


Abstract
The combination of incessant advances in sequencing technology producing large amounts of data and innovative bioinformatics approaches, designed to cope with this data flood, has led to new interesting results in the life sciences. Given the magnitude of sequence data to be processed, many bioinformatics tools rely on efficient solutions to a variety of complex string problems. These solutions include fast heuristic algorithms and advanced data structures, generally referred to as index structures. Although the importance of index structures is generally known to the bioinformatics community, the design and potency of these data structures, as well as their properties and limitations, are less understood. Moreover, the last decade has seen a boom in the number of variant index structures featuring complex and diverse memory-time trade-offs. This article brings a comprehensive state-of-the-art overview of the most popular index structures and their recently developed variants. Their features, interrelationships, the trade-offs they impose, but also their practical limitations, are explained and compared.
INTRODUCTION
Developments in sequencing technology continue to produce data at higher speed and lower cost. The resulting sequence data form a large fraction of the data processed in life sciences research. For example, de novo genome assembly joins relatively short DNA fragments together into longer contigs based on overlapping regions, whereas in RNA-seq experiments, cDNA is mapped to a reference genome or transcriptome. Further down the analysis pipeline, DNA and protein sequences are aligned to one another and similarity between aligned sequences is estimated to infer phylogenies (1). Although the type of sequences and applications varies widely, they all require basic string operations, most notably search operations. Given the sheer number and size of the sequences under consideration and the number of search operations required, efficient search algorithms are important components of genome analysis pipelines. For this reason, specialized data structures, generally bundled under the term ‘index structures’, are required to speed up string searching.
The use of specialized algorithms and data structures is motivated by the fact that the data flow has already surpassed the flow of advances in computer hardware and storage capabilities. However, although index structures are already widely used to speed up bioinformatics applications, they too are challenged by the recent data flood. Index structures require an initial construction phase and impose extra storage requirements. In return, they provide a wide variety of efficient string searching algorithms. Traditionally, this has led to a dichotomy between search efficiency and reduced memory consumption. However, recent advances in index structures have shown that compression and fast string searching can be achieved simultaneously using a combination of compression and indexing, thus solving this dichotomy (2).
There are many types of index structures. The most commonly known index structures are inverted indexes and lookup tables. These work in a similar way to the indexes found at the back of books. However, biological sequences generally lack a clear division in words or phrases, a prerequisite for inverted indexes to function properly. Two alternative index structures are used in bioinformatics applications. k-mer indexes divide sequences into substrings of fixed length k and are used, among others, in the BLAST (3) alignment tool. ‘Full-text indexes’, on the other hand, allow fast access to substrings of any length. Full-text indexes come at a greater memory and construction cost compared with k-mer indexes and are also far more complex. However, they contain much more information and allow for faster and more flexible string searching algorithms (4).
Full-text index structures are widely used crucial black box components of many bioinformatics applications. Their success is illustrated by the number of bioinformatics tools that currently use them. Examples are tools for short read mapping (5–9), alignment (10,11), repeat detection (12), error correction (13,14) and genome assembly (15–17). The memory and time performance of many of these tools are directly affected by the type and implementation of the index structure used. The choice for a tool impacts the choice of index structures and vice versa. However, the description of these tools in scientific literature often bypasses a detailed description about the specifications of the index structures used. Concepts such as suffix trees, suffix arrays or FM-indexes are introduced in general terms in bioinformatics courses, but most of the time, these index structures are applied as black boxes having certain properties and allowing certain operations on strings at a given time. This does injustice to the vast and rich literature available on index structures and does not present their complex design, possibilities and limitations. Moreover, most tools are designed using basic implementations of these index structures, without taking full advantage of the latest advances in indexing technology.
The goal of this article is 2-fold. On the one hand, we offer a comprehensive review of the basic ideas behind classical index structures, such as suffix trees, suffix arrays and Burrows–Wheeler-based index structures, such as the FM-indexes. No prior knowledge about index structures is required. On the other hand, we give an overview of the limitations of these structures as well as the research done in the last decade to overcome these limitations. Furthermore, in light of recent advances made in both sequencing technology as well as computing technology, we give prospects on future developments in index structure research.
Overview
This article is structured according to the following outline. The first main section introduces basic concepts and notations which are used throughout the article. This section also clarifies the relationship between computer science string algorithms and sequence analysis applications. Furthermore, it explains some algorithmic performance measures which have to be taken into account when dealing with advanced data structures. Readers well acquainted with data structures and algorithms may easily skip this section.
The second section reviews some of the most popular index structures currently in use. These include suffix trees, enhanced and compressed suffix arrays and FM-indexes, which are based on the Burrows–Wheeler transform. Both representational and algorithmic aspects of basic search operations of these index structures are discussed using a running example. Furthermore, the features of these different index structures are compared on an abstract level and their interrelation is made clear.
The next section gives an overview of current state-of-the-art main (RAM) memory index structures, with a focus on memory-time trade-offs. Several memory saving techniques are discussed, including compression techniques utilized in ‘compressed index structures’. The aim of this section is to provide insight into the complexity of the design of these compressed index structures, rather than to give their full details. It is shown how their design is composed of auxiliary data structures that govern the performance of the main index structure. On a larger scale, practical results from the bioinformatics literature illustrate the performance gain and limitations of search algorithms. Furthermore, a comparison between index structures, together with an extensive literature list, acts as a taxonomy for the currently known main memory full-text index structures.
While main memory index structures are the main focus of the second section, the design, limitations and improvements of external memory index structures are also discussed. The difference between index structures for internal and external memory is most prominent in their use of compression techniques, which are (still) less important in external memory. However, because harddisk access is much slower than main memory access, data structure layout and access patterns are much more important.
The second biggest bottleneck of index structure usage is the initial construction phase, which is covered in the final section. Both main memory as well as secondary memory construction algorithms are reviewed. The main conceptual ideas used for construction of the index structures discussed in previous sections are provided together with examples of the best results of construction algorithms found in the literature.
Finally, a summary of the findings presented in this article and some prospects on future directions of the research on index structures and its impact on bioinformatics applications is given. These prospects include variants and extensions of classical index structures, designed to answer specific biological queries, such as the search for structural RNA patterns, but also the use of new computing paradigms, such as the Google MapReduce framework (18).
IMPORTANT CONCEPTS
Index structures originate from the field of theoretical computer science. This section introduces some important concepts for readers not familiar with the field. Readers with a background in data structures and algorithms may skip this section, except for the notations introduced at the end of this section.
Strings versus sequences
The term ‘sequence’ is used for different concepts in the field of computer science and biology. What is called a sequence in biology is usually a ‘string’ in standard computer science parlance. The distinction between strings and sequences becomes especially prominent in computer science when introducing the concepts of substrings and subsequences. The former refer to contiguous intervals from larger strings, whereas the latter do not necessarily need to be contiguous intervals from the original string. As index structures work with substrings and to avoid ambiguity, we will stick to the standard computer science term string throughout this article, unless we explicitly want to stress the biological origin of the sequence.
String matching
Key components of genome analysis include statistical methods for scoring and comparing string hypotheses and string matching algorithms for efficient string comparisons. However, the former component falls beyond the scope of this review as our main focus lies on string matching algorithms studied in the field of computer science. This again gives rise to a terminology barrier between the two research fields. For nearly all index structures discussed in this review, efficient algorithms for exact and inexact string matching exist. These algorithms allow fast queries into sequence databases, similarity searches between sequences and DNA/RNA mapping. Inexact string matching is usually implemented using a backtracking algorithm on the suffix tree or a seed-and-extend approach. The latter approach may use maximal exact matches or other types of shared substrings. Maximal exact matches are examples of identical substrings shared between multiple strings and are frequently used as seeds in sequence alignment or in tools that determine sequence similarity (10). Searching for all maximal exact matches in an efficient way requires strong index structures that are fully expressive, i.e. allow for all suffix tree operations in constant time (19).
Index structures reaching full expressiveness are able to handle a multitude of string searching problems such as locating several types of repeats, finding overlapping strings and finding the longest common substring. These string matching algorithms are, among others, used in genome assembly (finding repeats and overlaps), error correction of sequencing reads (repeats), fast identification of DNA contaminants (longest common substring) and genealogical DNA testing (short tandem repeats).
In addition, some index structures are geared toward specific applications. ‘Affix index structures’, for example, allow bidirectional string searching. As a result, they can be used for searching RNA structure patterns (20) and for short read mapping (6). ‘Weighted suffix trees’ (21) can be used to find patterns in biological sequences that contain weights such as base probabilities, but are also applied in error correction (13). ‘Geometric suffix trees’ (22) have been used to index 3D protein structures. ‘Property suffix trees’ have additional data structures to efficiently answer property matching queries. This can be useful, for example, in retrieving all occurrences of patterns that appear in a repetitive genomic structure (23).
Theoretical complexity
As is the case for other data structures, the performance of algorithms working on index structures is usually expressed in terms of their theoretical complexity, indicated by the ‘big- notation’. Although a theoretical measure of the worst-case scenario, it contains valuable practical information about the qualitative and quantitative performance of algorithms and data structures. For example, some index structures contain an alphabet-dependency, whereas others do not. Thus, alphabet-independent index structures theoretically perform string searches equally well on DNA sequences (4 different characters) as on protein sequences (20 different characters). The qualitative information of the theoretical complexity usually categorizes the dependency of input parameters in terms of logarithmic, linear, quasilinear, quadratic or exponential dependency. Intuitively, this means that even if several algorithms nearly have the same execution time or memory requirements for a given input sequence, the execution time and memory requirements of some algorithms will grow much faster than those of others when the input size increases. In practice, quasilinear algorithms [complexity
notation’. Although a theoretical measure of the worst-case scenario, it contains valuable practical information about the qualitative and quantitative performance of algorithms and data structures. For example, some index structures contain an alphabet-dependency, whereas others do not. Thus, alphabet-independent index structures theoretically perform string searches equally well on DNA sequences (4 different characters) as on protein sequences (20 different characters). The qualitative information of the theoretical complexity usually categorizes the dependency of input parameters in terms of logarithmic, linear, quasilinear, quadratic or exponential dependency. Intuitively, this means that even if several algorithms nearly have the same execution time or memory requirements for a given input sequence, the execution time and memory requirements of some algorithms will grow much faster than those of others when the input size increases. In practice, quasilinear algorithms [complexity  (nlog n)] are sometimes much faster than linear algorithms [complexity
(nlog n)] are sometimes much faster than linear algorithms [complexity  (n)], because of the lower order terms and constants involved. These are usually omitted in the big-
(n)], because of the lower order terms and constants involved. These are usually omitted in the big- notation. In general, however, the big-
notation. In general, however, the big- notation is a good guideline for algorithm and data structure performance. Furthermore, this measure of algorithm and data structure efficiency is timeless and is not dependent on hardware, implementation and data specifications, as opposed to benchmark test results which can be misleading and may quickly become obsolete over time.
notation is a good guideline for algorithm and data structure performance. Furthermore, this measure of algorithm and data structure efficiency is timeless and is not dependent on hardware, implementation and data specifications, as opposed to benchmark test results which can be misleading and may quickly become obsolete over time.
Computer memory
Practical performance of index structures is not only governed by their algorithmic design, but also by the hardware that holds the data structure. Computer memory in essence is a hierarchical structure of layers, ordered from small, expensive, but fast memory to large, cheap and slow memory types. The hierarchy can roughly be divided into ‘main memory’, most notably RAM memory and caches, and secondary or ‘external memory’, which usually consists of hard disks or in the near future solid-state disks. Most index structures and applications are designed to run in main memory, because this allows for fast ‘random access’ to the data, whereas hard disks are usually 105–106 times slower for random access (24). As the price of biological data currently decreases much faster than the price of RAM memory and bioinformatics projects are becoming much larger, comparing more data than ever before, algorithms and data structures designed for cheaper external memory become more important (25). These external memory algorithms usually read data from external memory, process the information in main memory and output the result again to disk. As mentioned above, these ‘input/output’ (I/O) operations are very expensive. As a result, the algorithmic design needs to minimize these operations as much as possible, for example by keeping key information that is needed frequently into main memory. This technique, known as ‘caching’, is also used by file systems. File systems usually load more data into main memory than requested because it is physically located close to the requested data and may be predicted to become needed in the near future. The physical ‘locality’ of data organized by index structures is thus of great importance. Moreover, data that is often logically requested in sequential order, should also be physically ordered sequentially, because sequential disk access is almost as fast as random access in main memory. More information about index structure design for the different memory settings is found in ‘Popular index structures’, ‘Time-memory trade-offs’ and ‘Index structures in external memory’ sections.
Notations
The following notations are used throughout the rest of the text. Let the finite, totally ordered alphabet Σ be an array of size |Σ| (|·| will be used to denote the size of a string, set or array). The DNA-alphabet, for example, has size four and is given by Σ = {A,C,G,T}. Furthermore, let Σk and Σ*, respectively, be the set of all strings composed of k characters from Σ and the set of all strings composed of zero or more characters from Σ. The empty string will be denoted as ε. Let S ∈ Σn. All indexes in this article are zero-based. For every 0 ≤ i ≤ j < n, S[i] denotes the character at position i in S, S[i .. j] denotes a substring that starts at position i and ends at position j and S[i .. j ] = ε for i > j. S[i..] is the i-th suffix of S and S[..i] is the i-th prefix of S and S[−1..] = S[.. n] = ε. Likewise, A[i .. j] denotes an interval in an array A and the comma separator is used in 2D arrays, e.g. M[i, j] denotes the matrix element of M at the i-th row and j-th column.
S represents the indexed string which is usually very large, i.e. a chromosome or complete genome. Another string P denotes a pattern, which is searched in S. The length of P is m and usually m ≪ n holds, unless stated otherwise. For example, P can be a certain pattern, a sequencing read or a gene. The lexicographical order relation between two elements of Σ* is represented as <. The ‘longest common prefix’ LCP(S, P) of two strings S and P is the prefix S[..k], such that S[..k] = P[..k] and S[k + 1] ≠ P[k + 1].
As a final remark, note that all logarithms in this article have base two, unless stated otherwise.
POPULAR INDEX STRUCTURES
Index structures are data structures used to preprocess one or more strings to speed up string searches. As the examples in this section will illustrate, the types of searches can be quite diverse, yet some index structures manage to achieve an optimal performance for a broad class of search problems. The ultimate goal of index structures is to quickly capture maximal information about the string to be queried and to represent this information in a compact form. It turns out that both requirements often conflict in practice, with different types of index structures providing alternative trade-offs between speed and memory consumption. However, the speedup achieved over classical string searching algorithms often makes up for the extra construction and memory costs.
The type of index structures discussed here are ‘full-text index structures’. Unlike natural language, biological sequences do not show a clear structure of words and phrases, making popular ‘word-based’ index structures such as inverted files (26) and B-trees (27) less suited for indexing genomic sequences. Instead, full-text indexes that store information about all variable length substrings are better suited to analyze the complex nature of genome sequences.
The three most commonly used full-text index structures in bioinformatics today are suffix trees, suffix arrays and FM-indexes. The raison d'être of the latter two is the high-memory requirements of suffix trees. In this section, it is shown how those smaller indexes actually are reduced suffix trees and can be enhanced with auxiliary information to achieve complete suffix tree functionality.
Suffix trees
Suffix trees have become the archetypical index structure used in bioinformatics. Introduced by Weiner (28), who also gave a linear time construction algorithm, they are said to efficiently solve a myriad of string processing problems (29). Complex string problems such as finding the longest common substring can be solved in linear time using suffix trees. The suffix tree of a string S contains information about all suffixes of that string and gives access to all prefixes of those suffixes, thus effectively allows fast access to all substrings of the string S.
The suffix tree ST(S) is formally defined as the radix tree (30), i.e. a compact string search tree data structure, built from all suffixes of S. The edges of ST(S) are labeled with substrings of S and the leaves are numbered 0 to n − 1. The one-to-one correspondence between leaf i of ST(S) and suffix i of S is found by concatenating all edge labels on the path from the root to the leaf: the concatenated string ending in leaf i equals suffix S[i..]. Moreover, internal nodes correspond to the LCP of suffixes of S, such that labels of all outgoing edges from an internal node start with a different character and every internal node has at least two children. This last property allows to distinguish suffix trees and non-compact suffix ‘tries’ whose nodes can have single children because edge label lengths are all equal to one. In order for the above properties to hold for a string S, the last character of S has to uniquely appear in S. In practice, this problem is solved by appending a special end-character $ to the end of string S, with $∉Σ and $ < c, ∀c ∈ Σ. This special end-character plays the same role as the virtual end-of-string symbol used in regular expressions (also represented as $ in that context). Hereafter, for every indexed string S it is assumed S[n − 1] = $ or, equivalently, S ∈ Σ*$ holds. As a running example, the suffix tree ST(S) for the string S = ACATACAGATG$ is given in Figure 1.
Figure 1.

Suffix tree for string S = ACATACAGATG, where $ is the special end-character. Each number i inside a leaf represents suffix S[i..] of the string S. Dashed arrows correspond to suffix links. Edges are arranged in lexicographical order. For the sake of brevity, only the first characters followed by two dots and the special end-character $ are shown for edge labels that spell out the rest of the suffix corresponding to the leaf the edge is connected with.
The ‘label’ ℓ(v) of a node v of ST(S) is defined as the concatenation of edge labels on the path from the root to the node. From this definition it follows that ℓ(root) = ϵ. The ‘string depth’ of v is defined as |ℓ(v)|. The ‘suffix link’ sl(v) of an internal node v with label cw (c ∈ Σ and w ∈ Σ*) is the unique internal node with label w. Suffix links are represented as dashed lines in Figure 1.
Most suffix tree algorithms boil down to (partial or full) top-down or bottom-up traversals of the tree, or the following of suffix links (19). These different types of traversals are further illustrated using some classical string algorithms.
In the exact string matching problem, all positions of a substring P have to be found in string S. Exact string matching is an important problem on its own and is also used as a basis for more complex string matching problems. Since P is a substring of S if and only if P is a prefix of some suffix of S, it follows that matching every character of P along a path in ST(S) (starting at the root) gives the answer to the existential question. This algorithm thus requires a partial top-down traversal of ST(S) and has a time complexity of  (m). Since suffixes of S are grouped by common prefixes in ST(S), the set of leaves in the subtree below the path that spells out P represents all locations where P occurs in S. This set is denoted as occ(P, S) and can be obtained in
(m). Since suffixes of S are grouped by common prefixes in ST(S), the set of leaves in the subtree below the path that spells out P represents all locations where P occurs in S. This set is denoted as occ(P, S) and can be obtained in  (|occ(P, S)|) time. As an example, consider matching pattern P = AC to the running example in Figure 1. The algorithm first finds the edge with label A going down from the root and then continues down the tree along the edge labeled CA. After matching the character C, the algorithm decides that P is a substring of S. Furthermore, occ(P, S) = {0, 4} and thus P = S[0..1] = S[4..5]. This classical example already demonstrates the true power of suffix trees: the time complexity for matching k patterns of length m to a string of length n is
(|occ(P, S)|) time. As an example, consider matching pattern P = AC to the running example in Figure 1. The algorithm first finds the edge with label A going down from the root and then continues down the tree along the edge labeled CA. After matching the character C, the algorithm decides that P is a substring of S. Furthermore, occ(P, S) = {0, 4} and thus P = S[0..1] = S[4..5]. This classical example already demonstrates the true power of suffix trees: the time complexity for matching k patterns of length m to a string of length n is  (n + km). String matching algorithms that preprocess pattern P instead of string S [Boyer–Moore (31) and Knuth–Morris–Pratt (32), among others] require
(n + km). String matching algorithms that preprocess pattern P instead of string S [Boyer–Moore (31) and Knuth–Morris–Pratt (32), among others] require  (k(n + m)) time to solve the same problem. Since k and n are usually very large in most bioinformatics applications, for example in mapping millions (=k) short (=m) reads to the human genome (=n), this speedup is significant.
(k(n + m)) time to solve the same problem. Since k and n are usually very large in most bioinformatics applications, for example in mapping millions (=k) short (=m) reads to the human genome (=n), this speedup is significant.
Bottom-up traversals through suffix trees are mainly required for the detection of highly similar patterns, such as common substrings or (approximate) repeats. This follows from the fact that internal nodes of ST(S) represent the LCP of suffixes in their subtree. Internal nodes with maximal string depth correspond to suffixes with the largest LCP, which makes it easy to find maximal repeats and LCPs using a full bottom-up search of ST(S). In detail, the longest common substring of two strings S1 and S2 of lengths n1 and n2 is found by first building a suffix tree for the concatenated string S1S2, called a ‘generalized suffix tree’ (GST), and then traversing the GST twice. During an initial top-down traversal, string depths are stored at the internal nodes [if this information is gathered during construction of ST(S1S2), the top-down traversal can be skipped]. A consecutive bottom-up traversal determines whether leaves in the subtree of an internal node all originate from S1, S2 or both. This information can percolate up to parent nodes. In case leaves from both S1 and S2 have the current node as their ancestor, the corresponding suffixes have a common prefix. Since every internal node is visited at most once during each traversal, and calculations at every internal node can be done in constant time, this algorithm requires  (n1 + n2) time. The details of the algorithm can be found in (29). Maximal repeats, such as calculated in Vmatch (http://www.vmatch.de/), are found in a similar fashion. A maximal repeat is a substring of length l > 0 that occurs at least at two positions i1 < i2 in S and that is both left-maximal (S[i1 − 1] ≠ S[i2 − 1]) and right-maximal (S[i1 + l] ≠ S[i2 + l]). Labels of the internal nodes of ST(S) represent all repeated substrings that are right-maximal. There are, however, node labels that correspond to repeats that are not left-maximal. Similar to finding the longest common substring, a bottom-up traversal of ST(S) uses information in the leaves to check left-maximality and forwards this information to parent nodes. As an example, the maximal repeats in the running example (Figure 1) are ACA, AT, A and T. The first internal node v visited by a bottom-up traversal has ℓ(v) = ACA and v has two leaves: 0 and 4. Since leaf 0 is a child of v, left-maximality is guaranteed for v and every parent of v. The internal node w with label ℓ(w) = CA has leaves 5 and 1 as children, but because S[5 − 1] = S[1 − 1] = A, ℓ(w) = CA is not a maximal repeat.
(n1 + n2) time. The details of the algorithm can be found in (29). Maximal repeats, such as calculated in Vmatch (http://www.vmatch.de/), are found in a similar fashion. A maximal repeat is a substring of length l > 0 that occurs at least at two positions i1 < i2 in S and that is both left-maximal (S[i1 − 1] ≠ S[i2 − 1]) and right-maximal (S[i1 + l] ≠ S[i2 + l]). Labels of the internal nodes of ST(S) represent all repeated substrings that are right-maximal. There are, however, node labels that correspond to repeats that are not left-maximal. Similar to finding the longest common substring, a bottom-up traversal of ST(S) uses information in the leaves to check left-maximality and forwards this information to parent nodes. As an example, the maximal repeats in the running example (Figure 1) are ACA, AT, A and T. The first internal node v visited by a bottom-up traversal has ℓ(v) = ACA and v has two leaves: 0 and 4. Since leaf 0 is a child of v, left-maximality is guaranteed for v and every parent of v. The internal node w with label ℓ(w) = CA has leaves 5 and 1 as children, but because S[5 − 1] = S[1 − 1] = A, ℓ(w) = CA is not a maximal repeat.
A final way of traversing suffix trees is by following suffix links. Suffix links can both be used in suffix tree construction and algorithms for searching maximal exact matches or matching statistics. Intuitively, suffix links maintain a sliding window when matching a pattern to the suffix tree. Furthermore, suffix links act as a memory-efficient alternative to GSTs. As constructing, storing and updating suffix trees is a costly operation, the utilization of suffix links offers an important trade-off. The following algorithm demonstrates how suffix links enable a quick comparison between all suffixes of string S1 and the suffix tree ST(S2) of another string S2. Suppose the first suffix S1[0..] has been compared up to a node v with ℓ(v) = S2[0..i]. After following sl(v) = w, the second suffix S1[1..] is already matched to ST(S2) up to w, with ℓ(w) = S2[1..i]. In this way, i = |ℓ(w)| characters do not have to be matched again for this suffix. This process can be repeated until all suffixes of S1 are matched to ST(S2). Hence, the maximal exact matches between S1 and S2 can be found again in  (n1 + n2) time, but using less memory to store only the suffix tree of S2 plus its suffix links.
(n1 + n2) time, but using less memory to store only the suffix tree of S2 plus its suffix links.
Given enough fast memory, suffix trees are probably the best data structure ever invented to support string algorithms. For large-scale bioinformatics applications, however, memory consumption really becomes a bottleneck. Although the memory requirements of suffix trees are asymptotically linear, the constant factor involved is quite high, i.e. up to 10 (33) to 20 times (34) higher than the amount of memory required to store the input string. However, state-of-the-art suffix tree implementations are able to handle sequences of human chromosome size (10). During the last decade, a lot of research focused on tackling this memory bottleneck, resulting in many suffix tree variants that show interesting memory versus time trade-offs.
Suffix arrays
The most successful and well-known variants of suffix trees are the so-called suffix arrays (35). They are made up of a single array containing a permutation of the indexes of string S, making them extremely simple and elegant. In terms of performance, expressiveness is traded for lower memory footprint and improved locality. Suffix arrays in general only require four times the amount of storage needed for the input string, can be constructed in linear time and can exactly match all occurrences of pattern P in string S in  (mlogn+|occ(P, S)|) time using a binary search.
(mlogn+|occ(P, S)|) time using a binary search.
Suffix array SA(S) stores the lexicographical ordering of all suffixes of string S as a permutation of its index positions: S[SA[i − 1]..] < S[SA[i]..], 0 < i < n. The last column of Table 1 shows the lexicographical ordering for the running example. SA(S) itself can be found in the second column. The uniqueness of the lexicographical order is determined by the fact that all suffixes have different lengths, and the use of the special end-character $ < c, c ∈ Σ. By definition, S[SA[0]] always equals the string $. The relationship between suffix trees and suffix arrays becomes clear when traversing suffix trees depth-first and giving priority to edges with lexicographically smaller labels. Leaf numbers encountered in this order spell out the suffix array. All edges were lexicographically ordered on purpose in Figure 1, so that leaf numbers, read from left to right, form SA(S) as found in Table 1. Exact matching of substring P is done using two binary searches on SA(S). These binary searches locate PL = min{k|P ≤ S[SA[k]]} and PR = max{k|P ≥ S[SA[k]]}, which form the boundaries of the interval in SA(S) where occ(P, S) is found. Note that counting the occurrences requires  (mlogn) time, but finding occ(P, S) only requires an additional
(mlogn) time, but finding occ(P, S) only requires an additional  (|occ(P, S)|) time.
(|occ(P, S)|) time.
Table 1.
Arrays used by enhanced suffix arrays (columns 2–5), compressed suffix arrays (columns 2, 6 and 7) and FM-indexes (columns 8 – 14) for string S = ACATACAGATG$
| ESA |
CSA |
FM-index ‘rank’ |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| i | SA | LCP | child | sl | SA−1 | Ψ | BWT | $ | A | C | G | T | LF | S[SA[i]..] |
| 0 | 11 | −1 | 2 | 2 | G | 0 | 0 | 0 | 1 | 0 | 8 | $ | ||
| 1 | 4 | 0 | 6 | [0..11] | 7 | 6 | T | 0 | 0 | 0 | 1 | 1 | 10 | ACAGATG$ |
| 2 | 0 | 3 | 2 | [6..7] | 4 | 7 | $ | 1 | 0 | 0 | 1 | 1 | 0 | ACATACAGATG$ |
| 3 | 6 | 1 | 4 | [0..11] | 10 | 9 | C | 1 | 0 | 1 | 1 | 1 | 6 | AGATG$ |
| 4 | 2 | 1 | 5 | 1 | 10 | C | 1 | 0 | 2 | 1 | 1 | 7 | ATACAGATG$ | |
| 5 | 8 | 2 | 3 | [10..11] | 6 | 11 | G | 1 | 0 | 2 | 2 | 1 | 9 | ATG$ |
| 6 | 5 | 0 | 8 | 3 | 3 | A | 1 | 1 | 2 | 2 | 1 | 1 | CAGATG$ | |
| 7 | 1 | 2 | 7 | [1..5] | 9 | 4 | A | 1 | 2 | 2 | 2 | 1 | 2 | CATACAGATG$ |
| 8 | 10 | 0 | 10 | 5 | 0 | T | 1 | 2 | 2 | 2 | 2 | 11 | G$ | |
| 9 | 7 | 1 | 9 | [0..11] | 11 | 5 | A | 1 | 3 | 2 | 2 | 2 | 3 | GATG$ |
| 10 | 3 | 0 | 8 | 1 | A | 1 | 4 | 2 | 2 | 2 | 4 | TACAGATG$ | ||
| 11 | 9 | 1 | 11 | [0..11] | 0 | 8 | A | 1 | 5 | 2 | 2 | 2 | 5 | TG$ |
From left to right: index position, suffix array, LCP array, child array, suffix link array, inverse suffix array, Ψ-array, BWT text, ‘rank’ array, LF-mapping array and suffixes of string S. FM-indexes also require an array C(S).
Although conceptually simple, suffix arrays are not just reduced versions of suffix trees (36,37). Optimal solutions for complex string processing problems can be achieved by algorithms on suffix arrays without simulating suffix tree traversals. An example is the all pairs suffix–prefix problem in which the maximal suffix–prefix overlap between all ordered pairs of k strings of total length n can be determined by both suffix trees (29) and suffix arrays (37) in  (n + k2) time.
(n + k2) time.
Enhanced suffix arrays
Suffix arrays are not that information-rich compared with suffix trees, but require far less memory. They lack LCP information, constant time access to children and suffix links, which makes them less fit to tackle more complex string matching problems. Abouelhoda et al. (19) demonstrated how suffix arrays can be embellished with additional arrays to recover the full expressivity of suffix trees. These so-called ‘enhanced suffix arrays’ consist of three extra arrays that, together with a suffix array, form a more compact representation of suffix trees that can also be constructed in  (n) time. Furthermore, the next paragraphs demonstrate how the extra arrays of enhanced suffix arrays enable efficient simulation of all traversal types of suffix trees (19).
(n) time. Furthermore, the next paragraphs demonstrate how the extra arrays of enhanced suffix arrays enable efficient simulation of all traversal types of suffix trees (19).
A first array LCP(S) supports bottom-up traversals on suffix array SA(S). It stores LCP lengths of consecutive suffixes from the suffix array, i.e. LCP[i] = | LCP(S[SA[i − 1]..],S[SA[i]..])|, 0 < i < n. By definition, LCP[0] = −1. An example LCP array for the running example is shown in the third column of Table 1. Originally, Manber and Myers (35) utilized LCP arrays to speed up exact substring matching on suffix arrays to achieve an  (m+logn+|occ(P, S)|) time bound. Recently, Grossi (36) proved that the
(m+logn+|occ(P, S)|) time bound. Recently, Grossi (36) proved that the  (m + logn + |occ(P, S)|) time bound for exact substring matching can be reached by using only S, SA(S) and
(m + logn + |occ(P, S)|) time bound for exact substring matching can be reached by using only S, SA(S) and 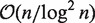 sampled LCP array entries. Furthermore, it is possible to encode those sampled LCP array entries inside a modified version of SA(S) itself. However, the details of this technique are rather technical and fall beyond the scope of this review. Later, Kasai et al. (38) showed how all bottom-up traversals of suffix trees can be mimicked on suffix arrays in linear time by traversing LCP arrays. In fact, LCP(S) represents the tree topology of ST(S). Recall that internal nodes of suffix trees group suffixes by their LCPs. In enhanced suffix arrays, internal nodes are represented by ‘LCP intervals’ ℓ-[i .. j]. Formally, an interval ℓ -[i .. j], 0 ≤ i < j < n is an LCP interval with ‘LCP value’ ℓ if for every i < k ≤ j: LCP[k] ≥ ℓ and there exists i < k ≤ j: LCP[k] = ℓ and LCP[i] < ℓ and LCP[j + 1] < ℓ. The LCP interval 0-[0..n − 1] is defined to correspond to the root of ST(S). Intuitively, an LCP interval is a maximal interval of minimal LCP length that corresponds to an internal node of ST(S). As an illustration, LCP interval 1-[1..5] with LCP value 1 of the example string S in Table 1 corresponds to internal node v with label ℓ(v) = A in Figure 1. Similarly, subinterval relations among LCP intervals relate to parent–child relationships in suffix trees. Abouelhoda et al. (19) have shown that the boundaries between LCP subintervals of LCP interval ℓ-[i .. j] are given by the ‘ℓ-indexes’ for which it holds that LCP[k] = ℓ, i < k ≤ j. Singleton intervals correspond to leaves in the suffix tree and non-singleton intervals correspond to internal nodes. Consider, for example, the LCP interval 1-[1..5] in the running example. Its ℓ-indexes are 3 and 4. The resulting subintervals are LCP intervals 3-[1..2] and 2-[4..5] and singleton interval [3..3]. The above definitions thus generate a virtual suffix tree called the ‘LCP interval tree’. Note that the topology of this tree is not stored in memory, but is traversed using the arrays SA(S) and LCP(S).
sampled LCP array entries. Furthermore, it is possible to encode those sampled LCP array entries inside a modified version of SA(S) itself. However, the details of this technique are rather technical and fall beyond the scope of this review. Later, Kasai et al. (38) showed how all bottom-up traversals of suffix trees can be mimicked on suffix arrays in linear time by traversing LCP arrays. In fact, LCP(S) represents the tree topology of ST(S). Recall that internal nodes of suffix trees group suffixes by their LCPs. In enhanced suffix arrays, internal nodes are represented by ‘LCP intervals’ ℓ-[i .. j]. Formally, an interval ℓ -[i .. j], 0 ≤ i < j < n is an LCP interval with ‘LCP value’ ℓ if for every i < k ≤ j: LCP[k] ≥ ℓ and there exists i < k ≤ j: LCP[k] = ℓ and LCP[i] < ℓ and LCP[j + 1] < ℓ. The LCP interval 0-[0..n − 1] is defined to correspond to the root of ST(S). Intuitively, an LCP interval is a maximal interval of minimal LCP length that corresponds to an internal node of ST(S). As an illustration, LCP interval 1-[1..5] with LCP value 1 of the example string S in Table 1 corresponds to internal node v with label ℓ(v) = A in Figure 1. Similarly, subinterval relations among LCP intervals relate to parent–child relationships in suffix trees. Abouelhoda et al. (19) have shown that the boundaries between LCP subintervals of LCP interval ℓ-[i .. j] are given by the ‘ℓ-indexes’ for which it holds that LCP[k] = ℓ, i < k ≤ j. Singleton intervals correspond to leaves in the suffix tree and non-singleton intervals correspond to internal nodes. Consider, for example, the LCP interval 1-[1..5] in the running example. Its ℓ-indexes are 3 and 4. The resulting subintervals are LCP intervals 3-[1..2] and 2-[4..5] and singleton interval [3..3]. The above definitions thus generate a virtual suffix tree called the ‘LCP interval tree’. Note that the topology of this tree is not stored in memory, but is traversed using the arrays SA(S) and LCP(S).
Fast top-down searches of suffix trees not only require their tree topology, but also constant time access to child nodes. For an LCP interval ℓ-[i .. j], this means constant time access to its ℓ-indexes. This information can be precomputed in linear time for the entire LCP interval tree and stored in another array of enhanced suffix arrays, the ‘child array’. The first ℓ-index is either stored in i or j [the exact location can be determined in constant time (19)] and the next ℓ-index is stored at the location of the previous ℓ-index. The child array for the running example is given in the fourth column of Table 1. As an example, again consider LCP interval 1-[1..5]. The first ℓ-index (3) is stored at position 5 and the second ℓ-index (4) is stored at position 3. Since child[4] = 5 is equal to the right boundary of the interval (which cannot equal ℓ by definition), 4 is the last ℓ-index. The child array allows enhanced suffix arrays to simulate top-down suffix tree traversals.
As a final step towards complete suffix tree expressiveness, suffix arrays can be enhanced with ‘suffix link arrays’ that store suffix links as pointers to other LCP intervals. These pointers are stored at the position of the first ℓ-index of an LCP interval because no two LCP intervals share the same position as their first ℓ-index (19). This property and the suffix link array for the running example can be checked in Table 1.
With three extra arrays added, enhanced suffix arrays support all operations and traversals on suffix trees using the same time complexity. However, the simple modular structure allows memory savings if not all traversals are required for an application. Furthermore, array representations generally show better locality than most standard suffix tree representations, which is important when converting the index to disk, but also improves cache usage in memory (39). Practical implementation improvements have further reduced memory consumption (40) of enhanced suffix arrays and have speeded up substring matching for larger alphabets (41). In practice, several state-of-the-art bioinformatics tools make use of enhanced suffix arrays for finding repeated structures in genomes (Vmatch), short read mapping (5) and genome assembly (16). If memory is a concern, enhanced suffix arrays occupy about the same amount of memory as regular suffix trees and are thus equally inapplicable for large strings. Suffix arrays (without enhancement) are preferred for exact substring matching in very large strings.
Compressed suffix arrays
Although suffix arrays are much more compact than suffix trees, their memory footprint is still too high for extremely large strings. The main reason stems from the fact that suffix arrays (and suffix trees) store pointers to string positions. The largest pointer takes  (log n) bits, which means that suffix arrays require
(log n) bits, which means that suffix arrays require  (nlog n) bits of storage. This is large compared with
(nlog n) bits of storage. This is large compared with  (nlog|Σ|) bits needed for storing uncompressed strings. A demand for smaller indexes that remain efficient gave rise to the development of ‘succinct indexes’ and ‘compressed indexes’. Succinct indexes require
(nlog|Σ|) bits needed for storing uncompressed strings. A demand for smaller indexes that remain efficient gave rise to the development of ‘succinct indexes’ and ‘compressed indexes’. Succinct indexes require  (n) bits of space, whereas the memory requirements of compressed indexes is in the order of magnitude of the compressed string (42).
(n) bits of space, whereas the memory requirements of compressed indexes is in the order of magnitude of the compressed string (42).
Many types of compressed suffix arrays (43) have already been proposed [see Navarro and Mäkinen for a recent review (42)]. They are usually centered around the idea of storing ‘suffix array samples’, complemented with a good compressible ‘neighbor array’ Ψ(S). To understand the role of the array Ψ(S), the concept of ‘inverse suffix arrays’ SA−1(S) is introduced for which holds that SA−1[SA[i]] ≡ SA[SA−1[i]] = i. Ψ(S) can then be defined as Ψ[i] ≡ SA−1[SA[i] + 1 mod(n − 1)] for 0 ≤ i < n. This definition closely resembles that of suffix links and it will thus come as no surprise that in practice Ψ can be used to recover suffix links (44). Consequently, the array Ψ can be used to recover suffix array samples from a sparse representation of SA(S). This is illustrated using the running example string from Table 1. Assume that only SA[0], SA[6] and SA[11] are stored and that the value of SA[10] is unknown. Note that Ψ[10] = 1 and SA[1] = 4 = 3 + 1, i.e. the requested value plus one. A sampled value of SA(S) is reached by repeatedly calculating Ψ[Ψ[..Ψ[10]]] = Ψk[10]. In the example k = 2, because Ψ[Ψ[10]] = 6. Consequently, SA[10] = SA[6] − k = 5 − 2 = 3. A more detailed discussion about compressed suffix arrays is given in the next section.
The Burrows–Wheeler transform
Several compressed index structures, most notably the FM-index (45), are based on the Burrows–Wheeler transform (46) BWT(S). This reversible permutation of the string S is also known to lie at the core of compression tools such as the fast ‘bzip2’ compression tool.
The Burrows–Wheeler transform does not compress a string itself, rather it enables an easier and stronger compression of the original string by exploiting regularities found in the string. Unlike SA(S) that is a permutation of the index positions of S, BWT(S) is a permutation of the characters of S. As a result, BWT(S) only occupies  (nlog|Σ|) bits of memory in contrast to
(nlog|Σ|) bits of memory in contrast to  (nlog n) bits needed for storing SA(S). As it contains the original string itself, the Burrows–Wheeler transform does not require an additional copy of S for string searching algorithms. Index structures having this property are called ‘self-indexes’.
(nlog n) bits needed for storing SA(S). As it contains the original string itself, the Burrows–Wheeler transform does not require an additional copy of S for string searching algorithms. Index structures having this property are called ‘self-indexes’.
Intuitively, the Burrows–Wheeler transformation orders the characters of S by the context following the characters. Thus, characters followed by similar substrings will be close together. A simple way to formally define BWT(S) uses a conceptual n × n matrix M whose rows are formed by the characters of the lexicographically sorted n cyclic shifts of S. BWT(S) is the string represented by the last column of M, or BWT[i] ≡ M[i, n − 1], 0 ≤ i < n. Note that the rows of M up to the character $ also represent the suffixes in lexicographical order, or, equivalently, in suffix array order. Thus, the first column of M equals the first characters of the suffixes in suffix array order, from which follows that BWT(S) can also be defined as BWT[i] ≡ S[SA[i] − 1 modn], 0 ≤ i < n, where the modulo operator is used for the case SA[i] = 0. From this definition it immediately follows that BWT(S) can be constructed in linear time using SA(S). BWT(S) for the running example can be found in Table 1, column 8, whereas the complete matrix M is given in Table 2.
Table 2.
Conceptual matrix M containing the lexicographically ordered n cyclic shifts of S = ACATACAGATG$
| i | S[SA[i]] | BWT[i] | offset[i] | LF[i] | |
|---|---|---|---|---|---|
| 0 | $ | ACATACAGAT | G | 0 | 8 |
| 1 | A | CAGATG$ACA | T | 0 | 10 |
| 2 | A | CATACAGATG | $ | 0 | 0 |
| 3 | A | GATG$ACATA | C | 0 | 6 |
| 4 | A | TACAGATG$A | C | 1 | 7 |
| 5 | A | ATG$ACATAC | G | 1 | 9 |
| 6 | C | AGATG$ACAT | A | 0 | 1 |
| 7 | C | ATACAGATG$ | A | 1 | 2 |
| 8 | G | $ACATACAGA | T | 1 | 11 |
| 9 | G | ATG$ACATAC | A | 2 | 3 |
| 10 | T | ACAGATG$AC | A | 3 | 4 |
| 11 | T | G$ACATACAG | A | 4 | 5 |
M[0..11,0] contains the lexicographically ordered characters of S and M[0..11,11] equals BWT(S). The last two columns are required for the inverse transformation. offset[i] stores the number of times BWT[i] has appeared earlier in BWT(S). The last column LF[i] contains pointers used during the inverse transformation algorithm: if S[i] = BWT[j], then BWT[LF[j]] = S[i − 1].
The inverse transformation that reconstructs S from BWT(S) is key to uncompression algorithms and the string matching algorithm utilized in compressed index structures. It recovers S back-to-front and is based on a few simple observations. First, although BWT(S) only stores the last column of M, the first column of M is easily retrieved from BWT(S) because it is the lexicographical ordering of the characters of S [and thus also BWT(S)]. Moreover, the first column of M can be represented in compact form as an array C(S) that stores the number of characters in S that are lexicographically smaller than character c ∈ Σ. More precisely: 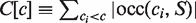 , ci ∈ Σ. For the running example, C(S) = [0,1,6,8,10] can be retrieved from Table 2. A second observation is that BWT(S) stores the order of characters preceding the suffixes in suffix array order. As a result, if the character at position i (S[i]) has been decoded and the lexicographical order of suffix S[i..] is known to be j, character S[i − 1] is found in BWT[j]. Finally, the most important observation that allows for the retrieval of S from BWT(S) is that identical characters preserve their relative order in the first and last columns of M. To see the correctness of this observation, let BWT[i] = BWT[j] = c for i < j. The lexicographical ordering of the cyclic permutations means that the suffix in row i of M corresponding to SA[i] is lexicographically smaller than the suffix in row j corresponding to SA[j]. From cS[SA[i]..] < cS[SA[j]..] it then follows that the location of character c corresponding to BWT[i] precedes the location of character c corresponding to BWT[j] in the first column of M. The relative order of identical characters in BWT(S) is captured in the array offset(S): offset[i] stores the number of times that character BWT[i] occurs in BWT(S) before position i, i.e. offset[i] ≡ |occ(BWT[i],BWT[..i − 1])|, 0 < i < n. Given a position i in BWT(S), the corresponding character in the first column of M can then be found at position LF[i] = C[BWT[i]] + offset[i]. The array LF(S) is called the ‘last-to-first column mapping’.
, ci ∈ Σ. For the running example, C(S) = [0,1,6,8,10] can be retrieved from Table 2. A second observation is that BWT(S) stores the order of characters preceding the suffixes in suffix array order. As a result, if the character at position i (S[i]) has been decoded and the lexicographical order of suffix S[i..] is known to be j, character S[i − 1] is found in BWT[j]. Finally, the most important observation that allows for the retrieval of S from BWT(S) is that identical characters preserve their relative order in the first and last columns of M. To see the correctness of this observation, let BWT[i] = BWT[j] = c for i < j. The lexicographical ordering of the cyclic permutations means that the suffix in row i of M corresponding to SA[i] is lexicographically smaller than the suffix in row j corresponding to SA[j]. From cS[SA[i]..] < cS[SA[j]..] it then follows that the location of character c corresponding to BWT[i] precedes the location of character c corresponding to BWT[j] in the first column of M. The relative order of identical characters in BWT(S) is captured in the array offset(S): offset[i] stores the number of times that character BWT[i] occurs in BWT(S) before position i, i.e. offset[i] ≡ |occ(BWT[i],BWT[..i − 1])|, 0 < i < n. Given a position i in BWT(S), the corresponding character in the first column of M can then be found at position LF[i] = C[BWT[i]] + offset[i]. The array LF(S) is called the ‘last-to-first column mapping’.
The above observations allow the back-to-front recovery of S from BWT(S) utilizing a zig-zag algorithm. Starting in row i0 of BWT(S) containing character $, the position of the previous character of S is found in row LF[i0] = i1. The next preceding character is found on row i2 = LF[i1] in BWT(S), and so on. Thus, to find the row of the next preceding character, the algorithm looks horizontally in Table 2 and the actual character is retrieved from the BWT column on that row in Table 2. Note that neither M nor its first column are ever used explicitly during the algorithm. They only serve to understand the procedure for the inverse transformation. In practice, C(S) and offset(S) are first constructed from BWT(S). During each step, LF[ik] is calculated using C(S) and offset(S) and BWT[LF[ik]] is returned as the preceding character. As an example, M, offset(S) and LF(S) for the running example can be found in Table 2 and C(S) is given above. S[SA[0]] = $ is preceded by the character BWT[i0] = G in the running example. Consequently, G$ is the lexicographical first suffix that starts with G, which translates into offset[i0] = 0. The first row of M whose corresponding suffix starts with G has row number C[G] = 8. Adding the number of suffixes that also start with G, but are lexicographically smaller than G$ (=0), returns the position in BWT(S) of the next character that will be decoded. BWT[8 + 0] = BWT[LF[0]] = T = S[9]. In the next step, S[8] is retrieved by computing LF[8] = 11 and BWT[11] = A. Eventually, S is retrieved in  (n) time using the LF-mapping.
(n) time using the LF-mapping.
The Burrows–Wheeler transform by itself only permutes strings without compressing them. It is, however, easier to compress BWT(S) than the original string S, as the order of the characters in BWT(S) is determined by similar contexts following the characters, analogous to the way suffixes are grouped by LCPs in suffix trees. An immediate consequence is that run-length encoding, which encodes runs of identical characters by their length, shows good compression results for BWT(S). Apart from run-length encoding (45,47), move-to-front lists (45), wavelet trees (42,47,48) and several entropy encoders, such as Huffman codes (49,50), have also been used successfully to compress BWT(S). For a complete overview on compression techniques based on the Burrows–Wheeler transform, we refer to the book of Adjeroh et al. (51).
Analogous to suffix arrays, BWT(S) can be used to find exact matches of substrings by applying binary search. Similar to compressed suffix arrays, binary searching BWT(S) requires auxiliary data structures, including Ψ(S) and (sampled) SA(S) (51), resulting in compressed suffix arrays. Given the relation between BWT(S) and SA(S), BWT(S) can also be utilized for constructing other compressed suffix arrays (52). Moreover, suffix trees, suffix arrays and other non-self-indexes require a copy of the indexed string S, which can be replaced by a compressed form of BWT(S) to reduce space.
FM-indexes
Another search method for exact string matching can be applied to Burrows–Wheeler transformed strings, using ideas from the inverse transformation algorithm. This method is referred to as ‘backward searching’ and forms the basic search mechanism of ‘FM-indexes’ (45). FM-index is the short name given by Ferragina and Manzini to their full-text self-indexes that require ‘minute amount of space’. The space requirement is proportional to and sometimes even smaller than that of the indexed string. FM-indexes can be constructed in  (n) time and all occurrences of pattern P can be located in
(n) time and all occurrences of pattern P can be located in  (m + |occ(P, S)|log n) time. Note that finding |occ(P, S)| only requires
(m + |occ(P, S)|log n) time. Note that finding |occ(P, S)| only requires  (m) time, which makes that FM-indexes have theoretical optimal time and space requirements for counting the number of occurrences of a pattern in a string.
(m) time, which makes that FM-indexes have theoretical optimal time and space requirements for counting the number of occurrences of a pattern in a string.
The backward search algorithm employed by FM-indexes requires BWT(S), C(S) and a 2D n × |Σ| array rank(S) [In many papers, rank(S) is referred to as Occ(S), but to avoid confusion with occ(P, S), the name ‘rank’ is used]. This array is defined as rank[i, c] ≡ |occ(c, BWT[..i])|, 0 ≤ i < n, c ∈ Σ. For the running example, rank(S) is shown as columns 9 – 13 in Table 1. The role of rank(S) is similar to the role offset(S) plays in the inverse transformation of BWT(S). However, while offset(S) only stores information on the number of occurrences of one character for each index position, rank(S) contains this information for all the characters in the alphabet in all index positions. The extra information contained in rank(S) compared with offset(S) gives it the advantage of granting random access to LF(S). Furthermore, rank(S) is easier to compress than offset(S) or LF(S) (51).
During the course of the search algorithm, P is matched from right to left. For every step i, 0 ≤ i < m, an interval BWT[si .. ei] is maintained that contains all occurrences of P[m − i..]. Initially, [s0 .. e0] ≡ [0..n − 1], and after m steps [sm .. em] contains the suffix array interval corresponding to occ(P, S). Given [si .. ei] and c = P[m − i − 1], the next interval is found using the formulas si+1 = C[c] + rank[c, si − 1] and ei+1 = C[c] + rank[c, ei + 1] − 1. Here, array C(S) is used to locate the interval of suffixes starting with c in SA(S) and array rank(S) is used to find the number of suffixes starting with c that are lexicographically smaller and larger than the ones prefixed by cP[m − i..]. As an example of backward searching, again consider matching P = CA to the running example in Table 1. Initially, the backward search interval is [0..11]. Since C[A] = 1 and C[C] = 6, the backward search interval narrows down to [s1..e1] = [1..5] in the next step, which corresponds to the suffix array interval containing suffixes starting with A. Note that BWT[3] = BWT[4] = C, so there are two suffixes starting with A that are preceded by C. Consequently, s2 = C[C] + rank[0, C] = 6 + 0 = 6 and e2 = C[C] + rank[5, C] − 1 = 6 + 2 − 1 = 7. The answer |occ(P, S)| = 7 − 6 + 1 = 2 is found in  (m) time. rank[0, C] = 0 means that there are no suffixes starting with C located in SA[0..0] and rank[5, C] = 2 means that there are 2 suffixes starting with C located in SA[0..5]. Also note the resemblance between LF-mapping and backward search: s2 also could have been found as the first occurrence of C in BWT[1..5], which is 3: LF[3] = 6 = s2. Likewise, e2 could have been found as the last occurrence of C in BWT[1..5]. However, instead of locating these occurrences, note that offset[3] = rank[3, C] − 1 = rank[1, C] − 1. Thus, the offset(S) values are stored in rank(S) at the boundaries of every interval, allowing search intervals to be narrowed down in constant time. As a result, the reverse search algorithm of the FM-index simulates a top-down search in a suffix ‘trie’, i.e. a suffix tree where every edge label contains only a single character.
(m) time. rank[0, C] = 0 means that there are no suffixes starting with C located in SA[0..0] and rank[5, C] = 2 means that there are 2 suffixes starting with C located in SA[0..5]. Also note the resemblance between LF-mapping and backward search: s2 also could have been found as the first occurrence of C in BWT[1..5], which is 3: LF[3] = 6 = s2. Likewise, e2 could have been found as the last occurrence of C in BWT[1..5]. However, instead of locating these occurrences, note that offset[3] = rank[3, C] − 1 = rank[1, C] − 1. Thus, the offset(S) values are stored in rank(S) at the boundaries of every interval, allowing search intervals to be narrowed down in constant time. As a result, the reverse search algorithm of the FM-index simulates a top-down search in a suffix ‘trie’, i.e. a suffix tree where every edge label contains only a single character.
After backward searching has terminated, occ(P, S) is still unknown. Using LF-mapping, this set can be retrieved from the interval BWT[sm .. em]. One possibility is to count the number of backward searches it takes to reach character $ for every sm ≤ i ≤ em. However, this would require too much time. To achieve better performance, FM-indexes mark additional positions with suffix array values in BWT(S). The number of suffix array values stored constitutes a time-space trade-off. Recall that LF[i] returns the position in SA(S) of suffix S[SA[i] − 1..]. Thus SA[LF[i]] = SA[i] − 1, such that LF(S) can be used to find the next smaller suffix array value. The ability of LF(S) to find smaller suffix array values is used as an argument to classify FM-indexes as compressed suffix arrays (45). Moreover, LF(S) and Ψ(S) are each others' inverse: SA[LF[i]] = SA[i] − 1 and SA[Ψ[i]] = SA[i] + 1, hence LF[Ψ[i]] = Ψ[LF[i]] = i.
FM-indexes combine fast string matching with low memory requirements. Their original design (45) compresses BWT(S) using move-to-front lists, run-length encoding and a variable-length prefix code. In the original paper, rank(S) was compressed using the ‘Four-Russians’ technique (53). Roughly speaking, this technique comes down to subdividing the problem into small enough subproblems and indexing all solutions to these small problems in a global table. The subdivision into smaller subproblems is done by recursively splitting arrays into equally sized blocks and storing answers to queries relative to the larger parent block. Other compression methods have been proposed that show better performance in practice (49) or that give different space-time trade-offs (47,48,50,54,55).
Since they allow fast pattern matching while having small memory requirements, FM-indexes have become a very popular tool for different types of genome analyses. Compressed full-text index structures are mainly used for exact string matching, but algorithms for inexact string matching exist (51,56). FM-indexes have started to become used as part of de novo genome assembly algorithms (17) and are supporting popular tools for mapping reads to reference sequences such as Bowtie (7), BWA (8) and SOAP2 (9).
TIME-MEMORY TRADE-OFFS
The increase in sequencing data requires efficient algorithms and data structures to form the backbone of computational tools for storing, processing and analyzing these sequences. Without the use of index structures, many algorithms that rely on string searching would become unfeasible due to a long execution time. However, index structures also incur a memory overhead to sequence analysis.
Over the last decade, much energy has been put into decreasing the memory consumption of index structures. The proposals differ in the performance overhead incurred by lowering the memory footprint. Some index structures suffer from a logarithmic slowdown, while others allow for the tuning of the space-time trade-off. There are indexes that have been especially designed for certain types of data, whereas others are tweaked for particular hardware architectures. An example of a data-specific property influencing index structure performance is the alphabet size of the sequences. Another major factor that allows classifying index structures is their expressiveness. Suffix trees are considered to have full expressiveness (29), supporting a large variety of string algorithms. Conversely, the bulk of recent compressed self-index structures are limited to performing mainly (in)exact string matching. These string matching self-indexes are often compared on the basis of four criteria: their performance of extracting a random substring of S, calculating |occ(P, S)| and occ(P, S) and their size. An overview of the memory taken by several index structures discussed in this section can be found in Table 3. This table represents memory requirements both in general terms of number of bits required per indexed character, as well as in terms of its size for indexing full genomes. Note, however, that the list of index structures in Table 3 is not complete nor gives a full overview of the memory-time trade-offs. For example, external memory index structures were omitted, but can be found in ‘Index structures in external memory’ section. Additionally, peak memory requirements during construction can be much higher than the figures described here (see ‘Construction’ section). Furthermore, index structures contain parameters that allow manual tuning of the memory-time trade-off. Finally, because the expressiveness differs greatly between index structures, Table 3 does not include any time-related results. Partial results for some algorithms can be found elsewhere (39,54,57,58).
Table 3.
Representative memory requirements for different index structure implementations, expressed both as bits per indexed character (column 2) and estimated size in megabytes for several known genomes (columns 3–5)
| Name index structure | Bits/char | Size for genome in MB |
Reference | ||
|---|---|---|---|---|---|
| Yeast | Fruit fly | Human | |||
| 2-bit encoded string | 2 | 3 | 35 | 775 | NCBIa |
| CSA Grossi et al. | 2.4 | 4 | 42 | 931 | (59,60) |
| FM-index | 3.36 | 5 | 59 | 1302 | (45,39) |
| SSA (best) | 4 | 6 | 70 | 1551 | (47,57) |
| CST Russo et al.b | 5 | 8 | 87 | 1939 | (61,62) |
| CSA Sadakane (best) | 5.6 | 8 | 98 | 2171 | (63,64) |
| LZ-index (best) | 6.64 | 10 | 116 | 2574 | (57) |
| byte encoded string | 8 | 12 | 139 | 3102 | NCBIa |
| CST Navarrob | 12 | 18 | 209 | 4653 | (62) |
| SSA (worst) | 20 | 30 | 349 | 7754 | (47,57) |
| CST Sadakaneb | 30 | 45 | 523 | 11 632 | (44,62) |
| LZ-index (worst) | 35.2 | 53 | 614 | 13 648 | (65,39) |
| Suffix array | 40 | 60 | 697 | 15 509 | (35) |
| Enhanced SA | 72 | 109 | 1255 | 27 916 | (19) |
| WOTD suffix tree | 76 | 115 | 1325 | 29 467 | (33) |
| ST McCreight | 232 | 350 | 4045 | 89 952 | (34,33) |
Column 6 contains references to the original theoretical proposals and an additional reference to the articles from which these practical estimates originate. For ease of comparison purposes, the index structures are sorted by increasing memory requirements. As a reference, the original (non-indexed) sequence is also included (bold), both stored using 2-bit encoding and byte encoding.
aGenome sizes were taken from the NCBI genome information pages http://www.ncbi.nlm.nih.gov/genome of Saccharomyces cerevisiae (yeast), Drosophila melanogaster (fruit fly) and Homo Sapiens (human).
bMean of the interval of possible memory requirements given in (62).
The remainder of this section focuses on the basic principles behind these index structures and the memory-time trade-offs induced by design choices and confounding factors such as application and data types.
Uncompressed index structures
Choosing appropriate data structures for implementing the different components of suffix trees forms a basic step in lowering their memory requirements. These components include nodes, edges, edge labels, leaf numbers and suffix links. The topological information of ST(S) and the edge labels are traditionally stored as pointers, resulting in suffix trees that require  (n) words of usually 32 bits. Note that for very large strings (n > 232 ≈ 4·109) 32 bits is insufficient for storing the pointers, thus larger representations are required. This factor is often overlooked when presenting theoretical results.
(n) words of usually 32 bits. Note that for very large strings (n > 232 ≈ 4·109) 32 bits is insufficient for storing the pointers, thus larger representations are required. This factor is often overlooked when presenting theoretical results.
There is only one major  (|Σ|)-sized memory-time trade-off in this traditional representation. This trade-off comes from the data structure that handles access to child vertices. Most implementations make use of—roughly ordered from high-memory requirements to low access time—static arrays, dynamic arrays (39), hash tables, linked lists and layouts with only pointers toward the first child and next sibling. Furthermore, mixed data structures that represent vertices with different numbers of children have also been proposed (66). Note that for DNA sequences, |Σ| is very small, turning array implementations into a workable solution. Also note that algorithms that perform full suffix tree traversals, such as repeat finding and many other string problems (29), do not suffer from a performance loss when implemented with more memory-efficient data structures.
(|Σ|)-sized memory-time trade-off in this traditional representation. This trade-off comes from the data structure that handles access to child vertices. Most implementations make use of—roughly ordered from high-memory requirements to low access time—static arrays, dynamic arrays (39), hash tables, linked lists and layouts with only pointers toward the first child and next sibling. Furthermore, mixed data structures that represent vertices with different numbers of children have also been proposed (66). Note that for DNA sequences, |Σ| is very small, turning array implementations into a workable solution. Also note that algorithms that perform full suffix tree traversals, such as repeat finding and many other string problems (29), do not suffer from a performance loss when implemented with more memory-efficient data structures.
In practice, suffix trees and suffix arrays require between 34n and 152n bits of memory. The suffix tree implementations described by Kurtz (66) perform very well and are implemented in the latest release of MUMmer (10), an open-source sequence analysis tool. The implementation in MUMmer allows indexing DNA sequences up to 250 Mbp on a computer with 4 GB of memory. Single human chromosomes are thus well within reach of standard suffix trees. Another implementation by Giegerich et al. (33) is even smaller, but lacks suffix links. Enhanced suffix arrays (19) also reach full expressiveness of suffix trees, as described in the previous section. When carefully implemented, they require anything between 40n and 72n bits. Enhanced suffix arrays use a linked list to represent the vertices of the tree. However, the  (|Σ|) performance penalty for string matching can be reduced to
(|Σ|) performance penalty for string matching can be reduced to  (|logΣ|) (41). Furthermore, enhanced suffix arrays form the basis of the Vmatch program that finds different types of exact and approximate repeats in sequences of several hundreds of Mbp in a few seconds. Moreover, according to a comparison between several implementations of suffix trees and enhanced suffix arrays (39), enhanced suffix arrays show the best overall performance for both the memory footprint and the traversal times. Finally, their modular design allows replacing some arrays by a compressed counterpart to further reduce space.
(|logΣ|) (41). Furthermore, enhanced suffix arrays form the basis of the Vmatch program that finds different types of exact and approximate repeats in sequences of several hundreds of Mbp in a few seconds. Moreover, according to a comparison between several implementations of suffix trees and enhanced suffix arrays (39), enhanced suffix arrays show the best overall performance for both the memory footprint and the traversal times. Finally, their modular design allows replacing some arrays by a compressed counterpart to further reduce space.
Sparse indexes
An intuitive solution for decreasing index structure memory requirements is sparsification or sampling of suffixes or array indexes. ‘Sparse suffix trees’ (67) and ‘sparse suffix arrays’ (68) adopt the idea of utilizing a sparse set of suffixes, whereas compressed suffix arrays and trees sample values in Ψ(S), C(S), rank(S) and other arrays involved in their design. As a consequence of sparsification, more string comparisons and sequential string searches are required. This, however, gives the opportunity to optionally tweak the size of the index structure based on the available memory. Although compressed index structures have received more attention in bioinformatics applications, sparse suffix arrays have been successfully used for exact pattern matching, retrieval of maximal exact matches (69) and read alignment (70). Furthermore, splitting indexes over multiple sparse index structures has been used for index structures that reside on disk (71) and for distributed query processing (72).
Word-based index structures are special cases of sparse index structures which only sample one suffix per word. Although word-based index structures are most popular in the form of inverted files, word-based suffix trees (73,74) and suffix arrays (68) also exist. Although it is possible to divide biological sequences into ‘words’, word-based index structures are generally designed to answer pattern matching queries on natural language data. On natural language data, Transier and Sanders (75) found that inverted files outperformed full-text indexes by a wide margin. Unfortunately, the inverted files were not compared against word-based implementations of suffix trees and suffix arrays. A somewhat dual approach was taken by Puglisi et al. (76), who adapted inverted files to become full-text indexes able to perform substring queries. They found compressed suffix arrays to generally outperform inverted files for DNA sequences, but the opposite conclusion was drawn for protein sequences. It turns out that compressed suffix arrays perform relatively better compared with inverted files when searching for patterns having fewer occurrences. Note that both comparative studies were performed in primary memory.
Compressed index structures
Compressed and succinct index structures are currently the most popular forms of index structures used in bioinformatics. Index structures such as compressed suffix arrays and FM-indexes are gradually built into state-of-the-art read mapping tools and other bioinformatics applications. Where traditional index structures require  (nlog n) bits of storage, succinct index structures require
(nlog n) bits of storage, succinct index structures require  (n) bits and the memory footprint of compressed index structures is defined relative to the ‘empirical entropy’ (77) of a string. Furthermore, these self-indexes contain S itself, thus saving again
(n) bits and the memory footprint of compressed index structures is defined relative to the ‘empirical entropy’ (77) of a string. Furthermore, these self-indexes contain S itself, thus saving again  (n) bits. Theoretically, this means that the size of compressed index structures can become a fraction of S itself. In practice, however, DNA and protein sequences do not compress very well (2,70). For this reason, the size of compressed index structures is roughly similar to the size of storing S using a compact bit representation. The major disadvantage of compressed index structures is the logarithmic increase in computation time for many string algorithms. This is, however, not the case for all string algorithms. For example, calculating |occ(P, S)| can still be done in
(n) bits. Theoretically, this means that the size of compressed index structures can become a fraction of S itself. In practice, however, DNA and protein sequences do not compress very well (2,70). For this reason, the size of compressed index structures is roughly similar to the size of storing S using a compact bit representation. The major disadvantage of compressed index structures is the logarithmic increase in computation time for many string algorithms. This is, however, not the case for all string algorithms. For example, calculating |occ(P, S)| can still be done in  (m) time for some compressed indexes. These internal differences between compressed index structures result from their complex nature, as they combine ideas from classical index structures, compression algorithms, coding strategies and other research fields. In the following paragraphs, the conceptual differences of state-of-the-art compressed index structures are surveyed, illustrated with theoretical and practical comparisons wherever possible. A more technical review is found in (42).
(m) time for some compressed indexes. These internal differences between compressed index structures result from their complex nature, as they combine ideas from classical index structures, compression algorithms, coding strategies and other research fields. In the following paragraphs, the conceptual differences of state-of-the-art compressed index structures are surveyed, illustrated with theoretical and practical comparisons wherever possible. A more technical review is found in (42).
Auxiliary data structures
Understanding the organization details and properties of compressed index structures requires prior knowledge of the auxiliary data structures involved in their design. Compressed indexes consist of many auxiliary structures that influence their memory-time trade-off, and have properties that dictate their expressiveness and performance for certain types of data. Representation of these auxiliary structures forms an active field of research. What follows is a brief summary of several commonly used auxiliary structures, not including the rather technical implementation details.
Almost all compressed index structures make use of bit vectors B to support random access and rank(B) and select(B) queries. Intuitively, rank(B) queries count the number of zeroes or ones before a certain index in the vector. Dual to this, select(B) queries return the position in B of the i-th zero or one. They often play a role in granting random access to a compressed or permutated string. Their usefulness, however, goes further than being mere building blocks of compressed index structures. For example, they can also be used to succinctly represent de Bruijn graphs (15), a typical data structure used in de novo genome assembly. Formally, rank(B) is represented as a 2D array defined by rank[i, c] ≡ |occ(c, B[..i])|, 0 ≤ i < |B|, c ∈ {0, 1}, similar to rank(S) for FM-indexes. select(B) is defined as select[i, c] ≡ j iff i = rank[j, c], 0 ≤ i < |occ(c, B)|, c ∈ {0, 1}. These data structures and their generalizations to non-binary strings strongly influence the memory-time trade-off of compressed index structures (48). As an example, the array rank(S) used in FM-indexes takes up to half of its size. As is the case for other data structures, there is no single optimal implementation for every application, but many proposals exist (78–80). The performance also depends on the restrictions imposed by the compressed index structure or the properties of the data, such as the sparsity of the original bit vector. From extremely sparse to more balanced, the best implementations require 0.2n bits (80) (1% ones), 0.8n bits (79) (20% ones) and 1.4n bits (80) (50% ones).
The above results for bit vectors have been generalized to non-binary strings (48,79), as worked with in many applications, including FM-indexes. A simple idea toward such a generalization is to create |Σ| bit vectors Bc, with Bc[j] = 1 iff S[j] = c. However, this entails an overhead both in time (random access to S) and memory. A careful implementation allows eliminating this overhead (48), but ‘wavelet trees’ (59) form an even more elegant solution.
Wavelet trees are balanced binary trees with |Σ| leaves. Every node v in the tree represents a subsequence S′ of S formed by the concatenation of all characters that belong to some interval Σ[i .. j]. The two children of v are the subsequences formed by the concatenation of all characters of S′ that belong to 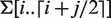 and
and 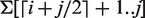 respectively. Vertex v itself is represented by a bit vector B of size |S′| that is defined as B[i] = 0 iff
respectively. Vertex v itself is represented by a bit vector B of size |S′| that is defined as B[i] = 0 iff 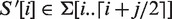 . Furthermore, B is preprocessed as to resolve rank(B) and select(B) in constant time. The wavelet tree for BWT(S) of the running example is shown in Figure 2, and has the same functionality as BWT(S) and rank(S). From this figure, BWT[9] can be found as follows. The root bit vector learns that BΣ[9] = 0, meaning that BWT[9] is a character from the first half of the alphabet. Since BΣ[9] is the sixth occurrence of 0 in BΣ (rank[9,0] = 5), it corresponds to B$AC[5] (zero-based index). Repetition of this procedure for the vertices corresponding to S$AC = $CCAAAAA and S$A = $AAAAA yields BWT[9] = A. rank(S) queries can be resolved in a similar way. Further research on wavelet trees gave rise to Huffman-shaped wavelet trees (60) and non-binary wavelet trees (48). This elegant, but somewhat complex data structure, has become very popular in index structure design. As an example result, all maximal repeats occurring in the complete human genome could be found in <17 h on a desktop PC (81) with 8 GB internal memory using an index structure based on the Burrows–Wheeler transform combined with a sparse wavelet tree implementation of the LCP array. Similar tests using suffix trees or enhanced suffix arrays failed due to the memory bottleneck.
. Furthermore, B is preprocessed as to resolve rank(B) and select(B) in constant time. The wavelet tree for BWT(S) of the running example is shown in Figure 2, and has the same functionality as BWT(S) and rank(S). From this figure, BWT[9] can be found as follows. The root bit vector learns that BΣ[9] = 0, meaning that BWT[9] is a character from the first half of the alphabet. Since BΣ[9] is the sixth occurrence of 0 in BΣ (rank[9,0] = 5), it corresponds to B$AC[5] (zero-based index). Repetition of this procedure for the vertices corresponding to S$AC = $CCAAAAA and S$A = $AAAAA yields BWT[9] = A. rank(S) queries can be resolved in a similar way. Further research on wavelet trees gave rise to Huffman-shaped wavelet trees (60) and non-binary wavelet trees (48). This elegant, but somewhat complex data structure, has become very popular in index structure design. As an example result, all maximal repeats occurring in the complete human genome could be found in <17 h on a desktop PC (81) with 8 GB internal memory using an index structure based on the Burrows–Wheeler transform combined with a sparse wavelet tree implementation of the LCP array. Similar tests using suffix trees or enhanced suffix arrays failed due to the memory bottleneck.
Figure 2.

Wavelet tree for indexing string S = GT$CCGAATAAA. Only the binary strings are stored in practice. Subsequences of S are shown only to ease the interpretation. This figure does not include data structures for resolving rank and select queries for every bit vector. For this small example, however, the answer to these queries is straightforward.
Other important index structure building blocks are auxiliary tree representations. Index structures use various types of trees, but a common design problem is the representation of their topology. As an example, suffix tree topology is traditionally implemented using pointers, requiring  (nlogn) bits of memory. In contrast, a popular way to succinctly represent tree topology by a sequence of balanced parentheses (82) only requires 2n + o(n) bits of memory. This implementation represents nodes in the tree as a pair of parentheses ‘()’. The nested structure of the parentheses then represents the tree (83), similar to a reduced form of the known Newick Tree Format (84). More tree operations are generally supported in constant or near-constant time by succinct tree topology representations compared with classical pointer-based representations, which only supports top-down traversals in constant time. Node depth, subtree size and the lowest common ancestor of two nodes (85) are examples of properties that can be retrieved in constant time from succinct representations where pointer-based representations require additional data structures to achieve the same performance. In theory, this means that a highly expressive suffix tree topology can be stored using 4n + o(n) bits instead of 64n bits using 32-bit pointers. Note, however, that the o(n)-term may become large in practice and even surpass the higher order term. For biological sequences, tests (83) show that these representations require something in between 2.1 and 4.84 bits per node, which has to be multiplied by 2n nodes in the worst case.
(nlogn) bits of memory. In contrast, a popular way to succinctly represent tree topology by a sequence of balanced parentheses (82) only requires 2n + o(n) bits of memory. This implementation represents nodes in the tree as a pair of parentheses ‘()’. The nested structure of the parentheses then represents the tree (83), similar to a reduced form of the known Newick Tree Format (84). More tree operations are generally supported in constant or near-constant time by succinct tree topology representations compared with classical pointer-based representations, which only supports top-down traversals in constant time. Node depth, subtree size and the lowest common ancestor of two nodes (85) are examples of properties that can be retrieved in constant time from succinct representations where pointer-based representations require additional data structures to achieve the same performance. In theory, this means that a highly expressive suffix tree topology can be stored using 4n + o(n) bits instead of 64n bits using 32-bit pointers. Note, however, that the o(n)-term may become large in practice and even surpass the higher order term. For biological sequences, tests (83) show that these representations require something in between 2.1 and 4.84 bits per node, which has to be multiplied by 2n nodes in the worst case.
Retrieval of the lowest common ancestor of two nodes v1 and v2, mentioned in the previous paragraph, is a fundamental operation for inexact string matching algorithms (29). Denoted by LCA(v1, v2), it is defined as the unique node v3 for which holds that ℓ(v3) = ℓ(LCA(v1, v2)) ≡ LCP(ℓ(v1), ℓ(v2)). This operation is supported by a combination of LCP arrays and data structures for resolving range minimum queries (86). This directly follows from the definition of LCA(v1, v2). Range minimum query data structures return the positions of the smallest values in any interval of an array. In LCP arrays, they return the length of the LCP of any two suffixes. Furthermore, range minimum query data structures can replace the child array in enhanced suffix arrays (40), because ℓ-indexes are the positions of minimal values in LCP intervals.
Compressed suffix arrays
Compression of suffix arrays is based on storing a sparse representation of SA(S) and storing Ψ(S) in compressed form. Ψ(S) has the property that it is increasing in areas of SA(S) that point to suffixes starting with the same character (87), which makes it compressible. The first real compressed suffix array was designed by Grossi and Vitter (43,87). They built on a hierarchical decomposition of SA(S) that halves the size of SA(S) in every level by removing values pointing to odd suffixes and dividing even suffix array values by two. Ψ(S) is stored in every level for odd suffix array values. rank(S) and select(S) data structures are used to retrieve the parity of suffixes on every level of the hierarchy and in an encoding of Ψ(S) (43,88). The number of levels stored in this representation is a parameter that tunes the memory-time trade-off.
Sadakane (63) further improved the above implementation by incorporating the compressed string into the index structure. A basic version of this self-index does not allow direct access to SA[i], but instead allows access to S[SA[i]], which is sufficient for pattern matching and finding |occ(P, S)|. Direct access to SA[i] and SA−1[i] and random access to S is achieved by incorporating the hierarchical structure by Grossi and Vitter. Sadakane's compressed suffix array was implemented (64) and constructed for the human genome. The index required ∼5.6n bits of memory, resulting in an overall memory footprint of <2 GB. Additionally, Sadakane designed a backward search algorithm, similar to that used by FM-indexes, for counting patterns (89). This strategy is much faster than the traditional binary search used by suffix arrays.
Other compressed suffix array designs incorporated wavelet trees (59). In practice, an example implementation (60) required 2.4n bits of memory for real DNA sequences.
A different solution to lower the memory requirements of suffix arrays was used for ‘compact suffix arrays’ (90). Here, the compression is based on self-repetitions, so-called runs, in SA(S). These are suffix array intervals [i .. i + ℓ] for which another interval [j .. j + ℓ] exists such that SA[i + k] = SA[j + k] + 1 for 0 ≤ k ≤ ℓ. In practice, compact suffix arrays take up more memory than existing compressed suffix arrays, but are also faster. It was shown that the number of self-repetitions in SA(S) is related to the number of equal-characters runs in BWT(S) (91), which can be compressed by run-length encoding. In terms of compression, however, this technique was superseded by other FM-indexes (47).
The above compressed suffix arrays are especially geared toward pattern matching. Some compressed index structures (42) are able to find |occ(P, S)| in  (m) time, but in practice they all require at least
(m) time, but in practice they all require at least  (|occ(P, S)|logn) time for retrieving the actual occurrences of pattern P. Furthermore, locating the patterns requires a lot of random accesses to the index structures, resulting in degrading performance due to cache misses. This becomes even more severe when ported to secondary memory (92). This also holds for FM-indexes, as discussed further. González and Navarro (92) designed locally compressed suffix arrays to cope with this problem. Their index structures are based on sampling exact suffix array values, differentially encoding SA(S) and encoding this array using dictionaries. However, these index structures are not self-indexes and have to be incorporated into existing compressed suffix arrays or FM-indexes. In practice, the speed for locating patterns is indeed much faster, even compared with the Lempel–Ziv index structures described further. However, their compression rate is not that high, as it requires up to 85% of the size of regular suffix arrays for DNA sequences and 70% for protein sequences.
(|occ(P, S)|logn) time for retrieving the actual occurrences of pattern P. Furthermore, locating the patterns requires a lot of random accesses to the index structures, resulting in degrading performance due to cache misses. This becomes even more severe when ported to secondary memory (92). This also holds for FM-indexes, as discussed further. González and Navarro (92) designed locally compressed suffix arrays to cope with this problem. Their index structures are based on sampling exact suffix array values, differentially encoding SA(S) and encoding this array using dictionaries. However, these index structures are not self-indexes and have to be incorporated into existing compressed suffix arrays or FM-indexes. In practice, the speed for locating patterns is indeed much faster, even compared with the Lempel–Ziv index structures described further. However, their compression rate is not that high, as it requires up to 85% of the size of regular suffix arrays for DNA sequences and 70% for protein sequences.
A practical performance comparison between compressed suffix arrays and plain suffix arrays was made by Sadakane and Shibuya (64). They both tested for the application of approximate string matching. Compressed suffix arrays required one sixth of the memory typically needed by plain suffix arrays, but were 2 – 20 times slower.
FM-indexes
As previously stated, FM-indexes are compressed full-text indexes based on the Burrows–Wheeler transform. Different memory-time trade-offs are reached for FM-indexes by using different techniques for compressing BWT(S) and rank(S). As a reminder, in the original proposal (45,55), BWT(S) is compressed by applying move-to-front transformation, run-length compression and a version of Elias-γ prefix codes (93). rank(S) is encoded by cutting the array in blocks and using the Four-Russians technique. In the original practical implementation (49), the dictionary used for the Four-Russians technique is replaced by a linear scan of a bit vector.
The above representation of FM-indexes is heavily dependent on the alphabet size. A simple way to reduce this dependence is to use a wavelet tree over BWT(S) and use any representation of rank(S) for bit vectors in every internal node (42). Huffman-shaped wavelet trees are used by ‘succinct suffix arrays’ (47). In a recent practical survey (54), this implementation shows the best known practical time-memory trade-offs for the most used basic operations on compressed index structures when applied to DNA and protein sequences. Although its memory footprint is somewhat higher (4n–20n bits) than that of the standard FM-index, it is 20 times faster than its classical counterpart (39). Compared to suffix trees, however, it is 20 times slower. There exist even smaller FM-indexes, such as ‘run-length FM-indexes’ (47) that apply run-length compression to BWT(S) prior to building a wavelet tree. A more recent proposal by Ferragina et al. (48), the ‘alphabet-friendly FM-indexes’, theoretically supersedes all previous FM-index implementations. In practice (54), however, the alphabet-friendly FM-index is superseded by the succinct suffix array for biological sequences. Only for strings with a large alphabet and small high-order entropy (making them highly compressible), such as natural language strings or XML files, alphabet-friendly FM-indexes outperform other FM-indexes.
Another possibility for lowering the memory dependence of FM-indexes was explored by Grabowski et al. (50). They first Huffman-encoded S and then applied the Burrows–Wheeler transform. They require sampling some characters from S additionally to the sampling of SA(S). Their best implementation slightly outperforms succinct suffix arrays on biological sequences and requires 3.28n bits of memory on average.
Note that locating patterns using FM-indexes is done by sampling suffix array values, which turns out to be rather slow in practice. A memory-time trade-off is imposed by the sampling rate. Improvements on the pattern locating performance can be made by using more complex sampling strategies, different from basic evenly spaced sampling (49,54). An alternative is to incorporate another index structure that supports fast locating of patterns (55,92).
Lempel-Ziv index structures
Similar to the above compressed full-text index structures, Lempel–Ziv indexes (94) are mainly designed for pattern matching. Unlike the above compressed index structures, however, Lempel–Ziv indexes are not based on suffix arrays or the Burrows–Wheeler transform. Instead, they build on the dictionary-based Lempel–Ziv (95) compression technique. Briefly, the LZ78 (96) compression is achieved by traversing S and replacing substrings of S with tuples (w, c), where w is a word from the dictionary and c ∈ Σ. Assume that at some point, S[..i − 1] has been compressed and the next tuple in the compressed string is (w, c). w equals the code word for the longest prefix of S[i..], say S[i .. j], that is already part of the dictionary and c = S[j + 1]. Furthermore, S[i .. j + 1] is added to the dictionary. Note that there are other variants of Lempel–Ziv compression, similar to the technique described here, which are omitted for the sake of brevity.
Due to space limitations, details on the structure and search algorithms of Lempel–Ziv indexes are omitted, but can be found elsewhere (42). What is important to note about their structure, however, is that Lempel–Ziv indexes contain many building blocks: compressed or sparse (suffix) tree data structures to compactly represent the dictionaries of forward and reverse code words, data structures for linking those trees and several other auxiliary data structures that answer rank(S) queries and data structures to answer orthogonal range queries. As a direct consequence, further improvements in these building blocks will improve the performance of Lempel–Ziv indexes. Compared with other compressed index structures, Lempel–Ziv index structures require more memory than other self-indexes on average and they are not competitive for counting occurrences of patterns [ (m2) time]. They, however, excel at retrieving the exact set of all occurrences occ(P, S).
(m2) time]. They, however, excel at retrieving the exact set of all occurrences occ(P, S).
Lempel–Ziv indexes have been turned into self-indexes by Navarro (65), who also designed an efficient implementation (97). Further improvements in counting occurrences were made by Ferragina and Manzini (55), who attached FM-indexes to Lempel–Ziv indexes. Other approaches (98,99) have minimized the redundancy caused by an overload of building blocks and have experimented with new auxiliary data structures. Recent tests (54,57,99) show that those new implementations have made Lempel–Ziv indexes more competitive compared with compressed suffix arrays and FM-indexes, but succinct suffix arrays are still reported to have better memory-time trade-offs. In the near future, however, Lempel–Ziv indexes could outperform other indexes for highly compressible strings because all building blocks of Lempel–Ziv index structures can be compressed, while other compressed indexes contain sampled suffix array values, which are incompressible (98).
Compressed suffix trees
The above compressed index structures were mainly designed for exact string matching. As such, they do not reach the full expressiveness of suffix trees. Examples of this expressiveness have been previously given as illustration of the different traversal types of suffix trees. In recent years, efforts have been made to increase the flexibility of compressed index structures either by designing index-specific algorithms or by implementing additional auxiliary data structures. Analogous to enhanced suffix arrays, the main auxiliary data structures used for augmenting compressed suffix arrays are succinct representations of LCP arrays (89), data structures for top-down tree traversals and suffix link support. As an example, the combination of Burrows Wheeler index structures and wavelet trees for succinct LCP arrays was used for locating all maximal repeats in the whole human genome (81). Ohlebush et al. (100), among others, noted that the backward search mechanism mimics top-down suffix ‘trie’ traversal. Using additional data structures to simulate suffix links, they calculated maximal exact matches between DNA sequences, using less memory than, for example, MUMmer (10).
Instead of developing application-specific compressed index structures, several ‘compressed suffix trees’ (44) or ‘compressed enhanced suffix arrays’ (101) have been designed that even surpass the expressiveness of classical suffix trees. Furthermore, because compressed suffix trees extend compressed self-indexes, they are self-indexes themselves. The difference between these structures and the compressed suffix arrays and FM-indexes on which they are built, is their ability to directly implement suffix tree algorithms using these structures. Although the extra data structures increase their memory footprint, compressed suffix trees are still smaller than classical suffix arrays. Furthermore, space-time trade-offs can be tuned to a certain extent, similar to the sparsification parameter in compressed suffix arrays and FM-indexes.
Over the last years, several compressed suffix tree designs have been proposed. These can be classified by their choice of auxiliary data structures, especially the representation of the suffix tree topology (102). They either use sequences of balanced parentheses or implicit representation by LCP intervals. Additional building blocks are succinct representations of LCP arrays and data structures for performing lowest common ancestor queries, which in turn support suffix links. As an example, the first compressed suffix tree reaching full expressiveness was given by Sadakane (44). It consists of a compressed suffix array, succinct LCP array, balanced parentheses representation for suffix tree topology and additional data structures for solving range minimum queries. In practice, an engineered version (58) of this compressed suffix tree required 25n–35n bits of memory and was able to index the complete human genome using only 8.5 GB. Compared with classical suffix trees, this compressed variant is two orders of magnitude slower on average. Nevertheless, compressed suffix trees are still much faster than brute force algorithms. Furthermore, many auxiliary data structures used in the design offer a memory-time trade-off which can be optimized for the available memory. Advancements made in representing auxiliary data structures have led to index structures with even smaller memory requirements (85). The smallest compressed suffix tree we know of (61) requires only 4n–6n bits of memory and is based on sampling the suffix tree. This low memory footprint, however, is paid for by giving up performance, and it is several orders of magnitude slower than Sadakane's compressed suffix tree (62). Another compressed suffix tree proposed by Fischer et al. (103) has a memory-time trade-off which lies between the two previously mentioned compressed suffix trees. Cánovas and Navarro (62) engineered an implementation of this compressed suffix tree and compared the impact of different LCP array implementations on the compressed suffix tree. Depending on the implementation of the LCP arrays used, the compressed suffix tree requires between 8n and 16n bits of memory. A compressed enhanced suffix array reaching full expressiveness is given by Ohlebusch and Gog (101). However, it does not support lowest common ancestor queries. Prospects are that space-time trade-offs of compressed index structures will keep improving due to improvements in auxiliary data structures, especially improvements in compressed suffix arrays and compressed LCP arrays.
Index structures in external memory
The solution for the memory bottleneck suffered by (main memory) index structures are index structures in external or secondary memory, such as hard disks. This paradigm shift is necessary when even the smallest compressed index structures cannot be stored in main memory. This limit is usually reached when even a compressed form of S cannot be stored in main memory. Secondary or external memory has the advantages of low cost, abundance and the persistence given to index structures. However, random access to secondary memory (disk) is much slower than random access to primary memory (RAM). In practice, this difference can be up to five orders of magnitude (24). Since index structures, such as suffix trees, intrinsically access data structures and input strings in a random manner, this leads to the so-called ‘I/O bottleneck’. Several techniques are used to minimize the effect of this bottleneck, both in hardware and in algorithm and data structure design. Solid-State disks, for example, are one order of magnitude faster than classical hard disks. Also, sequential disk access is almost as fast as random access on RAM. Another solution is to limit the number of I/O operations altogether by, for example, decreasing the size of the index structure. Buffering is another strategy commonly employed, as well as improving locality of information that is closely connected. To achieve this locality, redundancy is often introduced in the data structure, which is opposite to the space-saving techniques seen in main memory indexes. These techniques are not only applied for designing the spatial layout of index structures, but also for their traversal algorithms. In this section, existing index structures for external memory are reviewed with an emphasis on the high-level strategies employed. Other, more technical, reviews on this topic can be found elsewhere (25,71,104).
Suffix arrays
Both suffix trees and suffix arrays perform poorly when naively implemented in secondary memory. Since of their simple design, however, suffix arrays are easier to implement on disk. The basic idea is to use levels of sparse suffix arrays in faster memory to guide searches in the full suffix array stored on disk. Baeza-Yates et al. (105) proposed a two-level index structure. They also augmented the sparse suffix array, stored in RAM, with exact prefixes of the suffixes represented in the sparse suffix array. This has the advantage that no random access to S is needed for matching in the sparse suffix array. Tests revealed that this implementation is five times faster than a naive implementation (106) of a single-level suffix array on disk. Later, Sinha et al. (106) replaced sparse suffix arrays by pruned suffix ‘tries’ for the first level of the hierarchy. Again, labels on the pruned suffix ‘trie’ are explicitly stored instead of pointers to S. Sinha et al. also improved the second level of the hierarchy by storing SA(S), LCP(S) and substrings of S, to minimize random access to S. Note that in primary memory, redundancy is eliminated, whereas in secondary memory it is introduced to increase performance. Tests showed that this method is five times faster than the two-level method of Baeza-Yates et al. and requires ∼10 times less non-sequential I/O operations for pattern matching.
A larger number of levels is used in the design of ‘string B-trees’ (107). These index structures act as conceptual B-trees (27) over suffix arrays. Similar to B-trees, internal nodes are B-ary and the final suffix array values are found in the leaves. To speed up the search through the B-tree, each internal node v contains a ‘Patricia tree or blind tree’ for the suffixes in v. Blind trees are suffix tree variants for which edge labels are stored as the first character of the label and its length. Pattern matching in blind trees consists of two phases. A first phase, similar to pattern matching in suffix trees, finds candidate positions according to the matched characters on the edges of the tree. A second phase explicitly compares the pattern to the candidate substrings in S. This type of edge labeling followed by a blind search can also be applied to all external memory suffix tree implementations to minimize random access to S. This data structure has the advantage that pattern matching is theoretically I/O optimal and updates are supported due to its B-tree nature. Furthermore, succinct cache-oblivious string B-trees have been developed (108). Note that string B-trees are not suffix trees and thus do not reach full expressiveness. Another disadvantage is that the blind search method used is impractical for inexact string matching (109).
Distribution of suffix arrays has also been proposed (72). This allows processing batches of queries in parallel by dividing SA(S) in intervals or by interleaving suffix array values. This interleaving can be done by grouping every k-th suffix to a single computing unit or by grouping the suffixes of a substring of S together in one node, thus minimizing access to S. Although these designs look promising, we have no knowledge of any recent performance results for string matching algorithms on biological data using any of the above external memory suffix arrays.
Suffix trees
Because of the underlying tree data structure, efficient implementation on disk is more difficult for suffix trees than for suffix arrays. Although many papers about external memory suffix trees exist, most of them focus on construction in external memory. Less attention has been given to optimizing suffix tree layout for traversals and even fewer performance tests are available for algorithms that make use of external memory suffix trees. The most important factor in designing external memory representations of suffix trees is the grouping of nodes into blocks and the layout of these blocks onto disk. Other important aspects are node and edge label representations. For locality reasons, array-based representations are superior to other implementations (110) and nodes contain more information than their primary memory counterparts, while edge labels can be compactly represented by their first character and length as in blind trees. An example of this strategy is one of the earliest external suffix trees, the ‘compact Patricia tree’ (111), which uses a topology representation similar to the balanced parentheses representation.
A very intuitive external memory suffix tree layout is that of partitioning by prefixes. The suffix tree is split into an upper root-block and blocks containing the subtrees of a given prefix. This layout is similar to the two-level hierarchical layout for suffix arrays. For top-down traversals of the suffix tree, it works well in practice. Furthermore, this layout is created naturally during construction (109,110). A disadvantage, however, is its scalability. Although these indexes can be constructed for the human genome (112), larger sequences or data sets suffer from either a large growth in the size of the partitions or an exponential growth in the number of partitions. Moreover, data skewness results in decreasing performance, as some partitions are much larger than others. In theory, a multi-level hierarchical structure could alleviate the scalability problem and data skewness has already been tackled by using variable length prefixes (113,114). Another weakness of external memory suffix trees are suffix links. These links imply a lot of random access and are thus optional (113,114) or completely omitted (109,112). On the other hand, some authors (113) claim that the use of suffix links in external memory improves performance of some search algorithms, such as finding maximal exact matches. Clifford (115) designed ‘distributed suffix trees’, which contain a local version of suffix links, called ‘sparse suffix links’. These links point to the local root if the normal suffix link would point to a node in a different partition. Clifford points out that prefix partitioning allows traversals on the suffix tree to be run in parallel on the distributed subtrees. Furthermore, he claims that most bioinformatics applications do not require traversals that require communications between the different prefix-partitioned parts. Thus prefix-partitioning enables the parallelization of most search algorithms on suffix trees.
For exact pattern matching, prefix partitioned suffix trees work well. For other queries, however, transforming the tree layout to an already constructed prefix-partitioned suffix tree has been proposed. The goal of changing layouts is to increase scalability and improve the locality of the nodes. For pattern matching, however, the new layout could increase the number of I/O operations. Different techniques have been proposed to achieve this goal. Clark and Munro (111) focused on minimizing the number of blocks required to store suffix trees using a greedy bottom-up algorithm. ‘STELLAR’ (116), on the other hand, focused on improving locality of nodes for both parent–child links as well as suffix links. Other layouts introduce redundancy of data by having the subtrees stored in blocks on disk overlap (117,118). Although the redundancy introduced increases the memory footprint of the index structures, it improves locality of the nodes and improves the scalability of the index structures. Care has to be taken, however, not to destroy some of the expressiveness of suffix trees, including LCP values and suffix links.
In practice, the largest indexed single DNA sequence found in the literature contains 12 billion base pairs (119). Although no extensive performance results for string algorithms on this index were given, disk-based index structures are known to be several times faster than non-indexed methods for string matching on the scale of the human genome. Compared with string B-trees, disk-based suffix trees require a similar number of I/O operations (104) for pattern matching. Furthermore, Halachev et al. (120) showed that for protein data, pattern matching on disk-based suffix trees can be almost as fast as pattern matching on enhanced suffix arrays. As an example of other applications, a disk-based enhanced suffix array has been used to locate repeats in human chromosomes (12).
Compressed index structures
Data compression and indexing are very important in computational biology, although they seem to be opposites at first sight. With the rise of compressed index structures, this dichotomy can be considered solved (2) for the RAM model. However, designing a disk-based version of these indexes is non-trivial, because compressed suffix arrays and FM-indexes perform many random accesses and show a poor locality (92). Nevertheless, some compressed index structures for external memory do exist.
Mäkinen et al. (121) designed a secondary memory version of the compressed suffix array by Sadakane (88) using a multi-level hierarchical structure. They also designed a distributed compressed suffix array. External memory variants of FM-indexes have been developed by González and Navarro (122). They proposed external memory versions for auxiliary data structures for calculating rank(B) and select(B) and proposed a two-level hierarchy for storing rank(S). Different structures were designed for representing BWT(S) on disk, all having different trade-offs depending on the size of the available main memory. For fast locating, they adopted the locally compressed suffix array designed for fast locating (92). Arroyuelo and Navarro (123) designed an external memory Lempel–Ziv index based on the Lempel–Ziv index structure proposed by Navarro (65). A recent article by Russo et al. (124) shows how parallel and distributed compressed suffix arrays can efficiently answer more advanced queries such as longest common substrings. Furthermore, they designed parallel and distributed compressed suffix trees.
Although the idea of reducing space in external memory to reduce the number of I/O-operations is interesting, it is not known how this affects performance in practice. Some tests on natural language data suggest that compressed index structures are competitive in practice, although they are somewhat slower than string B-trees (122).
Recently, Chien et al. (125) proposed a new transformation, called the ‘geometric Burrows–Wheeler transform’, which connects index structures with range searching. It translates characters of a string into 2D points and vice versa and uses the vast research on 2D range queries to answer pattern matching queries. To achieve a succinct representation, sparsification is used by grouping substrings in meta characters. For external memory purposes it uses a string B-tree to find ranges in the sparse suffix array, while 2D search can be done using a wavelet tree. Tests (104) show that these compressed index structures are smaller compared with other external memory index structures, but they require more I/O operations. Another application opened by these index structures is the possibility to answer relevance queries (104). As an example, it would be possible to retrieve only the top k most similar sequences in a database.
CONSTRUCTION
Before index structures can be used, they first have to be constructed. Although construction is fast in theory, it is not always the case in practice. The current bottlenecks in constructing disk-based index structures for very large strings are memory limitations in the working space, cache misses and a high number of random accesses to secondary memory. The working space is the amount of memory required by the construction algorithm, which is usually higher than the memory required by the final index. Apart from dealing with these issues, some research has focused on parallelizing construction algorithms. In this section, an overview of existing construction algorithms for various index structures is given, illustrated with practical results found in the literature. Note that the figures in this section represent some of the historical breakthroughs in index structure construction, and are not meant as a comparison between the cited implementations. As a general reference, reported index structure construction times for the human genome, or for sequences in the same order of magnitude, were in the range of a few hours on desktop computers and in the range of minutes on clusters and specialized hardware.
Suffix trees
Historically, suffix tree construction goes back to Weiner (28), who gave a first  (n) algorithm. Later, Ukkonen (126) gave a simpler
(n) algorithm. Later, Ukkonen (126) gave a simpler  (n) algorithm, which has the nice property of being online, i.e. a new string can be added to the suffix tree by appending it to the back of the previous strings. The WOTD suffix tree by Giegerich et al. (33) comes with a lazy construction algorithm, in the sense that suffix tree nodes are added the first time that a traversal algorithm requires these nodes. Thus, suffix trees can also be efficiently used for smaller applications that do not require information about the whole tree. The suffix links that are a by-product of Ukkonen's algorithm have very nice features, as discussed in ‘Popular index structures’ Section, but they are omitted in other construction algorithms. To retrieve these suffix links, some post-processing algorithms exist (127). Although the above mentioned suffix tree construction algorithms only scale up to chromosome level, they form the basis for many external memory construction algorithms. Although a main memory suffix tree for the whole human genome was constructed by Kurtz (66), most main memory index structure construction algorithms focus on suffix arrays and compressed index structures.
(n) algorithm, which has the nice property of being online, i.e. a new string can be added to the suffix tree by appending it to the back of the previous strings. The WOTD suffix tree by Giegerich et al. (33) comes with a lazy construction algorithm, in the sense that suffix tree nodes are added the first time that a traversal algorithm requires these nodes. Thus, suffix trees can also be efficiently used for smaller applications that do not require information about the whole tree. The suffix links that are a by-product of Ukkonen's algorithm have very nice features, as discussed in ‘Popular index structures’ Section, but they are omitted in other construction algorithms. To retrieve these suffix links, some post-processing algorithms exist (127). Although the above mentioned suffix tree construction algorithms only scale up to chromosome level, they form the basis for many external memory construction algorithms. Although a main memory suffix tree for the whole human genome was constructed by Kurtz (66), most main memory index structure construction algorithms focus on suffix arrays and compressed index structures.
Suffix arrays
Originally, linear time suffix array construction required the construction of the suffix tree (29). During the last decade, however, many direct suffix array construction algorithms have been proposed. A taxonomy of existing suffix array construction algorithms is given by Puglisi et al. (128). Since suffix array construction consists of sorting all suffixes of S, many algorithms are based on known sorting algorithms. One of the most popular algorithms is the recursive  (n) KS3 algorithm of Kärkkäinen and Sanders (129). It can be modified to a parallel and external memory version, called DC3 (130), which can construct SA(S) for the whole human genome using only 1 GB RAM and for which a Message Passing Interface (MPI) version exists that has indexed the human genome in only a few minutes (on specialized hardware) (131). However, it was noted elsewhere that DC3 is unable to index strings longer than 4 Gb (132). Other algorithms try to minimize the working space in internal memory. So-called ‘lightweight’ (133,134) construction algorithms have a working space that approaches the theoretical minimum. Furthermore, according to extensive tests on biological sequences made by Mori (among others, http://code.google.com/p/libdivsufsort/), they are the fastest construction algorithms in practice. Another trick utilized is to only sort suffixes up to a certain LCP value, leading to ‘partial suffix arrays’. Although the expressiveness of partial suffix arrays is unclear, they have already been applied for error correction of sequencing reads (14). For the construction of enhanced suffix arrays, efficient LCP array construction algorithms have been developed (38) and
(n) KS3 algorithm of Kärkkäinen and Sanders (129). It can be modified to a parallel and external memory version, called DC3 (130), which can construct SA(S) for the whole human genome using only 1 GB RAM and for which a Message Passing Interface (MPI) version exists that has indexed the human genome in only a few minutes (on specialized hardware) (131). However, it was noted elsewhere that DC3 is unable to index strings longer than 4 Gb (132). Other algorithms try to minimize the working space in internal memory. So-called ‘lightweight’ (133,134) construction algorithms have a working space that approaches the theoretical minimum. Furthermore, according to extensive tests on biological sequences made by Mori (among others, http://code.google.com/p/libdivsufsort/), they are the fastest construction algorithms in practice. Another trick utilized is to only sort suffixes up to a certain LCP value, leading to ‘partial suffix arrays’. Although the expressiveness of partial suffix arrays is unclear, they have already been applied for error correction of sequencing reads (14). For the construction of enhanced suffix arrays, efficient LCP array construction algorithms have been developed (38) and  (n) algorithms exist for the construction of the other tables (19,127).
(n) algorithms exist for the construction of the other tables (19,127).
Compressed index structures
Working space is even more important for compressed full-text index structures. Compressed suffix arrays, FM-indexes and regular suffix arrays can easily be obtained from one another. However, suffix array construction requires 40n – 48n bits of memory, whereas FM-indexes can be stored in only 2n bits. Despite this, lightweight suffix array construction algorithms (134) are used by Burrows–Wheeler-based read mapping tools, such as BWA (8). Direct and lightweight construction of compressed index structures is therefore an important issue. A gap between theory and practice existed for several years, but several practical results have been reported recently. For example, a lightweight Burrows–Wheeler construction algorithm by Kärkkäinen (135) requires only 8n bits of working space for DNA sequences (which is equal to the size of a normal text string) and was implemented in the short read mapping tool Bowtie (7). Other direct construction algorithms include the parallel algorithm of Sirén (136) and the lightweight construction algorithms in both internal and external memory settings of Ferragina et al. (132). The former has the added value of being able to merge existing compressed suffix arrays, and the latter have very low working spaces. Moreover, a parallel BWT(S) construction algorithm (137) based on the Google MapReduce (18) framework has recently indexed the human genome in ∼10 min on the Amazon Elastic Compute Cloud. Finally, a lightweight construction algorithm for Lempel–Ziv indexes (138) has been reported that is competitive with construction algorithms for other compressed full-text indexes.
External memory suffix tree construction
Most work on external memory index structures has been done on construction algorithms, which have been extensively reviewed by Barsky et al. (71). To summarize their results, external memory allows for larger sequences to be indexed, but the scalability of the algorithms is limited by the number of random accesses to S and the suffix tree under construction. This means that the practical performance of many construction algorithms is limited to sequences which are smaller than the size of the available main memory. As an exception, the B2ST algorithm (119) was able to index DNA sequences of 12 Gb in <8 h, making this algorithm the first to partially overcome the above-mentioned bottlenecks. Furthermore, the authors believe the algorithm will scale up to sequences of 60 Gb.
CONCLUSION
In this review, we have shown the importance of data structures for processing and searching in strings, known as index structures. Many current sequence analysis tools heavily rely upon index structures for handling large amounts of data, which is currently a major concern to bioinformaticians. In the first main section, details concerning the most commonly used index structures were presented. The details given in this review are often omitted in articles describing tools and applications. However, we believe that these details are important to fully grasp the possibilities and limitations of these sequence analysis tools.
We have made a basic classification of existing index structures and explained the memory-time trade-offs related to these data structures. Since the number of available index structures is vast, we were only able to skim over the technical details involved in the design of these data structures. However, the interested reader was guided to more in-depth work in the literature. Note that the index structures discussed in this review mainly are all-purpose full-text index structures, although some focused on exact pattern matching. There are, however, other index structures specially designed for specific applications, as discussed in the first section of this review.
Furthermore, both main purpose full-text index structures and specialized index structures will always be hampered with space-time trade-offs. Several index structures allow tuning this trade-off by setting a sparsification parameter. This optimization of the available main memory is required because of the large difference in speed between internal and external memory. In some cases, the available main memory does not suffice and external memory index structures have to be used. Moreover, we saw that the performance of external memory index structures highly depends on the application for which the index structure is used. There is still a lot of work to be done on increasing the performance of disk-based index structures.
Construction of index structures in external memory has seen more investigation and clearly shows that the use of current index structures is limited to sequences that fit in main memory. Main memory construction algorithms are limited by the available work space for which the demand is several times higher than the memory required for the final index structure.
In the future, algorithms and data structures will have to be improved further to keep up with the rapidly evolving sequencing technology and the growing amount of data in general. To tackle the bottlenecks related to index structures mentioned here, new directions for their design have to be investigated (2). As a final note, we give some prospects for research on index structures for bioinformatics applications. Currently, the biggest issue in index structure research is closing the gap between theory and practice, which is illustrated by the fact that many theoretically superior index structures do not outperform simpler designs in practice. More engineering work has to be done to improve the practical performance of these index structures. These implementations should be grouped under a common interface in libraries and benchmarked using different types of (biological) sequences. One such library-project is the ‘Pizza&Chili website’ [two mirrors at http://pizzachili.di.unipi.it and http://pizzachili.dcc.uchile.cl], which bundles full-text compressed index structures for use in exact pattern matching. Another library containing several index structures, but also focusing on biological applications, is the SeqAn library (139).
Another significant topic for further research is the adaptation of index structures to modern hardware, such as multi-core CPUs (140,141) and solid-state disks. Recently, even more specialized hardware has been considered, including Graphical Processing Units (GPUs) (11) and GPFAs (142). Alternatively, large computer clusters, local or on the cloud, could allow for massive parallelization of index structures. Some applications have already been ported to these new platforms, including read mapping and SNP finding (143) using cloud computing, sequence alignment (11) on GPUs and suffix array construction (137) using Google's MapReduce (18). However, these techniques and implementations are very novel and further research will have to indicate their scope and potential.
For applications which require maintenance of the index structure, such as sequence databases or updating an existing index of the human genome, dynamic index structures are required. Historically, this is challenging due to the intrinsic interrelationship of suffixes, where insertion of a single character in a string can change the lexicographical order of many suffixes. However, some index structures that allow addition and removal of whole strings (61,107) and single characters (144,145) can be found in the literature. Moreover, several index structures were recently proposed for processing a set of very similar strings (146,147), where the size of the index structure only depends on a single reference sequence in the collection, rather than the combined size of all sequences in it.
Given these developments, index structures will continue to increase the performance of bioinformatics applications while coping with the continuous growth in sequence sizes.
FUNDING
Work of M.V. supported by the Agency for Innovation by Science and Technology, Flemish Government, Belgium [SB-101609]. All authors acknowledge the support of Ghent University (Multidisciplinary Research Partnership “Bioinformatics: from nucleotides to networks”). Funding for open access charge: Agency for Innovation by Science and Technology, Flemish Government, Belgium.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors wish to thank Martijn Devisscher, Ken Heyndrickx and Joachim De Schrijver whose feedback has been invaluable in opening up this article for the largest possible audience. The authors also like to acknowledge the members of the Nucleotides to Networks next-generation sequencing discussion group, in particular Yao-Cheng Lin and Lieven Sterck, for their helpful comments in improving the readability of the article. In addition, the authors would also like to thank the editor and the anonymous reviewers for their valuable comments and suggestions to improve the quality of the manuscript. Due to space constraints, it was impossible to cite all relevant publications in this review; our sincere apologies and appreciation to all colleagues whose important work is not cited.
REFERENCES
- 1.Felsenstein J. Inferring Phylogenies. Sunderland, Mass: Sinauer Associates; 2004. [Google Scholar]
- 2.Ferragina P. Proceedings of the 18th Annual European Symposium on Algorithms. Liverpool, UK: 2010. Data structures: time, I/Os, entropy, joules! pp. 1–16. [Google Scholar]
- 3.Altschul S, Gish W, Miller W, Myers E, and Lipman D. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 4.Flicek P, Birney E. Sense from sequence reads: methods for alignment and assembly. Nat. Meth. 2009;6:S6–S12. doi: 10.1038/nmeth.1376. [DOI] [PubMed] [Google Scholar]
- 5.Hoffmann S, Otto C, Kurtz S, Sharma C, Khaitovich P, Vogel J, Stadler P, Hackermüller J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009;5:e1000502. doi: 10.1371/journal.pcbi.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lam T, Li R, Tam A, Wong S, Wu E, Yiu S. 2009 IEEE International Conference on Bioinformatics and Biomedicine. Washington, DC, USA: 2009. High thoughput short read alignment via bi-directional BWT; pp. 31–36. [Google Scholar]
- 7.Langmead B, Trapnell C, Pop M, Salzberg S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li R, Yu C, Li Y, Lam T, Yiu S, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 10.Kurtz S, Phillippy A, Delcher A, Smoot M, Shumway M, Antonescu C, Salzberg S. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schatz M, Trapnell C, Delcher A, Varshney A. High-throughput sequence alignment using graphics processing units. BMC Bioinformatics. 2007;8:474. doi: 10.1186/1471-2105-8-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Askitis N, Sinha R. RepMaestro: scalable repeat detection on disk-based genome sequences. Bioinformatics. 2010;26:2368–2374. doi: 10.1093/bioinformatics/btq433. [DOI] [PubMed] [Google Scholar]
- 13.Schröder J, Schröder H, Puglisi S, Sinha R, Schmidt B. SHREC: a short-read error correction method. Bioinformatics. 2009;25:2157–2163. doi: 10.1093/bioinformatics/btp379. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Z, Yin J, Zhan Y, Xiong W, Li Y, Liu F. Proceedings of the Joint International Conference Frontiers in Algorithmics and Algorithmic Aspects in Information and Management. Jinhua, China: 2011. PSAEC: an improved algorithm for short read error correction using partial suffix arrays; pp. 220–232. [Google Scholar]
- 15.Conway T, Bromage A. Succinct data structures for assembling large genomes. Bioinformatics. 2011;27:479–486. doi: 10.1093/bioinformatics/btq697. [DOI] [PubMed] [Google Scholar]
- 16.Hernandez D, Francois P, Farinelli L, Osteras M, Schrenzel J. De novo bacterial genome sequencing: millions of very short reads assembled on a desktop computer. Genome Res. 2008;18:802–809. doi: 10.1101/gr.072033.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simpson J, Durbin R. Efficient construction of an assembly string graph using the FM-index. Bioinformatics. 2010;26:i367–i373. doi: 10.1093/bioinformatics/btq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dean J, Ghemawat S. Proceedings of the 6th Symposium on Operating System Design and Implementation. San Francisco, California, USA: 2004. MapReduce: simplified data processing on large clusters; pp. 137–150. [Google Scholar]
- 19.Abouelhoda M, Kurtz S, Ohlebusch E. Replacing suffix trees with enhanced suffix arrays. Discrete Algor. 2004;2:53–86. [Google Scholar]
- 20.Meyer F, Kurtz S, Backofen R, Will S, Beckstette M. Structator: fast index-based search for RNA sequence-structure patterns. BMC Bioinformatics. 2011;12:214. doi: 10.1186/1471-2105-12-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iliopoulos C, Makris C, Panagis Y, Perdikuri K, Theodoridis E, Tsakalidis A. The weighted suffix tree: an efficient data structure for handling molecular weighted sequences and its applications. Fund. Infor. 2006;71:259–277. [Google Scholar]
- 22.Shibuya T. Geometric suffix tree: Indexing protein 3-D structures. J. ACM. 2010;57:15. [Google Scholar]
- 23.Hon W, Patil M, Shah R, Thankachan S. Proceedings of the 2011 Data Compression Conference. Snowbird, Utah, USA: 2011. Compressed property suffix trees; pp. 123–132. [Google Scholar]
- 24.Jacobs A. The pathologies of big data. Commun. ACM. 2009;52:36–44. [Google Scholar]
- 25.Vitter J. External memory algorithms and data structures: dealing with massive data. ACM Comput. Surv. 2001;33:209–271. [Google Scholar]
- 26.Blumer A, Blumer J, Haussler D, McConnell R, Ehrenfeucht A. Complete inverted files for efficient text retrieval and analysis. J. ACM. 1987;34:578–595. [Google Scholar]
- 27.Bayer R, McCreight E. Organization and Maintenance of large ordered indexes. Acta Infor. 1972;1:173–189. [Google Scholar]
- 28.Weiner P. Proceedings of the 14th IEEE Symposium on Switching and Automata Theory. Iowa City, Iowa, USA: 1973. Linear pattern matching algorithm; pp. 1–11. [Google Scholar]
- 29.Gusfield D. Algorithms on Strings, Trees, and Sequences. 11th edn. Cambridge, UK and New York, USA: Cambridge University Press; 1997. [Google Scholar]
- 30.Morrison D. PATRICIA practical algorithm to retrieve information coded in alphanumeric. J. ACM. 1968;15:514–534. [Google Scholar]
- 31.Boyer R, Moore J. A fast string searching algorithm. Commun. ACM. 1977;20:762–772. [Google Scholar]
- 32.Knuth D, Morris J, Pratt V. Fast pattern matching in strings. SIAM J. Comput. 1977;6:323–350. [Google Scholar]
- 33.Giegerich R, Kurtz S, Stoye J. Efficient implementation of lazy suffix trees. Softw. Pract. Exp. 2003;33:1035–1049. [Google Scholar]
- 34.McCreight E. A space-economical suffix tree construction algorithm. J. ACM. 1976;23:262–272. [Google Scholar]
- 35.Manber U, Myers E. Suffix arrays: a new method for on-line string searches. SIAM J. Comput. 1993;22:935–948. [Google Scholar]
- 36.Grossi R. A quick tour on suffix arrays and compressed suffix arrays. Theor. Comput. Sci. 2011;412:2964–2973. [Google Scholar]
- 37.Ohlebusch E, Gog S. Efficient algorithms for the all-pairs suffix-prefix problem and the all-pairs substring-prefix problem. Inf. Process. Lett. 2010;110:123–128. [Google Scholar]
- 38.Kasai T, Lee G, Arimura H, Arikawa S, Park K. Proceedings of the 12th Symposium on Combinatorial Pattern Matching. Jerusalem, Israel: 2001. Linear-time longest-common-prefix computation in suffix arrays and its applications; pp. 181–192. [Google Scholar]
- 39.Grimsmo N. 2007. On performance and cache effects in substring indexes. Report IDI-TR-2007-04, Norwegian University of Science and Technology, Norway. [Google Scholar]
- 40.Fischer J, Heun V. Proceedings of the 1st Symposium on Combinatorics, Algorithms, Probabilistic and Experimental Methodologies. Hangzhou, China: 2007. A new succinct representation of RMQ-information and improvements in the enhanced suffix array; pp. 459–470. [Google Scholar]
- 41.Kim D, Kim M, Park H. Linearized suffix tree: an efficient index data structure with the capabilities of suffix trees and suffix arrays. Algorithmica. 2008;52:350–377. [Google Scholar]
- 42.Navarro G, Mäkinen V. Compressed full-text indexes. ACM Comput. Surv. 2007;39:2:1–2:61. [Google Scholar]
- 43.Grossi R, Vitter J. Compressed suffix arrays and suffix trees with applications to text indexing and string matching. SIAM J. Comput. 2005;35:378–407. [Google Scholar]
- 44.Sadakane K. Compressed suffix trees with full functionality. Theor. Comput. Syst. 2007;41:589–607. [Google Scholar]
- 45.Ferragina P, Manzini G. Proceedings of the 41st IEEE Symposium on Foundations of Computer Science. Redondo Beach, California, USA: 2000. Opportunistic data structures with application; pp. 390–398. [Google Scholar]
- 46.Burrows M, Wheeler D. A block-sorting lossless data compression algorithm. Technical Report 124. DEC SRC; 1994. [Google Scholar]
- 47.Mäkinen V, Navarro G. Succinct suffix arrays based on run-length encoding. Nordic J. Comput. 2005;12:40–66. [Google Scholar]
- 48.Ferragina P, Manzini G, Mäkinen V, Navarro G. Compressed representations of sequences and full-text indexes. ACM Trans. Algor. 2007;3:20. [Google Scholar]
- 49.Ferragina P, Manzini G. Proceedings of the 12th ACM-SIAM Symposium on Discrete Algorithms. Washington, DC, USA: 2000. An experimental study of an opportunistic index; pp. 269–278. [Google Scholar]
- 50.Grabowski S, Navarro G, Przywarski R, Salinger A, Mäkinen V. A simple alphabet-independent FM-index. Int. J. Founda. of Comput. Sci. 2006;17:1365–1384. [Google Scholar]
- 51.Adjeroh D, Bell T, Mukherjee A. The Burrows-Wheeler Transform: Data Compression, Suffix Arrays, and Pattern Matching. New York: Springer; 2008. [Google Scholar]
- 52.Hon W, Sadakane K, Sung W. Breaking a time-and-space barrier in constructing full-text indices. SIAM J. Comput. 2009;38:2162–2178. [Google Scholar]
- 53.Arlazarov V, Dinic E, Kronrod M, Faradzev I. On economic construction of the transitive closure of a directed graph. Dokl. Akad. Nauk SSSR. 1970;194:487–488. [Google Scholar]
- 54.Ferragina P, González R, Navarro G, Venturini R. Compressed text indexes: From theory to practice. ACM J. Exp. Algor. 2009;13:1.12–1.31. [Google Scholar]
- 55.Ferragina P, Manzini G. Indexing compressed text. J. ACM. 2005;52:552–581. [Google Scholar]
- 56.Russo L, Navarro G, Oliveira A, Morales P. Approximate string matching with compressed indexes. Algorithms. 2009;2:1105–1136. [Google Scholar]
- 57.Arroyuelo D, Navarro G. Practical approaches to reduce the space requirement of Lempel-Ziv based compressed text indices. ACM J. Exp. Algor. 2010;15:1.5:1.1–1.5:1.54. [Google Scholar]
- 58.Välimäki N, Mäkinen V, Gerlach W, Dixit K. Engineering a compressed suffix tree implementation. ACM J. Exp. Algor. 2009;14:4.2–4.23. [Google Scholar]
- 59.Grossi R, Gupta A, Vitter J. Proceedings of the 14th ACM-SIAM Symposium on Discrete Algorithms. Baltimore, Maryland, USA: 2003. High-order entropy-compressed text indexes; pp. 841–850. [Google Scholar]
- 60.Foschini L, Grossi R, Gupta A, Vitter J. When indexing equals compression: experiments with compressing suffix arrays and applications. ACM Trans. Algor. 2006;2:611–639. [Google Scholar]
- 61.Russo L, Navarro G, Oliveira A. Fully-compressed suffix trees. ACM Trans. Algor. 2011;7:53. [Google Scholar]
- 62.Cánovas R, Navarro G. Proceedings of the 9th Symposium on Experimental Algorithms. Ischia Island, Naples, Italy: 2010. Practical compressed suffix trees; pp. 94–105. [Google Scholar]
- 63.Sadakane K. New text indexing functionalities of the compressed suffix arrays. J. Algor. 2003;48:294–313. [Google Scholar]
- 64.Sadakane K, Shibuya T. Indexing huge genome sequences for solving various problems. Genome Inform. 2001;12:175–183. [PubMed] [Google Scholar]
- 65.Navarro G. Indexing text using the Ziv-Lempel trie. J. Discrete Algor. 2004;2:87–114. [Google Scholar]
- 66.Kurtz S. Reducing the space requirement of suffix trees. Softw. Pract. Exp. 1999;29:1149–1171. [Google Scholar]
- 67.Kärkkäinen J, Ukkonen E. Proceedings of the 2nd Conference on Computing and Combinatorics. Hong Kong, China: 1996. Sparse suffix trees; pp. 219–230. [Google Scholar]
- 68.Ferragina P, Fischer J. Proceedings of the 18th conference on Combinatorial Pattern Matching. London, Ontario, Canada: 2007. Suffix arrays on words; pp. 328–339. [Google Scholar]
- 69.Khan Z, Bloom J, Kruglyak L, Singh M. A practical algorithm for finding maximal exact matches in large sequence datasets using sparse suffix arrays. Bioinformatics. 2009;25:1609–1616. doi: 10.1093/bioinformatics/btp275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kulekci M, Hon W, Shah R, Vitter J, Xu B. Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine. Hong Kong, China: 2010. PSI-RA: a parallel sparse index for read alignment on genomes; pp. 663–668. [Google Scholar]
- 71.Barsky M, Stege U, Thomo A. A survey of practical algorithms for suffix tree construction in external memory. Softw. Pract. Exp. 2010;40:965–988. [Google Scholar]
- 72.Marín M, Navarro G. Proceedings of the 10th conference on String Processing and Information Retrieval. Manaus, Brazil: 2003. Distributed query processing using suffix arrays; pp. 311–325. [Google Scholar]
- 73.Andersson A, Larsson N, Swanson K. Suffix trees on words. Algorithmica. 1999;23:246–260. [Google Scholar]
- 74.Inenaga S, Takeda M. Proceedings of the 17th conference on Combinatorial Pattern Matching. Barcelona, Spain: 2006. On-line linear-time construction of word suffix trees; pp. 60–71. [Google Scholar]
- 75.Transier F, Sanders P. Proceedings of the 10th Workshop on Algorithm Engineering and Experimentation. San Francisco, California, USA: 2008. Compressed inverted indexes for in-memory search engines; pp. 3–12. [Google Scholar]
- 76.Puglisi S, Smyth W, Turpin A. Proceedings of the 13th conference on String Processing and Information Retrieval. Glasgow, UK: 2006. Inverted files versus suffix arrays for locating patterns in primary memory; pp. 122–133. [Google Scholar]
- 77.Manzini M. An analysis of the Burrows-Wheeler transform. J. ACM. 2001;48:407–430. [Google Scholar]
- 78.Raman R, Raman V, Rao S. Proceedings of the 13th Annual ACM-SIAM Symposium on Discrete Algorithms. San Francisco, California, USA: 2002. Succinct indexable dictionaries with applications to encoding k-ary trees and multisets; pp. 233–242. [Google Scholar]
- 79.Claude F, Navarro G. Proceedings of the 15th annual Symposium on String Processing and Information Retrieval. Melbourne, Australia: 2008. Practical Rank/Select queries over arbitrary sequences; pp. 176–187. [Google Scholar]
- 80.Okanohara D, Sadakane K. Proceedings of the 9th Workshop on Algorithm Engineering and Experiments. New Orleans, Louisiana, USA: 2007. Practical entropy-compressed rank/select dictionary; pp. 60–70. [Google Scholar]
- 81.Külekci M, Vitter J, Xir B. Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine. Hong Kong, China: 2010. Time-and space-efficient maximal repeat finding using the Burrows-Wheeler transform and wavelet trees; pp. 622–625. [DOI] [PubMed] [Google Scholar]
- 82.Jacobson G. Proceedings of the 30th IEEE Symposium on Foundations of Computer Science. Research Triangle Park, North Carolina, USA: 1989. Space-efficient static trees and graphs; pp. 549–554. [Google Scholar]
- 83.Arroyuelo D, Cánovas R, Navarro G, Sadakane K. Proceedings of the 11th Workshop on Algorithm Engineering and Experiments. Austin, Texas, USA: 2010. Succient trees in practice; pp. 84–97. [Google Scholar]
- 84.Archie J, Day W, Felsenstein J, Maddison W, Meacham C, Rohlf F, Swofford D. The Newick Tree Format. 1986 http://evolution.genetics.washington.edu/phylip/newicktree.html (May 2012, date last accessed) [Google Scholar]
- 85.Gog S, Fischer J. Proceedings of the 2010 Data Compression Conference. Snowbird, Utah, USA: 2010. Advantages of shared data structures for sequences of balanced parentheses; pp. 406–415. [Google Scholar]
- 86.Berkman O, Vishkin U. Recursive star-tree parallel data structure. SIAM J. Comput. 1993;22:221–242. [Google Scholar]
- 87.Grossi R, Vitter J. Proceedings of the 32nd Annual ACM Symposium on Theory of Computing, 32. Portland, Oregon, USA: 2000. Compressed suffix arrays and suffix trees with applications to text indexing and string matching; pp. 397–406. [Google Scholar]
- 88.Sadakane K. Proceedings of the 11th International Symposium on Algorithms and Computation. Taipei, Taiwan: 2000. Compressed text databases with efficient query algorithms based on the compressed suffix array; pp. 410–421. [Google Scholar]
- 89.Sadakane K. Proceedings of the 13th ACM-SIAM symposium on Discrete algorithms. San Francisco, California, USA: 2002. Succinct representations of LCP information and improvements in the compressed suffix array; pp. 225–232. [Google Scholar]
- 90.Mäkinen V. Compact suffix array - a space-efficient full-text index. Fund. Inform. 2003;56:191–210. [Google Scholar]
- 91.Mäkinen V, Navarro G. Proceedings of the 15th Symposium on Combinatorial Pattern Matching. Istanbul, Turkey: 2004. Compressed compact suffix arrays; pp. 420–433. [Google Scholar]
- 92.González R, Navarro G. Proceedings of the 18th Symposium on Combinatorial Pattern Matching. London, Ontario, Canada: 2007. Compressed text indexes with fast locate; pp. 216–227. [Google Scholar]
- 93.Elias P. Universal codeword sets and representations of integers. IEEE Trans. Inf. Theory. 1975;21:194–203. [Google Scholar]
- 94.Kärkkäinen J, Ukkonen E. Proceedings of the 3rd South American Workshop on String Processing. Recife, Brazil: 1996. Lempel-Ziv parsing and sublinear-size index structures for string matching; pp. 141–155. [Google Scholar]
- 95.Lempel A, Ziv J. On the complexity of finite sequences. IEEE Trans. Inf. Theory. 1976;22:75–81. [Google Scholar]
- 96.Ziv J, Lempel A. Compression of individual sequences via variable length coding. IEEE Trans. Inf. Theory. 1978;24:530–536. [Google Scholar]
- 97.Navarro G. Implementing the LZ-index: theory versus practice. ACM J. Exp. Algor. 2009;13:1.2. [Google Scholar]
- 98.Arroyuelo D, Navarro G, Sadakane K. Stronger Lempel-Ziv Based compressed text indexing. Algorithmica. 2012;62:54–101. [Google Scholar]
- 99.Russo L, Oliveira A. A compressed self-index using a Ziv-Lempel dictionnary. Inform. Retrieval. 2008;11:359–388. [Google Scholar]
- 100.Ohlebusch E, Gog S, Kügel A. Proceedings of the 17th Annual Symposium on String Processing and Information Retrieval. Los Cabos, Mexico: 2010. Computing matching statistics and maximal exact matches on compressed full-text indexes; pp. 347–358. [Google Scholar]
- 101.Ohlebusch E, Gog S. Proceedings of the 16th Symposium on String Processing and Information Retrieval. Saariselkä, Finland: 2009. A compressed enhanced suffix array supporting fast string matching; pp. 51–62. [Google Scholar]
- 102.Ohlebusch E, Fischer J, Gog S. Proceedings of the 17th Symposium on String Processing and Information Retrieval. Los Cabos, Mexico: 2010. CST++ pp. 322–333. [Google Scholar]
- 103.Fischer J, Mäkinen V, Navarro G. Faster entropy-bounded compressed suffix trees. Theor. Comput. Sci. 2009;410:5354–5364. [Google Scholar]
- 104.Hon W, Shah R, Vitter J. Proceedings of the 21st Symposium on Combinatorial Pattern Matching. New York, USA: 2010. Compression, indexing, and retrieval for massive string data; pp. 260–274. [Google Scholar]
- 105.Baeza-Yates R, Barbosa E, Ziviani N. Hierarchies of indices for text retrieval. J. Inf. Syst. 1996;21:497–514. [Google Scholar]
- 106.Sinha R, Puglisi S, Moffat A, Turpin A. Proceedings of the 2008 ACM SIGMOD International Conference on Management of Data. Vancouver, British Columbia, Canada: 2008. Improving suffix array locality for fast pattern matching on disk; pp. 661–672. [Google Scholar]
- 107.Ferragina P, Grossi R. The string B-tree: a new data structure for string search in external memory and its applications. J. ACM. 1999;46:236–280. [Google Scholar]
- 108.Ferragina P, Grossi R, Gupta A, Shah R, Vitter JS. Proceedings of the 27th ACM SIGMOD-SIGACT-SIGART Symposium on Principles of Database Systems. Vancouver, British Columbia, Canada: 2008. On searching compressed string collections cache-obliviously; pp. 181–190. [Google Scholar]
- 109.Hunt E, Atkinson M, Irving R. Database indexing for large DNA and protein sequence collections. The VLDB J. 2002;11:256–271. [Google Scholar]
- 110.Bedathur S, Haritsa J. Proceedings of the 20th International Conference on Data Engineering. Boston, Massachusetts, USA: 2004. Engineering a fast online persistent suffix tree construction; pp. 720–731. [Google Scholar]
- 111.Clark D, Munro J. Proceedings of the Seventh Annual ACM-SIAM Symposium on Discrete Algorithms. Atlanta, Georgia, USA: 1996. Efficient suffix trees on secondary storage (extended Abstract) pp. 383–391. [Google Scholar]
- 112.Tian Y, Tata S, Hankins R, Patel J. Practical methods for constructing suffix trees. VLDB J. 2005;14:281–299. [Google Scholar]
- 113.Phoophakdee B, Zaki M. Proceedings of the 2007 ACM SIGMOD International Conference on Management of Data. Beijing, China: 2007. Genome-scale disk-based suffix tree indexing; pp. 833–844. [Google Scholar]
- 114.Ghoting A, Makarychev K. I/O efficient algorithms for serial and parallel suffix tree construction. ACM Trans. Database Sys. 2010;35:25. [Google Scholar]
- 115.Clifford R. Distributed suffix trees. J. Discrete Algor. 2005;3:176–197. [Google Scholar]
- 116.Bedathur S, Haritsa J. Proceedings of the 12th International Conference on High Performance Computing. Goa, India: 2005. Search-Optimized suffix-tree storage for biological applications; pp. 29–39. [Google Scholar]
- 117.Brodal G, Fagerberg R. Proceedings of the 17th Annual ACM-SIAM Symposium on Discrete Algorithms. Miami, Florida, USA: 2006. Cache-oblivious string dictionaries; pp. 581–590. [Google Scholar]
- 118.Barsky M, Stege U, Thomo A, Upton C. Proceeding of the 17th ACM Conference on Information and Knowledge Management. Napa Valley, California, USA: 2008. A new method for indexing genomes using on-disk suffix trees; pp. 649–658. [Google Scholar]
- 119.Barsky M, Stege U, Thomo A, Upton C. Suffix trees for inputs larger than main memory. Inf. Syst. 2011;36:644–654. [Google Scholar]
- 120.Halachev M, Shiri N, Thamildurai A. Proceedings of the 1st International Conference on Bioinformatics Research and Development. Berlin, Germany: 2007. Efficient and scalable indexing techniques for biological sequence data; pp. 464–479. [Google Scholar]
- 121.Mäkinen V, Navarro G, Sadakane K. Proceedings of the 15th International Symposium on Algorithms and Computation. Hong Kong, China: 2004. Advantages of backward searching - efficient secondary memory and distributed implementation of compressed suffix arrays; pp. 681–692. [Google Scholar]
- 122.González R, Navarro G. A compressed text index on secondary memory. J. Comb. Math. Comb. Comput. 2009;71:127–154. [Google Scholar]
- 123.Arroyuelo D, Navarro G. Proceedings of the 18th Symposium on Combinatorial Pattern Matching. London, Ontario, Canada: 2007. A Lempel-Ziv text index on secondary storage; pp. 83–94. [Google Scholar]
- 124.Russo L, Navarro G, Oliveira A. Proceedings of the 21st Conference on Combinatorial Pattern Matching. New York, USA: 2010. Parallel and distributed compressed indexes; pp. 348–360. [Google Scholar]
- 125.Chien Y, Hon W, Shah R, Vitter J. Proceedings of the 2008 Data Compression Conference. Snowbird, Utah, USA: 2008. Geometric Burrows-Wheeler transform: linking range searching and text indexing; pp. 252–261. [Google Scholar]
- 126.Ukkonen E. On-line construction of suffix trees. Algorithmica. 1995;14:249–260. [Google Scholar]
- 127.Maaß M. Computing suffix links for suffix trees and arrays. Inf. Proces. Lett. 2007;101:250–254. [Google Scholar]
- 128.Puglisi S, Smyth W, Turpin A. A taxonomy of suffix array construction algorithms. ACM Comput. Surv. 2007;39:1–31. [Google Scholar]
- 129.Kärkkäinen J, Sanders P. Proceedings of the 30th International Conference on Automata Languages and Programming. Eindhoven, The Netherlands: 2003. Simple linear work suffix array construction; pp. 943–955. [Google Scholar]
- 130.Kärkkäinen J, Sanders P, Burkhardt S. Linear work suffix array construction. J. ACM. 2006;53:918–936. [Google Scholar]
- 131.Kulla F, Sanders P. Scalable parallel suffix array construction. Parallel Comput. 2007;33:605–612. [Google Scholar]
- 132.Ferragina P, Gagie T, Manzini G. Proceedings of the 9th Latin American Symposium on Theoretical Informatics. Oaxaca, Mexico: 2010. Lightweight data indexing and compression in external memory; pp. 697–710. [Google Scholar]
- 133.Manzini G, Ferragina P. Engineering a lightweight suffix array construction algorithm. Algorithmica. 2004;40:33–50. [Google Scholar]
- 134.Nong G, Zhang S, Chan W. Two efficient algorithms for linear time suffix array construction. IEEE Trans. Comput. 2011;60:1471–1484. [Google Scholar]
- 135.Kärkkäinen J. Fast BWT in small space by blockwise suffix sorting. Theor. Comput. Sci. 2007;387:249–257. [Google Scholar]
- 136.Sirén J. Proceedings of the 16th International Symposium on String Processing and Information Retrieval. Saariselkä, Finland: 2009. Compressed suffix arrays for massive data; pp. 63–74. [Google Scholar]
- 137.Menon R, Bhat G, Schatz M. Proceedings of the Second International Workshop on MapReduce and its Applications. San Jose, California, USA: 2011. Rapid parallel genome indexing with MapReduce; pp. 51–58. [Google Scholar]
- 138.Arroyuelo D, Navarro G. Space-Efficient Construction of Lempel-Ziv compressed text indexes. Inf. Comput. 2011;209:1070–1102. [Google Scholar]
- 139.Döring A, Weese D, Rausch T, Reinert K. SeqAn an efficient, generic C++ library for sequence analysis. BMC Bioinformatics. 2008;9:11. doi: 10.1186/1471-2105-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Loh W, Moon Y, Lee W. A fast divide-and-conquer algorithm for indexing human genome sequences. IEICE Trans. Inf. Systems. 2011;94:1369–1377. [Google Scholar]
- 141.Tsirogiannis D, Koudas N. Proceedings of the 13th International Conference on Extending Database Technology. Lausanne, Switzerland: 2010. Suffix tree construction algorithms on modern hardware; pp. 263–274. [Google Scholar]
- 142.Fernandez E, Najjar W, Lonardi S. Proceedings of the 19th Annual IEEE International Symposium on Field-Programmable Custom Computing Machines. Salt Lake City, Utah, USA: 2011. String matching in hardware using the FM-Index; pp. 218–225. [Google Scholar]
- 143.Langmead B, Schatz M, Lin J, Pop M, Salzberg S. Searching for SNPs with cloud computing. Genome Biol. 2009;10:R134. doi: 10.1186/gb-2009-10-11-r134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Salson M, Lecroq T, Léonard M, Mouchard L. A four-stage algorithm for updating a Burrows-Wheeler transform. Theor. Comput. Sci. 2009;410:4350–4359. [Google Scholar]
- 145.Salson M, Lecroq T, Léonard M, Mouchard L. Dynamic extended suffix arrays. J. Discrete Algor. 2010;8:241–257. [Google Scholar]
- 146.Mäkinen V, Navarro G, Sirén J, Välimäki N. Storage and retrieval of highly repetitive sequence collections. J. Comput. Biol. 2010;17:281–308. doi: 10.1089/cmb.2009.0169. [DOI] [PubMed] [Google Scholar]
- 147.Kuruppu S, Puglisi S, Zobel J. Proceedings of Australasian Computer Science Conference. Perth, Australia: 2011. Optimized relative Lempel-Ziv compression of genomes; pp. 91–98. [Google Scholar]