

SUMMARY
Individuals with metabolic syndrome are at high risk of developing chronic kidney disease (CKD) through unclear pathogenic mechanisms. Obesity and diabetes are known to induce glucolipotoxic effects in metabolically relevant organs. However, the pathogenic role of glucolipotoxicity in the aetiology of diabetic nephropathy is debated. We generated a murine model, the POKO mouse, obtained by crossing the peroxisome proliferator-activated receptor gamma 2 (PPARγ2) knockout (KO) mouse into a genetically obese ob/ob background. We have previously shown that the POKO mice showed: hyperphagia, insulin resistance, hyperglycaemia and dyslipidaemia as early as 4 weeks of age, and developed a complete loss of normal β-cell function by 16 weeks of age. Metabolic phenotyping of the POKO model has led to investigation of the structural and functional changes in the kidney and changes in blood pressure in these mice. Here we demonstrate that the POKO mouse is a model of renal disease that is accelerated by high levels of glucose and lipid accumulation. Similar to ob/ob mice, at 4 weeks of age these animals exhibited an increased urinary albumin:creatinine ratio and significantly increased blood pressure, but in contrast showed a significant increase in the renal hypertrophy index and an associated increase in p27Kip1 expression compared with their obese littermates. Moreover, at 4 weeks of age POKO mice showed insulin resistance, an alteration of lipid metabolism and glomeruli damage associated with increased transforming growth factor beta (TGFβ) and parathyroid hormone-related protein (PTHrP) expression. At this age, levels of proinflammatory molecules, such as monocyte chemoattractant protein-1 (MCP-1), and fibrotic factors were also increased at the glomerular level compared with levels in ob/ob mice. At 12 weeks of age, renal damage was fully established. These data suggest an accelerated lesion through glucolipotoxic effects in the renal pathogenesis in POKO mice.
INTRODUCTION
The prevalence of obesity as a consequence of the convergence between sedentary lifestyle, changes in dietary habits and genetic predisposition has increased at an alarming rate in recent years. Obese individuals are at greater risk of developing hypertension, heart disease, insulin resistance, type 2 diabetes and metabolic syndrome. Furthermore, diabetic nephropathy develops in approximately one third of individuals with both diabetes and obesity. Recent studies have shown that the progression of diabetic nephropathy to end-stage renal disease (ESRD), which ultimately requires kidney dialysis or transplant, is costly to the health sector, and treatments are limited to individuals with late-stage diabetic nephropathy. Late-stage diabetic nephropathy has been recognized as the single largest contributor to the cost of medical care for obese individuals with type 2 diabetes (Nichols et al., 2011). However, the onset and course of diabetic nephropathy can be largely ameliorated by the introduction of several interventions that, if implemented before the progression to these late stages, would decrease the likelihood of subsequent medical complications associated with ESRD.
In obese individuals, the accumulation of excess lipids in tissues other than adipose tissue contributes to organ damage through a process named lipotoxicity (Virtue and Vidal-Puig, 2010). The understanding of this toxic process is complex, but can be explained by considering the hypothesis of adipose tissue expandability (Medina-Gomez et al., 2007; Virtue and Vidal-Puig, 2008; Vidal-Puig and Unger, 2010). This hypothesis states that adipose tissue has a defined limit of expansion for any given individual. During the development of obesity, as an individual gains weight, a point will eventually be reached when the adipose tissue can no longer store more lipids. Once adipose tissue storage capacity is exceeded, then net lipid flux will increase to non-adipose organs and lipids will begin to be deposited ectopically. Ectopic lipid accumulation in cells such as myocytes, hepatocytes and β-cells causes deleterious effects such as insulin resistance and apoptosis. In recent years, evidence has emerged suggesting that the renal accumulation of lipids and their toxic effects can also lead to kidney dysfunction (Kambham et al., 2001; Rutledge et al., 2010). More specifically, it has been reported that saturated fatty acids induce insulin resistance in podocytes, key cells whose roles include maintaining the integrity of the glomerular filtration barrier during normal kidney function (Lennon et al., 2009), and in proximal tubular cells, causing significant cellular dysfunction and, ultimately, cell death via both apoptosis and necrosis (Katsoulieris et al., 2010). In the same way, it has been shown that treatment with inhibitors of β-hydroxymethyl glutaryl Co A (HMG-CoA) reductase, the key enzyme in the regulation of cholesterol synthesis, leads to an improvement in proteinuria and preserves kidney function in individuals with chronic kidney disease, suggesting a role of lipids in promoting renal injury (Bianchi et al., 2003; Tonelli et al., 2003; Lee et al., 2005).
Peroxisome proliferator-activated receptor gamma (PPARγ) is a member of the nuclear hormone receptor (NHR) superfamily that regulates much of the adipogenic programme. Although at least four different mRNAs have been shown to be transcribed from the PPARγ gene, these transcripts encode two separate protein isoforms, PPARγ1 and PPARγ2. Whereas PPARγ1 is expressed widely, PPARγ2 is found almost exclusively in adipose tissue under normal conditions. Recently, PPARγ has been increasingly recognized as a key player not only in the pathogenesis of metabolic syndrome, but also in derived renal complications. In addition, all three PPAR isoforms (α, γ, δ) have been identified as therapeutic targets in the treatment of certain conditions relating to metabolic syndrome. Synthetic PPARγ ligands, such as thiazolidinediones (TZDs) and fibrates, have been shown to improve glycaemic control in individuals with type 2 diabetes and to lower serum triglyceride levels in hyperlipidaemic individuals, respectively (Ruan et al., 2008). Moreover, it has been shown that treatment with PPARγagonists can ameliorate diabetic kidney disease through different mechanisms, involving inhibition of mesangial cell growth, reduction of the mesangial matrix and the production of cytokines (Kiss-Toth and Roszer, 2008). Several murine models with a partial lack of PPARγfunction have been generated and studied, including insulin-sensitive PPARγheterozygous mice. In this mouse the high-fat diet (HFD)-induced renal injury, systemic metabolic abnormalities, renal accumulation of lipids and changes in renal lipid metabolism were attenuated (Kume et al., 2007).
We have generated the POKO mouse model by ablation of PPARγ2 in an obese ob/ob background (Medina-Gomez et al., 2007; Medina-Gomez et al., 2009). POKO mice possess around 10–20% higher adipose tissue mass than wild-type (WT) mice but are less than half the weight of an ob/ob mouse. These mice are more insulin resistant than ob/ob mice from a young age, despite having less fat. In addition, the POKO mouse becomes diabetic and hyperlipidaemic at 16 weeks of age with increased toxic reactive lipid species in different tissues, behaving like a mouse model of lipotoxicity and metabolic syndrome (Medina-Gomez et al., 2007; Medina-Gomez et al., 2009).
Our aim in this study was to analyze the early mechanisms underlying the development of renal pathology that is associated with the development of metabolic syndrome. For this purpose, we used the POKO mouse as an experimental lipotoxic model and have studied the progression of kidney disease in isolated kidneys from 4- and 12-week-old mice. At 4 weeks of age these animals exhibited a similar phenotype toob/ob mice, including significantly increased blood pressure and an increased urinary albumin:creatinine ratio, but in contrast demonstrated a significant increase in renal hypertrophy index and increased p27Kip1 expression. Moreover, in spite of the fact that both ob/ob and POKO mice showed insulin resistance in kidney, an alteration of lipid metabolism and glomeruli damage associated with significantly increased renal fibrosis and inflammation were already present in POKO kidneys at this early age. Our data suggest that an accelerated renal lesion, through glucolipotoxic effects, is associated with metabolic syndrome and insulin resistance in POKO mice. Therefore, we propose that the POKO model is a good model of renal disease accelerated by both effects of hyperglycaemia and lipid accumulation.
RESULTS
POKO mice exhibit renal hypertrophy and incipient renal injury at an early age
We have shown that POKO mice were more hyperglycaemic and insulin resistant than ob/ob mice at 4 weeks of age (Medina-Gomez et al., 2007; Medina-Gomez et al., 2009). Table 1 shows body weights and urine and blood parameters of the four experimental genotypes (WT, PPARγ2 KO, ob/ob and POKO) at 4 and 12 weeks of age. At 4 weeks, as already described, the body weights of POKO and ob/ob mice were similar and both were higher compared with WT and PPARγ2 KO mice. Blood glucose levels were indistinguishable within the four genotypes at 3 weeks of age (data not shown); however, as expected, POKO mice became hyperglycaemic and hypertriglyceridaemic much more quickly than did their ob/ob littermates at 4 weeks of age after both were weaned to a chow diet. Despite high levels of glucose and triglycerides, POKO mice showed similar plasma levels of lipoproteins compared with ob/ob mice. At this age, the blood pressure of POKO and ob/ob mice was significantly higher than that of WT and PPARγ2 KO mice.
Table 1.
Characteristics of the four groups of mice at 4 and 12 weeks of age

Renal hypertrophy has been postulated to cause a progressive reduction in renal function (Yoshida et al., 1989). The renal hypertrophy index (RHI) was calculated by normalizing the kidney weight to the total body weight of 4-week-old mice. At this age, POKO mice exhibited a significantly increased RHI compared with that of ob/ob mice. In line with this hypertrophy, we showed a significant increase in the level of the G1 cyclin kinases activity modulator p27Kip1 protein in the kidneys of POKO mice compared with ob/ob kidneys at 4 weeks of age (Fig. 1A). Of note, the glomeruli size in kidneys from POKO mice was already significantly larger than in ob/ob mice at this age, and these two genotypes had a larger glomeruli size compared with WT and PPARγ2 KO mice (Table 1). Furthermore, proteinuria, measured as a ratio of albumin:creatinine in the urine, was higher in POKO and ob/ob mice compared with WT and PPARγ2 KO mice, although these differences were not significant between POKO and ob/ob mice at this age.
Fig. 1.
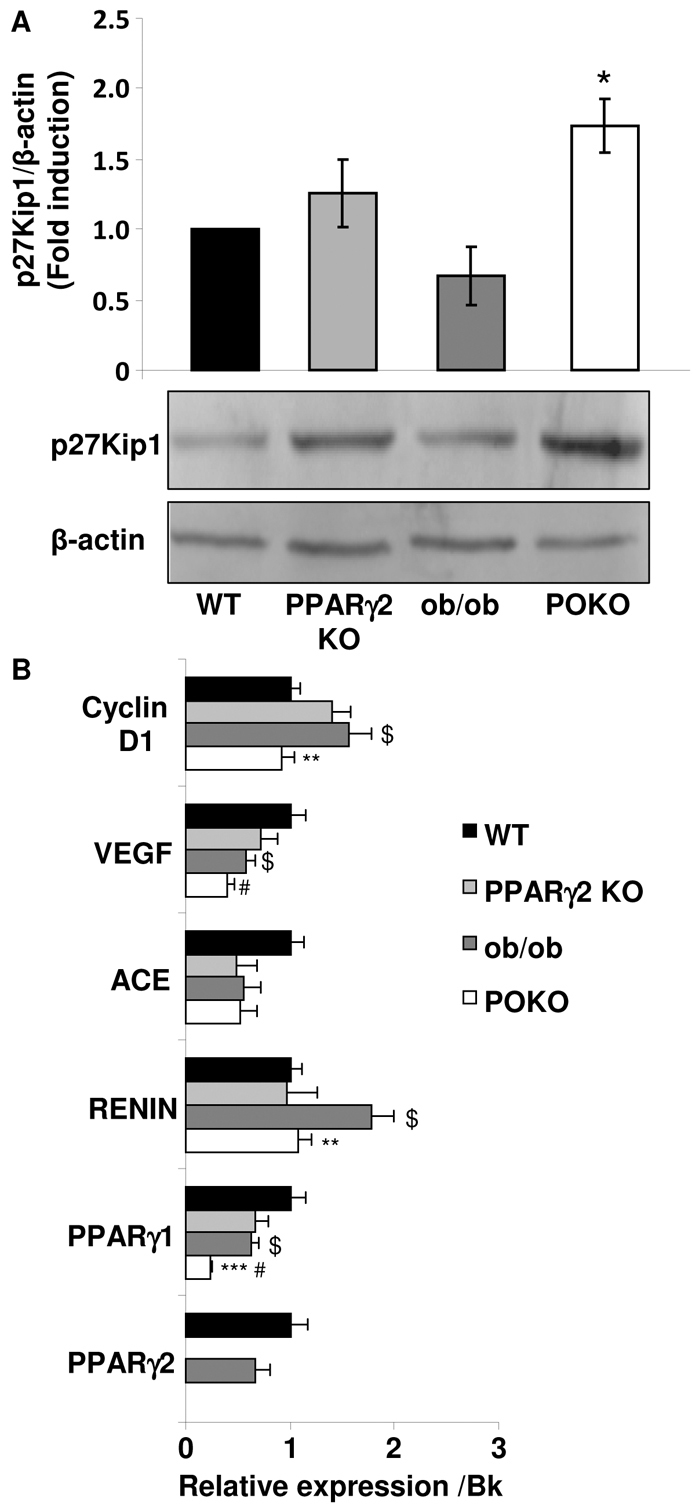
Incipient kidney renal injury in POKO mice at 4 weeks of age. (A) Representative immunoblot for p27Kip1 of renal protein from 4-week-old male WT, PPARγ2 KO, ob/ob and POKO mice. Levels were normalized to β-actin. Each value is the optical intensity of each band as a fold induction vs the WT control group (n=6). Fold induction is shown in graphic. *P<0.05 POKO vs ob/ob. (B) Total kidney mRNA levels of different genes from 4-week-old-male mice of each group. Data are means±s.e.m.; n=8–9 in each group; **P<0.01, ***P<0.001 POKO vs ob/ob; #P<0.05 POKO vs WT; $P<0.05 ob/ob vs WT. Normalized levels with BestKeeper (Bk).
Older POKO mice were significantly lighter than ob/ob mice. At 12 weeks of age, the RHI remained higher in POKO compared with ob/ob mice. Moreover, glomeruli size and proteinuria increased in the POKO and ob/ob genotypes with age compared with WT and PPARγ2 KO mice (Table 1). Although not significant, proteinuria in POKO mice also showed a trend to be higher than in ob/ob mice.
At 12 weeks of age, blood pressure remained similar between the POKO and ob/ob mice and both were higher compared with the WT and PPARγ2 KO genotypes. Despite weighing less at this age, POKO mice were significantly more hyperglycaemic compared with ob/ob mice. After monitoring mice under standardized conditions in metabolic cages, the volume of urine collected from POKO mice was higher compared with ob/ob mice, and POKO mice had significantly higher levels of glucose in urine.
Given the hypertrophy of the kidney in POKO mice at an early age, and higher proteinuria, together with high levels of glucose in urine, we expected to see altered kidney functionality in these mice. Detailed renal analysis at 4 weeks of age using electron microscopic analysis (Fig. 2) showed that the ultrastructure of kidney was altered in POKO mice compared with ob/ob and WT mice, with specific abnormal architectures being identifiable. At this age, extensive loss of foot process structure of the podocytes was visible in POKO mice, which also showed a significantly thicker glomerular basement membrane (GBM) compared with that in WT mice, without displaying differences in the thickness of the lamina densa. Along with the podocyte foot process broadening, we also noticed the density of interpodocyte slit pores along the GBM was also decreased, as has been reported previously in type 2 diabetes (Pagtalunan et al., 1997; White and Bilous, 2004).
Fig. 2.
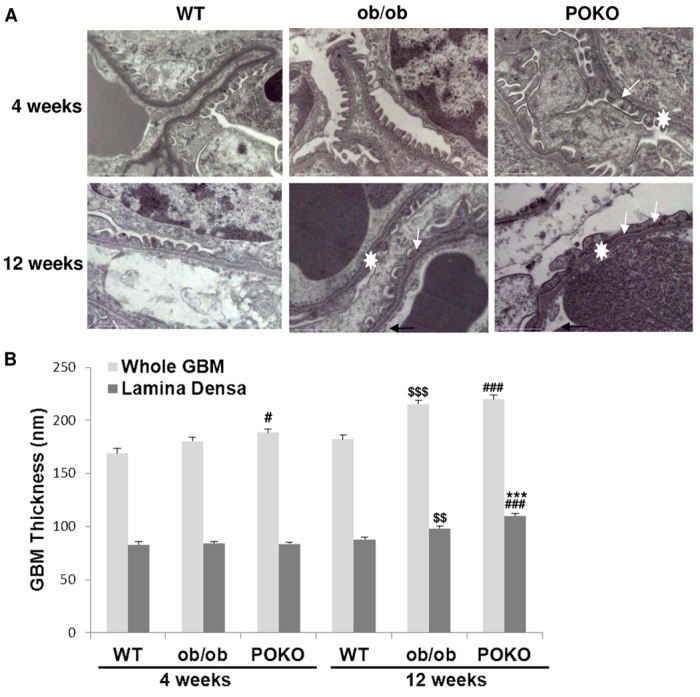
Ultrastructural changes in the glomeruli of the POKO mice. (A) Transmission electron microscopy of glomeruli of 4-week-old POKO mice with broadening of foot processes (arrows). Foot processes in POKO mice are completely lost by 12 weeks (arrows). Original magnification: 20,000×. (B) GBM thickening in POKO mice when measuring the lamina densa (n=3–4). GBM thicknesses (white star in A) were measured across the whole GBM and across the lamina densa. Data are means±s.e.m.; n=6 in each group; ***P<0.001 POKO vs ob/ob; #P<0.05, ###P<0.001 POKO vs WT; $$P<0.01, $$$P<0.001 ob/ob vs WT.
Kidney damage was confirmed in older POKO mice. They displayed aggravated changes in podocytes, with a more altered interdigitating foot process pattern than in ob/ob mice. At 12 weeks of age they showed higher lamina densa thickness compared with that of ob/ob mice and we also found an overall enlargement of glomerular vessels in POKO kidneys.
Fig. 1B shows gene expression at 4 weeks in kidney from the four genotypes. We did not find increased gene expression of PPARγ2 in ob/ob compared with WT kidneys, unlike in other tissues such as liver (Medina-Gomez et al., 2007), but PPARγ1 mRNA levels were significantly decreased in kidneys from POKO mice compared with the other genotypes. We observed that mRNA expression of both renin and cyclin-D1 was significantly decreased in POKO mice compared with ob/ob mice at 4 weeks of age. Although we did not find significant differences in vascular endothelial growth factor (VEGF) and angiotensin converter enzyme (ACE) mRNA expression between ob/ob and POKO mice, levels of mRNA of these two genes were lower in POKO mice compared with the other genotypes. Low levels of VEGF have been associated with the presence of glomerulosclerosis in older Zucker diabetic fatty (ZDF) rats (Hoshi et al., 2002). All these data suggest an incipient kidney injury in POKO mice at this early age.
Kidneys from POKO mice showed altered glucose metabolism and insulin resistance
At 4 weeks of age, mRNA expression of pyruvate carboxylase (PC) was higher in POKO mice compared with ob/ob mice (Fig. 3A). Despite similar expression of glucose transporter 2 (GLUT2) between the ob/ob and POKO genotypes, there was a trend of decreased insulin receptor substrate 2 (IRS-2) and glucose transporter 4 (GLUT4) expression in 4-week-old POKO mice compared with ob/ob, suggesting that insulin signalling might be altered in kidney from POKO mice (Fig. 3A). Expression of the nephrin gene, which is crucial for the action of insulin in glomerular podocytes (Coward et al., 2007), showed a trend to be decreased in kidney from POKO and ob/ob mice compared with WT, although without reaching significance. Podocin mRNA expression (Shono et al., 2007) followed a similar pattern within the four genotypes as nephrin. However, the expression of adiponectin was lower in the kidney of POKO and ob/ob compared with WT and PPARγ2 KO mice, and, interestingly, POKO mice showed significantly lower expression of this adipokine compared with ob/ob kidneys.
Fig. 3.
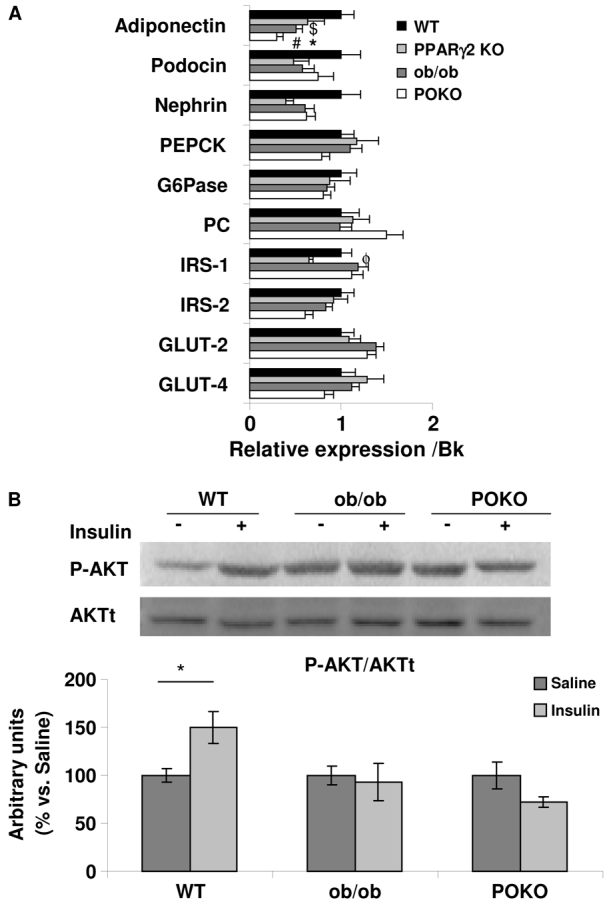
Insulin resistance in POKO kidneys. (A) Total kidney mRNA levels of glucose metabolism genes from 4-week-old male WT, PPARγ2 KO, ob/ob and POKO mice. Data are means±s.e.m.; n=8–9. *P<0.05 POKO vs ob/ob; #P<0.05 POKO vs WT; $P<0.05 ob/ob vs WT; ϕP<0.05 PPARγ2 KO vs WT. Normalized levels with BestKeeper (Bk). (B) Representative immunoblot for pAKT(Ser473) from WT, ob/ob and POKO 4-week-old-male mice treated and non treated with insulin. Levels were normalized to total protein kinase B (AKTt). Each value is the relative optical intensity of each band normalized as a percentage of the saline-treated group. Values are represented in graphic (n=5–8). Data are means±s.e.m. *P<0.05 WT saline vs WT insulin. G6Pase, glucose 6-phosphatase; PEPCK, phosphoenolpyruvate carboxykinase; IRS, insulin receptor substrate.
To further study insulin-stimulated signalling in kidneys, mice were fasted overnight, thereby lowering insulin to basal levels, before being administered either with insulin or saline via an intraperitoneal route. We used phospho-AKT(Ser473) to assess the PI3K pathway, which has been shown to occur predominantly in the podocyte (Welsh et al., 2010) (Fig. 3B). At 5 minutes post insulin injection, normal insulin-responsive kidneys were shown in WT mice. However, insulin-stimulated AKT phosphorylation was abrogated in ob/ob and in POKO kidneys, further confirming their insulin resistance.
Altered lipid metabolism counteracts insulin resistance in POKO kidneys
Ectopic lipid accumulation has been shown to impair insulin signalling and be responsible, at least in part, for insulin resistance in non-adipose tissues. Thus, we wondered whether lipid metabolism presented any alteration in the kidney of these mice and could have an effect on insulin signalling in POKO kidneys. First we analyzed the lipid distribution by Oil-Red-O staining and by electron microscopy. As shown in Fig. 4A, at 4 weeks of age lipid droplets could be observed at both the glomerular and tubular level in the kidneys of POKO mice, whereas they were only observed at the tubular level in kidneys from ob/ob mice. A detailed view of lipid droplets at the glomerular level in POKO kidneys is shown in the electron microscopic pictures of Fig. 4B. At 12 weeks of age, the localization of lipid droplets at both the tubular and glomerular level was similar in POKO and ob/ob kidneys (data not shown).
Fig. 4.
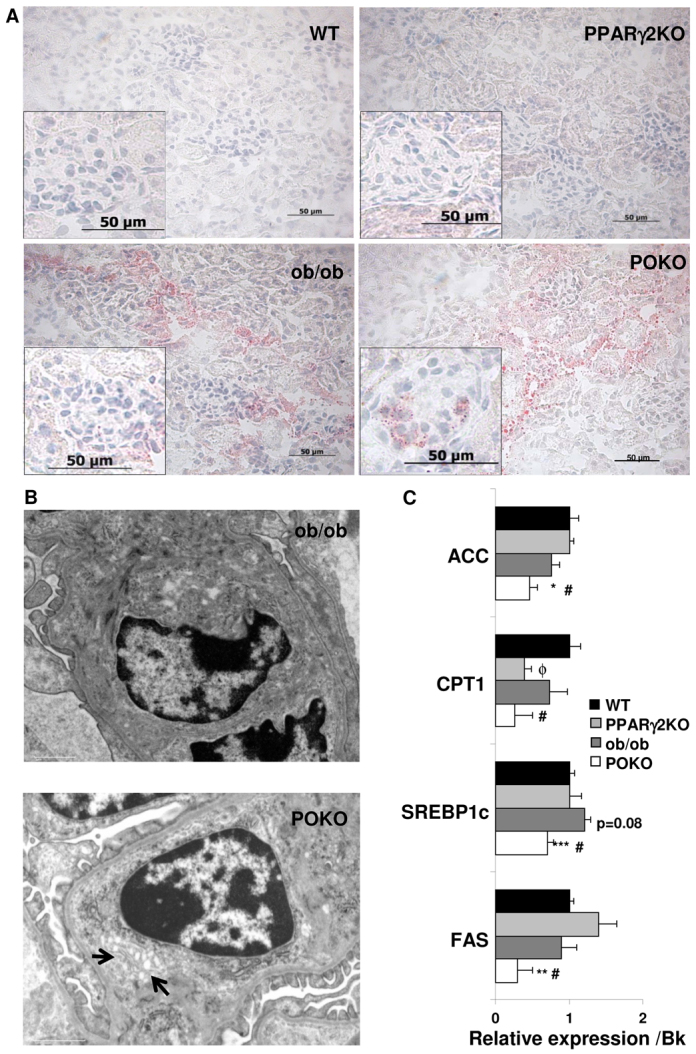
Altered lipid metabolism in POKO kidneys. (A) Representative Oil-Red-O staining in the kidney from WT, PPARγ2 KO, ob/ob and POKO mice at 4 weeks. Original magnification: 400× (n=4). (B) Intraglomerular electron micrographs of ob/ob mice (12,000×) and POKO mice (15,000×) showing accumulation of lipid droplets (arrows). (C) Total kidney mRNA levels of lipid metabolism genes from 4-week-old male WT, PPARγ2 KO, ob/ob and POKO mice. Data are means±s.e.m.; n=8–9. *P<0.05, **P<0.01, ***P<0.001 POKO vs ob/ob; #P<0.05 POKO vs WT; ϕP<0.05 PPARγ2 KO vs WT. Normalized levels with BestKeeper (Bk).
When we analyzed the mRNA levels of key genes involved in lipid metabolism (Fig. 4C), we observed that mRNA expression of sterol regulatory elements binding protein 1c (SREBP1c) was significantly lower in POKO mice compared with ob/ob mice. SREBP1c is a transcription factor that regulates the transcriptional activity of the enzymes that are involved in lipogenesis, such as fatty acid synthase (FAS) and acyl-CoA carboxylase (ACC). Consistently with lower SREBP1c levels, mRNA levels of ACC and FAS were lower in POKO compared with ob/ob mice. Furthermore, at 4 weeks of age we also found that mRNA expression of carnitine palmitoyltransferase 1 (CPT-1) was lower in POKO and PPARγ2 KO mice compared with WT mice (Fig. 4C).
POKO kidneys have decreased short-chain TAG but increased DAG, ceramides and other reactive lipid species associated with insulin resistance
After seeing an altered lipid metabolism in POKO mice, lipidomic analysis using liquid-chromatography–mass-spectrometry (LC/MS) was performed in extracts of kidneys from the four genotypes (Fig. 5A). Kidney from POKO mice had decreased short-chain triacylglycerols (TAGs) compared with kidney from ob/ob mice. Conversely, long- and medium-chain TAG concentration was increased in the kidney of POKO mice compared with their ob/ob littermates. As the TAG chain length increased (moving down the image, Fig. 5A), the levels increased in POKO compared with ob/ob mice (Spearman correlation R=0.56, P-value=1.55×10−8).
Fig. 5.
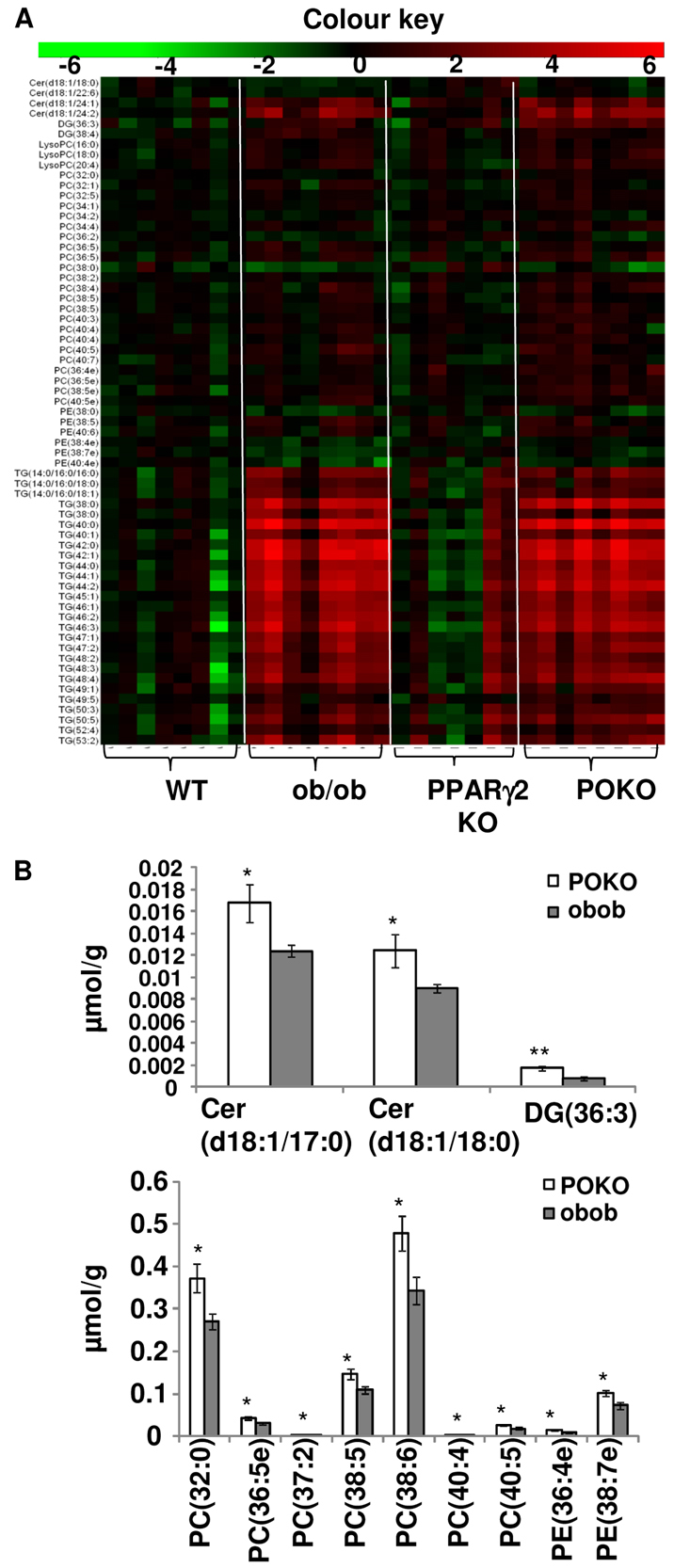
Lipidomic profiling of kidney. (A) The heat map represents log(2) values of the data normalized with the mean of the WT genotype. Lipids with ANOVA P-values<0.05 are shown. 4-week-old male WT, PPARγ2 KO, ob/ob and POKO mice (n=7–8). (B) Differentially regulated lipids between POKO and ob/ob genotypes. P-values *P<0.05, **P<0.005. DG, diacylglycerol; Cer, ceramides; PC, phosphatidylcholine; PE, phosphatidylethanolamine.
Interestingly, we found that there was also an increased concentration of other reactive lipid species, such as ceramides, diacylglycerols (DAGs), phosphatidylcholines (PCs), phosphatidylethanolamines (PEs) and plasmalogens, in the kidney of POKO mice compared with that of ob/ob mice (Fig. 5B). The kidney of POKO mice had significantly increased levels of two ceramide species (with 18:1/17:0 and 18:1/18:0 fatty acid chains), DAG (36:3), PC (36:5e), PE (36:4e) and PE (38:7e) plasmalogens species.
Increased markers of inflammation and profibrotic factors in the kidney of POKO mice
Fig. 6A shows immunohistochemistry of renal injury and inflammation markers in the kidney of the four experimental genotypes at 4 weeks of age. In the kidney of POKO mice, TGFβ was localized at glomeruli; this contrasts to kidney of ob/ob mice, in which TGFβ localization was at the tubular level. Monocyte chemoattractant protein-1 (MCP-1) immunoreactivity was shown in the kidneys of ob/ob and POKO mice. Interestingly, POKO kidney showed increased parathyroid hormone-related protein (PTHrP), a profibrotic and hypertrophy marker, showing staining localized at the glomerular level.
Fig. 6.
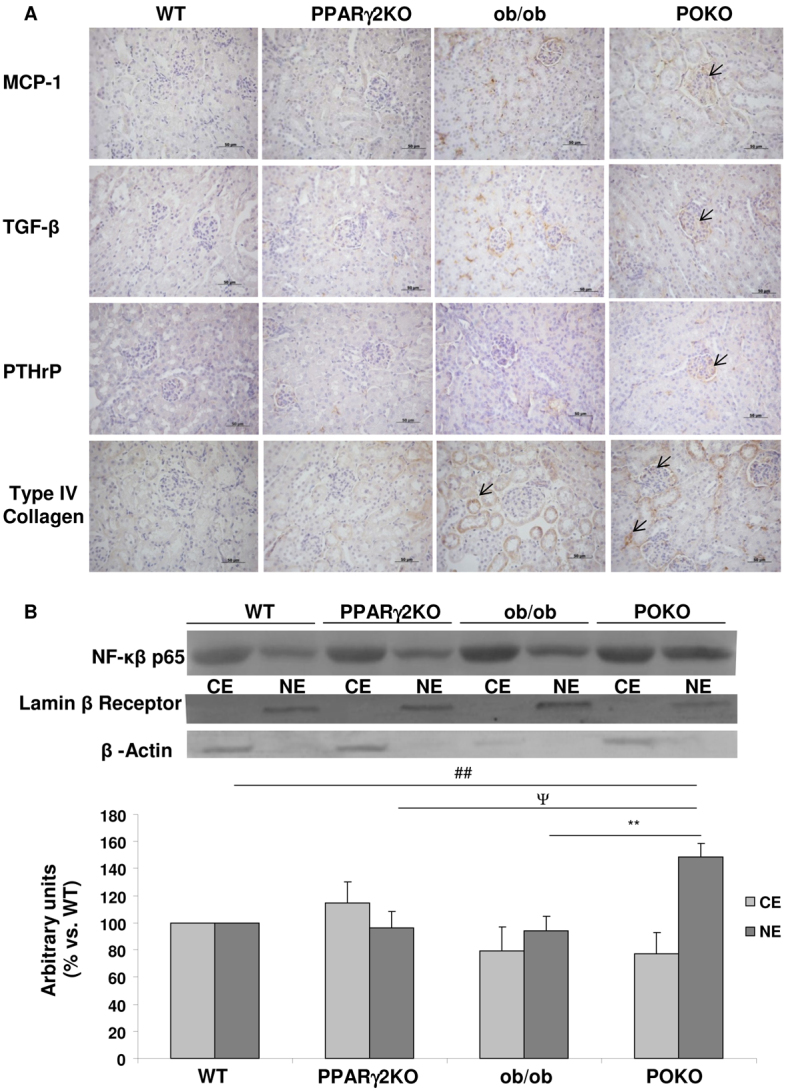
Inflammation markers and renal injury in the four genotypes. (A) Immunostaining for MCP-1, TGFβ, PTHrP and type IV collagen in the kidney from male WT, PPARγ2 KO, ob/ob and POKO mice at 4 weeks (n=4–5). Original magnification: 400×; scale bars: 50 μm. (B) Representative immunoblot for cytosolic (CE) and nuclear (NE) p65 NFκB protein in renal extracts from WT, PPARγ2 KO, ob/ob and POKO 4-week-old male mice. Levels were normalized to β-actin in the cytosolic fraction and lamin-β receptor in the nuclear fraction. Each value is the relative optical intensity of each band normalized as a percentage of that of the WT group. Values are represented in graphic (n=5–7). Data are means±s.e.m. **P<0.01 POKO vs ob/ob; ##P<0.01 POKO vs WT; ΨP<0.05 POKO vs PPARγ2 KO.
The mRNA expression levels of uncoupling protein 2 (UCP-2), antiapoptotic protein (Bcl-2), cell-surface marker for murine macrophages (F4/80; also known as Emr1), differentiation cluster 68 (CD68) and tumour necrosis factor-α (TNFα) were not significantly altered among genotypes. Of interest, expression of transcription factor C/EBP homologous protein (CHOP) was decreased in POKO compared with ob/ob kidney, although not significantly, and cyclooxygenase-2 (COX-2) and TNFα expression had a trend to be increased in POKO compared with ob/ob mice (supplementary material Fig. S1).
Furthermore, we measured levels of cytosolic and nuclear p65 nuclear factor kappa-B (NFκB) protein by western blot in the four genotypes. POKO kidney extracts exhibited a higher translocation of p65 to the nucleus compared with that in the other genotypes, suggesting an upregulation of NFκB activation in the inflammatory process (Fig. 6B).
Faster renal disease with increased extracellular matrix proteins at the glomerular level in POKO kidneys
Immunostaining of type IV collagen in the four genotypes at 4 weeks of age is shown in Fig. 6. Interestingly, the localization of collagen is at the tubular level in ob/ob kidneys, but it is detected at both the glomerular and tubular levels in POKO mice at 4 weeks.
Following a similar pattern, periodic acid-Schiff (PAS) and Masson’s trichrome staining also showed incipient glomerular and tubule-interstitial fibrosis in the kidneys of POKO mice, but fibrosis was only observed in kidneys of ob/ob mice at the tubular level at 4 weeks of age (Fig. 7A). In mice at 12 weeks of age, fibrosis seemed to be localized at both the glomerular and tubular-interstitial levels in these two genotypes (Fig. 7B).
Fig. 7.

Fibrotic markers in the progression of the renal disease in the POKO mice. Representative photomicrographs for PAS staining and Masson’s trichrome staining of kidney sections from male WT, PPARγ2 KO, ob/ob and POKO mice at 4 (A) and 12 (B) weeks. Glomerular fibrosis is shown (arrows) at different ages. Original magnification: 400×. n=4–5.
DISCUSSION
Metabolic syndrome is a group of risk factors for atherosclerotic, cardiovascular and renal diseases that are associated with obesity, type 2 diabetes, insulin resistance and lack of physical activity. The most common abnormalities of renal structure in obese individuals with type 2 diabetes include podocyte disorders, mesangial expansion and glomerulomegaly. These alterations are accompanied by functional abnormalities, such as renal hyperperfusion, increased filtration and albuminuria. Understanding the mechanisms that produce the constellation of these clinical and pathological alterations that define diabetic nephropathy in humans remains incomplete. More relevantly, not all obese individuals with diabetes develop kidney failure and enter into a dialysis programme at the later stages of renal disease. Two of the main issues explaining the lack of resources for mechanistic investigations are: (1) diabetic nephropathy is a slowly progressive disease and (2) most of the rodent models produce some but not all of the features of human diabetic nephropathy over prolonged periods. A mouse model of renal disease in a context of metabolic syndrome that resembles the human features in an accelerated, uniform and robust way is a valuable resource, allowing the testing of potential therapeutic interventions in the early stages of this disease.
Previously, our results from the POKO mouse, deficient in both the PPARγ2 isoform and in leptin, defined this murine model as a model of early lipotoxicity (Medina-Gomez et al., 2007; Medina-Gomez et al., 2009). POKO and ob/ob mice have a similar positive energy balance, with hyperphagia and low energy expenditure, but because POKO mice lack PPARγ2, they are unable to expand adipose tissue and, as a result, they developed lipotoxicity and became markedly more insulin resistant and diabetic than their ob/ob littermates at a very early age. In this study we show that POKO mice presented accelerated renal dysfunction at the early age of 4 weeks. POKO mice exhibited renal hypertrophy with defects in proliferation markers and an alteration of lipid metabolism, despite their similar weight and blood pressure compared with ob/ob mice. Imbalance in the glucose metabolism of the POKO mice compared with ob/ob mice shows an incipient insulin resistance in kidney, a mechanism that could be implicated in renal failure induced by glucolipotoxicity. POKO mice showed faster progression towards renal disease, consistent with increased inflammation and profibrotic markers in glomeruli compared with ob/ob mice. We suggest that, in the context of glucolipotoxicity, the local inflammatory phenotype in addition to the associated insulin resistance could contribute to kidney injury in POKO mice.
In addition to the well-established therapeutic role of PPARγ for treating type 2 diabetes, favourable renal effects of this nuclear receptor have also been shown. In heterozygous PPARγ-deficient mice fed with a HFD, renal injury and renal lipid accumulation was attenuated. However, therapy with the ligands of PPARγ, TZDs, has been associated with a marked reduction in albuminuria, reduced glomerular hyperfiltration and mesangial expansion, together with decreased inflammation and lower levels of profibrotic markers in the kidney of individuals with type 2 diabetes. Furthermore, TZDs also ameliorated the progression of glomerulosclerosis in the non-diabetic model of 5/6 nephrectomy (Ma et al., 2001). The possibility of direct renal effects is increased by the PPARγ presence in renal glomeruli, cultured mesangial cells and proximal tubular cells. It has already been shown that PPARγ expression is decreased in kidney from diabetic rats on a HFD (Kumar Sharma et al., 2011), but increased PPARγ expression was also correlated with less severe overall sclerosis, induced in non-diabetic puromycin-aminonucleoside-treated rats (Yang et al., 2006), and a protective effect against cyclosporine A (CsA)-induced pancreatic and renal injury (Chung et al., 2005). Here we show that both isoforms γ1 and γ2 of PPARγare expressed in kidney. Whereas PPARγ2 expression was not significantly induced in ob/ob kidneys compared with WT kidneys from mice at 4 weeks of age, PPARγ1 was decreased in ob/ob and POKO compared with WT kidneys. More interestingly, POKO kidneys showed lower expression of PPARγ1 than ob/ob kidneys. The expression of both isoforms of PPARγ was very low in POKO kidneys, although it would be speculative to associate the kidney dysfunction in these animals with the decreased expression of PPARγ compared with ob/ob kidneys. A kidney-specific knockout mouse for PPARγ would help in the investigation into the role of this nuclear receptor in the alteration of renal lipid metabolism and certain aspects of kidney dysfunction during the development of metabolic syndrome.
Here, we show that the POKO mouse is a model with robust and uniform kidney injury that develops accelerated features of diabetic nephropathy at the age of 4 weeks in a context of glucolipotoxicity, characterized by higher plasma levels of glucose and triglycerides than their ob/ob littermates. Also at 4 weeks, glomerular hypertrophy was present in the kidney of POKO but absent in ob/ob mice despite the similar body weight, blood pressure and marked proteinuria in both models. p27Kip1 is a cyclin-dependent kinase (CDK) inhibitor that binds to various CDK-cyclin complexes, inhibiting their activity and preventing progression of the cell cycle. Cells arrested in the G1 phase produce more protein and extracellular matrix (ECM) components, and enlarge (Wolf, 1999). p27Kip1 protein was increased in the kidney of POKO mice at 4 weeks of age and this increase was consistent with an increase in ECM components in the glomerulus of the kidney at this early age. Other characteristic features already detectable at this age were the initiation of podocyte loss and the thickening of the glomerular basement membrane, which was persistent throughout life. Although ob/ob kidneys also show a progressive decline in renal function, the absence of these changes suggests that the complications are more attributed to the development of an obese phenotype with age. The glomerular hypertrophy in POKO mice could be associated with induced apoptosis, as has already been shown in diabetic nephropathy, and decreased expression of cyclin-D1 (Muller et al., 2009; Romero et al., 2010; Jung et al., 2011). Renal and/or glomerular VEGF mRNA has been shown to be increased in diverse experimental models of type 2 diabetes, such as ZDF rats (Chou et al., 2002), Otsuka Long-Evans Tokushima fatty (OLETF) rats (Tsuchida et al., 1999) and db/db mice (Ziyadeh et al., 2000). However, in these models, VEGF mRNA expression rose early in the course of diabetes. At a later age, when glomerulosclerosis was most pronounced, renal VEGF mRNA levels were reduced in ZDF rats (Hoshi et al., 2002). In our models, we found significantly decreased VEGF gene expression in the kidney of POKO compared with ob/ob mice at 4 weeks of age, suggesting that these mice could already have signs of glomerulosclerosis. In fact, we found increased profibrotic factors and increased ECM in POKO kidneys at the age of 4 weeks at the glomerular level. Although we did not find significant differences in other systems that participate in diabetic kidney disease and diabetic albuminuria, such as ACE, we identified lower renin mRNA expression in POKO compared with ob/ob mice. In conditions of type 2 diabetes and obesity, the renin-angiotensin system is normally increased (Wahba and Mak, 2007). The lower expression of renin in POKO kidney that we have observed here could be explained by the fact that PPARγ is known to stimulate renin gene transcription, acting through PPARγ-binding sequences in renin promoter (Desch et al., 2010b; Desch et al., 2010a).
In this study, we observed that POKO mice have an altered lipid metabolism in kidney. Our results of gene expression showed that de novo fatty acid synthesis was decreased in POKO compared with ob/ob kidneys; however, fatty acid oxidation was also decreased. Lipids are fundamental constituents of all living cells. Most cells have complex machineries to regulate the import, synthesis, storage and utilization of lipids. However, lipid accumulation in cells that are not equipped with the molecular tools to handle large amounts of lipid loads, as in settings of lipotoxicity, have been associated with cellular dysfunction and injury. Moorhead et al. initially suggested the lipid nephrotoxicity hypothesis, proposing that an increase in hepatic synthesis of lipoproteins could aggravate renal disease (Moorhead et al., 1982; Rutledge et al., 2010). Previously, our data indicated that the altered lipid composition of islets was a relatively late event in the context of lipotoxicity, which is preceded by lipid changes in the serum, liver, adipose tissue and muscle. In this study we found that, in this context, the kidney could be one of the first organs affected by lipid accumulation. The localization of the excess of lipids varies between animal models, and is likely to have different mechanisms and consequences within different structures in the kidney. Here we show by Oil-RedO staining and more specifically by electron microscopy images the existence of lipid accumulation in the glomeruli of POKO kidneys at an early age. These glomerular lipid deposits are similar to the fatty streaks of atherosclerotic lesions and are injurious to underlying glomerular structures, leading to glomerulosclerosis (Lee et al., 1991; Lee, 2011). The principal determinant of this ectopic lipid accumulation in POKO glomeruli with an altered lipid metabolism could be due to the excessive intracellular free fatty acid (FFA) and triglycerides content. This might lead to the accumulation of potentially toxic metabolites such as long-chain triglycerides, DAGs, PCs and ceramides, which was clearly observed by our lipidomic profile because it has already been shown that there is a marked increase in the formation of three choline-containing phospholipids (phosphatidylcholine, lysophosphatidylcholine and sphingomyelin) in the diabetic kidney (Suzuki et al., 1987). The increased toxic lipid profile in glomeruli of POKO kidneys might cause the upregulation of growth factors, including TGFβ, proinflammatory cytokines and adhesion molecules, including MCP-1, specifically in glomeruli of our mouse model. Targeting of individual chemokines and adhesion molecules has also proven effective in inhibiting progressive renal macrophage infiltration (Tang et al., 1996; Lloyd et al., 1997). Although we did not detect any significant increase in macrophage infiltration at this early age, the upregulation of MCP-1 and TGFβ is supportive of a role of these molecules in causing renal injury in the inductive phase of experimental kidney disease in young POKO mice. These molecules could enter into a vicious circle that, in a paracrine manner, ultimately stimulates higher expression of cytokines, provoking an increased inflammatory state such as has already been reported in the case of TGFβ that stimulates the podocyte to express MCP-1, via sequential signalling of the TGFβ type I receptor and PI3K (Lee et al., 2009). Moreover, an increase in palmitic acid has been associated with increased levels of endoplasmic reticulum (ER) stress and oxidative stress in renal proximal tubular cells (Katsoulieris et al., 2010). In these terms, more studies would need to be performed to check whether the toxic lipid accumulation in POKO glomeruli could cause similar effects.
Although multiple factors contribute to the metabolic syndrome, insulin resistance seems to be a central pathophysiological process behind the development of complications associated with metabolic syndrome. Disruption of normal insulin signalling, hyperinsulinaemia, insulin resistance or absolute insulin deficiency might play a significant role in the pathogenesis of obesity complications. Moreover, increased insulin resistance has been reported in individuals with type 2 diabetes (Groop et al., 1993) and non-diabetic obese individuals (Kambham et al., 2001), who are prone to renal lesions, but also in individuals with mild renal impairment, commonly known as renal insulin resistant syndrome (Becker et al., 2005). The association between ceramides and insulin resistance has been reported previously in insulin-sensitive cells (Powell et al., 2004; Chavez et al., 2005; Sabin et al., 2007). We previously showed that the insulin resistance in POKO mice was associated with hypertriglyceridaemia at 4 weeks of age. Insulin resistance in adipose tissue was demonstrated by the extremely low levels of GLUT4 protein in POKO adipose tissue when compared with GLUT4 levels in adipose tissue from ob/ob mice (Medina-Gomez et al., 2007; Medina-Gomez et al., 2009). Although we have not shown significant differences in the gene expression of GLUT4 or IRS-2, key genes involved in insulin signalling in podocytes, in kidney of the POKO mouse, we observed that the expression of these genes followed the same pattern, with a further decrease in POKO compared with ob/ob mice. AKT phosphorylation was abrogated in both POKO and ob/ob kidneys, suggesting a severe degree of insulin resistance in the kidney of both genotypes. To determine the development of podocyte-specific insulin resistance, we measured the expression of other genes that might be associated. Nephrin is essential to allow insulin-dependent GLUT4 vesicles to translocate to the plasma membrane in podocytes, and regulation of nephrin expression in the early phases of diabetic nephropathy has previously been described (Coward et al., 2007). Nephrin is thought to prevent protein loss through the maintenance of the slit diaphragm between adjacent podocyte foot processes, serving as a signal molecule and regulating the actin cytoskeleton of the podocyte through Nck adaptor proteins, which could in turn affect proper trafficking of receptors and glucose transporter to the cell membrane (Jones et al., 2006; Jones et al., 2009). However, we did not find significant differences in nephrin mRNA expression between the four genotypes. Podocin also participates in the maintenance of the slit diaphragm (Huber et al., 2001). In addition to serving as a structural protein of the slit diaphragm of normal podocytes, podocin might also serve as a scaffold that links tight junction proteins to the actin cytoskeleton in nephrotic foot processes (Shono et al., 2007). We found a similar expression pattern between podocin mRNA and nephrin in the four genotypes. Therefore, the insulin resistance that is present in kidneys from POKO and ob/ob mice could not be explained by lower expression of these molecules.
Interestingly, POKO kidney showed lower expression of adiponectin than found in ob/ob kidneys. Low levels of adiponectin are associated with insulin resistance but also have been associated with inflammation in ESRD, and it has been proposed that adiponectin is a key regulator of albuminuria, probably acting through the AMPK pathway to modulate oxidant stress in podocytes (Sharma et al., 2008). Although we did not find altered expression of genes such as UCP-2, Bcl-2 or Chop, we observed a tendency towards increased Cox-2 and TNFα expression in both PPARγ-deficient mice. NFκB is one of the cross-talk points of multiple signal transduction pathways, which plays a key role in inflammatory responses. We observed increased nuclear NFκB translocation in POKO mice compared with the rest of genotypes. Our results of increased MCP-1 together with increased profibrotic markers such as TGFβ and activation of the NFκB pathway suggest that the inductive phase in inflammation that mediates renal injury is taking place in the kidneys of young POKO mice.
The number of therapeutic options that slow the progression of diabetic kidney disease to ESRD remains limited. Therefore, comprehensive investigation into the early signalling events in new mouse models, which contribute to the understanding of this increasingly prevalent disease, might identify novel avenues for treatment and prevention in humans. From our results in POKO mice at 4 weeks of age, we define the POKO mouse as a good model for the study of the initiation and progression of renal disease associated with metabolic syndrome and glucolipotoxicity. This is a mouse model that resembles features from human diabetic nephropathy. In this study, POKO mice exhibited renal hypertrophy and an alteration of lipid metabolism with defects in proliferation markers, despite having similar weight and blood pressure compared with ob/ob mice at an early age. Imbalance in glucose and lipid metabolism of POKO mice compared with ob/ob mice showed an incipient insulin resistance associated with decreased adiponectin expression in kidney, a mechanism that could be implicated in renal failure induced by glucolipotoxicity. POKO mice showed faster progression of renal disease with increased inflammation and profibrotic markers associated with the lipotoxicity and insulin resistance in glomeruli compared with ob/ob mice.
METHODS
Generation of mice homozygous for PPARγ2 KO and leptin deficiency (ob/ob)
Mice heterozygous for a disruption in exon B1 of PPARγ2 on a 129Sv background (PPARγ2+/−) were crossed with heterozygous ob/ob (Lepob/Lep+) mice on a C57Bl/6 background to obtain mice heterozygous for both the PPARγ2 ablation and the leptin point mutation (PPARγ2+/− Lepob/Lep+). These mice were crossed to obtain the four experimental genotypes: WT (PPARγ2+/+ Lep+/Lep+), PPARγ2 KO (PPARγ2−/− Lep+/Lep+), ob/ob (PPARγ2+/+ Lepob/Lepob) and POKO (PPARγ2−/− Lepob/Lepob). Genotyping for deletion of PPARγ2 and the point mutation in the ob gene was performed by PCR using standard protocols (Hirasawa et al., 1997; Medina-Gomez et al., 2005).
Animal care
Animals were housed at a density of four animals per cage in a temperature-controlled room (20–22°C) with 12-hour light-dark cycles. Mice of the four experimental genotypes were placed at weaning (3 weeks of age) on a normal chow diet (10% of calories derived from fat; D12450B, Research Diets). Food and water were available ad libitum unless noted. All animal protocols used in this study were approved by the Ethics Committee of the Universidad Rey Juan Carlos.
Blood pressure measurements, metabolic cages and collection of samples
Blood pressure (BP) of conscious mice was measured using a tail-cuff sphygmomanometer (LE 5001, LETICA) in appropriate holding cages at 4 and 12 weeks of age.
At 4 and 12 weeks, mice were placed in metabolic balance cages for 24 hours. Urine was collected to measure urinary albumin excretion (UAE) and also glucose using already published protocols (Medina-Gomez et al., 2007). After the experimental period, the mice were killed. The right kidney was removed, weighed and frozen in liquid nitrogen for subsequent total RNA and protein extraction; the left kidney was fixed in 4% buffered p-formaldehyde for morphological and immunochemistry studies. One-half of each kidney was embedded in paraffin for histology and immunohistochemistry, and the other half was frozen for Oil-RedO staining. Serum was collected for biochemical measurements (Medina-Gomez et al., 2007; Medina-Gomez et al., 2009). Lipoproteins in serum were determined by autoanalyzer (Advia 2400, Siemens Healthcare).
Total RNA extraction and quantitative RT-PCR
Total RNA was isolated from either mouse kidney homogenate obtained using TriReagent (Sigma-Aldrich). Total RNA was quantified by Nanodrop 1000 Spectrophotometer (Thermo Scientific). A total of 500 μg of RNA was reverse transcribed into cDNA using the High Capacity cDNA Promega Kit according to manufacturer’s instructions (5 minutes 65°C and 1 hour 37°C). PCR was performed in duplicate for each sample using adequate dilution of cDNA as template for different genes (see supplementary material Table S1 for SYBR Green primers and TaqMan probes).β-actin and 36B4 were used as housekeeping genes. The amplification was carried out in an ABI PRISM 7000 Sequence Detection System (Applied Biosystems) by using the following conditions: 2 minutes 50°C, 10 minutes 95°C, 40 cycles (15 seconds 95°C, 1 minute 60°C). Reagents were from Applied Biosystems (for TaqMan probes) and SYBR Green (for SYBR Green primers). To calculate the relative quantity of gene expression, we used a standard curve method using the untreated samples as calibrator. To validate housekeeping genes we used the BestKeeper software tool (http://www.gene-quantification.info/) (Pfaffl et al., 2004).
Insulin stimulation
Mice were fasted overnight for 14 hours followed by intraperitoneal injection with human insulin Actrapid (Novo Nordisk) 10 U/kg body weight or saline for studying insulin signalling and action. After 5 minutes of injection, kidneys were harvested and frozen in liquid nitrogen for protein extraction.
Total protein extraction, cytosolic and nuclear fraction, and western blotting
A quarter of the kidney was homogenized in RIPA lysis buffer, supplemented with protease inhibitors cocktail (Sigma-Aldrich) and sodium orthovanadate for total protein extraction. Nuclear extract was prepared as described previously (Starkey et al., 2006). Protein was determined by the Bradford’s method (Bio-Rad Laboratories, Inc.), using BSA as standard.
The kidney samples were subjected to SDS-PAGE on 10 or 12% polyacrylamide gels. Proteins were then electrophoretically transferred to polyvinylidene difluoride filters. After transferring, the filters were blocked with 5% nonfat dry milk in TBS-Tween 20 (TBS-T) followed by incubation with one of the following primary antibodies: 40 μg of protein were used to determine p27 (SantaCruz Biotechnology, Inc.), 60 μg of protein were used in p65 NFκB (C-20) (SantaCruz Biotechnology, Inc.) and AKT1/2 (SantaCruz Biotechnology, Inc.) and 30 μg of protein were used for phospho-AKT(Ser473) (Cell Signaling Technology) overnight. After three washes in TBS-T, membranes were probed with the appropriate secondary antibody conjugated to alkaline phosphatase (GE Healthcare). The blots were visualized by using a chemifluorescent detection system (ECF, GE Healthcare) and scanned by Typhoon (GE Healthcare).
The membrane was re-probed with rabbit anti-β-actin antibody (Sigma-Aldrich) and anti-lamin-β receptor (Abcam) to correct for small differences in loading. Using the ImageJ 1.45 software (National Institutes of Health, Bethesda, MD), the protein band density was measured. The amount of protein under control conditions was assigned a relative value of 100%.
Lipid profiling
Approximately 5 mg of kidney samples were weighed and diluted with 50 μl of 0.9% NaCl. Samples were spiked with 10 μl of an internal standard reference compounds mixture (Laaksonen et al., 2006) and subsequently extracted with 200 μl of chloroform:methanol (2:1) solvent. Samples were homogenized for 2 minutes at 20 Hz with Retsch grinding mill by adding three grinding balls. After incubating for 30 minutes at room temperature, approximately 100 μl of the lower layer was separated by centrifugation at 8000 g for 3 minutes at room temperature. A total of 10 μl of labelled standard mixture (three stable-isotope-labelled reference compounds) was added to the lipid extracts.
Immunohistochemistry
Fixed renal tissue sections (4 μm) were dehydrated by graded ethanol and xylene, and then embedded in paraffin. The sections were deparaffinized and rehydrated. Sections were incubated with anti-MCP-1 (SantaCruz Biotechnology, Inc.), anti-TGFβ (SantaCruz Biotechnology, Inc.), anti-PTHrP (SantaCruz Biotechnology, Inc.) and anti-type-IV-collagen (SantaCruz Biotechnology, Inc.). Sections were incubated with a biotinylated anti-IgG (Vector Laboratories) and incubated with the avidin-biotin-peroxidase complex (Vector Laboratories). 3,3′-diaminobenzidine (DAB) substrate (Sigma-Aldrich) was used as the chromogen. The tissue sections were counterstained with Harris haematoxylin. Some kidney samples were incubated without primary antibody as negative controls.
The stained kidney sections were imaged with a light microscope Zeiss Standard 25. Sections were scored semiquantitatively in a blinded manner by two independent observers: staining intensity negative (0), mild (1+), moderate (2+) or strong (3+). The final score was the mean of the two evaluations.
PAS staining, Masson’s trichrome staining, glomerular measure and Oil-red-O staining
Paraffin sections underwent PAS staining and Masson’s trichrome staining (Cook, 1973). Glomerular area measure was performed in PAS-stained sections. Every section contained three to five glomeruli; only glomeruli containing visible vascular pole were measured. Glomerular images were digitized using a Canon Powershot A640 camera attached to a light microscope. After digitalization, glomerular tufts were traced and the areas were calculated using image analysis software (AxioVision Software 4.6, Carl Zeiss). The mean of each area measured was calculated.
Frozen sections were also used for Oil-red-O staining, by which the renal accumulation of neutral lipids was evaluated, as previously reported (Cook, 1973).
Electron microscopy
Kidney tissue was fixed in 2.5% glutaraldehyde and postfixed in 1% osmium tetroxide. The preserved tissue is dehydrated with an increasing concentration of ethanol and embedded in epoxy resin. The specimen was thin-sectioned and the ultrathin sections were stained in an aqueous solutions of uranyl acetate followed by lead citrate and examined under a JEOL GEM 1010 transmission electron microscope (TEM).
TRANSLATIONAL IMPACT.
Clinical issue
Individuals with metabolic syndrome are at high risk of developing kidney disease. Kidney disease progresses slowly and eventually causes end-stage renal failure requiring dialysis or kidney transplant, imposing high health-care costs. Although several animal models have been developed to understand the crucial steps that precipitate renal disease, most rodent models produce only some features of human diabetic nephropathy, and not in the context of metabolic syndrome.
Results
Here, the authors define a mouse model for studying the early pathological changes in the kidney that result from increased fat and glucose levels (glucolipotoxicity). They use the POKO mouse, obtained by crossing the insulin-resistant ob/ob mouse (which lacks leptin) with mice lacking nuclear receptor peroxisome proliferator-activated receptor gamma-2 (PPARγ2; known to have a role in adipogenesis and insulin sensitivity). At 4 weeks of age, POKO mice show renal lipid accumulation that is associated with changes in glucose metabolism and renal function. At this early age, pathogenic effectors, such as inflammation and fibrosis, that characterize the advanced stages of renal disease are observed. By 12 weeks of age, renal damage is severe, suggesting that kidney damage is accelerated in POKO mice owing to glucolipotoxicity.
Implications and future directions
Previously developed models of kidney disease are limited in their ability to address issues such as the early effects of high glucose levels or a high-fat diet and the associated factors that cause pathological changes in the kidney. These new findings establish the POKO mouse as a good model for understanding the early events leading to kidney disease associated with metabolic syndrome. This model will enable further investigations of mechanisms by which the kidney initially starts to fail owing to glucolipotoxicity and ultimately progresses to end-stage renal failure. Further work should reveal the molecular effectors that cause kidney damage in the early phase and suggest biomarkers that might be useful for managing renal disease in the context of metabolic syndrome.
For ultrastructural evaluation, electron micrographs of six glomeruli per kidney were randomly taken at both 3000× and 30,000× magnification for each mouse. At the lower magnification, an overview of the glomerulus was obtained. At the higher magnification, GBM and lamina densa thicknesses were obtained from measurements at three different sites of cross-sectioning, with the aid of Motic Images Plus 2.0 software.
Statistical analysis
Results are expressed as mean±s.e.m. throughout the text. Statistical differences and interactions were evaluated through a two-way lack of leptin and lack of PPARγ2 factorial analysis of variance (ANOVA). When statistically significant differences resulted at the interaction level, Student’s t-test was carried out to compare the effects. Differences were considered statistically significant at P<0.05.
Acknowledgments
We thank Sergio Ferreiro, Davinia Hernandez and Alejandro Gomez for their work in the animal facility. We thank Maria Conejero for her help with TEM, Jose Antonio Mas for his help in genotyping and Ricardo J. Bosch for his supporting help. We thank the funding bodies that have supported research within our laboratory leading to this work.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
C.M.-G., A.I. and G.M.-G. conceived and designed the experiments. C.M.-G., A.I., V.V., M.C., Y.V., I.V., K.B., F.C. and G.M.-G. performed the experiments. C.M.-G., A.I., V.V., M.R., M.O. and G.M.-G. analyzed the data. C.M., A.I., M.C., M.R., A.V.-P. and G.M.-G. wrote the paper.
FUNDING
This work was supported by the Ramon y Cajal programme from Ministerio de Ciencia e Innovacion (MICINN) [BFU2009-10006 to G.M.-G. and BFU2008-04901-C03-03 to M.R.]; Universidad Rey Juan Carlos-Comunidad de Madrid [CCG10-URJC/BIO-560 to A.I.]; Comunidad de Madrid [S2010/BMD-2423 to G.M.-G. and M.R.]; L’Oreal “Women for Science” to G.M.-G.; and FP6 Hepadip, Diabetes UK and the MRC programme grant and MRC CORD to A.V.-P.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.009266/-/DC1
REFERENCES
- Becker B., Kronenberg F., Kielstein J. T., Haller H., Morath C., Ritz E., Fliser D. (2005). Renal insulin resistance syndrome, adiponectin and cardiovascular events in patients with kidney disease: the mild and moderate kidney disease study. J. Am. Soc. Nephrol. 16, 1091–1098 [DOI] [PubMed] [Google Scholar]
- Bianchi S., Bigazzi R., Caiazza A., Campese V. M. (2003). A controlled, prospective study of the effects of atorvastatin on proteinuria and progression of kidney disease. Am. J. Kidney Dis. 41, 565–570 [DOI] [PubMed] [Google Scholar]
- Chavez J. A., Holland W. L., Bar J., Sandhoff K., Summers S. A. (2005). Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J. Biol. Chem. 280, 20148–20153 [DOI] [PubMed] [Google Scholar]
- Chou E., Suzuma I., Way K. J., Opland D., Clermont A. C., Naruse K., Suzuma K., Bowling N. L., Vlahos C. J., Aiello L. P., et al. (2002). Decreased cardiac expression of vascular endothelial growth factor and its receptors in insulin-resistant and diabetic states: a possible explanation for impaired collateral formation in cardiac tissue. Circulation 105, 373–379 [DOI] [PubMed] [Google Scholar]
- Chung B. H., Li C., Sun B. K., Lim S. W., Ahn K. O., Yang J. H., Choi Y. H., Yoon K. H., Sugawara A., Ito S., et al. (2005). Rosiglitazone protects against cyclosporine-induced pancreatic and renal injury in rats. Am. J. Transplant. 5, 1856–1867 [DOI] [PubMed] [Google Scholar]
- Cook H. C. (1973). A histochemical characterization of malignant tumour mucins as a possible aid in the identification of metastatic deposits. Med. Lab. Technol. 30, 217–224 [PubMed] [Google Scholar]
- Coward R. J., Welsh G. I., Koziell A., Hussain S., Lennon R., Ni L., Tavare J. M., Mathieson P. W., Saleem M. A. (2007). Nephrin is critical for the action of insulin on human glomerular podocytes. Diabetes 56, 1127–1135 [DOI] [PubMed] [Google Scholar]
- Desch M., Schubert T., Schreiber A., Mayer S., Friedrich B., Artunc F., Todorov V. T. (2010a). PPARgamma-dependent regulation of adenylate cyclase 6 amplifies the stimulatory effect of cAMP on renin gene expression. Mol. Endocrinol. 24, 2139–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desch M., Schreiber A., Schweda F., Madsen K., Friis U. G., Weatherford E. T., Sigmund C. D., Sequeira Lopez M. L., Gomez R. A., Todorov V. T. (2010b). Increased renin production in mice with deletion of peroxisome proliferator-activated receptor-gamma in juxtaglomerular cells. Hypertension 55, 660–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groop L. C., Widen E., Ferrannini E. (1993). Insulin resistance and insulin deficiency in the pathogenesis of type 2 (non-insulin-dependent) diabetes mellitus: errors of metabolism or of methods? Diabetologia 36, 1326–1331 [DOI] [PubMed] [Google Scholar]
- Hirasawa T., Ohara T., Makino S. (1997). Genetic typing of the mouse ob mutation by PCR and restriction enzyme analysis. Exp. Anim. 46, 75–78 [DOI] [PubMed] [Google Scholar]
- Hoshi S., Shu Y., Yoshida F., Inagaki T., Sonoda J., Watanabe T., Nomoto K., Nagata M. (2002). Podocyte injury promotes progressive nephropathy in zucker diabetic fatty rats. Lab. Invest. 82, 25–35 [DOI] [PubMed] [Google Scholar]
- Huber T. B., Kottgen M., Schilling B., Walz G., Benzing T. (2001). Interaction with podocin facilitates nephrin signaling. J. Biol. Chem. 276, 41543–41546 [DOI] [PubMed] [Google Scholar]
- Jones N., Blasutig I. M., Eremina V., Ruston J. M., Bladt F., Li H., Huang H., Larose L., Li S. S., Takano T., et al. (2006). Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 440, 818–823 [DOI] [PubMed] [Google Scholar]
- Jones N., New L. A., Fortino M. A., Eremina V., Ruston J., Blasutig I. M., Aoudjit L., Zou Y., Liu X., Yu G. L., et al. (2009). Nck proteins maintain the adult glomerular filtration barrier. J. Am. Soc. Nephrol. 20, 1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung D. S., Lee S. H., Kwak S. J., Li J. J., Kim D. H., Nam B. Y., Kang H. Y., Chang T. I., Park J. T., Han S. H., et al. (2011). Apoptosis occurs differentially according to glomerular size in diabetic kidney disease. Nephrol. Dial. Transplant. 27, 259–266 [DOI] [PubMed] [Google Scholar]
- Kambham N., Markowitz G. S., Valeri A. M., Lin J., D’Agati V. D. (2001). Obesity-related glomerulopathy: an emerging epidemic. Kidney Int. 59, 1498–1509 [DOI] [PubMed] [Google Scholar]
- Katsoulieris E., Mabley J. G., Samai M., Sharpe M. A., Green I. C., Chatterjee P. K. (2010). Lipotoxicity in renal proximal tubular cells: relationship between endoplasmic reticulum stress and oxidative stress pathways. Free Radic. Biol. Med. 48, 1654–1662 [DOI] [PubMed] [Google Scholar]
- Kiss-Toth E., Roszer T. (2008). PPARgamma in Kidney Physiology and Pathophysiology. PPAR Res. 2008, 183108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume S., Uzu T., Araki S., Sugimoto T., Isshiki K., Chin-Kanasaki M., Sakaguchi M., Kubota N., Terauchi Y., Kadowaki T., et al. (2007). Role of altered renal lipid metabolism in the development of renal injury induced by a high-fat diet. J. Am. Soc. Nephrol. 18, 2715–2723 [DOI] [PubMed] [Google Scholar]
- Laaksonen R., Katajamaa M., Paiva H., Sysi-Aho M., Saarinen L., Junni P., Lutjohann D., Smet J., Van Coster R., Seppanen-Laakso T., et al. (2006). A systems biology strategy reveals biological pathways and plasma biomarker candidates for potentially toxic statin-induced changes in muscle. PLoS ONE 1, e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. Y., Chung C. H., Khoury C. C., Yeo T. K., Pyagay P. E., Wang A., Chen S. (2009). The monocyte chemoattractant protein-1/CCR2 loop, inducible by TGF-beta, increases podocyte motility and albumin permeability. Am. J. Physiol. Renal. Physiol. 297, F85–F94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. S. (2011). Mechanisms and consequences of hypertriglyceridemia and cellular lipid accumulation in chronic kidney disease and metabolic syndrome. Histol. Histopathol. 26, 1599–1610 [DOI] [PubMed] [Google Scholar]
- Lee H. S., Lee J. S., Koh H. I., Ko K. W. (1991). Intraglomerular lipid deposition in routine biopsies. Clin. Nephrol. 36, 67–75 [PubMed] [Google Scholar]
- Lee T. M., Lin M. S., Tsai C. H., Chang N. C. (2005). Add-on and withdrawal effect of pravastatin on proteinuria in hypertensive patients treated with AT receptor blockers. Kidney Int. 68, 779–787 [DOI] [PubMed] [Google Scholar]
- Lennon R., Pons D., Sabin M. A., Wei C., Shield J. P., Coward R. J., Tavare J. M., Mathieson P. W., Saleem M. A., Welsh G. I. (2009). Saturated fatty acids induce insulin resistance in human podocytes: implications for diabetic nephropathy. Nephrol. Dial. Transplant. 24, 3288–3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd C. M., Minto A. W., Dorf M. E., Proudfoot A., Wells T. N., Salant D. J., Gutierrez-Ramos J. C. (1997). RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J. Exp. Med. 185, 1371–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L. J., Marcantoni C., Linton M. F., Fazio S., Fogo A. B. (2001). Peroxisome proliferator-activated receptor-gamma agonist troglitazone protects against nondiabetic glomerulosclerosis in rats. Kidney Int. 59, 1899–1910 [DOI] [PubMed] [Google Scholar]
- Medina-Gomez G., Virtue S., Lelliott C., Boiani R., Campbell M., Christodoulides C., Perrin C., Jimenez-Linan M., Blount M., Dixon J., et al. (2005). The link between nutritional status and insulin sensitivity is dependent on the adipocyte-specific peroxisome proliferator-activated receptor-gamma2 isoform. Diabetes 54, 1706–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Gomez G., Gray S. L., Yetukuri L., Shimomura K., Virtue S., Campbell M., Curtis R. K., Jimenez-Linan M., Blount M., Yeo G. S., et al. (2007). PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet. 3, e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Gomez G., Yetukuri L., Velagapudi V., Campbell M., Blount M., Jimenez-Linan M., Ros M., Oresic M., Vidal-Puig A. (2009). Adaptation and failure of pancreatic beta cells in murine models with different degrees of metabolic syndrome. Dis. Model. Mech. 2, 582–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhead J. F., Chan M. K., El-Nahas M., Varghese Z. (1982). Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet 2, 1309–1311 [DOI] [PubMed] [Google Scholar]
- Muller M., Gagiannis S., Nawroth P. P., Brune M., Schilling T. (2009). Activation of the receptor for parathyroid hormone and parathyroid hormone related protein induces apoptosis via the extrinsic and intrinsic signaling pathway. Int. J. Mol. Med. 24, 373–380 [DOI] [PubMed] [Google Scholar]
- Nichols G. A., Vupputuri S., Lau H. (2011). Medical care costs associated with progression of diabetic nephropathy. Diabetes Care 34, 2374–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagtalunan M. E., Miller P. L., Jumping-Eagle S., Nelson R. G., Myers B. D., Rennke H. G., Coplon N. S., Sun L., Meyer T. W. (1997). Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Invest. 99, 342–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M. W., Tichopad A., Prgomet C., Neuvians T. P. (2004). Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-based tool using pair-wise correlations. Biotechnol. Lett. 26, 509–515 [DOI] [PubMed] [Google Scholar]
- Powell D. J., Turban S., Gray A., Hajduch E., Hundal H. S. (2004). Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem. J. 382, 619–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero M., Ortega A., Izquierdo A., Lopez-Luna P., Bosch R. J. (2010). Parathyroid hormone-related protein induces hypertrophy in podocytes via TGF-beta(1) and p27(Kip1): implications for diabetic nephropathy. Nephrol. Dial. Transplant. 25, 2447–2457 [DOI] [PubMed] [Google Scholar]
- Ruan X., Zheng F., Guan Y. (2008). PPARs and the kidney in metabolic syndrome. Am. J. Physiol. Renal. Physiol. 294, F1032–F1047 [DOI] [PubMed] [Google Scholar]
- Rutledge J. C., Ng K. F., Aung H. H., Wilson D. W. (2010). Role of triglyceride-rich lipoproteins in diabetic nephropathy. Nat. Rev. Nephrol. 6, 361–370 [DOI] [PubMed] [Google Scholar]
- Sabin M. A., Stewart C. E., Crowne E. C., Turner S. J., Hunt L. P., Welsh G. I., Grohmann M. J., Holly J. M., Shield J. P. (2007). Fatty acid-induced defects in insulin signalling, in myotubes derived from children, are related to ceramide production from palmitate rather than the accumulation of intramyocellular lipid. J. Cell Physiol. 211, 244–252 [DOI] [PubMed] [Google Scholar]
- Sharma A. K., Bharti S., Ojha S., Bhatia J., Kumar N., Ray R., Kumari S., Arya D. S. (2011). Up-regulation of PPARgamma, heat shock protein-27 and -72 by naringin attenuates insulin resistance, beta-cell dysfunction, hepatic steatosis and kidney damage in a rat model of type 2 diabetes. Br. J. Nutr. 106, 1713–1723 [DOI] [PubMed] [Google Scholar]
- Sharma K., Ramachandrarao S., Qiu G., Usui H. K., Zhu Y., Dunn S. R., Ouedraogo R., Hough K., McCue P., Chan L., et al. (2008). Adiponectin regulates albuminuria and podocyte function in mice. J. Clin. Invest. 118, 1645–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shono A., Tsukaguchi H., Yaoita E., Nameta M., Kurihara H., Qin X. S., Yamamoto T., Doi T. (2007). Podocin participates in the assembly of tight junctions between foot processes in nephrotic podocytes. J. Am. Soc. Nephrol. 18, 2525–2533 [DOI] [PubMed] [Google Scholar]
- Starkey J. M., Haidacher S. J., LeJeune W. S., Zhang X., Tieu B. C., Choudhary S., Brasier A. R., Denner L. A., Tilton R. G. (2006). Diabetes-induced activation of canonical and noncanonical nuclear factor-kappaB pathways in renal cortex. Diabetes 55, 1252–1259 [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Fausto A., Hruska K. A., Avioli L. V. (1987). Stimulation of phosphatidylcholine biosynthesis in diabetic hypertrophic kidneys. Endocrinology 120, 595–601 [DOI] [PubMed] [Google Scholar]
- Tang W. W., Qi M., Warren J. S. (1996). Monocyte chemoattractant protein 1 mediates glomerular macrophage infiltration in anti-GBM Ab GN. Kidney Int. 50, 665–671 [DOI] [PubMed] [Google Scholar]
- Tonelli M., Moye L., Sacks F. M., Kiberd B., Curhan G. (2003). Pravastatin for secondary prevention of cardiovascular events in persons with mild chronic renal insufficiency. Ann. Intern. Med. 138, 98–104 [DOI] [PubMed] [Google Scholar]
- Tsuchida K., Makita Z., Yamagishi S., Atsumi T., Miyoshi H., Obara S., Ishida M., Ishikawa S., Yasumura K., Koike T. (1999). Suppression of transforming growth factor beta and vascular endothelial growth factor in diabetic nephropathy in rats by a novel advanced glycation end product inhibitor, OPB-9195. Diabetologia 42, 579–588 [DOI] [PubMed] [Google Scholar]
- Vidal-Puig A., Unger R. H. (2010). Special issue on lipotoxicity. Biochim. Biophys. Acta 1801, 207–208 [DOI] [PubMed] [Google Scholar]
- Virtue S., Vidal-Puig A. (2008). It’s not how fat you are, it’s what you do with it that counts. PLoS Biol. 6, e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtue S., Vidal-Puig A. (2010). Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome – an allostatic perspective. Biochim. Biophys. Acta 1801, 338–349 [DOI] [PubMed] [Google Scholar]
- Wahba I. M., Mak R. H. (2007). Obesity and obesity-initiated metabolic syndrome: mechanistic links to chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2, 550–562 [DOI] [PubMed] [Google Scholar]
- Welsh G. I., Hale L. J., Eremina V., Jeansson M., Maezawa Y., Lennon R., Pons D. A., Owen R. J., Satchell S. C., Miles M. J., et al. (2010). Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 12, 329–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K. E., Bilous R. W. (2004). Structural alterations to the podocyte are related to proteinuria in type 2 diabetic patients. Nephrol. Dial. Transplant. 19, 1437–1440 [DOI] [PubMed] [Google Scholar]
- Wolf G. (1999). Molecular mechanisms of renal hypertrophy: role of p27Kip1. Kidney Int. 56, 1262–1265 [DOI] [PubMed] [Google Scholar]
- Yang H. C., Ma L. J., Ma J., Fogo A. B. (2006). Peroxisome proliferator-activated receptor-gamma agonist is protective in podocyte injury-associated sclerosis. Kidney Int. 69, 1756–1764 [DOI] [PubMed] [Google Scholar]
- Yoshida Y., Fogo A., Ichikawa I. (1989). Glomerular hemodynamic changes vs. hypertrophy in experimental glomerular sclerosis. Kidney Int. 35, 654–660 [DOI] [PubMed] [Google Scholar]
- Ziyadeh F. N., Hoffman B. B., Han D. C., Iglesias-De La, Cruz M. C., Hong S. W., Isono M., Chen S., McGowan T. A., Sharma K. (2000). Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. USA 97, 8015–8020 [DOI] [PMC free article] [PubMed] [Google Scholar]