


Abstract
Bis-(3′–5′)-cyclic dimeric guanosine monophosphate (c-di-GMP) modulates the transition between planktonic and biofilm life styles. In response to c-di-GMP, the enhancer binding protein FleQ from Pseudomonas aeruginosa derepresses the expression of Pel exopolysaccharide genes required for biofilm formation when a second protein, FleN is present. A model is that binding of c-di-GMP to FleQ induces its dissociation from the pelA promoter allowing RNA polymerase to access this site. To test this, we analyzed pelA DNA footprinting patterns with various combinations of FleQ, FleN and c-di-GMP, coupled to in vivo promoter activities. FleQ binds to two sites called box 1 and 2. FleN binds to FleQ bound at these sites causing the intervening DNA to bend. Binding of c-di-GMP to FleQ relieves the DNA distortion but FleQ remains bound to the two sites. Analysis of wild type and mutated versions of pelA-lacZ transcriptional fusions suggests that FleQ represses gene expression from box 2 and activates gene expression in response to c-di-GMP from box 1. The role of c-di-GMP is thus to convert FleQ from a repressor to an activator. The mechanism of action of FleQ is distinct from that of other bacterial transcription factors that both activate and repress gene expression from a single promoter.
INTRODUCTION
Bis-(3′–5′)-cyclic dimeric guanosine monophosphate (c-di-GMP) is an intracellular secondary messenger that modulates the transition between planktonic and biofilm lifestyles in many gram-negative bacteria (1–8). In addition, c-di-GMP modulates virulence in some bacteria (9,10). High intracellular c-di-GMP is often associated with the production of biofilms and the formation of adhesive organelles such as pili or stalks, whereas reduced intracellular c-di-GMP is often correlated with increased motility and virulence. C-di-GMP is synthesized by diguanylate cyclases (DGC) with GGDEF motifs, degraded by phosphodiesterases (PDE) with EAL or HD-GYP motifs and bound by a wide variety of effectors (11–15). Proteins containing PilZ domains (16–19), or harboring degenerate GGDEF or EAL domains (20,21) bind c-di-GMP. C-di-GMP is also a riboswitch ligand (22). The binding of c-di-GMP to these effectors impacts at different cellular and molecular levels including enzymatic activity, protein/protein interaction, transcription and translation. The mechanisms involved in c-di-GMP control at each of these levels are just coming into focus.
Several transcriptional regulators have been described that respond to c-di-GMP (23–26). One of these, FleQ from the opportunistic pathogen Pseudomonas aeruginosa (27), binds c-di-GMP to regulate the expression of genes involved in biofilm formation. These include the pel and psl operons, which direct the synthesis of Pel and Psl exopolysaccharides, and the cdrA gene coding for an adhesin essential for Psl exopolysaccharide structure (27,28). FleQ represses the expression of these genes in the absence of c-di-GMP whereas in the presence of c-di-GMP the repression is relieved (27). FleQ is also the master regulator of flagella gene expression in P. aeruginosa (29,30). It activates the expression of the flhA and fliLMNOPQ genes involved in flagellar export, the flhF-fleN genes involved in regulation and localization of the flagellar apparatus, the fleSR genes encoding a two-component system that regulates other flagella genes and the fliEFG operon encoding structural components of the flagellar basal body and motor switch complex (30,31). Transcriptome analysis showed that expression of most of the FleQ-regulated flagellar genes is downregulated when intracellular c-di-GMP is high (27,32). However, the effect of c-di-GMP on flagella gene expression is modest and clear evidence that the effect observed is directly mediated by FleQ is missing.
FleQ is an enhancer binding protein (EBP), belonging to the NtrC family of bacterial transcription factors. It contains an N-terminal FleQ domain, a central AAA+/ATPase σ54-interaction domain and a C-terminal helix-turn-helix DNA-binding domain. EBPs typically bind >100 nt upstream of the transcription start site of a target promoter and then interact with a σ54-RNA polymerase complex by a DNA looping mechanism. RNA polymerase open complex formation is driven by ATP hydrolysis catalyzed by the EBP. A specific motif, GG and GC, located at positions −24 and −12 relative to the transcription start site, is recognized by the σ54-RNA polymerase complex. Whereas, expression of all FleQ-regulated operons involved in flagella biosynthesis depends on σ54 (30,31), the expression of FleQ regulated genes involved in biofilm formation does not, suggesting a different mechanism of regulation (30). FleQ works in concert with another protein, FleN, which is a putative ATPase with a deviant Walker A motif (33). Mutation of fleN leads to an upregulation of FleQ-dependant flagella genes (27) and fleN mutants are multiflagellate (34). FleN is required for full expression of pel, psl and cdr operons in the presence of high levels of c-di-GMP (27). FleN alone does not bind to DNA but FleQ and FleN interact and a FleQ/FleN/DNA complex has been observed to form at flhA and pel promoters in gel shift assays (27,35).
Hickman and Harwood (27) suggested based on gel shift data, that FleQ represses pel gene expression by a simple roadblock mechanism in which FleQ sits at the pelA promoter and prevents RNA polymerase from binding. They further suggested that the binding of c-di-GMP to FleQ induces dissociation of FleQ from the pelA promoter allowing the RNA polymerase to access DNA and form an active transcription complex. To test this hypothesis and to further probe the mechanism FleQ and FleN action in response to c-di-GMP, we analyzed pelA promoter DNA footprinting patterns with various combinations of FleQ, FleN and c-di-GMP, coupled to in vivo promoter activities. Our results show that the pelA promoter contains two FleQ binding sites on either side of its transcription start site. In addition, we provide evidence that FleQ is not only a repressor of pel gene expression in the absence of c-di-GMP but also an activator of pel expression in the presence of c-di-GMP. It appears that the two FleQ binding sites at the pel promoter are independently dedicated to activation and repression.
MATERIALS AND METHODS
Strains, media and compounds
Pseudomonas aeruginosa and E. coli strains were routinely grown on LB medium at 37°C. Antibiotics were added at the following concentrations: 60 μg/ml gentamycin or tetracycline for P. aeruginosa and 10 μg/ml gentamycin or tetracycline for E. coli. C-di-GMP and ATPγS were purchased from BIOLOG Life Science Institute (Bremen, Germany). Pseudomonas aeruginosa strains PAO1ΔwspF, PAO1ΔfleQ, PAO1Δpel, PAO1ΔpelΔpsl, PAO1ΔpelΔpslΔfleN, PAO1ΔpelΔpslΔfleQ and PAO1ΔpelΔpslΔfleQΔfleN were constructed using standard protocols for allelic exchange in P. aeruginosa. Deletions of pel, psl and wspF were constructed as previously described (1,32,36). To construct the chromosomal deletion of fleQ and fleN, DNA flanking the fleQ or the fleN genes was PCR-amplified and cloned into pEX19Gm (37). The resulting plasmid was used to transform E. coli S17-1, and was then mobilized into different P. aeruginosa strains. Transconjugants were selected on M63 media with succinate (10 mM) and containing gentamycin, followed by recovery of deletion mutants on LB plates containing 5% sucrose. Candidate deletion mutants were screened by PCR. Plasmids pJN1120 and pJN105 were electroporated into strains PAO1ΔpelΔpsl, PAO1ΔpelΔpslΔfleQ PAO1ΔpelΔpslΔfleN and PAO1ΔpelΔpslΔfleQΔfleN by a standard electroporation method (27,38,39).
DNase I footprinting
DNase I footprint analysis was performed using a non-radiochemical capillary electrophoresis method (40). Two DNA fragments containing the pelA promoter were used in this study. The fragments were generated by PCR using two 6-FAM (6-carboxyfluorescein phosphoramidate) primers: pelA-R1 ACTCGATGGGTCGAAGGATGTTAC, which paired to the RNA 49- to 73-nt downstream from the pelA translation start site or pelA-R2, TCGGAAGCATGGATGCGGTTTC, which paired to the RNA 43- to 65-nt upstream from the pelA translation start site and the primer pelA-F TTCCTCGCACGCAACTGAAG, which paired 479- to 499-nt upstream from the pelA translation start site, leading to a PCR products labeled with FAM of 572 and 456 bases, respectively. The template for the PCR reaction was PAO1 chromosomal DNA except when different mutations of the pel promoter were studied; in these cases the templates were the plasmids mini-CTX-lacZ-pelAmut1, 2, 3 or 4 and the primer pelA-R2 was necessarily used. The footprint assays were performed as follows: FleQ and/or FleN proteins were incubated with ATP (10 µM) or c-di-GMP (100 µM or 1 mM) and binding buffer [10 mM Tris-HCl, pH 7.8, 50 mM KCl, 8 mM Mg acetate, 50 ng/µl bovine serum albumin (BSA) and 5% glycerol] for 10 min before adding the labeled PCR fragment (0.45 pmol) and incubating for 30 min. Reaction mixtures were treated for 2 min with DNase I (0.3 units, Promega). The samples were phenol extracted and ethanol precipitated. Fragments were analyzed with an Applied Biosystems 3730×l Genetic Analyzer (Genewiz, Inc or Genomic Resources at Fred Hutchinson Cancer Research Center). DNA fragment sizes were determined using ABI Peak Scanner software.
RNA extraction and primer extension
Pseudomonas aeruginosa strains were grown in LB until the culture reached an OD600 of 0.5–0.7. Cells were then harvested and total RNA was extracted and purified by using an RNeasy Mini kit (Qiagen). Reverse transcription was performed using SuperScript III reverse transcriptase (Invitrogen). Briefly, 0.4 nM of 6-FAM primers pelA-R1 or pelA-R2 (see the sequences above or Figure 1), were incubated with 15 µg of total RNA for 20 min at 58°C and then incubated at room temperature for 10 min, the SuperScript III reagents for cDNA synthesis were then added as specified by the manufacturer and the reaction mixtures were incubated for 1 h at 55°C. Synthesized cDNA was purified with a Qiagen PCR purification kit, concentrated by ethanol precipitation and then analyzed as described above. Primer extension analysis was performed at least two times with two different primers to confirm the transcription start site.
Figure 1.

Nucleotide sequence of the pel promoter region. The two FleQ binding sites are boxed and the repeated sequences are in bold. The identity of various promoter mutations is shown above the sequence. The transcription start site is indicated by a thick arrow and the putative −10 and −35 elements are underlined. The translation start site ATG is in bold. The asterisk indicates the location of the hypersensitive site. The two thin arrows indicate the two primers pelA-R1 and pelA-R2 used for primer extension and DNAse I footprinting experiments.
Protein purification and limited proteolysis
FleQ and FleN proteins were purified from E. coli strain ER2566 carrying pFleQ-Int1 or pFleN-Int1, as previously described (27). The limited proteolysis experiments were done on 8 µg of each purified protein with three different conditions: FleQ plus FleN, FleQ plus BSA and FleN plus BSA. Oligonucleotides (50 µM) corresponding to both strands of the FleQ binding sites (positions −61 to +20 relative to the transcription start site) were mixed together in 10 mM Tris–HCl (pH 7.3), 1 mM EDTA and 185 mM NaCl, and annealed by incubation at 95°C for 5 min followed by chilling on ice. When indicated, DNA fragments equimolar to FleQ were added. In a final volume of 45 µl, 16 µg of protein was incubated with or without ATP (10 µM) or c-di-GMP (1 mM) for 10 min before adding trypsin at a protein/enzyme mass ratio of 10/1 for FleQ digestion (1.6 µg of trypsin) or 4/1 for FleN digestion (4 µg of trypsin). The reactions were stopped by removing 11 µl from the proteolysis mix at 2, 10, 30 and 60 min intervals and then plunging each sample into a cold tubes containing 1.5 µl of 100 mM PMSF (phenylmethylsulfonyl fluoride), a protease inhibitor. SDS loading buffer was added and the samples were subsequently incubated for 10 min at 95°C. The samples were then analyzed on SDS–PAGE followed by transfer to Hybond™-ECL™ nitrocellulose membrane and detected with FleQ or FleN antibodies. Each experiment was repeated at least three times and gave the same profile of proteolysis.
Construction of chromosomal pelA-lacZ fusions and measurement of promoter activities in vivo
Chromosomal promoter-lacZ reporter fusions were constructed using the mini-CTX-lacZ vector (41). A 537-bp PCR product, encompassing the region between 29 bases downstream and 508 bases upstream of the translational start site of the pelA gene, was generated using primers pPelfullfor (5′-CGGCGAATTCCTGGTGCGGTTCCTCGCACGCAAC-3′) and pPelfullrev (5′-GATCGGATCCACGGCGATTCCTTTCTTGCTG-3′). The fragment was then cloned into a mini-CTX-lacZ vector (Y. Irie and M. Parsek, unpublished data). Mutations in the pelA promoter (Figure 1) were generated by an overlapping PCR procedure with two sets of divergent and complementary primers, each containing the expected substitutions and the convergent primers pPelfullfor and pPelfullrev. Primer sequences are available upon request. The fragments were cloned into the mini-CTX-lacZ vector. The resulting plasmids (mini-CTX-lacZ-pelAwt, mut1, 2, 3 or 4), bearing the wild type or mutated pelA promoters, were used to transform E. coli S17-1, and then introduced at the attB site of the P. aeruginosa chromosome by mating. Transconjugants were selected on M63 medium with succinate and containing tetracycline. Strains were grown overnight aerobically in LB at 37°C and β-galactosidase activities of whole cells were measured by the method of Miller (42). Results are the average of three independent experiments.
RESULTS
Identification of the pelA promoter region
The pel operon encoding pelABCDEFG genes directs the synthesis of an exopolysaccharide important for biofilm formation by P. aeruginosa (43). To determine the pelA transcription start site we isolated RNA from strains PAO1ΔfleQ, PAO1ΔwspF and PAO1Δpel and did primer extension analysis. Based on previous work, we expected to see pel transcripts in strain PAO1ΔfleQ, because in such a background pel genes are expressed at high levels (27). Similarly, a wspF deletion causes the DGC WspR to be activated, resulting in c-di-GMP accumulation, which induces pel expression (1,27). Strain PAO1Δpel was used as negative control. The pel deletion is 3.2 kb in size. It starts 355 bases upstream of the translation start site of pelA and encompasses most of pelA (32). One specific pelA transcript was detected with RNA isolated from strains PAO1ΔfleQ and PAO1ΔwspF but not detected with RNA isolated from strain PAO1Δpel (Figure 2). Other fragments were observed in all three conditions tested, and thus were not specific to the pelA transcript. The 6-FAM specific pel fragment identified with primer pelA-R1 (Figure 1) was 207 bp long, allowing us to map the pelA start site to a location 134-bp upstream of the ATG start codon (Figure 1). The same experiment performed with another primer, pelA-R2 (Figure 1), confirmed the location of the transcription start site. A putative −10 sequence (TATTTA), but no typical −24/−12 σ54-RNA polymerase binding site was found upstream of the transcription start site (Figure 1). This observation is in good agreement with previous data showing that pel operon expression does not depend on σ54 (30). A putative −35 region (TTAAAA) close to the consensus (TTGACA) of σ70 promoters was found in the pel promoter region but the spacing between the −10 and −35 sequences is 15 bases instead of the conventional spacer of 17 bases.
Figure 2.
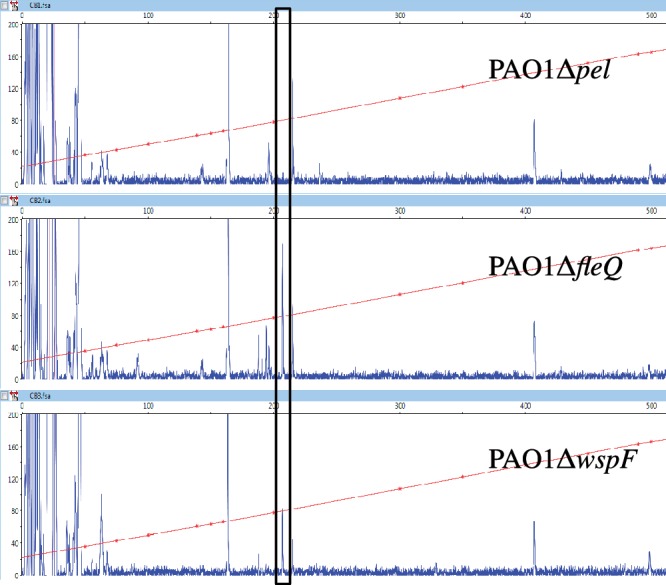
Identification of the transcription start site of pelA. Primer extension analysis of the pelA transcript was performed using a 6-FAM labeled primer (pelA-R1), which hybridizes to the RNA 49–73 bp downstream from the translation start site of pelA. Total RNA were isolated from PAO1Δpel, PAO1ΔfleQ and PAO1ΔwspF strains. The fluorescence intensity of FAM-labeled cDNA fragments (ordinate) is plotted against the sequence length of the fragment (abscissa). The horizontal line indicates the GS-500 internal size standard. The major transcript detected in strains PAO1ΔfleQ and PAO1ΔwspF but not detected in strain PAO1Δpel is boxed.
Identification of two FleQ binding sites on pel DNA
Binding sites for FleQ on the flagella regulon flhA, fliE, fliL and fleSR promoters have been reported, but no sequence conservation among these binding sites was observed (31). To determine the location of FleQ binding on the pelA promoter in vitro, DNaseI footprinting experiments were performed with a 5′FAM labeled probe of 572 bases corresponding to positions +207 to −365 relative to the transcription start site of the pelA gene. As the concentration of FleQ was increased, two protected regions appeared (Figure 3). Whatever the concentration of FleQ, both regions seemed to be occupied simultaneously, without apparent hierarchy between the two. The use of a fragment of 456 bases corresponding to position +91 to −365, relative to the transcription start site of the pelA gene, confirmed these results. The two protected regions are 19 bases long and separated by 34 bases. They are centered at position −48 (FleQ box 1) and +6 (FleQ box 2) relative to the transcription start site (Figure 1). It is noteworthy that FleQ box 2 overlaps the pelA transcription start site. Since FleQ has a AAA+ ATPase domain, we also tested the effect of ATP on FleQ binding to the pelA promoter and observed similar footprint patterns plus or minus ATP (data not shown). This result, together with previous gel shift assays results (27), confirms that the binding of FleQ to the pel promoter is independent of ATP.
Figure 3.

The pel promoter contains two FleQ boxes. 5′-FAM-labeled pel DNA (0.45 pmol) was incubated with or without FleQ (0.14, 0.28 or 0.56 μM, as indicated) and in the presence of 10 μM of ATP, then submitted to DNase I digestion (0.3 U) and analyzed by capillary electrophoresis. The fluorescence intensity of FAM-labeled DNA fragments (ordinate), in arbitrary units, is plotted against the sequence length of the fragment (abscissa), in bases, relative to the first base of the pelA-R1 primer. The whole electrophoregram is shown. The horizontal line indicates the GS-500 internal size standard. The two boxes labeled 1 and 2 indicate the two regions protected by FleQ.
Each FleQ box contains the sequence ATTGAC (Figure 1). To determine, in vitro, the functional role of the two FleQ boxes and the repeated sequences, we generated several mutated versions of the pelA promoter by substituting four bases inside the two FleQ boxes, and inside or outside the repeated sequence (Figure 1). DNase I footprinting experiments were performed on these variant DNA templates. Mutations, either outside (mut1) or inside (mut2) the ATTGAC motif in FleQ box 1, abolished the binding of FleQ to the FleQ box 1, but did not affect the binding of FleQ to FleQ box 2 (Figure 4). A mutation in the ATTGAC motif in FleQ box 2 (mut3) abolished the binding of FleQ to it but not to FleQ box 1. Mutation outside the repeated sequence of FleQ box 2 (mut4) did not abolish FleQ binding to either box. This result shows that the binding of FleQ to one binding site is independent of protein occupancy at the other binding site. The 19 bases of the FleQ boxes are not equally involved in the binding of FleQ but an intact repeated sequence seems to be required for the binding of FleQ to the pelA promoter.
Figure 4.

Effects of mutations in the pelA promoter on the binding of FleQ. 5′-FAM-labeled pel DNA (0.45 pmol) carrying different nucleotide substitutions as indicated in Figure 1 was incubated in the presence of ATP (10 μM), with or without FleQ (0.28 μM) and in the presence or absence of c-di-GMP (100 μM), as indicated. The sequence length of the fragment, in bases, is relative to the first base of the pelA-R2 primer. Part of the electrophoregram is shown. The two boxes labeled 1 and 2 indicate the two regions of FleQ binding, the dashed line of the box indicates which box is mutated.
FleN stimulates bending of pelA promoter DNA when FleQ and ATP or ADP are present
Previous gel shift data showed that FleN in conjunction with FleQ retarded the migration of a pelA promoter probe fragment more than did FleQ alone and we also know that FleN and FleQ interact with each other (27,35). In in vitro footprinting experiments with FleN and FleQ, we found that FleQ protected the same two regions on the pelA promoter, but in addition FleN induced the appearance of a DNase I hypersensitive site (Figure 5). The hypersensitive site was observed when ATP, ADP (data not shown) or ATPγS, a non-hydrolysable analogue of ATP, were included in the footprinting reaction mixtures. However, no hypersensitive site was observed in the presence of AMP or in the absence of any nucleotides. The DNase I hypersensitive site is located between the two FleQ binding boxes, centered at −20 relative to the pelA transcription start site (Figure 1). Neither the protected regions nor hypersensitive sites were detected when DNase I footprinting experiments were carried out with FleN alone. DNase I hypersensitive sites are often indicative of flexible DNA regions, able to bend or to loop. FleN likely causes the pelA promoter to distort in the presence of FleQ and this distortion requires ATP but not its hydrolysis. DNase I footprinting experiments were also performed with FleQ and FleN on the four variant pel promoters. The hypersensitive site was observed when mut4 was the template but not when mut1, 2 or 3 were the templates (data not shown). This suggests that the DNA bending mediated by FleN occurs only when FleQ is bound to both FleQ boxes.
Figure 5.
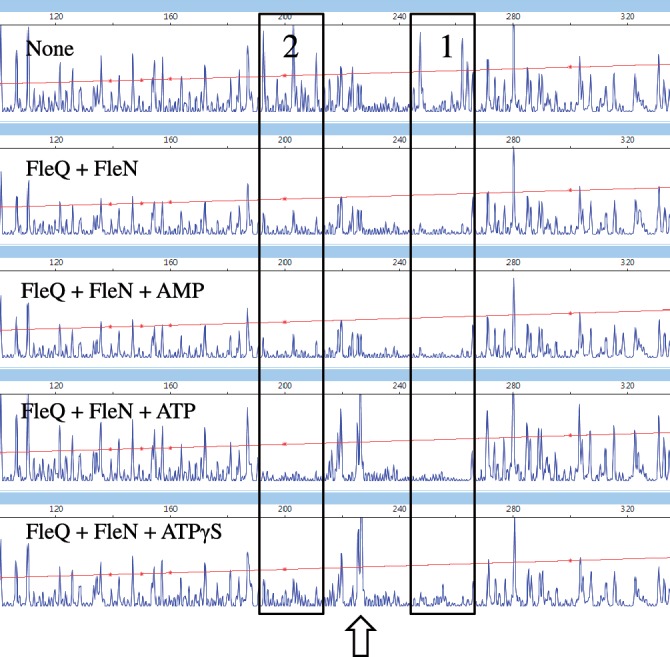
FleN causes the pel promoter to bend in conjunction with FleQ and ATP or ATPγS. Analysis of the 5′-FAM-labeled pel DNA (0.45 pmol) incubated with FleQ alone or with FleN at equimolar concentrations (0.28 μM) and in the presence and absence of ATP (10 μM), AMP (10 μM) or ATPγS (10 μM). The sequence length of the fragment (abscissa), in bases, is relative to the first base of the pelA-R1 primer. The two FleQ binding sites are boxed and the hypersensitive site is indicated by an arrow.
FleQ stays bound to the pelA promoter and the DNA bending mediated by FleN is relieved when c-di-GMP is added
It was previously suggested that the binding of c-di-GMP to FleQ induces a dissociation of FleQ from the pelA promoter (27). To test this hypothesis, we footprinted the pelA promoter in the presence of c-di-GMP. When FleQ was incubated with c-di-GMP prior to DNase I treatment, the two FleQ protected regions on the pelA promoter persisted on the electropherogram, indicating that FleQ remains bound to the pelA promoter (Figure 6). In addition, c-di-GMP did not influence the footprint patterns of FleQ on the mutated variant pelA promoters (Figure 4). When FleN, FleQ and c-di-GMP were incubated together prior to DNase I treatment, the two FleQ protected regions on the pelA promoter again persisted, but no hypersensitive site was detected (Figure 6). We therefore conclude that in the presence of c-di-GMP, FleQ remains on the pelA promoter but the bending mediated by FleN is relieved. It is noticeable that the footprint of FleQ on FleQ box 2 is slightly less pronounced in the presence of c-di-GMP, regardless of whether FleN is present. This may reflect a change in the affinity of FleQ for FleQ box 2 in the presence of c-di-GMP.
Figure 6.

Effect of c-di-GMP on FleQ and FleN binding to the pel promoter. Analysis of the 5′-FAM-labeled pel DNA (0.45 pmol) incubated with FleQ alone or with FleN at equimolar concentrations (0.28 μM), in the presence of ATP (10 μM) and in the presence or absence of c-di-GMP (100 μM). The sequence length of the fragment (abscissa), in bases, is relative to the first base of the pelA-R1 primer. The two FleQ binding sites are boxed and the hypersensitive site is indicated by an arrow.
Previous gel shift assay results showed that when FleQ was present, FleN induced a supershift of a pelA promoter fragment and this shift was then abrogated in the presence of c-di-GMP. This was interpreted to suggest that c-di-GMP binding to FleQ caused it to dissociate from the pelA promoter (27). An alternative explanation that is consistent with the footprinting data is that the supershift reflects bending of DNA induced by FleQ and FleN, and c-di-GMP alleviates this bending to abolish the supershift. Another possible explanation is that the gel shift assays measure complexes stable enough to be trapped upon migration on a gel; however, footprinting experiments identify even weakly bound protein/DNA complexes too unstable to be retained on a gel.
Analysis of FleQ and FleN interaction in the presence of ATP or c-di-GMP
The DNA bending stimulated by FleN occurs only when ATP is present and disappears when c-di-GMP is added (Figure 6). It could be that ATP is required for FleQ and FleN to interact and that c-di-GMP stimulates the dissociation of FleQ from FleN. Alternatively, conformational changes in FleQ or FleN due to the binding of ATP or c-di-GMP could be required to trigger DNA bending/unbending. To test these hypotheses, the interaction between FleQ and FleN was analyzed by limited proteolysis. Purified FleQ and FleN, together or separately (with BSA as control), were incubated with trypsin for different intervals of time in the presence or absence of ATP or c-di-GMP. The digestion products were then separated by SDS–PAGE, transferred to a membrane and revealed with FleQ or FleN antibodies. A fragment of 20 kDa was detected when FleQ was subjected to trypsin digestion for 30 min (Figure 7A and B, left panel). When c-di-GMP was also present in the incubation mixture, the 20 kDa FleQ fragment was not present, but a new proteolysis-resistant fragment of 30 kDa appeared (Figure 7A and B, right panel). This suggests that FleQ undergoes a conformational change when it binds c-di-GMP. In contrast, FleQ does not undergo an obvious conformational change upon ATP binding and ATP does not affect the conformational change of FleQ due to c-di-GMP binding. Inclusion of c-di-GMP did not alter the digestion profile of FleN (Figure 7D). When FleQ and FleN were incubated together with trypsin, FleQ is more resistant to proteolysis as exemplified by the persistence of the band corresponding to the intact FleQ protein (55 kDa) after 10 min of incubation (Figure 7C, all panels), whereas without FleN, the same band had disappeared by this time (Figure 7A). The same digestion profile of FleQ was observed with or without ATP (Figure 7C), indicating that FleQ and FleN interact irrespective of the presence of ATP. If c-di-GMP induces the dissociation of a FleQ/FleN complex, then the digestion profile of FleQ in the presence of FleN and c-di-GMP should be equivalent to the digestion profile of FleQ alone in the presence of c-di-GMP. This was not the case. Our results show that in the presence of c-di-GMP, the FleQ protein was still protected from trypsin digestion by FleN (Figure 7C). This result was confirmed when FleQ and FleN digests were revealed with FleN antibodies (compare Figure 7D and E). In order to get closer to in vivo conditions, we tested the effect of pel promoter DNA on FleQ and FleN interaction. FleN, plus or minus FleQ and c-di-GMP, was incubated with double stranded DNA encompassing the two FleQ binding sites prior to trypsin addition. The FleQ and FleN proteins interacted similarly with or without the DNA present (Figure 7F). After 60 min of incubation with trypsin, the FleN protein was almost entirely digested in the absence of FleQ whereas with FleQ, FleN was more resistant to proteolysis (Figure 7F).
Figure 7.

Effects of c-di-GMP and ATP on the kinetics of trypsin digestion of FleQ alone or FleQ and FleN. FleQ and BSA (A and B), FleN and BSA (D) or FleQ and FleN (C, E and F) were incubated in the presence or absence of c-di-GMP (1 mM), ATP (100 μM) or a DNA fragment encompassing the two FleQ binding sites (3.2 μM), as indicated and then subjected to proteolysis with trypsin. Times of incubation are indicated above the gel. Digestion products were separated by SDS–PAGE, transferred to a nitrocellulose membrane and detected with anti-FleQ antibodies (A, B and C) or with anti-FleN antibodies (D, E and F).
These results indicate that FleQ and FleN form a complex in the presence as well as in the absence of c-di-GMP or ATP. However, the trypsin digestion profile of FleQ in the presence of c-di-GMP and FleN (Figure 7C, right panel) was different from the digestion profile obtained in the absence of c-di-GMP (Figure 7C, middle panel), suggesting that a conformational change of a FleQ/FleN complex occurs in the presence of c-di-GMP. The distortion of DNA seen in footprints that depends on the presence of c-di-GMP or ATP is more likely to represent conformational changes related to their binding to FleQ and/or FleN rather than to effects related to the relief of interactions between the two proteins.
FleQ functions as a transcriptional repressor in the absence of c-di-GMP and as an activator when c-di-GMP is present
To study the in vivo participation of the two FleQ boxes in regulating pelA promoter activity, we constructed transcriptional fusions of lacZ to promoter fragments carrying mutations in the two FleQ boxes and introduced each of them at the attB site of strains PAO1ΔpelΔpsl (referred to here as the WT strain), PAO1ΔpelΔpslΔfleQ (referred to here as the fleQ strain), PAO1ΔpelΔpslΔfleN (referred as the fleN strain) and PAO1ΔpelΔpslΔfleQΔfleN (referred as the fleQfleN strain). The level of intracellular c-di-GMP was manipulated in these strains by expressing the DGC PA1120 from plasmid pJN1120 (27,44). The empty vector pJN105 was used as a control. The wild-type strain expressing the wild-type pelA promoter (PpelA-wt) had low β-galactosidase activity. As expected β-galactosidase activity was much higher in the strains deleted of fleQ or with a high intracellular level of c-di-GMP (Figure 8). These results confirm the previous report of FleQ-mediated repression of the pel genes at low intracellular c-di-GMP and derepression when c-di-GMP levels are high (27).
Figure 8.

FleQ acts as a repressor in the absence of c-di-GMP and as an activator in the presence of c-di-GMP. β-Galactosidase activities of wild type or mutated PpelA-lacZ fusions, as indicated on the left of the diagram, in PAO1ΔpelΔpsl (WT), PAO1ΔpelΔpslΔfleQ (fleQ), PAO1ΔpelΔpslΔfleN (fleN) or PAO1ΔpelΔpslΔfleQΔfleN (fleQfleN) strains carrying pJN105 (vector control) or pJN1120 (allowing the overexpression of the DGC encoded by PA1120). The white boxes indicate the FleQ boxes, the grey boxes, the repeated sequences observed in the FleQ boxes. Mutations in either boxes are indicated by an X and the transcription start site is indicated by an arrow. The β-galactosidase values are indicated on the right and expressed as means ± standard deviation.
In the wild-type strain carrying the vector control plasmid, the promoter-fusions carrying mutations inside FleQ box 1 (PpelA-mut1 and PpelA-mut2) had low, wild-type levels of β-galactosidase (Figure 8). Thus, in the absence of c-di-GMP, repression of pel genes does not require an intact FleQ box 1. The wild-type strain with the promoter carrying mutations outside of the repeated sequence of FleQ box 2 (PpelA-mut4) also had low β-galactosidase activity when c-di-GMP was low. In contrast, mutations in the repeated sequence of FleQ box 2 (PpelA-mut3) caused an increase of β-galactosidase activity (Figure 8). From the footprinting experiments, we know that a mutation inside the repeated sequence of FleQ box 2 (mut3) impairs the binding of FleQ to FleQ box 2, whereas a mutation in the FleQ box 2 but outside of the repeated sequence (mut4) does not (Figure 4). We can, therefore, conclude that the binding of FleQ to FleQ box 2 of the pel promoter is a necessary requirement for FleQ-mediated repression. When intracellular c-di-GMP was high (strains carrying pJN1120), an intact FleQ box 1 was required for PpelA-lacZ activation. Moreover, the activity of PpelA-mut3 was increased by nearly a factor two compared to the activity of PpelA-wt in high c-di-GMP cells (Figure 8). This increased activity depended on the presence of FleQ. From this we conclude that the role of c-di-GMP is not simply to derepress pelA expression but rather to switch FleQ from a repressor to an activator.
Previous results using RT-PCR showed that FleN was required for full expression of pel genes when c-di-GMP is high (27). The data presented in Figure 8 indicate that FleN participates in the FleQ-mediated activation of pel genes in response to c-di-GMP, but the effects are not as pronounced as previously reported. For an unknown reason, the expression of the PpelA-mut4 fusion did not seem to be affected by the deletion of fleN.
DISCUSSION
This work suggests that FleQ controls gene expression by a novel mechanism that involves binding to two sites in the promoter of the operon it controls. At one site (box 2), FleQ functions to repress gene expression, and at the other site (box 1), FleQ functions to activate gene expression in response to c-di-GMP. A revised model for pel regulation by FleQ can be proposed based on the data presented here (Figure 9). In the absence of c-di-GMP, FleQ binds to its two FleQ binding sites on the pel promoter (Figure 9A). FleN is found bound to FleQ (Figure 9B). In the presence of ATP, we propose that FleN forms dimers inducing a bending of the pelA promoter by bridging the bound FleQ proteins (Figure 9C). We think that this conformation could either impair the binding of the RNA polymerase or prevent RNA polymerase bound to the pelA promoter from forming an open complex, leading to pel repression. In the presence of c-di-GMP, FleQ undergoes a conformational change that probably induces a cascade of conformational changes in the FleQ/FleN/DNA complex such that the bending is relaxed. FleQ is switched to an activator (Figure 9D). We propose that the relief of the bending may either induce RNA polymerase binding or remodel RNA polymerase binding and the FleQ bound to FleQ box 1 favors transcription initiation. This model is speculative and still incomplete, but it is consistent with the ability of FleQ to mediate both repression and activation in response to c-di-GMP while bound at the same sites on DNA. The intracellular concentration of c-di-GMP in P. aeruginosa is subject to both temporal variations depending on the activity of DGCs and PDEs responding to different signals, to spatial variations due to specific subcellular localization of some DGCs (45,46) and the partitioning of c-di-GMP during cell division (47). Thus, having the necessary macromolecular components pre-assembled at the promoter could be a biological advantage in allowing cells to respond rapidly to c-di-GMP concentration changes to fine-tune gene expression.
Figure 9.

Model of pel regulation. FleQ binds to two FleQ boxes on the pel promoter (A). FleQ interacts with FleN in the absence of ATP (B) as well as in the presence of ATP, but in this case it induces a distortion of pel DNA (C). We propose that FleN forms a bridge between two FleQ bound to their binding sites. The binding of FleQ to FleQ box 2 is essential for repression. The binding of c-di-GMP to FleQ induces a conformational change of FleQ, probably propagated through FleN, which induces the relief of pel distortion and leads to pel expression (D). The binding of FleQ to FleQ box 1 is essential for activation.
Only a few bacterial transcription factors have been shown to both activate and repress gene expression from a single promoter and these have mechanisms of action that are distinct from that of FleQ. The well-described MerR protein both activates and represses expression of mer genes, coding for mercury resistance (48–50). MerR shares with FleQ that the binding of their effectors, mercury or c-di-GMP, switches the regulators from a repressor to an activator without changing the location or the occupancy of the binding sites. However, while MerR directs repression and activation from a single binding site by remodeling the spacing between the −10 and −35 region in response to mercury, FleQ has separate binding sites for each function and likely uses a different mechanism to stimulate RNA polymerase. Another example is the TyrR protein from E. coli. In the presence of tyrosine, TyrR, an EBP, forms hexamers and binds to adjacent high affinity and low affinity sites, leading to tyrP repression (51–54). In the presence of phenylalanine, TyrR forms dimers that bind only to the high affinity site, allowing interaction with the RNA polymerase, and activation of gene expression (52–55). Finally, LldR, a Gnt family member from E. coli, both activates and represses expression of the lldPRD operon, involved in lactate metabolism in response to lactate (56). Binding of LldR to two sites located on either side of the lldPRD transcription start site is required to repress transcription. Binding of a LldR/lactate complex to the site upstream of the transcription start site is required to activate gene expression (56). FleQ behaves somewhat differently in that it needs to bind to only one of its two binding sites (FleQ box 2) to repress pel expression (Figure 8). Also full activation of pel expression in response to c-di-GMP requires a second protein, FleN.
FleQ is an EBP that has the additional unusual property of regulating gene expression independently of RNA polymerase σ54 in some circumstances. Expression of biofilm-related genes, such as pel genes, occurs normally in a σ54 (rpoN) mutant (30). Also the pelA promoter encompasses putative −10 and −35 elements, suggesting that transcription is mediated by a σ70-RNA polymerase complex (Figure 1). Transcriptional control by an EBP in combination with σ70-RNA is uncommon but not unprecedented. TyrR from E. coli (52), PhhR from Pseudomonas putida (57), as well as NtrC (58) or HupR (59) from Rhodobacter capsulatus are such proteins. σ54 does mediate FleQ-dependant activation of flagella gene expression (30). Moreover, the σ54 binding domain and the catalytic ATP site of FleQ are perfectly well conserved whereas the abovementioned EBP-like proteins that work with σ70-RNA polymerase either lack or have a poorly conserved σ54 binding domain and/or catalytic site (51,60). FleQ is apparently a versatile protein able to function with two different kinds of RNA polymerase holoenzyme. At this point, we are uncertain as to how c-di-GMP may influence FleQ mediated regulation of flagella genes. Based on microarray data, flagella gene expression is down regulated by 1.5- to 2-fold in response to high c-di-GMP in P. aeruginosa. We have been unable to obtain reproducible DNaseI footprints of FleQ at one flagella promoter (fleSR) that we tested and thus have not evaluated effects of c-di-GMP on this interaction.
Recent studies analyzed four other transcriptional regulators that respond to c-di-GMP (23–26). In Xanthomonas campestris, c-di-GMP acts as a negative regulator of Clp, impairing the binding of Clp to DNA and the induction of genes important for virulence (25,61,62). In Vibrio cholerae, c-di-GMP induces the oligomerization of VpsT necessary for its binding to vpsL, the first gene of a Vibrio polysaccharide operon. C-di-GMP enhances the binding of MrkH from Klebsiella pneumoniae and Bcam1349 from Burkholderia cenocepacia to the promoter region of type 3 fimbriae and cellulose synthase genes, respectively (23,26). VpsT belongs to the LuxR family of regulators, whereas Clp and Bcam1349 are homologous to Crp (24,25,27). MrkH contains a PilZ domain but does not contain an obvious DNA binding domain (26). C-di-GMP binds to the W[F/L/M][T/S]R sequence of VpsT (24) and to the conserved cyclic nucleotide monophosphate binding domain of Clp (25,61,62). The c-di-GMP binding site of FleQ remains unknown but we have determined that its N-terminal domain is not involved in the detection of c-di-GMP (27). Alignments of a variety of EBPs and FleQ homologues revealed that a region of about 30 amino acids at the C-terminal margin of the AAA+-ATPase domain is conserved in FleQ homologues. A deletion of 20 amino acids that we made in this region abolished c-di-GMP binding but point mutations in that region did not (Baraquet and Harwood, unpublished data). Thus we cannot distinguish whether we removed the c-di-GMP binding site or whether the deletion caused a conformational change in FleQ that impaired the binding of c-di-GMP elsewhere in the protein.
A large number of activators and repressors induce distortion or bending of DNA upon binding (63), and the binding of effectors to such regulators can modulate DNA bending (64–70). Modulation of DNA bending by transcription factors can influence transcription by impairing or improving RNA polymerase binding. The region of the pel promoter located between the two FleQ boxes is distorted when FleQ, FleN and ATP are all present (Figure 5). Our data suggest that FleN, a non-DNA binding protein, induces the distortion probably by interacting with the DNA-binding protein FleQ. FleN is a putative ATPase with a deviant Walker A motif (33), belonging to a functionally diverse group including MinD and ParA (71), which have the common feature of undergoing ATP dependent dimerization (72,73). We hypothesize that FleN forms dimers upon ATP binding and then interacts with FleQ bound to DNA to form a bridge. In fact we observe bending only when FleQ can bind to its two FleQ binding sites simultaneously.
While DNA bending mediated by FleN appears prerequisite for pel expression the precise role of FleN is still unclear. FleN could simply have a structural role and the oligomerization of FleN upon ATP binding may be crucial to induce distortion of DNA via FleQ. It may also be that FleQ, FleN, ATP and DNA form a complex ready to sense c-di-GMP more efficiently than FleQ alone. Another hypothesis could be that FleN has a regulatory role, detecting ATP as an additional signal for pel expression. Indeed, continuous pel expression is not necessary for biofilm maintenance but for growing larger biofilms (74). This result implies that consumption of metabolic energy is required and ATP could signal the energetic status of cells.
FUNDING
National Institute of General Medical Sciences [GM56665 to C.S.H.]; National Institute of Allergy and Infectious Disease [AI077628 to M.R.P.]; National Science Foundation [MCB0822405 to M.R.P.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
Authors thank Ruben Ramphal for the generous gift of FleQ and FleN antisera and Yasuhiko Irie for providing them the mini-CTX-lacZ-pelAwt plasmid.
REFERENCES
- 1.Hickman JW, Tifrea DF, Harwood CS. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci. USA. 2005;102:14422–14427. doi: 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simm R, Morr M, Kader A, Nimtz M, Romling U. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol. Microbiol. 2004;53:1123–1134. doi: 10.1111/j.1365-2958.2004.04206.x. [DOI] [PubMed] [Google Scholar]
- 3.Tischler AD, Camilli A. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol. Microbiol. 2004;53:857–869. doi: 10.1111/j.1365-2958.2004.04155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolfe AJ, Visick KL. Get the message out: cyclic-Di-GMP regulates multiple levels of flagellum-based motility. J. Bacteriol. 2008;190:463–475. doi: 10.1128/JB.01418-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thormann KM, Duttler S, Saville RM, Hyodo M, Shukla S, Hayakawa Y, Spormann AM. Control of formation and cellular detachment from Shewanella oneidensis MR-1 biofilms by cyclic di-GMP. J. Bacteriol. 2006;188:2681–2691. doi: 10.1128/JB.188.7.2681-2691.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross P, Weinhouse H, Aloni Y, Michaeli D, Weinberger-Ohana P, Mayer R, Braun S, de Vroom E, van der Marel GA, van Boom JH, et al. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature. 1987;325:279–281. doi: 10.1038/325279a0. [DOI] [PubMed] [Google Scholar]
- 7.Lim B, Beyhan S, Meir J, Yildiz FH. Cyclic-diGMP signal transduction systems in Vibrio cholerae: modulation of rugosity and biofilm formation. Mol. Microbiol. 2006;60:331–348. doi: 10.1111/j.1365-2958.2006.05106.x. [DOI] [PubMed] [Google Scholar]
- 8.Aldridge P, Paul R, Goymer P, Rainey P, Jenal U. Role of the GGDEF regulator PleD in polar development of Caulobacter crescentus. Mol. Microbiol. 2003;47:1695–1708. doi: 10.1046/j.1365-2958.2003.03401.x. [DOI] [PubMed] [Google Scholar]
- 9.Cotter PA, Stibitz S. c-di-GMP-mediated regulation of virulence and biofilm formation. Curr. Opin. Microbiol. 2007;10:17–23. doi: 10.1016/j.mib.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Tamayo R, Pratt JT, Camilli A. Roles of cyclic diguanylate in the regulation of bacterial pathogenesis. Annu. Rev. Microbiol. 2007;61:131–148. doi: 10.1146/annurev.micro.61.080706.093426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hengge R. Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 2009;7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- 12.Jenal U, Malone J. Mechanisms of cyclic-di-GMP signaling in bacteria. Ann. Rev. Genet. 2006;40:385–407. doi: 10.1146/annurev.genet.40.110405.090423. [DOI] [PubMed] [Google Scholar]
- 13.Mills E, Pultz IS, Kulasekara HD, Miller SI. The bacterial second messenger c-di-GMP: mechanisms of signalling. Cell Microbiol. 2011;13:1122–1129. doi: 10.1111/j.1462-5822.2011.01619.x. [DOI] [PubMed] [Google Scholar]
- 14.Schirmer T, Jenal U. Structural and mechanistic determinants of c-di-GMP signalling. Nat. Rev. Microbiol. 2009;7:724–735. doi: 10.1038/nrmicro2203. [DOI] [PubMed] [Google Scholar]
- 15.Yan H, Chen W. 3′,5′-Cyclic diguanylic acid: a small nucleotide that makes big impacts. Chem. Soc. Rev. 2010;39:2914–2924. doi: 10.1039/b914942m. [DOI] [PubMed] [Google Scholar]
- 16.Amikam D, Galperin MY. PilZ domain is part of the bacterial c-di-GMP binding protein. Bioinformatics. 2006;22:3–6. doi: 10.1093/bioinformatics/bti739. [DOI] [PubMed] [Google Scholar]
- 17.Paul K, Nieto V, Carlquist WC, Blair DF, Harshey RM. The c-di-GMP binding protein YcgR controls flagellar motor direction and speed to affect chemotaxis by a “backstop brake” mechanism. Mol. Cell. 2010;38:128–139. doi: 10.1016/j.molcel.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryjenkov DA, Simm R, Romling U, Gomelsky M. The PilZ domain is a receptor for the second messenger c-di-GMP: the PilZ domain protein YcgR controls motility in enterobacteria. J. Biol. Chem. 2006;281:30310–30314. doi: 10.1074/jbc.C600179200. [DOI] [PubMed] [Google Scholar]
- 19.Merighi M, Lee VT, Hyodo M, Hayakawa Y, Lory S. The second messenger bis-(3′-5′)-cyclic-GMP and its PilZ domain-containing receptor Alg44 are required for alginate biosynthesis in Pseudomonas aeruginosa. Mol. Microbiol. 2007;65:876–895. doi: 10.1111/j.1365-2958.2007.05817.x. [DOI] [PubMed] [Google Scholar]
- 20.Lee VT, Matewish JM, Kessler JL, Hyodo M, Hayakawa Y, Lory S. A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol. Microbiol. 2007;65:1474–1484. doi: 10.1111/j.1365-2958.2007.05879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newell PD, Monds RD, O’Toole GA. LapD is a bis-(3′,5′)-cyclic dimeric GMP-binding protein that regulates surface attachment by Pseudomonas fluorescens Pf0-1. Proc. Natl. Acad. Sci. USA. 2009;106:3461–3466. doi: 10.1073/pnas.0808933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sudarsan N, Lee ER, Weinberg Z, Moy RH, Kim JN, Link KH, Breaker RR. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science. 2008;321:411–413. doi: 10.1126/science.1159519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fazli M, O’Connell A, Nilsson M, Niehaus K, Dow JM, Givskov M, Ryan RP, Tolker-Nielsen T. The CRP/FNR family protein Bcam1349 is a c-di-GMP effector that regulates biofilm formation in the respiratory pathogen Burkholderia cenocepacia. Mol. Microbiol. 2011;82:327–341. doi: 10.1111/j.1365-2958.2011.07814.x. [DOI] [PubMed] [Google Scholar]
- 24.Krasteva PV, Fong JC, Shikuma NJ, Beyhan S, Navarro MV, Yildiz FH, Sondermann H. Vibrio cholerae VpsT regulates matrix production and motility by directly sensing cyclic di-GMP. Science. 2010;327:866–868. doi: 10.1126/science.1181185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tao F, He YW, Wu DH, Swarup S, Zhang LH. The cyclic nucleotide monophosphate domain of Xanthomonas campestris global regulator Clp defines a new class of cyclic di-GMP effectors. J. Bacteriol. 2010;192:1020–1029. doi: 10.1128/JB.01253-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilksch JJ, Yang J, Clements A, Gabbe JL, Short KR, Cao H, Cavaliere R, James CE, Whitchurch CB, Schembri MA, et al. MrkH, a novel c-di-GMP-dependent transcriptional activator, controls Klebsiella pneumoniae biofilm formation by regulating type 3 fimbriae expression. PLoS Pathog. 2011;7:e1002204. doi: 10.1371/journal.ppat.1002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hickman JW, Harwood CS. Identification of FleQ from Pseudomonas aeruginosa as a c-di-GMP-responsive transcription factor. Mol. Microbiol. 2008;69:376–389. doi: 10.1111/j.1365-2958.2008.06281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borlee BR, Goldman AD, Murakami K, Samudrala R, Wozniak DJ, Parsek MR. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol. Microbiol. 2010;75:827–842. doi: 10.1111/j.1365-2958.2009.06991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arora SK, Ritchings BW, Almira EC, Lory S, Ramphal R. A transcriptional activator, FleQ, regulates mucin adhesion and flagellar gene expression in Pseudomonas aeruginosa in a cascade manner. J. Bacteriol. 1997;179:5574–5581. doi: 10.1128/jb.179.17.5574-5581.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dasgupta N, Wolfgang MC, Goodman AL, Arora SK, Jyot J, Lory S, Ramphal R. A four-tiered transcriptional regulatory circuit controls flagellar biogenesis in Pseudomonas aeruginosa. Mol. Microbiol. 2003;50:809–824. doi: 10.1046/j.1365-2958.2003.03740.x. [DOI] [PubMed] [Google Scholar]
- 31.Jyot J, Dasgupta N, Ramphal R. FleQ, the major flagellar gene regulator in Pseudomonas aeruginosa, binds to enhancer sites located either upstream or atypically downstream of the RpoN binding site. J. Bacteriol. 2002;184:5251–5260. doi: 10.1128/JB.184.19.5251-5260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Starkey M, Hickman JH, Ma L, Zhang N, De Long S, Hinz A, Palacios S, Manoil C, Kirisits MJ, Starner TD, et al. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J. Bacteriol. 2009;191:3492–3503. doi: 10.1128/JB.00119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koonin EV. A superfamily of ATPases with diverse functions containing either classical or deviant ATP-binding motif. J. Mol. Biol. 1993;229:1165–1174. doi: 10.1006/jmbi.1993.1115. [DOI] [PubMed] [Google Scholar]
- 34.Dasgupta N, Arora SK, Ramphal R. fleN, a gene that regulates flagellar number in Pseudomonas aeruginosa. J. Bacteriol. 2000;182:357–364. doi: 10.1128/jb.182.2.357-364.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dasgupta N, Ramphal R. Interaction of the antiactivator FleN with the transcriptional activator FleQ regulates flagellar number in Pseudomonas aeruginosa. J. Bacteriol. 2001;183:6636–6644. doi: 10.1128/JB.183.22.6636-6644.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirisits MJ, Prost L, Starkey M, Parsek MR. Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol. 2005;71:4809–4821. doi: 10.1128/AEM.71.8.4809-4821.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998;212:77–86. doi: 10.1016/s0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 38.Choi KH, Kumar A, Schweizer HP. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods. 2006;64:391–397. doi: 10.1016/j.mimet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 39.Newman JR, Fuqua C. Broad-host-range expression vectors that carry the L-arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene. 1999;227:197–203. doi: 10.1016/s0378-1119(98)00601-5. [DOI] [PubMed] [Google Scholar]
- 40.Wilson DO, Johnson P, McCord BR. Nonradiochemical DNase I footprinting by capillary electrophoresis. Electrophoresis. 2001;22:1979–1986. doi: 10.1002/1522-2683(200106)22:10<1979::AID-ELPS1979>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 41.Becher A, Schweizer HP. Integration-proficient Pseudomonas aeruginosa vectors for isolation of single-copy chromosomal lacZ and lux gene fusions. BioTechniques. 2000;29:948–950, 952. doi: 10.2144/00295bm04. [DOI] [PubMed] [Google Scholar]
- 42.Miller J. Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1972. [Google Scholar]
- 43.Friedman L, Kolter R. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. J. Bacteriol. 2004;186:4457–4465. doi: 10.1128/JB.186.14.4457-4465.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kulasakara H, Lee V, Brencic A, Liberati N, Urbach J, Miyata S, Lee DG, Neely AN, Hyodo M, Hayakawa Y, et al. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. Proc. Natl. Acad. Sci. USA. 2006;103:2839–2844. doi: 10.1073/pnas.0511090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guvener ZT, Harwood CS. Subcellular location characteristics of the Pseudomonas aeruginosa GGDEF protein, WspR, indicate that it produces cyclic-di-GMP in response to growth on surfaces. Mol. Microbiol. 2007;66:1459–1473. doi: 10.1111/j.1365-2958.2007.06008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Merritt JH, Ha DG, Cowles KN, Lu W, Morales DK, Rabinowitz J, Gitai Z, O′Toole GA. Specific control of Pseudomonas aeruginosa surface-associated behaviors by two c-di-GMP diguanylate cyclases. MBio. 2010;1:pii:e00183–10. doi: 10.1128/mBio.00183-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Christen M, Kulasekara HD, Christen B, Kulasekara BR, Hoffman LR, Miller SI. Asymmetrical distribution of the second messenger c-di-GMP upon bacterial cell division. Science. 2010;328:1295–1297. doi: 10.1126/science.1188658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown NL, Stoyanov JV, Kidd SP, Hobman JL. The MerR family of transcriptional regulators. FEMS Microbiol. Rev. 2003;27:145–163. doi: 10.1016/S0168-6445(03)00051-2. [DOI] [PubMed] [Google Scholar]
- 49.Lund PA, Brown NL. Regulation of transcription in Escherichia coli from the mer and merR promoters in the transposon Tn501. J. Mol. Biol. 1989;205:343–353. doi: 10.1016/0022-2836(89)90345-8. [DOI] [PubMed] [Google Scholar]
- 50.Lund PA, Ford SJ, Brown NL. Transcriptional regulation of the mercury-resistance genes of transposon Tn501. J. Gen. Microbiol. 1986;132:465–480. doi: 10.1099/00221287-132-2-465. [DOI] [PubMed] [Google Scholar]
- 51.Dixon MP, Pau RN, Howlett GJ, Dunstan DE, Sawyer WH, Davidson BE. The central domain of Escherichia coli TyrR is responsible for hexamerization associated with tyrosine-mediated repression of gene expression. J. Biol. Chem. 2002;277:23186–23192. doi: 10.1074/jbc.M112184200. [DOI] [PubMed] [Google Scholar]
- 52.Pittard AJ, Davidson BE. TyrR protein of Escherichia coli and its role as repressor and activator. Mol. Microbiol. 1991;5:1585–1592. doi: 10.1111/j.1365-2958.1991.tb01904.x. [DOI] [PubMed] [Google Scholar]
- 53.Yang J, Hwang JS, Camakaris H, Irawaty W, Ishihama A, Pittard J. Mode of action of the TyrR protein: repression and activation of the tyrP promoter of Escherichia coli. Mol. Microbiol. 2004;52:243–256. doi: 10.1111/j.1365-2958.2003.03965.x. [DOI] [PubMed] [Google Scholar]
- 54.Pittard J, Camakaris H, Yang J. The TyrR regulon. Mol. Microbiol. 2005;55:16–26. doi: 10.1111/j.1365-2958.2004.04385.x. [DOI] [PubMed] [Google Scholar]
- 55.Andrews AE, Lawley B, Pittard AJ. Mutational analysis of repression and activation of the tyrP gene in Escherichia coli. J. Bacteriol. 1991;173:5068–5078. doi: 10.1128/jb.173.16.5068-5078.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aguilera L, Campos E, Gimenez R, Badia J, Aguilar J, Baldoma L. Dual role of LldR in regulation of the lldPRD operon, involved in l-lactate metabolism in Escherichia coli. J. Bacteriol. 2008;190:2997–3005. doi: 10.1128/JB.02013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Herrera MC, Ramos JL. Catabolism of phenylalanine by Pseudomonas putida: the NtrC-family PhhR regulator binds to two sites upstream from the phhA gene and stimulates transcription with sigma70. J. Mol. Biol. 2007;366:1374–1386. doi: 10.1016/j.jmb.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 58.Bowman WC, Kranz RG. A bacterial ATP-dependent, enhancer binding protein that activates the housekeeping RNA polymerase. Genes. Dev. 1998;12:1884–1893. doi: 10.1101/gad.12.12.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dischert W, Vignais PM, Colbeau A. The synthesis of Rhodobacter capsulatus HupSL hydrogenase is regulated by the two-component HupT/HupR system. Mol. Microbiol. 1999;34:995–1006. doi: 10.1046/j.1365-2958.1999.01660.x. [DOI] [PubMed] [Google Scholar]
- 60.Davies KM, Skamnaki V, Johnson LN, Venien-Bryan C. Structural and functional studies of the response regulator HupR. J. Mol. Biol. 2006;359:276–288. doi: 10.1016/j.jmb.2006.02.072. [DOI] [PubMed] [Google Scholar]
- 61.Chin KH, Lee YC, Tu ZL, Chen CH, Tseng YH, Yang JM, Ryan RP, McCarthy Y, Dow JM, Wang AH, et al. The cAMP receptor-like protein CLP is a novel c-di-GMP receptor linking cell-cell signaling to virulence gene expression in Xanthomonas campestris. J. Mol. Biol. 2010;396:646–662. doi: 10.1016/j.jmb.2009.11.076. [DOI] [PubMed] [Google Scholar]
- 62.Leduc JL, Roberts GP. Cyclic di-GMP allosterically inhibits the CRP-like protein (Clp) of Xanthomonas axonopodis pv. citri. J. Bacteriol. 2009;191:7121–7122. doi: 10.1128/JB.00845-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perez-Martin J, de Lorenzo V. Clues and consequences of DNA bending in transcription. Annu. Rev. Microbiol. 1997;51:593–628. doi: 10.1146/annurev.micro.51.1.593. [DOI] [PubMed] [Google Scholar]
- 64.Ansari AZ, Bradner JE, O′Halloran TV. DNA-bend modulation in a repressor-to-activator switching mechanism. Nature. 1995;374:371–375. doi: 10.1038/374370a0. [DOI] [PubMed] [Google Scholar]
- 65.Ansari AZ, Chael ML, O′Halloran TV. Allosteric underwinding of DNA is a critical step in positive control of transcription by Hg-MerR. Nature. 1992;355:87–89. doi: 10.1038/355087a0. [DOI] [PubMed] [Google Scholar]
- 66.Chen XC, Feng J, Hou BH, Li FQ, Li Q, Hong GF. Modulating DNA bending affects NodD-mediated transcriptional control in Rhizobium leguminosarum. Nucleic Acids Res. 2005;33:2540–2548. doi: 10.1093/nar/gki537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guazzaroni ME, Krell T, Gutierrez del Arroyo P, Velez M, Jimenez M, Rivas G, Ramos JL. The transcriptional repressor TtgV recognizes a complex operator as a tetramer and induces convex DNA bending. J. Mol. Biol. 2007;369:927–939. doi: 10.1016/j.jmb.2007.04.022. [DOI] [PubMed] [Google Scholar]
- 68.Maddocks SE, Oyston PC. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology. 2008;154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 69.O’Halloran TV, Frantz B, Shin MK, Ralston DM, Wright JG. The MerR heavy metal receptor mediates positive activation in a topologically novel transcription complex. Cell. 1989;56:119–129. doi: 10.1016/0092-8674(89)90990-2. [DOI] [PubMed] [Google Scholar]
- 70.Parsek MR, Kivisaar M, Chakrabarty AM. Differential DNA bending introduced by the Pseudomonas putida LysR-type regulator, CatR, at the plasmid-borne pheBA and chromosomal catBC promoters. Mol. Microbiol. 1995;15:819–828. doi: 10.1111/j.1365-2958.1995.tb02352.x. [DOI] [PubMed] [Google Scholar]
- 71.Lutkenhaus J, Sundaramoorthy M. MinD and role of the deviant Walker A motif, dimerization and membrane binding in oscillation. Mol. Microbiol. 2003;48:295–303. doi: 10.1046/j.1365-2958.2003.03427.x. [DOI] [PubMed] [Google Scholar]
- 72.Hu Z, Lutkenhaus J. A conserved sequence at the C-terminus of MinD is required for binding to the membrane and targeting MinC to the septum. Mol. Microbiol. 2003;47:345–355. doi: 10.1046/j.1365-2958.2003.03321.x. [DOI] [PubMed] [Google Scholar]
- 73.Leonard TA, Butler PJ, Lowe J. Bacterial chromosome segregation: structure and DNA binding of the Soj dimer—a conserved biological switch. EMBO J. 2005;24:270–282. doi: 10.1038/sj.emboj.7600530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Colvin KM, Gordon VD, Murakami K, Borlee BR, Wozniak DJ, Wong GC, Parsek MR. The Pel polysaccharide can serve a structural and protective role in the biofilm matrix of Pseudomonas aeruginosa. PLoS Pathog. 2011;7:e1001264. doi: 10.1371/journal.ppat.1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]