


Abstract
We present a set of four parameters that in combination can predict DNA-binding residues on protein structures to a high degree of accuracy. These are the number of evolutionary conserved residues (Ncons) and their spatial clustering (ρe), hydrogen bond donor capability (Dp) and residue propensity (Rp). We first used these parameters to characterize 130 interfaces in a set of 126 DNA-binding proteins (DBPs). The applicability of these parameters both individually and in combination, to distinguish the true binding region from the rest of the protein surface was then analyzed. Rp shows the best performance identifying the true interface with the top rank in 83% cases. Importantly, we also used the unbound-bound test cases of the protein–DNA docking benchmark to test the efficacy of our method. When applied to the unbound form of the DBPs, Rp can distinguish 86% cases. Finally, we have applied the SVM approach for recognizing the interface region using the above parameters along with the individual amino acid composition as attributes. The accuracy of prediction is 90.5% for the bound structures and 93.6% for the unbound form of the proteins.
INTRODUCTION
Protein–DNA interactions are vital for gene expression and control. The growing number of protein–DNA complexes deposited in the Protein Data Bank (PDB) (1) has enabled systematic studies on characterization of the DNA-binding region that is crucial for recognition (2–6). Extensive analyses have been carried out on DNA-binding proteins (DBPs) in terms of amino acid composition (7), packing density of binding residues and B-factor (8), evolutionary conservation of amino acid residues and base-pairs constituting the interface regions, as well as evolutionary profiles of surface patches (4,9–12). Interactions are not only studied at specific amino acid—base level (13), but have also been extended to atom–atom non-covalent interactions from the corresponding protein and DNA components; van der Waals contacts are found to constitute two-thirds of all protein–DNA interactions (14). Electrostatic potential has been employed to characterize and predict protein–DNA binding region (15,16). All these observations suggest that the amino acids at the interface possess characteristics that distinguish them from residues elsewhere on protein surface. Using the concept of hotspots, Ahmad et al. (4) showed that a potential relationship exists among the free energy of binding, sequence conservation and structural cooperativity of conserved residues in protein–DNA recognition. They coupled parameters derived from the thermodynamics of binding together with measures of evolutionary conservation in their analysis and prediction.
Polar interactions have been shown to play a major role at the interface of protein–DNA complexes and thus contribute significantly to the binding. Water mediated hydrogen bonds constitute 15% of all protein–DNA interactions (14), almost at the same level as direct hydrogen bonds. Of all the interfacial water molecules, ∼6% bridge protein and DNA and 76% form hydrogen bond with either component, thereby solvating and stabilizing the protein and DNA separately (17). Owing to their large presence it has been believed that water molecules play a significant role in protein–DNA interaction contributing to the binding affinity, but its role in binding specificity is largely unknown (18–20).
Apart from the features mentioned above even nonspecific DNA–protein interaction modes exhibit some similarity to specific DNA–protein-binding modes, and this feature has also been implemented in prediction (21). Position specific scoring matrices (PSSM) have been employed for detecting DNA-binding residues from primary sequence (22) and in structures (12). Amongst a pool of DBPs and non-binding proteins, many groups tried to predict the DBPs as a whole and not just their binding regions (16,23,24), using mostly electrostatic potential and knowledge based energy functions. A server called PreDs predicts whether a protein is a DBP or not and additionally highlights its binding site as well (25). This method also exploited the electrostatic potential in addition to local and global curvatures at the protein surface. At present, there are many databases providing structural data of protein–nucleic acid complexes, base amino acid interactions, thermodynamic and conformational parameters (26,27). There have also been studies on some specific protein–DNA interactions, such as transcription factor–transcription binding sites (TF–TFBSs), leading to generalized advanced rules capturing features of biological variations in TF and TFBS sequence patterns (28).
Predicting the DNA-binding region, given the 3D structure of a protein, remains a challenging task. The differential characteristics at the binding region may suffice for the prediction of interaction sites from sequence as well as from the coordinates of the 3D structure of a protein; several algorithms have been implemented along this line over the years (8,12,29–33). In this work we have identified a number of important differential features residing at the interface in relation to the rest of the protein surface based on simple properties, such as conservation, clustering, residue propensity and probable hydrogen bond donors using a large dataset of 130 protein–DNA complexes. We have applied these properties both individually and in combination (using SVM—Support Vector Machines) to predict the binding sites in the bound as well as the unbound forms of the structures of DBPs.
MATERIALS AND METHODS
Dataset
Atomic coordinates of the protein–DNA complexes were obtained from the PDB (1). Out of the 126 protein–DNA complexes used in Biswas et al. (6), four PDB files (1k6o, 1jb7, 1t2k and 1k78) consisted of two different protein monomers interacting with DNA in spatially distinct regions—these were split into two separate protein–DNA complexes, but involving the same DNA, creating a dataset of 130 complexes. For homodimeric proteins (62 in number), only one subunit along with the associated DNA was used. For each of the protein–DNA complex, the interface residues were identified. Atoms/residues from both partners that lose >0.1 Å2 of surface area upon complexation constitute the protein interface (34). Accessibilities were calculated using the program NACCESS (35), which employs the Lee and Richards algorithm (36).
Definition of interface/patch parameters
Sequence conservation
Evolutionary sequence conservation was determined from multiple sequence alignment of homologous proteins extracted from the HSSP database of sequence-structure alignments (homology-derived secondary structure of proteins, http://swift.cmbi.kun.nl/swift/hssp) (37). The Shannon entropy of the aligned sequences at position i was estimated as:
| (1) |
where pk is the number fraction of residues of class k at the ith position, the amino acids being grouped into seven classes based on the similarity of environment in protein structures (38). The sequence entropy is a measure of the divergence at each position in the alignment—thus, the lower the value of s, the greater is the degree of sequence conservation.
Identification of conserved residues at the interface
The average sequence entropy for each interface with ‘n’ number of residues was calculated:
| (2) |
Interface residues with sequence entropy lower than the average (<s>int) were considered as conserved and their total number in each interface is denoted by Ncons.
Measurement of the extent of spatial clustering of conserved residues and the inclusion of the residue composition
The degree of spatial clustering of a set of residues can be measured as the average of the inverse distance between every possible pairs in that set (39),
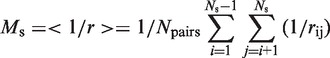 |
(3) |
where Ns is the number of residues in the set, Npairs is the number of unique pairs of residues in the set given by: Npairs = (Ns − 1).Ns/2; and, rij is the distance between the centers-of-mass of the two residues in question, i and j. The higher the value of Ms, the greater is the degree of spatial clustering of the residues in the set.
For each interface two Ms values were calculated, one only for the subset of conserved residues (Ms,cons) and another for the entire interface (Ms,int). The ratio (ρ) of Ms,cons to Ms,int enables comparison of the scattering of inter-residue distances between these two sets, which is actually an indicator of the extent of clustering of evolutionary conserved residues, having being used earlier for analyzing protein–protein binding sites (40).
| (4) |
ρ > 1.0 indicates that the subset of evolutionary conserved residues is clustered within the interface. This gives us a single overall numeric value representing whether or not (and to what extent) the conserved residues are clustered within the interface.
The amino acid composition of interface residues is known to differ significantly from that of the non-interface surface in protein–DNA complexes (2,3,6). Therefore, we calculated the average amino acid composition of conserved interface residues (averaged over the entire dataset) (Supplementary Table S1), and used these values to find the Euclidean distance (de) of the residue composition of the conserved subset in any surface patch. Amino acids were grouped into five classes such that the residues within a class have similar values of residue propensity for being in a protein–DNA interface (6). This class composition, rather than the individual compositions, was used in the calculation of de.
| (5) |
where, Ci is the average composition of the conserved residues belonging to the ith class for the interface taken over the entire dataset, ci is the corresponding value for any given surface patch (including the interface). This compositional disparity was combined with the degree of clustering of evolutionary conserved positions to get a score ρe.
| (6) |
The higher the clustering and the closer the composition of residues in a patch to the average value, the higher would be the score. This composite score enables us to combine two important discriminatory features of protein–DNA interfaces.
Potential hydrogen bond donors
Side-chain groups of positively charged amino acids such as arginine (PDB atom labels: NE, NH1, NH2), histidine (ND1, NE2) and lysine (NZ), as well as of asparagine (ND2), glutamine (NE2), tryptophan (NE1), serine (OG), threonine (OG1) and tyrosine (OH) with accessibility ≥10 Å2 were assumed to be capable of getting involved in hydrogen bonding with DNA and their number (Dp) in each interface/patch was calculated.
Residue propensity score
Finally, the amino acid composition was used to calculate residue propensity score (41) given by
| (7) |
where ni is the number of residue of type i and pi is its propensity to be in the interface.
Generation of surface patches and the evaluation of parameters
The surface patches were defined in two steps. First, the surface residues on each protein component were identified with the consideration of those with relative accessibility >5% (for homodimeric proteins residues located at the dimeric interface were excluded). Next each surface residue (represented by its center of mass) was taken as the central seed residue and a surface patch was constructed by including all neighboring surface residues contained within spheres of increasing radii—the patch size was allowed to increase until the number of residues contained in the patch matched with the total number of interface residues. Depending on its location a patch could be of two types, one being devoid of any interface residue, and the other type allowed a maximum of 10% of residues in common with the real interface. Hence a number of overlapping patches were generated comparable to the size of the interface in terms of residue numbers. All the parameters described above (Ncons, ρ, ρe, Dp and Rp) were computed for the real interface and for all possible surface patches of each protein. Values of each parameter were then used to arrange the surface patches in descending order and the true interface was ranked. The interface was ranked 1 if it occurred within the top 10% of surface patches. In a few cases where the number of generated patches was lower than 10, even if the interface had the highest value for a parameter it would not fall within the top 10%—a rank of 1 was assigned to these.
Training and test datasets used in model building by SVM
All 130 interfaces were screened for possible inclusion in the positive dataset. Those with very few homologs (less than eight, or when the sequences were all identical) failed to give proper Ms values and were excluded—this led to 119 positive cases. Negative examples were randomly picked from a consolidated list of all surface patches such that each complex structure provided at least one, but not more than two patches—this led to 153 negative examples. A total of 70% of the above set was randomly picked for creating the training dataset consisting of 83 positives and 107 negatives. The remaining 30% were used as test set (36 positives and 46 negatives). The SVM classifier was also applied to 47 unbound cases from the protein–DNA docking benchmark version 1.2 (42) for testing.
Parameter selection
Altogether 25 features were used as attributes for modeling the SVM classifier. The attributes were the fractional composition of each of the 20 amino acids, along with the five parameters (Ncons, ρ, ρe, Dp and Rp) enumerated earlier. These 25 parameters were then ranked by Weka version 3.4.11 evaluator—weak.attrubuteSelection.SVMAttributeEval (using 10-fold cross validation) (43).
SVM implementation
The freely downloadable LIBSVM package was used for the implementation of SVM with the C-SVC SVM type (SVM type for classification) and the widely used Radial Basis Function (RBF) kernel (44). Two parameters are required for optimizing the RBF–SVM classifier; γ, which determines the capacity of the RBF kernel and the regularization parameter, C. All the attributes in the training and test datasets were scaled in the range of −1 to 1.
SVM optimization
The penalty parameter C and the RBF kernel parameter γ were optimized using repeated grid search and leave-one-out cross-validation. In this cross-validation, a single instance of the training dataset was used as the test while all the other were used for training the classifier. The process was repeated for all the instances such that every instance was tested once individually. Matthews correlation coefficient (MCC) was used during cross-validation instead of percent accuracy, as the positive to negative ratio (83:107) is not one.
Performance measure
The performance was measured by prediction accuracy and MCC calculated as,
| (8) |
 |
(9) |
Tp, Fp, Tn and Fn represent the numbers of true positive, false positive, true negative and false negative, respectively. MCC takes into consideration true and false positives and negatives and is generally regarded as a balanced measure which can be used even if the classes are of very different sizes. The MCC is in essence a correlation coefficient between the observed and predicted binary classifications; it returns a value between −1 and +1. A coefficient of +1 represents a perfect prediction, 0 an average random prediction and −1 an inverse prediction. Unlike MCC, accuracy is sensitive to dataset imbalance. Also the sensitivity [Tp/(Tp + Fn)], specificity [Tn/(Tn + Fp)], precision [Tp/(Tp + Fp)] and F-measure [2 × precision × sensitivity / (precision + sensitivity)] of the model were determined.
RESULTS
In this work our goal was to characterize the nucleic acid binding region of DBPs with evolutionary and other structural features and study their application in distinguishing/identifying the DNA-binding region. Parameters defining the binding site of 130 protein–DNA complexes were compared to those derived from the rest of the protein surface. The performances of these features were tested, individually and in combination (using SVM) on several other datasets including the unbound form of the DBPs. We also tested the suitability of using these parameters in the identification of the binding site of RNA-binding proteins.
Clustering of conserved residue positions in protein–DNA interfaces
We first detected the conserved residues residing at the interface in 130 protein–DNA complexes—on average their number (Ncons) is 18 (Table 1). In an earlier study on protein–protein hetero-complexes, the degree of clustering of conserved interface residues had been measured by using the simple function Ms [Equation (3)] (40); the larger this value, the higher is the degree of clustering. The same concept has been employed here to a set of protein–DNA complexes: we calculated Ms for both the whole interface (Ms,int) and for the subset of conserved residues (Ms,cons). In 88.5% (108/122) cases (eight entries were found to have very few homologs and were thus excluded from the analysis), Ms,cons is found to have a value greater than Ms,int (Figure 1), indicating that the residues that are subjected to evolutionary pressure do remain clustered in the majority of the protein–DNA interfaces. The statistical significance of their difference and their average over the entire dataset and of their ratio, ρ [Equation (4)] are given in Table 1. In protein–protein complexes a ρ-value >1 was found in 86.7% cases (40). Furthermore, as was observed in case of the hetero-complexes (40), we also found that the subsets of evolutionary conserved residues in the interface were significantly more clustered compared to subsets of the same size consisting of randomly selected interface residues. The latter calculation was repeated by generating 1000 random subsets for each interface and the resulting average <Ms,random> was compared to Ms,cons. Ms,cons is higher than <Ms,random> in 88.5% cases (Supplementary Figure S1). An example of the clustering of conserved residues at the interface as compared to a few random surface patches are shown in Supplementary Figure S2.
Table 1.
Average values of interface parameters in protein–DNA complexes
| Parameters | Values |
|---|---|
| Number of complexes | 122a |
| <sint>b | 0.51 ± 0.28 |
| <scons>b | 0.18 ± 0.20 |
| Ms,cons | 0.09 ± 0.02c |
| Ms,int, [<Ms,random>] | 0.08 ± 0.02c, [0.08 ± 0.01]c |
| ρ | 1.11 ± 0.10 |
| ρe | 0.12 ± 0.08 |
| Rp | 0.71 ± 2.91 |
| Ncons | 18 ± 10 |
| Dp | 18 ± 8 |
aOf the 130 DBPs, 8 with only a few homologs were excluded.
b<sint> is defined for a structure [Equation (2)]. Here the value provided is the average over all the structures. Similarly, <scons> is the value for the conserved residues only.
cThe differences between Ms,int and Ms,cons (and between <Ms,random> and Ms,cons) are statistically significant at 1% level, P < 0.001.
Figure 1.

Plot of Ms,cons versus Ms,int (clustering of conserved residues versus that for all the residues in the interface).
Conservation and clustering to discriminate interface from other surface patches
All possible surface patches were generated for each protein as described in ‘Materials and Methods’ section. As was done for the interface, the conserved residues and the clustering of conserved residues were determined for each surface patch. The ρ-values of all the possible surface patches along with that of the interface were then explored to see to what extent this feature can be used to identify the true interface. Arranging the ρ-values in descending order, in ∼47% cases the ρ for the interface was among the top 10% of all the values, corresponding to a rank of 1 (on a scale of 1 to 10) (Supplementary Figure S3). Identification with this feature was slightly higher in case of homodimers and protein–protein complexes, ρ being ranked 1 in 54% and 49% cases respectively (40). In an attempt to improve the ranking we incorporated a measure of the similarity in the residue composition of the conserved residues in a patch and the corresponding average values over all the interfaces, expressed in terms of the Euclidean distance, de. The true interface with the minimum compositional variability would have the minimum de making the ratio of ρ to de, ρe [Equation (6)] the highest among all the patches. This improved our identification of interface by 7% to 54% (Figure 2b and Supplementary Figure S3) making it comparable to that observed for homodimers. An example of the improvement of discrimination in going from ρ to ρe is provided in Figure 3; although the interface had a high value of ρ, it was with ρe that the interface had the highest value. We then used conservation as the sole criterion (considering the number of conserved residues, Ncons). Interestingly, it gave a much better result. More than 70% of the interfaces could be identified with rank 1 (Table 2 and Figure 2).
Figure 2.

Distribution of the rank (on a scale of 1 to 10) of the known DNA-binding site relative to other patches on the surface of the protein using four different parameters. In (a) 77 structures are used with a strict definition of patches, in (b) 106 structures (where the patches may contain up to 10% interface residues).
Figure 3.

Distribution of five parameters calculated for all patches for the DNA complex of human topoisomerase I (PDB code, 1ej9). On each graph all the surface patches are represented in grey and the value for the known DNA-binding interface is indicated by an arrow. The parameters used are (a) ρ, (b) ρe, (c) Rp, (d) Dp and (e) Ncons.
Table 2.
Percentage of cases where the true interface is ranked #1 using different parameters applied to different datasets
| Parametera | This dataset [77, 106]b | Jones and Stawiskic [52, 65]b |
|---|---|---|
| ρe | 47, 54 | 50, 51 |
| Rp | 79, 83 | 81, 82 |
| Dp | 68, 70 | 67, 72 |
| Ncons | 70, 73 | 71, 68 |
aρ is omitted being already incorporated in ρe.
bThe first entry indicates the percentage of cases using stringent conditions (the surface patches devoid of any interface residue), the latter for patches that may contain up to 10% of interface residues.
Hydrogen bond donor
There are reports of high hydrogen bond density being present at protein–DNA interfaces (2–3). Besides protein–DNA interfaces are also enriched in positively charged residues with greater hydrogen bond donor capability (3,8,30). Therefore, we calculated the total number of hydrogen bond donors (Dp) and their accessibilities (both at the interface and the surface) (Table 3). Furthermore, we tried to find out if the application of a cut-off value on the accessibility (in the calculation of Dp) has any effect on the usefulness of the parameter. We observed that restricting to donors that have accessibility by ≥10 Å2 can best distinguish the true interface from the rest of the surface in comparison to all other cut-off values that we tested (0 or 1.5 or 20 Å2). Out of all the donors that are involved in hydrogen bonding with DNA in the complex, only 16% have accessibility <10 Å2 (Supplementary Figure S4). The average Dp was found to be 18 ± 8 (Table 1) at the interface, comparable to the value of 20 ± 12 reported by Stawiski et al. (16), even though we have excluded those with accessibility less than 10 Å2.
Table 3.
Average accessible surface area, <ASA> of all the donor groups in DNA-binding proteins
| Groups | Residues | <ASA> (Å2) in |
|||
|---|---|---|---|---|---|
| Interface |
Surfacea | ||||
| Before complexationa,b | After complexation | ||||
| NE | Arg | 10 ± 4 (10 ± 6) | 3 ± 3 | 7 ± 3 | |
| NH1 | Arg | 29 ± 10 (31 ± 15) | 11 ± 7 | 25 ± 9 | |
| NH2 | Arg | 35 ± 11 (34 ± 19) | 13 ± 10 | 31 ± 11 | |
| ND1 | His | 11 ± 8 (10 ± 9) | 3 ± 4 | 10 ± 5 | |
| NE2 | His | 15 ± 9 (17 ± 9) | 5 ± 6 | 13 ± 9 | |
| NZ | Lys | 35 ± 8 (32 ± 12) | 19 ± 9 | 33 ± 7 | |
| ND2 | Asn | 30 ± 12 (27 ± 17) | 12 ± 10 | 31 ± 10 | |
| NE1 | Trp | 12 ± 7 (9 ± 9) | 3 ± 4 | 7 ± 5 | |
| NE2 | Gln | 31 ± 15 (21 ± 19) | 12 ± 10 | 27 ± 9 | |
| OG | Ser | 17 ± 8 (17 ± 11) | 6 ± 5 | 14 ± 6 | |
| OG1 | Thr | 15 ± 7 (14 ± 10) | 5 ± 6 | 12 ± 6 | |
| OH | Tyr | 21 ± 11 (21 ± 15) | 7 ± 7 | 19 ± 9 | |
aThe difference between the accessibilities is significant at 0.1 to 5% level (P-value ranging from 0.001 to 0.05), except for ND1, NE2 (His and Gln), OH and ND2.
bThe values for the unbound form (from the protein–DNA docking benchmark) are given in parentheses, for comparison.
Dp could identify 68–70% of the true interfaces in our dataset with rank 1 (Figure 2). The performance was equally good (67–72%) when Dp was applied to the combined dataset of Jones and Stawiski (15,16) (Table 2). A noteworthy feature is that all the donor groups (with the exception of ND2 of Asn) have greater accessible surface area at the interfacial region before forming complex than at any other surface region (Table 3). Though we have not used accessibility directly in prediction this may be a distinctive feature. It may be mentioned that the average accessible surface area per residue of positive electrostatic patches in the nucleic acid (NA)-binding region was found to be slightly larger than that of non-NA-binding protein regions, though no statistical significance could be assigned to the observation (16).
Amino acid propensity
Amino acid composition/propensity markedly differs at the interface compared to that in the remaining surface due to the excess negative charges associated with DNA and high degree of hydrogen bonding across the interface (6). A residue propensity score, Rp [Equation (7)] that depends on the number of occurrence of a given residue in the interface and its propensity value was previously found to be useful in discriminating protein–protein interfaces from non-specific contacts in crystal lattice (41). When applied to protein–DNA complexes, Rp could identify 79–83% of the interfaces from among all the surface patches in our dataset (Figure 2), the best performer among all the parameters studied. Also Rp could identify 82% of the interfaces of Jones and Stawiski dataset with rank 1 from among all other surface patches (Table 2).
Analyzing the features on the unbound form of the protein–DNA complexes
We also tested each of the parameters individually on the unbound form of the proteins, as provided in the protein–DNA docking benchmark (42). The benchmark consists of 47 DNA–protein complexes, and structures are available for all the proteins in both their bound and unbound forms, with interface RMSD (conformational change of the protein–DNA interface was calculated by superimposition of all Cα and phosphate atoms at the interface) ranging from 0 to 8 Å; 12 structures have RMSD >5 Å. We mapped the protein chain of the unbound form on to the corresponding chain in the complex, the fitting being performed using the McLachlan (45) algorithm, as implemented in the program ProFit (46). The residues in the unbound form which are structurally equivalent to the residues located in the interface of the complex constitute the potential interface on the unbound form. Five cases were found to have very few homologs and were not analyzed. On average 17 residues were found to be conserved, which as expected is nearly the same as in the interface of the complex (Table 1). The average ρ was 1.13 ± 0.2 and 90% (38/42) cases had ρ > 1. The average number of hydrogen bond donors was found to be 15 ± 6, again quite similar to the bound form. Though the value of average Rp was rather low (−0.1 ± 2), it had a good discriminating power for the identification of the interface from random surface patches—86% of the cases had rank 1 (Figure 4). Dp could assign rank 1 to 67% of the interfaces. As compared to the bound form of the proteins (Figure 2), ρe seems to have performed better in identifying the true interface for the unbound form (54 versus 62%).
Figure 4.

Distribution of the rank (on a scale of 1 to 10) of the DNA-binding site in the unbound form (obtained by mapping the interface information from the bound structure) of 42 DNA-binding proteins taken from benchmark version 1.2, relative to other patches on the surface of the protein using four different parameters. Patches were identified using the strict definition.
Predicting the DNA-binding region
As summarized in Table 2 except for clustering based parameter ρe, all other parameters, considered alone, were good for discriminating the true interface in at least 70% cases from other surface regions in DBPs. The success rate is equally impressive when applied to an independent dataset due to Jones and Stawiski, Rp performs the best followed by Dp and Ncons. Indeed, Rp outperforms the prediction accuracy of the method by Jones (15) and Stawiski (16), especially in comparison with the enzyme dataset of Stawiski (Table 4). It may be mentioned that residue interface propensity was one among five parameters that were used by Jones et al. (15) who found that the one based on electrostatic score performed the best (and shown in Table 4). We then wanted to see the combined effect of the five parameters along with 20 additional descriptors (representing the residue composition in a given patch) by training a mathematical model, SVM. The binary classifier gives output as positive or negative to depict the DNA-binding and non-binding regions, respectively.
Table 4.
Comparison of the efficiency of the present method with other techniques
| Dataset (# of cases) | Reported prediction accuracy (%) | Accuracy (%) using |
|
|---|---|---|---|
| Rp | Dp | ||
| Jones (56) | 68 | 82a | 72a |
| Stawiski (54) | 81 | ||
| Stawiski enzyme data set (16) | 50 | 92b | 62b |
aThe present method was applied to the combined Jones and Stawiski datasets as given in Table 2.
bBased on 13 cases (three could not be used as no surface patch showed up).
SVM training and predictions
The SVM classifier was trained several times using combinations of different top ranked attributes and the values of γ and C were optimized to maximize the MCC value. These were subsequently used to predict the test dataset to assess the performance of the combination of attributes. Results presented in Table 5 show that the model which was trained with the top 15 attributes had the highest MCC and was subsequently used for testing. This model when applied to the test dataset performed quite well (Table 6); all the performance measures are better as compared to the model using all the attributes (Supplementary Table S2).
Table 5.
Summary of SVM modeling
| Attributes | C | γ | MCC |
|---|---|---|---|
| Top 5 | 15 | 0.013 | 0.7867 |
| Top 10 | 14 | 0.5 | 0.8393 |
| Top 15 | 7 | 0.021 | 0.8608 |
| All 25 | 3 | 0 | 0.8508 |
Table 6.
Performance of the model on our test set and the unbound cases in protein–DNA docking benchmark
| Test set | Accuracy | Specificity | Sensitivity/ Recall | Precision | F-measure |
|---|---|---|---|---|---|
| Our dataseta | 90.5 | 91.7 | 88.8 | 89.9 | 89.1 |
| protein–DNA docking benchmarkb | 93.6 | 92.8 | 95.2 | 86.9 | 90.9 |
aValues shown are average performance on 10 different randomly generated test sets.
b42 positives and 83 negatives.
In addition to the leave-one-out method we also optimized the kernel parameters using 5-fold cross validation—the training dataset was spilt into five subsets, where one of the subsets was used as the test set while the other four subsets were used for training the classifier. The trained classifier was then tested using the test set. The process was repeated five times using a different subset for testing, thereby ensuring that all subsets were used for both training and testing. The results were essentially the same, except that the model which was trained with all the 25 parameters had the highest MCC (0.8674).
Test on the unbound form of the protein–DNA benchmark
The trained SVM classifier was used for detecting the likely interface in the unbound DBPs taken from the docking benchmark (42), which contained 47 such structures. While the mapped interface on the unbound form constituted the positive examples, the negatives were picked up from the surface patches. Approximately two surface patches were randomly picked for each structure as negatives, making the negative to positive ratio as 2:1. The classifier gave very good result with only two Fn and six Fp predictions. The corresponding accuracy, specificity, sensitivity and other performance parameters are given in Table 6.
Application of the parameters on protein–RNA structures
Protein–RNA interaction is far less studied than the one involving protein and DNA, mainly due to its complexity and the lesser number of structures available. Dinucleotide-specific contacts were found to be different in case of RNA-binding proteins (RBPs) as compared to DBPs and could be used to predict targets of RBPs (47). Recently, Ahmad and Sarai extended their moment-based approach for predicting DBPs (48) to RBPs and found distinct patterns of net charge, dipole and quadruple moments (49). It is interesting to see how our four parameters used for the characterization of the protein–DNA interfaces perform in identification of the interfaces in protein–RNA complexes. Of the 51 complexes listed in Biswas et al. (50) 45 could be analyzed (the remaining did not have enough homologs). Comparison of the results (Figure 5) with those from protein–DNA complexes (Figure 2) indicates that the performance with Ncons for ranking the true interface as 1 remains nearly the same. However the performance for all other parameters deteriorated by ∼12% with Rp, 14–15% with Dp and 3–16% with ρe.
Figure 5.

Distribution of the rank (on a scale of 1 to 10) of the known RNA-binding site relative to other patches on the surface of the protein using four different parameters. In (a) 39 structures are used with a strict definition of patches, in (b) 45 structures (where the patches may contain up to 10% interface residues).
Testing the specificity of the model using a set of non-DBPs
To further validate the specificity of the model, we tested our SVM classifier solely on a negative dataset (84 cases) based on 42 weakly associated homodimeric proteins (51). We opted for these dimers as their interface size is comparable to that of the protein–DNA complexes discussed here (∼1600 versus ∼2000 Å2). For each protein two patches were defined—one corresponding to the dimeric interface and another randomly selected from the rest of the protein surface. The results showed only seven Fp among the interfaces and six from the surface patches. The number of false positives remained the same when the classifier was tested with the 42 positives randomly selected from the protein–DNA set and the negatives comprising of 42 examples of either the protein–protein interfaces or the random surface patches. Thus the classifier has the ability to distinguish the protein–DNA interface from the patches arising out of protein–protein binding region or a random surface of non-DBPs.
DISCUSSION
We have analyzed and used four different parameters (and one variant) individually for predictions of DNA-binding sites on the surface of protein structures. One of the parameters, ρ is based on clustering of conserved residues. Though it is known that the putative hotspots for DNA binding are those which occur as clusters of conserved residues (4), we have defined ρ in an analogous way to what was done for the analysis of protein–protein interfaces (40). In 88.5% of the protein–DNA interfaces ρ is >1 (Figure 1 and Table 1). The usefulness of the clustering parameter for the identification of the interface from any random surface patch can be improved by 7% by modifying ρ into ρe that incorporates a weighing factor depending on the variation of the amino acid composition of conserved residues of a given interface/patch from the corresponding average composition observed in all the interfaces. Another parameter to be used was based on the hydrogen bond donors. Interestingly, the accessible surface areas of such groups are found to be more in the interface than when these are located in the rest of the surface (Table 3). This is akin to what has been observed at the residue level in protein–protein interfaces (Guharoy et al., unpublished data). To improve the discriminatory power, only those groups with an accessible surface area of ≥10 Å2 were used for the calculation of Dp. An example of the values of the parameters at the interface (being ranked the highest in all but ρ) with respect to all other surface patches are shown in Figure 3.
Using a single parameter the best prediction (83%) was obtained using Rp (Figure 2), the residue propensity score, which also worked well for protein–protein interfaces (41). Rp is equally efficient when applied to the unbound form of DBPs, identifying 86% cases (Figure 4). This is indeed a very high quality prediction rate compared to the previous analysis by Jones et al. (15), which attained 68% correct prediction using a similar approach of patch analysis and the true interface ranking on the basis of electrostatic potential. We also applied our parameters to the combined dataset of Jones and Stawiski (15,16) and obtained 82% correct prediction using Rp and 72% using Dp (Table 4). We separately dealt with the 16 enzyme complexes in Stawiski’s dataset that were very poorly identified by them, and found that out of 13 complexes (surface patch did not show up in three cases) Rp and Dp could identify 12 and 8, respectively, of the interfaces correctly (Table 4).
There are now attempts to distinguish DNA from RNA binding surfaces (52,53). The parameter, Rp based on features of DNA-binding interfaces is ∼12% less successful in identifying the RNA-binding site (Figure 5). Dp is also less effective. Thus there are some differences in the residue propensity and the number of hydrogen bond donors from DNA and RNA, which could be exploited to distinguish between the two types of surface patches.
Finally, we built a SVM classifier with 15 attributes. The model had a very high MCC of 0.86 compared to all other earlier models and an accuracy of ∼90% (Tables 5 and 6). Other DNA-binding site prediction methods have reported MCC of 0.54 and 0.62 for the top two models with accuracy of 85% and 87%, respectively (21). SVM predictors developed by Kuznetsov et al. (11), which have used structural and evolutionary information in the form of PSSM, achieved a maximum MCC of 0.66 with 82% accuracy. Using the surface curvature and the electrostatic potential of the DNA-binding and non-binding sites, the web server PredDs (25) reported accuracy of 94%, with 86% sensitivity and 96% specificity—values comparable to ours, though our method appears to be more sensitive. This method also outperforms the available sequence-based prediction methods of DNA-binding sites, such as DP-Bind (22), DBSpred (7), DBS-PSSM (54) and BindN (55) in terms of their reported accuracy, sensitivity and specificity. A very recent method, metaDBSite that integrated results from other web-servers including a few of those mentioned above can predict solely on the basis of sequence information and reports a sensitivity of 77% (56). While the ultimate goal is to be able to predict the residues that bind DNA directly from amino acid sequence (57), a structure-based method, such as this can be incorporated to develop a more robust method of prediction. It may be mentioned that given the complexity of predicting the specificity of a protein for a DNA sequence, the structure is usually used to complement the results from sequence-based approach (58,59).
Identifying the binding region in the unbound form of the protein is a challenging task. Almost all earlier investigations exploited the bound complex in characterizing and identifying the DNA-binding site. A method named DISPLAR (30) used 14 unbound DBPs in testing and gave an accuracy of 77%. In this work we too started with the complex form in characterizing the binding site with different set of parameters, but tested them on the unbound form of the proteins available in protein–DNA docking benchmark (42). All the parameters performed well by ranking >60% of the interface regions correctly. In contrast to DISPLAR, our SVM model could identify the binding region with an accuracy of 93.6%.
CONCLUSION
We have developed five parameters based on the residue propensity, conservation and structural features of the binding region in DBPs, and analyzed their usefulness in identifying the interface from all possible surface patches. Using 15 attributes we have applied the SVM approach for the identification of the DNA-binding site on protein molecular surface and achieve results that are better or at least comparable to the existing algorithms.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1 and 2, Supplementary Figures 1–4 and Supplementary Reference [60].
FUNDING
Department of Science and Technology, India (Research grant to P.C.); Council of Scientific and Industrial Research (fellowships to A.P. and M.G.); Department of Biotechnology (fellowships to S.D. and S.S.). Funding for open access charge: Department of Science and Technology, India.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nadassy K, Wodak SJ, Janin J. Structural features of protein–nucleic acid recognition sites. Biochemistry. 1999;38:1999–2017. doi: 10.1021/bi982362d. [DOI] [PubMed] [Google Scholar]
- 3.Jones S, van Heyningen P, Berman HM, Thornton JM. Protein–DNA interactions: a structural analysis. J. Mol. Biol. 1999;287:877–896. doi: 10.1006/jmbi.1999.2659. [DOI] [PubMed] [Google Scholar]
- 4.Ahmad S, Keskin O, Sarai A, Nussinov R. Protein–DNA interactions: structural, thermodynamic and clustering patterns of conserved residues in DNA-binding proteins. Nucleic Acids Res. 2008;36:5922–5932. doi: 10.1093/nar/gkn573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sathyapriya R, Vijayabaskar MS, Vishveshwara S. Insights into protein- DNA interactions through structure network analysis. PLoS Comput. Biol. 2008;4:e1000170. doi: 10.1371/journal.pcbi.1000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biswas S, Guharoy M, Chakrabarti P. Dissection, residue conservation, and structural classification of protein-DNA interfaces. Proteins. 2009;74:643–654. doi: 10.1002/prot.22180. [DOI] [PubMed] [Google Scholar]
- 7.Ahmad S, Gromiha MM, Sarai A. Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information. Bioinformatics. 2004;20:477–486. doi: 10.1093/bioinformatics/btg432. [DOI] [PubMed] [Google Scholar]
- 8.Xiong Y, Liu J, Wei DQ. An accurate feature-based method for identifying DNA-binding residues on protein surfaces. Proteins. 2011;79:509–517. doi: 10.1002/prot.22898. [DOI] [PubMed] [Google Scholar]
- 9.Luscombe NM, Thornton JM. Protein-DNA interactions: amino acid conservation and the effects of mutations on binding specificity. J. Mol. Biol. 2002;320:991–1009. doi: 10.1016/s0022-2836(02)00571-5. [DOI] [PubMed] [Google Scholar]
- 10.Mirny LA, Gelfand MS. Structural analysis of conserved base pairs in protein-DNA complexes. Nucleic Acids Res. 2002;30:1704–1711. doi: 10.1093/nar/30.7.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuznetsov IB, Gou Z, Li R, Hwang S. Using evolutionary and structural information to predict DNA-binding sites on DNA-binding proteins. Proteins. 2006;64:19–27. doi: 10.1002/prot.20977. [DOI] [PubMed] [Google Scholar]
- 12.Xiong Y, Xia J, Zhang W, Liu J. Exploiting a reduced set of weighted average features to improve prediction of DNA-binding residues from 3D structures. PLoS One. 2011;6:e28440. doi: 10.1371/journal.pone.0028440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mandel-Gutfreund Y, Margalit H. Quantitative parameters for amino acid-base interaction: implications for prediction of protein-DNA binding sites. Nucleic Acids Res. 1998;26:2306–2312. doi: 10.1093/nar/26.10.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luscombe NM, Laskowski RA, Thornton JM. Amino acid-base interactions: a three-dimensional analysis of protein–DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones S, Shanahan HP, Berman HM, Thornton JM. Using electrostatic potentials to predict DNA-binding sites on DNA-binding proteins. Nucleic Acids Res. 2003;31:7189–7198. doi: 10.1093/nar/gkg922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stawiski EW, Gregoret LM, Mandel-Gutfreund Y. Annotating nucleic acid-binding function based on protein structure. J. Mol. Biol. 2003;326:1065–1079. doi: 10.1016/s0022-2836(03)00031-7. [DOI] [PubMed] [Google Scholar]
- 17.Reddy CK, Das A, Jayaram B. Do water molecules mediate protein-DNA recognition? J Mol. Biol. 2001;314:619–632. doi: 10.1006/jmbi.2001.5154. [DOI] [PubMed] [Google Scholar]
- 18.Woda J, Schneider B, Patel K, Mistry K, Berman HM. An analysis of the relationship between hydration and protein-DNA interactions. Biophys. J. 1998;75:2170–2177. doi: 10.1016/S0006-3495(98)77660-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarai A, Kono H. Protein-DNA recognition patterns and predictions. Annu. Rev. Biophys. Biomol. Struct. 2005;34:379–398. doi: 10.1146/annurev.biophys.34.040204.144537. [DOI] [PubMed] [Google Scholar]
- 20.Temiz NA, Camacho CJ. Experimentally based contact energies decode interactions responsible for protein-DNA affinity and the role of molecular waters at the binding interface. Nucleic Acids Res. 2009;37:4076–4088. doi: 10.1093/nar/gkp289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao M, Skolnick J. From nonspecific DNA-protein encounter complexes to the prediction of DNA-protein interactions. PLoS Comput. Biol. 2009;5:e1000341. doi: 10.1371/journal.pcbi.1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang S, Gou Z, Kuznetsov IB. DP-Bind: a web server for sequence-based prediction of DNA-binding residues in DNA-binding proteins. Bioinformatics. 2007;23:634–636. doi: 10.1093/bioinformatics/btl672. [DOI] [PubMed] [Google Scholar]
- 23.Zhao H, Yang Y, Zhou Y. Structure-based prediction of DNA-binding proteins by structural alignment and a volume-fraction corrected DFIRE-based energy function. Bioinformatics. 2010;26:1857–1863. doi: 10.1093/bioinformatics/btq295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao M, Skolnick J. DBD-Hunter: a knowledge-based method for the prediction of DNA-protein interactions. Nucleic Acids Res. 2008;36:3978–3992. doi: 10.1093/nar/gkn332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsuchiya Y, Kinoshita K, Nakamura H. PreDs: a server for predicting dsDNA-binding site on protein molecular surfaces. Bioinformatics. 2005;21:1721–1723. doi: 10.1093/bioinformatics/bti232. [DOI] [PubMed] [Google Scholar]
- 26.Kumar MD, Bava KA, Gromiha MM, Prabakaran P, Kitajima K, Uedaira H, Sarai A. ProTherm and ProNIT: thermodynamic databases for proteins and protein-nucleic acid interactions. Nucleic Acids Res. 2006;34:204. doi: 10.1093/nar/gkj103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tkachenko MY, Boryskina OP, Shestopalova AV, Tolstorukov MY. ProtNA-ASA: protein-nucleic acid structural database with information on accessible surface area. Int. J. Quantum Chem. 2010;110:230–232. [Google Scholar]
- 28.Chan TM, Wong KC, Lee KH, Wong MH, Lau CK, Tsui SKW, Leung KS. Discovering approximate-associated sequence patterns for protein–DNA interactions. Bioinformatics. 2011;27:471–478. doi: 10.1093/bioinformatics/btq682. [DOI] [PubMed] [Google Scholar]
- 29.Wang L, Brown SJ. BindN: a web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res. 2006;34:W243–W248. doi: 10.1093/nar/gkl298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tjong H, Zhou HX. Displar DISPLAR: an accurate method for predicting DNA-binding sites on protein surfaces. Nucleic Acids Res. 2007;35:1465–1477. doi: 10.1093/nar/gkm008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zakrzewska K, Bouvier B, Michon A, Blanchet C, Lavery R. Protein-DNA binding specificity: a grid-enabled computational approach applied to single and multiple protein assemblies. Phys. Chem. Chem. Phys. 2009;11:10712–10721. doi: 10.1039/b910888m. [DOI] [PubMed] [Google Scholar]
- 32.Langlois RE, Lu H. Boosting the prediction and understanding of DNA-binding domains from sequence. Nucleic Acids Res. 2010;38:3149–3158. doi: 10.1093/nar/gkq061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dror I, Shazman S, Mukherjee S, Zhang Y, Glaser F, Mandel-Gutfreund Y. Predicting nucleic acid binding interfaces from structural models of proteins. Proteins. 2011 doi: 10.1002/prot.23214. 12 October 2011 (doi: 10.1002/prot.23214; epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chakrabarti P, Janin J. Dissecting protein–protein recognition sites. Proteins. 2002;47:334–343. doi: 10.1002/prot.10085. [DOI] [PubMed] [Google Scholar]
- 35.Hubbard SJ. NACCESS: Program for Calculating Accessibilities. Department of Biochemistry and Molecular Biology. London, UK: University college of London; 1992. [Google Scholar]
- 36.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 37.Sander C, Schneider R. The HSSP database of protein structure–sequence alignments. Nucleic Acids Res. 1993;21:3105–3109. doi: 10.1093/nar/21.13.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guharoy M, Chakrabarti P. Conservation and relative importance of residues across protein-protein interfaces. Proc. Natl Acad. Sci. USA. 2005;102:15447–15452. doi: 10.1073/pnas.0505425102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schueler-Furman O, Baker D. Conserved residue clustering and protein structure prediction. Proteins. 2003;52:225–235. doi: 10.1002/prot.10365. [DOI] [PubMed] [Google Scholar]
- 40.Guharoy M, Chakrabarti P. Conserved residue clusters at protein-protein interfaces and their use in binding site identification. BMC Bioinformatics. 2010;11:286. doi: 10.1186/1471-2105-11-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bahadur RP, Chakrabarti P, Rodier F, Janin J. A dissection of specific and non-specific protein-protein interfaces. J. Mol. Biol. 2004;336:943–955. doi: 10.1016/j.jmb.2003.12.073. [DOI] [PubMed] [Google Scholar]
- 42.van Dijk M, Bonvin AMJJ. A protein–DNA docking benchmark. Nucleic Acids Res. 2008;36:e88. doi: 10.1093/nar/gkn386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frank E, Hall M, Trigg L, Holmes G, Witten IH. Data mining in bioinformatics using Weka. Bioinformatics. 2004;20:2479–2481. doi: 10.1093/bioinformatics/bth261. [DOI] [PubMed] [Google Scholar]
- 44.Chang CC, Lin CJ. LIBSVM: a library for support vector machines. 2001 Version-2.84 Publisher: Software available at http://www.csie.ntu.edu.tw/~cjlin/libsvm (April 2007, date last accessed) [Google Scholar]
- 45.McLachlan AD. Rapid comparison of protein structres. Acta Crsyt. 1982;A38:871–873. [Google Scholar]
- 46.Martin ACR, Porter CT. ProFit. software available at http://www.bioinf.org.uk/software/profit/ [Google Scholar]
- 47.Fernandez M, Kumagai Y, Standley DM, Sarai A, Mizuguchi K, Ahmad S. Prediction of dinucleotide-specific RNA-binding sites in proteins. BMC Bioinformatics. 2011;12(Suppl. 13):S5. doi: 10.1186/1471-2105-12-S13-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ahmad S, Sarai A. Moment-based prediction of DNA-binding proteins. J. Mol. Biol. 2004;341:65–71. doi: 10.1016/j.jmb.2004.05.058. [DOI] [PubMed] [Google Scholar]
- 49.Ahmad S, Sarai A. Analysis of electric moments of RNA-binding proteins: implications for mechanism and prediction. BMC Struct. Biol. 2011;11:8. doi: 10.1186/1472-6807-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Biswas S, Guharoy M, Chakrabarti P. Structural segments and residue propensities in protein-RNA interfaces. Bioinformation. 2008;2:422–427. doi: 10.6026/97320630002422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dey S, Pal A, Chakrabarti P, Janin J. The subunit interfaces of weakly associated homodimeric proteins. J. Mol. Biol. 2010;398:146–160. doi: 10.1016/j.jmb.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 52.Zhao H, Yang Y, Zhou Y. Structure-based prediction of RNA-binding domains and RNA-binding sites and application to structural genomics targets. Nucleic Acids Res. 2011;39:3017–3025. doi: 10.1093/nar/gkq1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shazman S, Elber G, Mandel-Gutfreund Y. From face to interface recognition: a differential geometric approach to distinguish DNA from RNA binding surfaces. Nucleic Acids Res. 2011;39:7390–7399. doi: 10.1093/nar/gkr395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmad S, Sarai A. PSSM-based prediction of DNA binding sites in proteins. BMC Bioinformatics. 2005;6:33–38. doi: 10.1186/1471-2105-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, Brown SJ. BindN: a web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res. 2006;34:W243–W248. doi: 10.1093/nar/gkl298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Si J, Zhang Z, Lin B, Schroeder M, Huang B. MetaDBSite: a meta approach to improve protein DNA-binding sites prediction. BMC Syst. Biol. 2011;5(Suppl. 1):S7. doi: 10.1186/1752-0509-5-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ofran Y, Mysore V, Rost B. Prediction of DNA-binding residues from sequence. Bioinformatics. 2007;23:i347–i353. doi: 10.1093/bioinformatics/btm174. [DOI] [PubMed] [Google Scholar]
- 58.Chen C-Y, Chien T-Y, Lin C-K, Lin C-W, Weng Y-Z, Chang DT-H. Predicting target DNA sequences of DNA-binding proteins based on unbound structures. PLoS One. 2012;7:e30446. doi: 10.1371/journal.pone.0030446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morozov AV, Havranek JJ, Baker D, Siggia ED. Protein-DNA binding specificity predictions with structural models. Nucleic Acids Res. 2005;33:5781–5798. doi: 10.1093/nar/gki875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.