


Abstract
Synonymous codons are unevenly distributed among genes, a phenomenon termed codon usage bias. Understanding the patterns of codon bias and the forces shaping them is a major step towards elucidating the adaptive advantage codon choice can confer at the level of individual genes and organisms. Here, we perform a large-scale analysis to assess codon usage bias pattern of pyrimidine-ending codons in highly expressed genes in prokaryotes. We find a bias pattern linked to the degeneracy of the encoded amino acid. Specifically, we show that codon-pairs that encode two- and three-fold degenerate amino acids are biased towards the C-ending codon while codons encoding four-fold degenerate amino acids are biased towards the U-ending codon. This codon usage pattern is widespread in prokaryotes, and its strength is correlated with translational selection both within and between organisms. We show that this bias is associated with an improved correspondence with the tRNA pool, avoidance of mis-incorporation errors during translation and moderate stability of codon–anticodon interaction, all consistent with more efficient translation.
INTRODUCTION
The degeneracy of the genetic code implies that most amino acids are encoded by a family of synonymous codons, usually with the first two nucleotides fixed and the third one varied. While synonymous codons encode the same amino acid, their relative usage varies dramatically between different species and between genes of the same species. The deviation from uniform codon distribution is referred to as codon usage bias (1,2). Many theories have been proposed to explain the forces shaping codon usage bias (3–7). For example, mutational biases influence the genome base composition, which in turn affects codon usage considerably (8). Asymmetry in DNA replication and repair of the leading and lagging DNA strands (9) creates further codon usage bias. Furthermore, it has been shown that certain types of codon usage bias are associated with gene expression levels (10–17). This association is often explained through selection towards translational efficiency (18–22), which regards optimization of both translation rate (23) and fidelity (24,25). Selection towards codon usage that promotes efficient translation has both local effects on specific genes (22,26) as well as global effects on the organism’s fitness. The latter is achieved by promoting rapid recycling of ribosomes, reducing costs wasted on correcting translation errors (27) and lowering the production of inactive and at times toxic proteins (28).
While the concept of selecting codons to enhance translation efficiency is easily comprehended, the underlying principles that determine the preferred codons are still under debate. Previous studies identified several codon properties as associated with expression-dependent selection: (i) robustness—preference of certain codons that reduce the impact of mutations and errors in translation (29,30), (ii) compatibility with the cellular tRNA pool—preference of codons matching the more abundant tRNA iso-acceptor, which will be translated faster and with fewer errors than alternative synonymous codons (10,15–17,19,21,23,31–37). Furthermore, it has been suggested that the tRNA pool itself is regulated to optimize gene expression under different growth conditions (38,39). However, differences in tRNA abundance cannot explain codon usage bias for synonymous codons that are translated by the same tRNA. This is often the case in prokaryotes, where due to the striking absence of tRNAs containing adenine (A) at the wobble position (40), synonymous codons ending with a pyrimidine (Y) residue [uracil (U) or cytosine (C)] are translated by a single tRNA containing guanine (G) at the wobble position. We took advantage of this phenomenon to explore the pattern of codon usage bias that seems independent of tRNA abundance, focusing on pyrimidine-ending codons (hereinafter N1N2Y3). Intriguingly, we have observed a clear association between the number of synonymous codons encoding an amino acid (degeneracy level) and the preference for U or C at the third codon position, which is emphasized in organisms where translational selection is operational in highly expressed genes. Specifically, we find that U is preferred over C at position Y3 in amino acids encoded by four codons, while C is preferred over U in amino acids encoded by two and three codons. We analyze and discuss these observations in light of existing and new theories regarding properties of codon–anticodon interaction.
MATERIALS AND METHODS
Genome data
We retrieved the genomic sequence and genome-related data of 1346 prokaryotes from the NCBI FTP site (ftp://ftp.ncbi.nih.gov/genomes/Bacteria, April 2011) including 1245 bacteria and 101 archaea. To reduce bias caused by inclusion of closely related organisms we randomly selected one organism per genus as representative. This resulted in a data set of 481 prokaryotes including 424 bacteria and 57 archaea (Supplementary Data set S1).
Codon-pair bias calculation
In each organism, for each of the codon-pairs of the N1N2Y3 type, we compared the counts of the two synonymous codons (ending with C and U) in the ribosomal genes to their expected counts based on the nucleotide distribution in the third codon position of the same genes (Supplementary Data set S1). The comparison was done using χ2 test with Yates correction. False discovery rate (FDR) correction was applied to all the tests of a specific organism.
Clustering procedure
χ2 test results were transformed into a numeric representation. Statistically significant preferences for C at Y3 (C-Bias) or U at Y3 (U-Bias) were assigned the values 1 and −1, respectively. Otherwise, for insignificant results or insufficiency of data to employ the test (N-Bias) a value of 0 was assigned. Results were arranged in a matrix where columns and rows indicated the codon-pairs and organisms, respectively. Hierarchical clustering was performed using a hamming distance metric.
Computation of organism genome bias—ENC′diff
ENC′ is a variant of the effective number of codons (ENC) index (41), accounting for background nucleotide composition (42). It takes the value of 61 when all codons are used at the frequency expected given the nucleotide composition, and its value decreases as codon usage deviates from the expected. The nucleotide composition of a gene was used as background. To obtain an estimate of translational selection at an organism scale, we computed ENC′diff for each organism according to Equation (1).
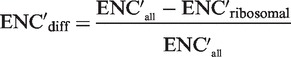 |
(1) |
ENC′diff is the difference between the averages of ENC′ of all genes and the genes encoding ribosomal proteins, normalized by the average ENC′ of all genes.
Fold rule scores
We defined two scores of codon bias; one at an organism scale and one at a gene scale. The scores take into account the type of bias found in N1N2Y3 codon-pairs and the degeneracy level of the amino acid the pair encodes (2-/3-fold in cases of amino acids encoded by pairs or a triplet of codons and 4-fold in cases of amino acids encoded by quartets of codons; codons for amino acids encoded by six codons were divided to a pair and a quartet of codons).
Organism fold rule score (OFRS)
A score describing the conformity of an organism to the fold rule in genes encoding ribosomal proteins.
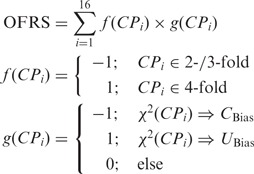 |
(2) |
where CPi is the i-th out of 16 N1N2Y3 codon-pairs, 4-fold and 2-/3-fold indicate the level of degeneracy, and χ2(CPi) indicates the type of bias observed in a χ2 test.
Gene fold rule score (GFRS)
A score describing the conformity of a gene to the fold rule.
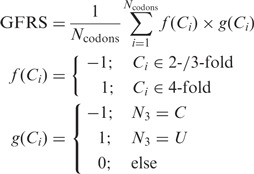 |
(3) |
where Ncodons is the number of codons in the gene, Ci is the i-th codon, 4-fold and 2-/3-fold indicate the level of degeneracy, and N3 is the nucleotide found at position N3 of the codon.
Gene expression data
Normalized mRNA expression profiles of nine organisms were extracted from the GEO database as detailed in the ‘Supplementary Methods’.
tRNA repertoire
We scanned the sequences of all non-coding RNAs identified in each organism using the program tRNAscan-SE (43). This comprised the repertoire of identified tRNAs and their respective anticodons.
RESULTS AND DISCUSSION
Association between codon usage bias and amino acid degeneracy
We compiled genome sequence data of 481 prokaryotes, one representative per genus (Supplementary Data set S1), to be included in the analysis. For each organism, we used genes encoding ribosomal proteins as proxy for highly expressed genes. We employed χ2 test to ask whether the 16 codon-pairs of the type N1N2Y3 exhibit codon preferences beyond those expected from the nucleotide content in the third codon positions of these genes (Supplementary Data set S1). A statistically insignificant result was designated N-Bias whereas a statistically significant result was designated C-Bias or U-Bias depending on bias direction.
To systematically analyze the patterns of bias, we transformed the results into a numeric representation (see ‘Materials and Methods’ section) and clustered the resulting matrix in both dimensions (Figure 1). Remarkably, the clustering clearly distinguishes between two groups of codon-pairs based on the level of degeneracy of the amino acid they encode. The codon-pair group that shows distinct C-Bias (marked by the cyan bar) contains all N1N2Y3 codon-pairs that encode two-fold degenerate amino acids, the codon-pair AUY that encodes the three-fold degenerate Ile and the two-fold component of the six-fold Ser. We refer to this group as the 2-/3-fold group. The second group, which shows distinct U-Bias (marked by the yellow bar), includes all the N1N2Y3 codon-pairs that encode the four-fold degenerate amino acids, both those that are strictly four-fold and the four-fold component of six-fold degenerate amino acids (Ser, Leu, Arg). We refer to this group as the 4-fold group. These results are consistent with and lend statistical support to similar observations by Ran and Higgs (44,45) in a smaller set of organisms. We define a fold rule, by which the preferred codon in codon-pairs belonging to the 2-/3-fold group is N1N2C3, whereas the preferred codon in codon-pairs belonging to the 4-fold group is N1N2U3.
Figure 1.

An association between the degeneracy level of amino acids and codon bias pattern in N1N2Y3 codon-pairs. The matrix of codon-pair biases in multiple organisms (Green: U-Bias, magenta: C-Bias, black: N-Bias) was clustered using a hamming distance metric. The clustering reveals a clear distinction between codon-pairs belonging to codon families with four-fold degeneracy (yellow bar) and those belonging to two- and three-fold degenerate families (cyan bar), where the former are statistically significantly biased towards the U-ending codon and the latter are statistically significantly biased towards the C-ending codon. This distinction is most pronounced in a subset of organisms (blue bar).
The fold rule is associated with translational selection
Approximately 25% of the organisms included in the analysis (marked by the blue bar in Figure 1) show a very clear demarcation between the two groups of codons. Of note, these organisms represent multiple clades throughout the taxonomic tree (Supplementary Figure S1). If this widespread phenomenon represents variance in translation efficiency of synonymous codons, we would expect to see an association between the level of translational selection and the extent of codon bias following the fold rule. To this end, we computed for each organism a measure of agreement with the fold rule and a measure of translational selection. The organism fold rule score (OFRS) quantifies the adherence of an organism to the fold rule (see ‘Materials and Methods’ section). This measure is the difference between the numbers of codon-pairs demonstrating statistically significant bias conforming to the fold rule and opposing it. To quantify the level of translational selection of an organism, we employed a measure similar to the one proposed by Rocha (5). We calculated ENC′diff, the normalized difference between the average ENC′ of all genes and the average ENC′ of ribosomal genes, where ENC′ is a measure of codon bias of a gene (see ‘Materials and Methods’ section). The stronger the selection towards efficient translation in highly expressed genes, the larger the difference between codon usage in ribosomal genes and all other genes, resulting in higher ENC′diff. The values of ENC′diff in our data set range from −0.084 in the archaeon Thermofilum pendens to 0.274 in the bacterium Leuconostoc mesenteroides. In 460 out of 481 organisms, the value of ENC′diff is positive, indicating that genes encoding ribosomal proteins use a less random set of codons as suggested by Rocha (5). We find a correlation between OFRS and ENC′diff (r = 0.582, P = 6.4 × 10−45; Figure 2A), suggesting that this fold-dependent codon bias pattern is associated with translational selection. To further establish this conclusion, for each codon-pair, we divided the organisms into three groups according to the type of the χ2 result (C-Bias, U-Bias and N-Bias). We compared the ENC′diff distribution between the C-Bias and U-Bias organism groups using the Mann–Whitney test (Supplementary Figure S2). In 2-/3-fold codon-pairs the C-Bias group has higher ENC′diff values than the U-Bias group. The only exception is the Cys-UGY codon-pair in which the U-Bias group is too small to produce a statistically significant result. In this case, however, it is possible to see that the C-Bias group has higher ENC′diff values than the N-Bias group (P = 9.8 × 10−4). For most 4-fold codon-pairs we see the opposite phenomenon, where the U-Bias group has higher ENC′diff values compared to the C-Bias group. One exception is the Val codon-pair GUY, which shows the same trend although statistically insignificantly. Still, comparison between the U- and N-Bias groups shows higher ENC′diff values in the U-Bias group (P = 4.1 × 10−6). The other exception is the Leu codon-pair CUY in which the U-Bias and C-Bias groups have similar ENC′diff distributions. These results suggest that codon bias in most N1N2Y3 codon-pairs is associated with selection towards translational efficiency.
Figure 2.

Organisms conforming to the fold rule demonstrate strong translational selection and fast growth. OFRS and ENC′diff were calculated as described in ‘Materials and Methods’ section and minimal generation times were taken from (46). (A) Association between OFRS and ENC′diff was evaluated by computing Pearson correlation coefficient. (B) Association between OFRS and minimal generation time was evaluated by computing Spearman correlation coefficient (since the generation time variable is not normally distributed). Organisms most compatible with the fold rule (marked by the blue bar in Figure 1) are colored dark gray and have relatively high ENC′diff and low minimal generation time values.
It has been suggested that selection for fast growth affects codon bias by promoting the use of translationally efficient codons (46,47). We evaluated the association between minimal generation-times taken from Vieira-Silva and Rocha (46) and OFRS values. Figure 2B demonstrates a strong correlation (rs = −0.654, P = 4.3 × 10−27) as would be expected if the preferred codons according to the fold rule indeed promote translational efficiency. Notably, for 17 of the organisms marked by the blue bar in Figure 1, there are generation-time values, and 14 of them show minimal generation-time of 2.5 h or less.
Highly expressed genes demonstrate fold-dependent codon bias
We showed an association between fold-related codon bias in genes encoding ribosomal proteins and translational selection strength at the organism level. Since the strength of translational selection is likely to vary between genes of the same organism depending on their expression level, we examined the relationship between gene codon preference and expression level. To this end, we computed a gene fold rule score (GFRS) that represents the general tendency of a gene to use N1N2C3 over N1N2U3 codons for 2-/3-fold codon-pairs and vice versa for 4-fold codon-pairs (see ‘Materials and Methods’ section). We computed the correlation between GFRS values and gene expression levels obtained from microarray results in nine prokaryotes, representing several major taxonomic clades and a range of ENC′diff values (Supplementary Table S1). Since GFRS is normalized by the total number of codons in the open reading frame (ORF), there might be bias towards high scores in short ORFs. This may bias the correlation, especially given the known inverse correlation between expression and ORF length (48). We therefore performed a partial Pearson correlation while controlling for ORF length. Four of the five organisms that have high ENC′diff (Shewanella oneidensis MR-1, Streptococcus mutans UA159, Escherichia coli K12 and Haemophilus influenzae Rd KW20) show a marked correlation between GFRS and gene expression in the 50% of genes with the highest expression levels but not in the 50% of genes with low expression levels, suggesting that translational selection is most prominent in genes expressed above a certain expression level threshold (Supplementary Figure S3). Bacillus subtilis 168, despite its high ENC′diff value, shows only a weak correlation between the GFRS and gene expression. It is, however, evident that B. subtilis genes with high GFRS other than those encoding ribosomal proteins (marked red), tend to be highly expressed. Organisms with low ENC′diff values (Rhodobacter sphaeroides 2.4.1, Pseudomonas aeruginosa PAO1, Thermus thermophilus HB8 and Nitrosomonas europaea ATCC19718) exhibited weaker correlations between GFRS and expression and less pronounced differences between weakly and highly expressed genes. These latter results are consistent with our hypothesis, as in the absence of translational selection, we would not expect a correlation between translation-related codon property and gene expression.
Possible mechanisms underlying fold-dependent codon bias
We have observed that highly expressed genes show a clear bias in N1N2Y3 codons that is associated with the degeneracy level of the encoded amino acid. Remarkably, these tendencies are mostly observed in organisms with high translational selection and seem to be associated with more efficient translation. In the next sections, we analyze several possible mechanisms in an attempt to understand the forces shaping this pattern of codon bias.
tRNA availability
We stated above that N1N2Y3 codon-pairs are translated by a single tRNA with G at the wobble position. However, over the years the wobble rules have been modified and expanded (49). It is now acknowledged that an unmodified U in the first position of the tRNA anticodon (U34) is capable of reading N1N2U3 codons and to a lesser extent N1N2C3 codons, in addition to reading the N1N2A3 (Watson–Crick base pairing) and N1N2G3 (conventional wobble) codons. Certain U34 modifications abolish the ability to read the N1N2C3 codon whereas others limit the wobble even further to only purine-ending codons (N1N2R3). While it is suggested that the price for such ‘superwobbling’ is reduced translational efficiency (50), in 4-fold codon-pairs it might tilt the balance towards U-Bias due to an increase in the available tRNA pool. We would expect C-Bias in codon-pairs of 4-fold amino acids only when the pool of G34N35N36 tRNA fitting both codons is larger than the pool of U34N35N36 tRNA, thereby minimizing the effect of tRNA availability. To this end, we analyzed the effect of the two tRNA species on codon bias across the organisms in our study. Since the actual tRNA pool is not available for most organisms, we used the number of tRNA genes as a proxy for tRNA levels. Figure 3 demonstrates that as the U34/G34 tRNA ratio increases, the bias leans in the direction of the N1N2U3 codon. Only in relatively low ratios, do we see C-Bias in 4-fold codon-pairs. This is most pronounced in Pro-CCY, Gly-GGY and Ser-UCY, where all organisms with U34/G34 tRNA ratio above 1 are U-biased. A similar trend is observed in Leu-CUY, Val-GUY, Thr-ACY and Ala-GCY. Since for Arg-CGY the major tRNA is A34C35G36 while U34C35G36 is very rarely used (mostly in archaea), it is impossible to determine how the U34/A(I)34 tRNA ratio will affect its codon bias. Ran and Higgs (45) also showed U-preference but claimed that it stems from a surprisingly well-translated non-standard U3:U34 base pairing. Since U34 tRNA simultaneously increases the tRNA pool of N1N2U3 codons and hypothetically improves their translation, we were unable to evaluate the selective significance of each factor.
Figure 3.

U-Bias in 4-fold codon-pairs is associated with the tRNA repertoire. Circles represent organisms with the indicated U34N35N36 (X-axis) and G34N35N36 (Y-axis) tRNA gene copy numbers. The only exception is the Arg codon-pair CGY for which the common tRNA is A34C35G36 (where A is modified to I). Circle size represents the number of organisms with the specific tRNA gene counts. Circle color represents the direction of bias in the codon-pair (Green: U-Bias; magenta: C-Bias). Circles along the diagonal indicate equal number of gene copies of the two tRNAs. Most organisms with C-Bias have a low U34/G34 ratio.
Ran and Higgs (44,45) further suggested that the C-preference observed in the 2-fold group results from a more rapid processing of the strongly interacting Watson–Crick C:G base pair by the ribosome than of the weakly interacting wobble G:U base pair. We hypothesized that the U34N35N36 tRNA may have an indirect contribution to this bias. Translation of N1N2Y3 codons by U34N35N36 tRNA would cause a major disruption of the genetic code in the 2-fold group. However, it is conceivable that the mechanisms restricting the specificity of the U34N35N36 tRNA to N1N2R3 codons are not absolute, leading to a certain rate of amino acid mis-incorporation. If such mistakes occur, we would expect selection to favor the use of N1N2C3 that is less likely to be misread by the U34N35N36 tRNA, as is indeed the case. Another expectation arising from such a mechanism is stronger selection when substitution of the correct amino acid with its N1N2R3 encoded neighbor is not well tolerated. Although it is known that two amino acids that share the first two nucleotides in their codons tend to have similar properties (51,52), some neighboring amino acids are more likely to be replaced during evolution than others, indicating that putative mistranslations between them are better tolerated. Table 1 shows the substitution scores given by a range of blocks substitution matrices (BLOSUM) (53), which can be used as an indication of how conservative a substitution is. We used BLOSUM70–BLOSUM90 matrices in order to focus on substitutions likely to happen in relatively similar proteins without changing or impairing their function. The most extreme cases are the Ser/Arg pair, which is substituted less than expected in all the BLOSUM matrices checked, and the Asp/Glu pair, which is substituted more than expected. As anticipated, the Ser 2-fold codon-pair shows C-Bias in 199 organisms and U-Bias in only one organism (C/U ratio 199), whereas the Asp codon-pair has almost the same number of organisms demonstrating C- and U-Bias (69 and 62, respectively, C/U ratio 1.1). The His/Gln pair, which is also substituted more than expected, has a relatively low C/U ratio for His (4.2), whereas the Phe/Leu and Asn/Lys pairs which are relatively neutral have a high C/U ratio for Phe and Asn (52.5 and 27, respectively), although not as high as Ser. These results lend further support to the possible role of error minimization in shaping codon bias in 2-/3-fold codon-pairs. Note that Tyr, Cys and Ile were not included in this analysis since there are no U34N35N36 tRNAs that can misread them.
Table 1.
Propensity towards C-Bias depends on the similarity between amino acids sharing the codon quartet
| Ser(AGY)–Arg(AGR) | Phe(UUY)–Leu(UUR) | Asn(AAY)–Lys(AAR) | His(CAY)–Gln(CAR) | Asp(GAY)–Glu(GAR) | |
|---|---|---|---|---|---|
| BLOSUM substitution valuesa | −1:−2 | 0 | 0 | 1 | 1:2 |
| C-Biasb | 199 | 210 | 189 | 140 | 69 |
| U-Biasb | 1 | 4 | 7 | 33 | 62 |
| N-Biasb | 41 | 27 | 45 | 68 | 110 |
| C/U Ratio | 199 | 52.5 | 27 | 4.2 | 1.1 |
aRange is the minimal and maximal substitution values observed in BLOSUM substitution matrices 70, 75, 80, 85 and 90
bCalculated from organisms with high ENC′diff values (upper 50%) for Ser, Phe, Asn, His and Asp.
Nucleotide bias
In a previous study of codon bias in the prokaryotic world, Hershberg and Petrov (54) showed that codons are biased beyond, but in the same direction of the general nucleotide content of the organism. Although there is no correlation between organism GC content and ENC′diff (r = 1.3 × 10−4, P = 1), we wished to explore the possibility that nucleotide content has an effect on the observed codon bias. When examining ENC′diff versus GC content, no consistent effect of GC content on the bias pattern could be observed (Supplementary Figure S4). However, when concentrating on organisms with high ENC′diff values (above 0.1), where translational selection is strong, it is possible to see a complicated pattern of GC content influence (Supplementary Figure S5 and discussion therein). We hypothesized that since GC content is mostly reflected in the N3 codon position (55), it might also be associated with the number of tRNAs that read the resulting codons. While GC content is not statistically significantly associated with the number of G34N35N36 tRNA genes (r = 0.07, P = 0.1), it is associated with U34N35N36 and C34N35N36 tRNA genes (r = −0.26, P = 5.8 × 10−9 and r = 0.63, P = 2.4 × 10−55, respectively). When evaluating how the GC content affects the ratio of U34/G34 (Supplementary Figure S6), we see that this ratio tends to be high (above 1) in organisms with low GC content and low (below 1) in organisms with high GC content. We have shown before that the U34/G34 ratio affects bias direction in 4-fold codon-pairs. We can therefore surmise that in organisms with high GC content, the tRNA pool available to the N1N2U3 codon is not significantly larger than that of the N1N2C3 codon and therefore, we see less cases of U-Bias. In 2-fold codon-pairs, the same reasoning may apply since a low U34/G34 ratio indicates a low probability of mistranslation and therefore reduced selection towards C-Bias. Interestingly, of the three codon-pairs in which there is no risk of mistranslation by the tRNA of the neighboring codon-pair (Cys-UGY, Ile-AUY and Tyr-UAY), only Cys demonstrates a small association with GC content. It therefore appears that the effect of GC content on the tRNA pool available for each codon is responsible for at least some of the observed bias.
Optimal stability of codon–anticodon interaction
Grosjean et al. (56–58) noticed that N1N2Y3 codon-pairs beginning with G or C in the first two positions were biased towards the N1N2U3 codon, while those beginning with A or U in the first two positions were biased towards the N1N2C3 codon. They noted that since A and U are weak (W) binders that form only two hydrogen bonds each, while C and G are strong (S) binders that can form three hydrogen bonds each, the bias yields codon–anticodon binding of intermediate strength, suggesting that too strong and too weak codon–anticodon binding is selected against, probably because it hampers translation efficiency. Grosjean’s optimal stability model was further supported by Gouy and Gautier (16), Sharp et al. (10) and Percudani and Ottonello (59). Our results regarding codon-pairs of the type WWY and SSY are in complete accordance with this theory, as all of the WWY pairs are of the 2-/3-fold C-Bias type and all the SSY pairs are of the 4-fold U-Bias type. Since the original optimal stability model does not address codon bias in SWY and WSY codon-pairs and yet they appear to be biased in the same direction as their fold counterparts, we hypothesized that there might be a more general stability model linking fold and bias for all N1N2Y3 codons.
In 1978, Lagerkvist noticed that the genetic code is arranged such that splitting of a codon quartet into more than one amino acid (or stop signal) happens only when the first two nucleotides common to the quartet (N1 and N2) are both weak or if one is weak and the other is strong and N2 is a purine (60). He suggested that in these cases the codon–anticodon interaction is weak and requires stabilization by base pairing in the third position through Watson–Crick or wobble interaction. As a result, at least one tRNA is needed to efficiently read each codon-pair (the G34N35N36 tRNA for the N1N2Y3 codons and the U34N35N36 tRNA for the N1N2R3 codons), and therefore two amino acids can share the quartet with little risk of codon mistranslation. In the other codon quartets, the first two nucleotides bind the anticodon with enough strength to compensate for a mismatch in the third position. In this case, since a single tRNA can read all four codons, the quartet must encode a single amino acid to avoid mistranslation. Lehmann and Libchaber (61) provided further support to this theory by showing, based on structural considerations, the effect of N2 on the stability of the interaction. A Y2 in the codon implies an R35 in the anticodon, which forms an anticodon-loop-stabilizing hydrogen-bond with the U33 tRNA nucleotide to allow efficient binding (62). They assigned each codon quartet a score based on the interaction of N1–N36 and N2–N35 where an A:U base pair contributes two hydrogen bonds, a C:G base pair contributes three hydrogen bonds, and a Y2 (reflecting an R35) contributes one hydrogen bond (Figure 4). We speculated that by combining their score with a measure of the stability of the N3–N34 base pair, we might arrive at a more accurate representation of codon–anticodon stability, which should provide a more general insight into codon usage bias. Hence, to the original score, we added the number of hydrogen bonds formed in the wobble position between the codon and G34N35N36 tRNA (the major tRNA responsible for translating N1N2Y3 codons). In the case of the Arg C1G2Y3 codon-pair, which is read in most organisms by the I34C35G36 tRNA, we added two hydrogen bonds to both codons. However, since the two hydrogen bonds stabilizing the I:C base pair are stronger than those stabilizing the I:U base pair (63), we considered the total score of the CGC codon as higher than the total score of CGU. The extended stability scores (ESS) are summarized in Table 2. Remarkably, in organisms under translational selection, codon bias follows the optimal stability rule. In the 2-/3-fold group where ESS is low (6 or 7 for N1N2U3 and 7 or 8 for N1N2C3) the ‘not too weak’ N1N2C3 is preferred, while in the 4-fold group where ESS is high (8 or 9 for N1N2U3 and 9 or 10 for N1N2C3), the ‘not too strong’ N1N2U3 is preferred (except for Leu). This notable choice of moderate stability appears to support the superiority of such an interaction over both weaker and stronger interactions.
Figure 4.

Hydrogen bonds stabilizing codon–anticodon interactions [an extension of the model suggested in (61)]. In black are the standard Watson–Crick interactions formed between N1:N36 and N2:N35, representing two or three hydrogen bonds each. In light gray is the loop-stabilizing bond between U33 and N35, which occurs only when N35 is a purine. In dark gray is the interaction formed between N3:N34, which represents two hydrogen bonds for U3:G34 and three hydrogen bonds for C3:G34.
Table 2.
Extended stability score of N1N2Y3 codon-pairs
| Amino acid | Codon pair | Fold | N1:N36 H-bonds | N2:N35 H-bonds | N2 Y/R | C3:G34 H-bonds | U3:G34 H-bonds | N1N2C3 Extended stability score | N1N2U3 Extended stability score |
|---|---|---|---|---|---|---|---|---|---|
| Asn | AAY | 2 | 2 | 2 | 0 | 3 | 2 | 7a | 6 |
| Tyr | UAY | 2 | 2 | 2 | 0 | 3 | 2 | 7a | 6 |
| Ser | AGY | 2 | 2 | 3 | 0 | 3 | 2 | 8a | 7 |
| Ile | AUY | 3 | 2 | 2 | 1 | 3 | 2 | 8a | 7 |
| His | CAY | 2 | 3 | 2 | 0 | 3 | 2 | 8a | 7 |
| Asp | GAY | 2 | 3 | 2 | 0 | 3 | 2 | 8a | 7 |
| Cys | UGY | 2 | 2 | 3 | 0 | 3 | 2 | 8a | 7 |
| Phe | UUY | 2 | 2 | 2 | 1 | 3 | 2 | 8a | 7 |
| Arg | CGYb | 4 | 3 | 3 | 0 | 2(+) | 2(−) | 8(+) | 8(−)a |
| Thr | ACY | 4 | 2 | 3 | 1 | 3 | 2 | 9 | 8a |
| Leu | CUY | 4 | 3 | 2 | 1 | 3 | 2 | 9 | 8 |
| Gly | GGY | 4 | 3 | 3 | 0 | 3 | 2 | 9 | 8a |
| Val | GUY | 4 | 3 | 2 | 1 | 3 | 2 | 9 | 8a |
| Ser | UCY | 4 | 2 | 3 | 1 | 3 | 2 | 9 | 8a |
| Pro | CCY | 4 | 3 | 3 | 1 | 3 | 2 | 10 | 9a |
| Ala | GCY | 4 | 3 | 3 | 1 | 3 | 2 | 10 | 9a |
aMarks the extended stability score of the preferred codon according to the observed fold rule.
bThe Arg-CGY codon-pair is read by the ACG (modified to ICG) tRNA. Since bonds formed within an I:C base pair are stronger than within an I:U pair, the relevant hydrogen bonds and the extended stability scores are marked with (+) and (−), respectively.
CONCLUDING REMARKS
It is widely accepted that in organisms undergoing translational selection the preferred codons used in highly expressed genes are those that are associated with more efficient translation and hence promote organism fitness. What determines the identity of preferred codons of an organism? Our study identifies a distinctive codon preference pattern in N1N2Y3 codon-pairs: preference of C at position N3 of 2-/3-fold codon-pairs and preference of U at position N3 of 4-fold codon-pairs. Of the factors examined in our study as possibly playing a role in this type of codon bias, the stability of codon–anticodon interaction was found to be associated with both C-Bias and U-Bias, while other codon properties were associated with only one type of bias (Table 3). Thus, the stability of the interaction in combination with other codon–anticodon properties seems to underlie the observed codon bias. To summarize, 2-/3-fold codon-pairs are biased towards the N1N2C3 codons that form a perfect Watson–Crick pairing with the available G34N35N36 tRNAs, create a moderately stable interaction with the tRNA instead of a weak one and reduce the risk of mistranslation. The preference of N1N2U3 codons in 4-fold codon-pairs is also supported by more than one explanation. First, since N1N2U3 codons exhibit moderate interaction with their G34N35N36 anticodons compared to N1N2C3 codons, they assure a not too sticky and presumably more efficient codon-anticodon interaction. Second, these codons can be read, although less efficiently, by U34N35N36 anticodons and therefore the ratio of U34/G34 tRNAs determines which codon has a larger tRNA pool and consequently affects the direction of bias. Since for most organisms and most codon-pairs this ratio is high, U-Bias is usually promoted. Interestingly, we have observed an influence of the GC content in organisms demonstrating translational selection in certain codon-pairs. We propose that the explanation may be linked in a non-trivial way to the tRNA repertoire that affects the tRNA pool in 4-fold codon-pairs and the chance of mistranslation in 2-fold codon-pairs. However, since this does not explain the C-Bias observed in the 2-/3-fold codon-pairs Cys-UGY, Ile-AUY and Tyr-UAY that are not at risk of mistranslation, it appears that essentially there may be two major forces affecting codon bias. One regards the frequency of the various possibilities of codon–anticodon interaction and the consequences of a correct or incorrect translation stemming from it, and the other regards the stability of this interaction and the way it influences translation efficiency. In order to better understand the very fundamental concept of translation selection that has implications on the evolution of all organisms, it would be necessary to perform carefully designed experiments that will differentiate between the contributions of the various forces to codon bias.
Table 3.
Summary of potential forces affecting codon bias in N1N2Y3 codon-pairs
| Amino acid | Codon pair | Preferred codona | Watson–Crickb | GCc | tRNA poold | Error minimizatione | Stabilityf | Extended stabilityg |
|---|---|---|---|---|---|---|---|---|
| Asn | AAY | C | + | − − | NR | + | + | + |
| Tyr | UAY | C | + | − − | NR | NR | + | + |
| Ser | AGY | C | + | − | NR | + | NR | + |
| Ile | AUY | C | + | − | NR | NR | + | + |
| His | CAY | C | + | − − | NR | + | NR | + |
| Asp | GAY | C | + | − − | NR | + | NR | + |
| Cys | UGY | C | + | + | NR | NR | NR | + |
| Phe | UUY | C | + | − | NR | + | + | + |
| Arg | CGY | U | NR | − | − | NR | + | + |
| Thr | ACY | U | − | ++ | + | NR | NR | + |
| Leu | CUY | C/U | NR | ++ | + | NR | NR | NR |
| Gly | GGY | U | − | − | ++ | NR | + | + |
| Val | GUY | U | − | + | + | NR | NR | + |
| Ser | UCY | U | − | ++ | ++ | NR | NR | + |
| Pro | CCY | U | − | + | ++ | NR | + | + |
| Ala | GCY | U | − | + | + | NR | + | + |
aCodon preference is determined by bias direction observed in organisms with high ENC’diff as seen in Supplementary Figure S2.
bAgreement with a prefect Watson–Crick base pairing. (+) The preferred codon forms a perfect Watson–Crick pairing with the available tRNA, (−) the preferred codon forms a wobble pairing with the tRNA, (NR) both codons form wobble pairing (due to A to I modification) or there is no preferred codon.
cGC effect on bias direction as seen in Supplementary Figure S5: (−) no effect, (− −) opposite effect, (+) weak effect, (++) strong effect.
dtRNA pool size effect on bias direction: (−) no effect, (+) weak effect, (++) strong effect, (NR) not relevant.
eIndication of error minimization: (+) observed, (NR) not relevant.
fAgreement with Grosjean’s stability model (56): (+) the preferred codon forms moderate interaction with the tRNA compared to its synonym, (NR) not relevant to SWY and WSY codon-pairs.
gAgreement with the Extended Stability model: (+) the preferred codon forms a moderate interaction with the tRNA compared to its synonym, (NR) there is no distinctly preferred codon.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1, Supplementary Figures 1–6, Supplementary Methods, Supplementary Dataset 1, and Supplementary References [64,65].
FUNDING
The Israel Science Foundation administered by the Israeli Academy for Sciences and Humanities [1171/09 awarded to H.M.] Ministry of Science and Technology Eshkol scholarship awarded (to N.W). Funding for open access charge: The Israel Science Foundation administered by the Israeli Academy for Sciences and Humanities [1171/09 awarded to H.M.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Norman Grover for his help in the statistical analysis, and Asaf peer, Yael Altuvia, Eric Westhof and Omri Wolf for their insightful comments.
REFERENCES
- 1.Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 2011;12:32–42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hershberg R, Petrov DA. Selection on codon bias. Annu. Rev. Genet. 2008;42:287–299. doi: 10.1146/annurev.genet.42.110807.091442. [DOI] [PubMed] [Google Scholar]
- 3.Xia X. Maximizing transcription efficiency causes codon usage bias. Genetics. 1996;144:1309–1320. doi: 10.1093/genetics/144.3.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wada A, Suyama A. Third letters in codons counterbalance the (G + C)-content of their first and second letters. FEBS Lett. 1985;188:291–294. [Google Scholar]
- 5.Rocha EP. Codon usage bias from tRNA's point of view: redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 2004;14:2279–2286. doi: 10.1101/gr.2896904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knight RD, Freeland SJ, Landweber LF. A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2001;2:RESEARCH0010. doi: 10.1186/gb-2001-2-4-research0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ermolaeva MD. Synonymous codon usage in bacteria. Curr. Issues Mol. Biol. 2001;3:91–97. [PubMed] [Google Scholar]
- 8.Palidwor GA, Perkins TJ, Xia X. A general model of codon bias due to GC mutational bias. PLoS One. 2010;5:e13431. doi: 10.1371/journal.pone.0013431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sueoka N. On the genetic basis of variation and heterogeneity of DNA base composition. Proc. Natl Acad. Sci. USA. 1962;48:582–592. doi: 10.1073/pnas.48.4.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharp PM, Tuohy TM, Mosurski KR. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986;14:5125–5143. doi: 10.1093/nar/14.13.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharp PM, Li WH. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’ codons. Nucleic Acids Res. 1986;14:7737–7749. doi: 10.1093/nar/14.19.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLachlan AD, Staden R, Boswell DR. A method for measuring the non-random bias of a codon usage table. Nucleic Acids Res. 1984;12:9567–9575. doi: 10.1093/nar/12.24.9567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Man O, Pilpel Y. Differential translation efficiency of orthologous genes is involved in phenotypic divergence of yeast species. Nat. Genet. 2007;39:415–421. doi: 10.1038/ng1967. [DOI] [PubMed] [Google Scholar]
- 14.Lithwick G, Margalit H. Hierarchy of sequence-dependent features associated with prokaryotic translation. Genome Res. 2003;13:2665–2673. doi: 10.1101/gr.1485203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holm L. Codon usage and gene expression. Nucleic Acids Res. 1986;14:3075–3087. doi: 10.1093/nar/14.7.3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gouy M, Gautier C. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 1982;10:7055–7074. doi: 10.1093/nar/10.22.7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennetzen JL, Hall BD. Codon selection in yeast. J. Biol. Chem. 1982;257:3026–3031. [PubMed] [Google Scholar]
- 18.Sorensen MA, Kurland CG, Pedersen S. Codon usage determines translation rate in Escherichia coli. J. Mol. Biol. 1989;207:365–377. doi: 10.1016/0022-2836(89)90260-x. [DOI] [PubMed] [Google Scholar]
- 19.Robinson M, Lilley R, Little S, Emtage JS, Yarranton G, Stephens P, Millican A, Eaton M, Humphreys G. Codon usage can affect efficiency of translation of genes in Escherichia coli. Nucleic Acids Res. 1984;12:6663–6671. doi: 10.1093/nar/12.17.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makoff AJ, Oxer MD, Romanos MA, Fairweather NF, Ballantine S. Expression of tetanus toxin fragment C in E. coli: high level expression by removing rare codons. Nucleic Acids Res. 1989;17:10191–10202. doi: 10.1093/nar/17.24.10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lavner Y, Kotlar D. Codon bias as a factor in regulating expression via translation rate in the human genome. Gene. 2005;345:127–138. doi: 10.1016/j.gene.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 22.Bulmer M. The selection-mutation-drift theory of synonymous codon usage. Genetics. 1991;129:897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varenne S, Buc J, Lloubes R, Lazdunski C. Translation is a non-uniform process. Effect of tRNA availability on the rate of elongation of nascent polypeptide chains. J. Mol. Biol. 1984;180:549–576. doi: 10.1016/0022-2836(84)90027-5. [DOI] [PubMed] [Google Scholar]
- 24.Ehrenberg M, Kurland CG. Costs of accuracy determined by a maximal growth rate constraint. Q. Rev. Biophys. 1984;17:45–82. doi: 10.1017/s0033583500005254. [DOI] [PubMed] [Google Scholar]
- 25.Precup J, Parker J. Missense misreading of asparagine codons as a function of codon identity and context. J. Biol. Chem. 1987;262:11351–11355. [PubMed] [Google Scholar]
- 26.Drummond DA, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nat. Rev. Genet. 2009;10:715–724. doi: 10.1038/nrg2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ibba M, Soll D. Quality control mechanisms during translation. Science. 1999;286:1893–1897. doi: 10.1126/science.286.5446.1893. [DOI] [PubMed] [Google Scholar]
- 28.Zaher HS, Green R. Quality control by the ribosome following peptide bond formation. Nature. 2009;457:161–166. doi: 10.1038/nature07582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Archetti M. Genetic robustness and selection at the protein level for synonymous codons. J. Evol. Biol. 2006;19:353–365. doi: 10.1111/j.1420-9101.2005.01029.x. [DOI] [PubMed] [Google Scholar]
- 30.Gilchrist MA, Shah P, Zaretzki R. Measuring and detecting molecular adaptation in codon usage against nonsense errors during protein translation. Genetics. 2009;183:1493–1505. doi: 10.1534/genetics.109.108209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.dos Reis M, Savva R, Wernisch L. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 2004;32:5036–5044. doi: 10.1093/nar/gkh834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J. Mol. Biol. 1981;151:389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 33.Ikemura T. Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. Differences in synonymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer RNAs. J. Mol. Biol. 1982;158:573–597. doi: 10.1016/0022-2836(82)90250-9. [DOI] [PubMed] [Google Scholar]
- 34.Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- 35.Fluitt A, Pienaar E, Viljoen H. Ribosome kinetics and aa-tRNA competition determine rate and fidelity of peptide synthesis. Comput. Biol. Chem. 2007;31:335–346. doi: 10.1016/j.compbiolchem.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanaya S, Yamada Y, Kudo Y, Ikemura T. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene. 1999;238:143–155. doi: 10.1016/s0378-1119(99)00225-5. [DOI] [PubMed] [Google Scholar]
- 38.Dong H, Nilsson L, Kurland CG. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 1996;260:649–663. doi: 10.1006/jmbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- 39.Berg OG, Kurland CG. Growth rate-optimised tRNA abundance and codon usage. J. Mol. Biol. 1997;270:544–550. doi: 10.1006/jmbi.1997.1142. [DOI] [PubMed] [Google Scholar]
- 40.Marck C, Grosjean H. tRNomics: analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features. RNA. 2002;8:1189–1232. doi: 10.1017/s1355838202022021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 42.Novembre JA. Accounting for background nucleotide composition when measuring codon usage bias. Mol. Biol. Evol. 2002;19:1390–1394. doi: 10.1093/oxfordjournals.molbev.a004201. [DOI] [PubMed] [Google Scholar]
- 43.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Higgs PG, Ran W. Coevolution of codon usage and tRNA genes leads to alternative stable states of biased codon usage. Mol. Biol. Evol. 2008;25:2279–2291. doi: 10.1093/molbev/msn173. [DOI] [PubMed] [Google Scholar]
- 45.Ran W, Higgs PG. The influence of anticodon-codon interactions and modified bases on codon usage bias in bacteria. Mol. Biol. Evol. 2010;27:2129–2140. doi: 10.1093/molbev/msq102. [DOI] [PubMed] [Google Scholar]
- 46.Vieira-Silva S, Rocha EP. The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet. 2010;6:e1000808. doi: 10.1371/journal.pgen.1000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharp PM, Emery LR, Zeng K. Forces that influence the evolution of codon bias. Phil. Trans. R. Soc. Lond. B Biol. Sci. 2010;365:1203–1212. doi: 10.1098/rstb.2009.0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jansen R, Gerstein M. Analysis of the yeast transcriptome with structural and functional categories: characterizing highly expressed proteins. Nucleic Acids Res. 2000;28:1481–1488. doi: 10.1093/nar/28.6.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cochella L, Green R. Wobble during decoding: more than third-position promiscuity. Nat. Struct. Mol. Biol. 2004;11:1160–1162. doi: 10.1038/nsmb1204-1160. [DOI] [PubMed] [Google Scholar]
- 50.Rogalski M, Karcher D, Bock R. Superwobbling facilitates translation with reduced tRNA sets. Nat. Struct. Mol. Biol. 2008;15:192–198. doi: 10.1038/nsmb.1370. [DOI] [PubMed] [Google Scholar]
- 51.Haig D, Hurst LD. A quantitative measure of error minimization in the genetic code. J. Mol. Evol. 1991;33:412–417. doi: 10.1007/BF02103132. [DOI] [PubMed] [Google Scholar]
- 52.Freeland SJ, Hurst LD. The genetic code is one in a million. J. Mol. Evol. 1998;47:238–248. doi: 10.1007/pl00006381. [DOI] [PubMed] [Google Scholar]
- 53.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hershberg R, Petrov DA. General rules for optimal codon choice. PLoS Genet. 2009;5:e1000556. doi: 10.1371/journal.pgen.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hildebrand F, Meyer A, Eyre-Walker A. Evidence of selection upon genomic GC-content in bacteria. PLoS Genet. 2010;6:e1001107. doi: 10.1371/journal.pgen.1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grosjean H, Sankoff D, Jou WM, Fiers W, Cedergren RJ. Bacteriophage MS2 RNA: a correlation between the stability of the codon: anticodon interaction and the choice of code words. J. Mol. Evol. 1978;12:113–119. doi: 10.1007/BF01733262. [DOI] [PubMed] [Google Scholar]
- 57.Fiers W, Grosjean H. On codon usage. Nature. 1979;277:328. doi: 10.1038/277328a0. [DOI] [PubMed] [Google Scholar]
- 58.Grosjean H, Fiers W. Preferential codon usage in prokaryotic genes: the optimal codon-anticodon interaction energy and the selective codon usage in efficiently expressed genes. Gene. 1982;18:199–209. doi: 10.1016/0378-1119(82)90157-3. [DOI] [PubMed] [Google Scholar]
- 59.Percudani R, Ottonello S. Selection at the wobble position of codons read by the same tRNA in Saccharomyces cerevisiae. Mol. Biol. Evol. 1999;16:1752–1762. doi: 10.1093/oxfordjournals.molbev.a026087. [DOI] [PubMed] [Google Scholar]
- 60.Lagerkvist U. “Two out of three”: an alternative method for codon reading. Proc. Natl Acad. Sci. USA. 1978;75:1759–1762. doi: 10.1073/pnas.75.4.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lehmann J, Libchaber A. Degeneracy of the genetic code and stability of the base pair at the second position of the anticodon. RNA. 2008;14:1264–1269. doi: 10.1261/rna.1029808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.von Ahsen U, Green R, Schroeder R, Noller HF. Identification of 2'-hydroxyl groups required for interaction of a tRNA anticodon stem-loop region with the ribosome. RNA. 1997;3:49–56. [PMC free article] [PubMed] [Google Scholar]
- 63.Janke EM, Riechert-Krause F, Weisz K. Low-temperature NMR studies on inosine wobble base pairs. J. Phys. Chem. B. 2011;115:8569–8574. doi: 10.1021/jp200840j. [DOI] [PubMed] [Google Scholar]
- 64.Letunic I, Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- 65.Letunic I, Bork P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39:W475–W478. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.