, suggesting its potential predictive role for cancer risk. The final tree structure identified two terminal nodes with significantly different risk. Compared to patients with the low-risk genetic profile, those with the high-risk genetic profiles characterized by node 3 had a 1.39-fold increased risk (95% CI, 1.02-1.90) and those in node 4 had a highly significant nearly 4.5-fold increase in risk (95% CI, 1.32-15.08) To provide internal validation of the tree structure generated by this analysis, we performed bootstrap analysis with 10,000 re-samplings. We found that these results were highly consistent (Figure 1 and Table 5).
Figure 1. Higher-order gene-gene interactions and RCC risk.
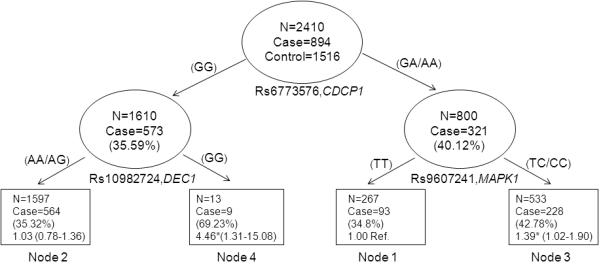
Values in each node denotes the case/control numbers with OR and 95% CI in parenthesis. *Significant at P < 0.05
Table 5.
Effect of terminal nodes derived from CART analysis on RCC risk
| Node group | Control (n %) | Case (n %) | OR (95% CI)* | P* value | Bootstrap (95% CI) |
|---|---|---|---|---|---|
| Low risk (node 1) | 174 (65.17) | 93 (34.83) | 1(reference) | ||
| Medium low risk (node 2) | 1033 (64.68) | 564 (35.32) | 1.03(0.78-1.36) | 0.84 | 0.78-1.37 |
| Medium high risk (node 3) | 305 (57.22) | 228 (42.78) | 1.39(1.02-1.90) | 3.65×10−2 | 1.02-1.89 |
| High risk (node 4) | 4 (30.77) | 9 (69.23) | 4.46 (1.32-15.08) | 1.63×10−2 | 1.47-18.87 |
| P for trend | 1.72×10−3 |
Note: Node groups are as shown in Figure 1.
Adjusted for age, sex, smoking status.
Discussion
The incidence of RCC continues to rise over the past two decades and has been thought to be due to an increased incidental detection by the radiographic identification [30]. It has been well established that the VHL-HIF1α pathway plays an important role in renal tumorigenesis [10,11]. We identified 429 miRNA binding site SNPs in 102 VHL-HIF1α pathway genes, and evaluated 53 tagging SNPs for their associations with RCC risk. Our results identified five tagging SNPs significantly associated with RCC risk. CART analysis further revealed potential high-order gene-gene interactions and categorized subjects into different risk groups according to their specific polymorphic signatures. Our study provides the first molecular epidemiological evidence supporting a connection between miRNA binding site SNPs within the VHL-HIF1α pathway and RCC risk.
The top two significant SNPs tagged binding site SNPs in MAPK1, which highlights the important function of this gene in RCC risk (Table 3). MAPK1 (also known as ERK2) has been found to be significantly active in advanced RCC and activity levels can predict the onset of metastasis with localized disease [31].It has been shown that HIF-1α Ser-641/643 phosphorylation by p42 MAPK (ERK2) promotes HIF-1α nuclear accumulation and transcriptional activity [32]. MAPK1 has also been demonstrated to be a critical mediator of EPAS/HIF-2α activation [33]. In addition, MAPKK inhibitor PD98059 has been shown to block HIF-1α’s transcriptional activity [34]. PolymiRTS database annotated the G allele of rs13943 as a potential binding site of hsa-miR-3145-5p, and the C allele as a potential binding site for hsa-miR-1288 and has-miR-3169. It follows that these binding sites and miRNAs changes are associated with MAPK1 activation changes which contribute to RCC risk. Alternatively, it is possible that these tagging SNPs may be linked to other causal variants in MAPK1.
In addition to MAPK1 variants, tagging SNP: rs6773576 in CDCP1 was also found significantly associated with RCC risk and with bootstrap P values < 0.05 for more than 800 of 1000 samplings (Table 3). It has been reported that induction of CDCP1 is regulated by HIF-1/2 and increases ccRCC migration by activation of PKCδ, and patient survival can be stratified by cell surface expression levels of CDCP1 [35]. Tagging SNP: rs6773576 tags miRNA binding site SNP: rs12947 in CDCP1 and PolymiRTS database predicted that the A allele of rs12947 is a potential binding site for hsa-miR-379, hsa-miR-380, hsa-miR-3924, hsa-miR-411, and hsa-miR-4495. However, G allele of rs12947 destroys these binding sites without creating a new site. Same as those in MAPK1, it is probably that the binding site and miRNAs changes are associated with CDCP1 level change or linked to other causal variants in CDCP1 that contribute to RCC risk.
We identified a significant gene-dosage effect for the five tagging SNPs that had significant main effects. Those with the highest number of risk genotypes had the highest risk of RCC, suggesting that additional variation within this key pathway involved in RCC development was detrimental and had a larger effect than any single variant. Furthermore, the magnitude of each individual SNP was modest, but cumulatively doubled the risk for individuals with four or five of these risk genotypes. This highlights the importance of assessing multiple SNPs within a shared pathway for risk assessment. Further studies will be needed to explore the biological basis for these findings and establish functional interactions that modulate RCC risk.
Again, within the framework of a pathway, we hypothesized that gene-gene interactions would further modulate risk of RCC. Indeed that is what we observed. Potential gene-gene interactions among three variants were observed with CDCP1: rs6773576 being the initial split in our CART analysis, suggesting that this variant was responsible for the most variation in risk. DEC1 is a hypoxia-regulated transcription factor that functions downstream of both CDCP1 and MAPK1. Individuals carrying the common genotype for CDCP1: rs6773576 and the variant genotype for DEC1: rs10982724 had significant almost 4.5-fold increase in risk, a much larger effect magnitude than that conferred by either variant individually. Further studies will be needed to explore the biological basis for these findings and establish functional interactions that modulate RCC risk.
In summary, we have performed analysis of miRNA binding site variants in VHL-HIF1α pathway genes and RCC risk. These data may offer some mechanistic assumptions, as well as providing genetic information for predicting those at risk and identify tumors in an early, curable stage. However, there are some limitations to this research. First, other SNPs in important regions (exons, introns, promoters, etc) were not included in this specific study. It is also possible that some miRNA binding site SNPs were not predicted by Targetscan and therefore were not included into our study. Although rare VHL mutations contribute to RCC risk, we did not have information on these mutations for our study participants. In addition, we limited our analysis in non-Hispanic Caucasians to control potential confounding by population stratification. Future replication studies in independent populations will be needed to verify some of our results. Finally, besides age, gender, and smoking status, other epidemiology risk factors for RCC have been identified or suspected as risk factors, such as overweight and obesity, kidney disease, hypertension, alcohol consumption, and dietary factors [36]. Due to lack of information regarding these factors in all of our study participants, we were unable to include them in our analysis. However, our previous study showed that such risk factors might not significantly affect the relationship between SNPs and RCC risk [37].
Nevertheless, the comprehensive query of the VHL-HIFα pathway polymorphisms and our large population (894 cases and 1516 controls) with detailed risk information provide substantial evidence for the involvement of these novel miRNA binding site SNPs as predictors or modulators of RCC risk. Together with other known epidemiology, clinical, and genetic factors, may contribute to a risk assessment model for identifying those at high risk.
Acknowledgements
This work was supported by a grant from the National Institutes of Health (R01 CA98897) and also by part of MD Anderson’s Cancer Center Support Grant CA016672. H.W. is a Halliburton Employees Fellow in Cancer Prevention.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev. 2008;34:193–205. doi: 10.1016/j.ctrv.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Grabowski J, Silberstein J, Saltzstein SL, Saenz N. Renal tumors in the second decade of life: results from the California Cancer Registry. J Pediatr Surg. 2009;44:1148–51. doi: 10.1016/j.jpedsurg.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 4.Howlader N, N.A., Krapcho M, Neyman N, Aminou R, Waldron W, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ, Cronin KA, Edwards BK. SEER Cancer Statistics Review. 2011:1975–2008. [Google Scholar]
- 5.Hainsworth JD, Sosman JA, Spigel DR, Edwards DL, Baughman C, Greco A. Treatment of metastatic renal cell carcinoma with a combination of bevacizumab and erlotinib. J Clin Oncol. 2005;23:7889–96. doi: 10.1200/JCO.2005.01.8234. [DOI] [PubMed] [Google Scholar]
- 6.Stadler WM. Targeted agents for the treatment of advanced renal cell carcinoma. Cancer. 2005;104:2323–33. doi: 10.1002/cncr.21453. [DOI] [PubMed] [Google Scholar]
- 7.van Houwelingen KP, van Dijk BA, Hulsbergen-van de Kaa CA, Schouten LJ, Gorissen HJ, Schalken JA, van den Brandt PA, Oosterwijk E. Prevalence of von Hippel-Lindau gene mutations in sporadic renal cell carcinoma: results from The Netherlands cohort study. BMC Cancer. 2005;5:57. doi: 10.1186/1471-2407-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McRonald FE, Morris MR, Gentle D, Winchester L, Baban D, Ragoussis J, Clarke NW, Brown MD, Kishida T, Yao M, Latif F, Maher ER. CpG methylation profiling in VHL related and VHL unrelated renal cell carcinoma. Mol Cancer. 2009;8:31. doi: 10.1186/1476-4598-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groulx I, Lee S. Oxygen-dependent ubiquitination and degradation of hypoxia-inducible factor requires nuclear-cytoplasmic trafficking of the von Hippel-Lindau tumor suppressor protein. Mol Cell Biol. 2002;22:5319–36. doi: 10.1128/MCB.22.15.5319-5336.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 11.Rathmell WK, Chen S. VHL inactivation in renal cell carcinoma: implications for diagnosis, prognosis and treatment. Expert Rev Anticancer Ther. 2008;8:63–73. doi: 10.1586/14737140.8.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- 13.Han JY, Oh SH, Morgillo F, Myers JN, Kim E, Hong WK, Lee HY. Hypoxia-inducible factor 1alpha and antiangiogenic activity of farnesyltransferase inhibitor SCH66336 in human aerodigestive tract cancer. J Natl Cancer Inst. 2005;97:1272–86. doi: 10.1093/jnci/dji251. [DOI] [PubMed] [Google Scholar]
- 14.Krishnamachary B, Zagzag D, Nagasawa H, Rainey K, Okuyama H, Baek JH, Semenza GL. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006;66:2725–31. doi: 10.1158/0008-5472.CAN-05-3719. [DOI] [PubMed] [Google Scholar]
- 15.Gunaratnam L, Morley M, Franovic A, de Paulsen N, Mekhail K, Parolin DA, Nakamura E, Lorimer IA, Lee S. Hypoxia inducible factor activates the transforming growth factor-alpha/epidermal growth factor receptor growth stimulatory pathway in VHL(-/-) renal cell carcinoma cells. J Biol Chem. 2003;278:44966–74. doi: 10.1074/jbc.M305502200. [DOI] [PubMed] [Google Scholar]
- 16.Haase VH. The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease. Kidney Int. 2006;69:1302–7. doi: 10.1038/sj.ki.5000221. [DOI] [PubMed] [Google Scholar]
- 17.Zimmer M, Doucette D, Siddiqui N, Iliopoulos O. Inhibition of hypoxia-inducible factor is sufficient for growth suppression of VHL-/- tumors. Mol Cancer Res. 2004;2:89–95. [PubMed] [Google Scholar]
- 18.Facchini G, Perri F, Caraglia M, Pisano C, Striano S, Marra L, Fiore F, Aprea P, Pignata S, Iaffaioli RV. New treatment approaches in renal cell carcinoma. Anticancer Drugs. 2009;20:893–900. doi: 10.1097/CAD.0b013e32833123d4. [DOI] [PubMed] [Google Scholar]
- 19.Collins S, McKiernan J, Landman J. Update on the epidemiology and biology of renal cortical neoplasms. J Endourol. 2006;20:975–85. doi: 10.1089/end.2006.20.975. [DOI] [PubMed] [Google Scholar]
- 20.Wysocki PJ. mTOR in renal cell cancer: modulator of tumor biology and therapeutic target. Expert Rev Mol Diagn. 2009;9:231–41. doi: 10.1586/erm.09.8. [DOI] [PubMed] [Google Scholar]
- 21.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–14. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robins H, Press WH. Human microRNAs target a functionally distinct population of genes with AT-rich 3′ UTRs. Proc Natl Acad Sci U S A. 2005;102:15557–62. doi: 10.1073/pnas.0507443102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chow TF, Youssef YM, Lianidou E, Romaschin AD, Honey RJ, Stewart R, Pace KT, Yousef GM. Differential expression profiling of microRNAs and their potential involvement in renal cell carcinoma pathogenesis. Clin Biochem. 2010;43:150–8. doi: 10.1016/j.clinbiochem.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 24.Horikawa Y, Wood CG, Yang H, Zhao H, Ye Y, Gu J, Lin J, Habuchi T, Wu X. Single nucleotide polymorphisms of microRNA machinery genes modify the risk of renal cell carcinoma. Clin Cancer Res. 2008;14:7956–62. doi: 10.1158/1078-0432.CCR-08-1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin J, Horikawa Y, Tamboli P, Clague J, Wood CG, Wu X. Genetic variations in microRNA-related genes are associated with survival and recurrence in patients with renal cell carcinoma. Carcinogenesis. 2010;31:1805–12. doi: 10.1093/carcin/bgq168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bao L, Zhou M, Wu L, Lu L, Goldowitz D, Williams RW, Cui Y. PolymiRTS Database: linking polymorphisms in microRNA target sites with complex traits. Nucleic Acids Res. 2007;35:D51–4. doi: 10.1093/nar/gkl797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu X, Ye Y, Kiemeney LA, Sulem P, Rafnar T, Matullo G, Seminara D, Yoshida T, Saeki N, Andrew AS, Dinney CP, Czerniak B, Zhang ZF, Kiltie AE, Bishop DT, Vineis P, Porru S, Buntinx F, Kellen E, Zeegers MP, Kumar R, Rudnai P, Gurzau E, Koppova K, Mayordomo JI, Sanchez M, Saez B, Lindblom A, de Verdier P, Steineck G, Mills GB, Schned A, Guarrera S, Polidoro S, Chang SC, Lin J, Chang DW, Hale KS, Majewski T, Grossman HB, Thorlacius S, Thorsteinsdottir U, Aben KK, Witjes JA, Stefansson K, Amos CI, Karagas MR, Gu J. Genetic variation in the prostate stem cell antigen gene PSCA confers susceptibility to urinary bladder cancer. Nat Genet. 2009;41:991–5. doi: 10.1038/ng.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu X, Scelo G, Purdue MP, Rothman N, Johansson M, Ye Y, Wang Z, Zelenika D, Moore LE, Wood CG, Prokhortchouk E, Gaborieau V, Jacobs KB, Chow WH, Toro JR, Zaridze D, Lin J, Lubinski J, Trubicka J, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Jinga V, Bencko V, Slamova A, Holcatova I, Navratilova M, Janout V, Boffetta P, Colt JS, Davis FG, Schwartz KL, Banks RE, Selby PJ, Harnden P, Berg CD, Hsing AW, Grubb RL, 3rd, Boeing H, Vineis P, Clavel-Chapelon F, Palli D, Tumino R, Krogh V, Panico S, Duell EJ, Quiros JR, Sanchez MJ, Navarro C, Ardanaz E, Dorronsoro M, Khaw KT, Allen NE, Bueno-de-Mesquita HB, Peeters PH, Trichopoulos D, Linseisen J, Ljungberg B, Overvad K, Tjonneland A, Romieu I, Riboli E, Stevens VL, Thun MJ, Diver WR, Gapstur SM, Pharoah PD, Easton DF, Albanes D, Virtamo J, Vatten L, Hveem K, Fletcher T, Koppova K, Cussenot O, Cancel-Tassin G, Benhamou S, Hildebrandt MA, Pu X, Foglio M, Lechner D, Hutchinson A, Yeager M, Fraumeni JF, Jr., Lathrop M, Skryabin KG, McKay JD, Gu J, Brennan P, Chanock SJ. A genome-wide association study identifies a novel susceptibility locus for renal cell carcinoma on 12p11.23. Hum Mol Genet. 2011;21:456–62. doi: 10.1093/hmg/ddr479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sauerbrei W, Schumacher M. A bootstrap resampling procedure for model building: application to the Cox regression model. Stat Med. 1992;11:2093–109. doi: 10.1002/sim.4780111607. [DOI] [PubMed] [Google Scholar]
- 30.Hollingsworth JM, Miller DC, Daignault S, Hollenbeck BK. Rising incidence of small renal masses: a need to reassess treatment effect. J Natl Cancer Inst. 2006;98:1331–4. doi: 10.1093/jnci/djj362. [DOI] [PubMed] [Google Scholar]
- 31.Campbell L, Nuttall R, Griffiths D, Gumbleton M. Activated extracellular signal-regulated kinase is an independent prognostic factor in clinically confined renal cell carcinoma. Cancer. 2009;115:3457–67. doi: 10.1002/cncr.24389. [DOI] [PubMed] [Google Scholar]
- 32.Mylonis I, Chachami G, Samiotaki M, Panayotou G, Paraskeva E, Kalousi A, Georgatsou E, Bonanou S, Simos G. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J Biol Chem. 2006;281:33095–106. doi: 10.1074/jbc.M605058200. [DOI] [PubMed] [Google Scholar]
- 33.Conrad PW, Freeman TL, Beitner-Johnson D, Millhorn DE. EPAS1 trans-activation during hypoxia requires p42/p44 MAPK. J Biol Chem. 1999;274:33709–13. doi: 10.1074/jbc.274.47.33709. [DOI] [PubMed] [Google Scholar]
- 34.Hur E, Chang KY, Lee E, Lee SK, Park H. Mitogen-activated protein kinase kinase inhibitor PD98059 blocks the trans-activation but not the stabilization or DNA binding ability of hypoxia-inducible factor-1alpha. Mol Pharmacol. 2001;59:1216–24. doi: 10.1124/mol.59.5.1216. [DOI] [PubMed] [Google Scholar]
- 35.Razorenova OV, Finger EC, Colavitti R, Chernikova SB, Boiko AD, Chan CK, Krieg A, Bedogni B, LaGory E, Weissman IL, Broome-Powell M, Giaccia AJ. VHL loss in renal cell carcinoma leads to up-regulation of CUB domain-containing protein 1 to stimulate PKC{delta}-driven migration. Proc Natl Acad Sci U S A. 2011;108:1931–6. doi: 10.1073/pnas.1011777108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ljungberg B, Campbell SC, Choi HY, Jacqmin D, Lee JE, Weikert S, Kiemeney LA. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60:615–21. doi: 10.1016/j.eururo.2011.06.049. [DOI] [PubMed] [Google Scholar]
- 37.Wu X, Scelo G, Purdue MP, Rothman N, Johansson M, Ye Y, Wang Z, Zelenika D, Moore LE, Wood CG, Prokhortchouk E, Gaborieau V, Jacobs KB, Chow WH, Toro JR, Zaridze D, Lin J, Lubinski J, Trubicka J, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Jinga V, Bencko V, Slamova A, Holcatova I, Navratilova M, Janout V, Boffetta P, Colt JS, Davis FG, Schwartz KL, Banks RE, Selby PJ, Harnden P, Berg CD, Hsing AW, Grubb RL, 3rd, Boeing H, Vineis P, Clavel-Chapelon F, Palli D, Tumino R, Krogh V, Panico S, Duell EJ, Quiros JR, Sanchez MJ, Navarro C, Ardanaz E, Dorronsoro M, Khaw KT, Allen NE, Bueno-de-Mesquita HB, Peeters PH, Trichopoulos D, Linseisen J, Ljungberg B, Overvad K, Tjonneland A, Romieu I, Riboli E, Stevens VL, Thun MJ, Diver WR, Gapstur SM, Pharoah PD, Easton DF, Albanes D, Virtamo J, Vatten L, Hveem K, Fletcher T, Koppova K, Cussenot O, Cancel-Tassin G, Benhamou S, Hildebrandt MA, Pu X, Foglio M, Lechner D, Hutchinson A, Yeager M, Fraumeni JF, Jr., Lathrop M, Skryabin KG, McKay JD, Gu J, Brennan P, Chanock SJ. A genome-wide association study identifies a novel susceptibility locus for renal cell carcinoma on 12p11.23. Hum Mol Genet. 2012;21:456–62. doi: 10.1093/hmg/ddr479. [DOI] [PMC free article] [PubMed] [Google Scholar]