


Abstract
The pre-translational modification of messenger ribonucleic acids (mRNAs) by alternative promoter usage and alternative splicing is an important source of pleiotropy. Despite intensive efforts, our understanding of the functional implications of this dynamically created diversity is still incomplete. Using the available knowledge of interaction modules, particularly within intrinsically disordered regions (IDRs), we analysed the occurrences of protein modules within alternative exons. We find that regions removed or included by pre-translational variation are enriched in linear motifs suggesting that the removal or inclusion of exons containing these interaction modules is an important regulatory mechanism. In particular, we observe that PDZ-, PTB-, SH2- and WW-domain binding motifs are more likely to occur within alternative exons. We also determine that regions removed or included by alternative promoter usage are enriched in IDRs suggesting that protein isoform diversity is tightly coupled to the modulation of IDRs. This study, therefore, demonstrates that short linear motifs are key components for establishing protein diversity between splice variants.
INTRODUCTION
The major pre-translational mechanisms for expanding the repertoire of gene function are alternative splicing, known to occur in at least 86% of human genes (1) and alternative promoter usage, known to occur in 30–50% of human genes (2), with other mechanisms such as ribonucleic acid (RNA) editing (3) also contributing to the diversification of the human proteome. Alternative gene products increase the signalling and regulatory complexity of the proteome in both temporal- and tissue-specific manner (4). This observed proteomic complexity enabled by the one- to many relationship between most genes and their protein products raises the question of how these isoforms confer distinct functionality.
A potential explanation for this functional diversity at the protein level is modulation of domain–domain interactions (5); however, proteome-wide studies indicate that the removal of a globular domain is a relatively rare event (6,7). Instead, studies have shown that intrinsically disordered regions (IDRs) are preferentially found within alternative exons (8,9). This enrichment for IDRs does not explain the functional diversity found between many alternative protein products of a gene. For example, alternative splicing is known to determine the binding properties, stability, subcellular localisation and post-translational modifications (PTMs) of a large number of proteins (4,10). As short linear-motif (SLiM) interaction modules are enriched within IDRs (11,12), we hypothesised that the removal or addition of SLiM-containing exons could confer distinct functions to a splice variant, as these interaction modules are associated with a diverse array of cellular processes (11,13). These include promoting transport [e.g. the nuclear localisation signal (NLS)], directing cleavage (e.g. caspase-3 scission sites), acting as sites for PTMs (e.g. phosphorylation sites), mediating ligand binding (e.g. the PxxP SH3-binding motif) and marking proteins for degradation (e.g. KEN-box motif) (14).
SLiMs (∼3–10 amino acids in length) are typically associated with low-affinity interactions [generally in the 1–150 μM range (14)], predisposing them to reversible and transient associations (11). Although the context of a motif is important for binding (15), the majority of the binding affinity and specificity arises from a limited number of amino acids, 2–5 on average (11). This ensures that only a few stochastic mutations are required to convergently evolve a functional motif. For example, in neuronal cells, a single mutation of an innocuously exposed TQG sequence can create a TQT dynein-binding motif resulting in the synaptic transport of the protein (16). In this manner, motifs can arise by convergent evolution (11), with stochastic mutations more likely to occur in regions with high substitution rates, such as IDRs (17) and alternative exons (18). The presence of motifs within alternative exons can, in turn, create novel functionality for splice variants. For example, the inclusion of an alternative exon 3 amino acids in length creates a dynein-binding motif in a splice variant of myosin Va, enabling splice variant-specific cargo recognition (19). The presence of SLiMs in alternative exons can also create splice variants with novel cellular localisations, as occurs in human 8-oxoguanine deoxyribonucleic acid (DNA) glycosylase when an alternative exon containing an NLS targeting motif is removed, leading to the exclusion of the splice variant from the nucleus (20).
In this article, we investigate whether the removal or addition of exons containing SLiMs is a common regulatory mechanism used by the cell. We analyse the experimentally validated SLiM instances annotated in the Eukaryotic Linear Motif (ELM) (13) and Domino (21) resources, along with other functional units (globular domains, phosphorylation sites, transmembrane regions and signal peptides), for their presence in sequences altered between known protein isoforms (AltSeqs). We demonstrate that SLiMs are enriched within AltSeqs and confirm that partial domains are under-represented within AltSeqs (6). We also demonstrate that exons excluded or included by alternative promoter usage are enriched with IDRs demonstrating that these unstructured regions of proteins are a recurring property of non-constitutive exons.
MATERIALS AND METHODS
Data sets
SLiM instances are extracted from the ELM resource (version 08/2011) (13), a database of manually annotated experimentally verified SLiMs. These SLiMs are divided by ELM into different functional classes (160 in total), each describing a unique molecular function. Each ELM class is described by a regular expression defined using experimentally validated SLiM instances. These 1595 instances represent a gold standard for SLiM annotation and were collected independently of whether they were present in alternative exons.
An additional data set is also derived from the Domino peptide interaction database (version 10/2009) (21) to validate the results produced using data from the ELM resource. Domino annotates high-quality experimental data on globular domain-peptide interactions independently of ELM and, therefore, can be used as a cross-validation data set. A total of 848 protein isoforms produced from 274 genes are extracted from the Domino resource with peptides shorter than 30 amino acids. A minimal length of 30 amino acids is chosen, as this is shorter than all known linear-motif interaction domains (shortest WW domain) (22). Five linear motifs classes, whose interactions have been analysed in greater detail by high-throughput (HTP) studies and/or curated by experimental annotation databases are investigated in detail. These linear-motif instances bind to PDZ (23), PTB (24), SH2 (13,25), SH3 (26) and WW (13,27) domains and together create a dataset of 408 motif instances within 302 genes.
As additional annotation, for each canonical protein sequence with a known motif instance, globular domains are extracted from Pfam v25 (28), phosphorylation sites from the low-throughput annotation of Phospho.ELM (03/2011) (29) and functional elements (transmembrane domains and signal peptides) from UniProt annotation. These features are mapped onto the canonical protein sequence as defined by UniProt.
Isoform data are retrieved from UniProt (05/2011) (30), a manually annotated, non-redundant protein sequence database. This resource curates annotated protein splice variants of genes only if there is experimental evidence that it exists or has at least one messenger RNA (mRNA) with correct intron/exon boundaries. It, therefore, represents a high-quality resource of validated protein isoforms. The analyses use the canonical isoform as chosen by UniProt. All protein products of a gene are extracted from the UniProt resource for protein sequences with at least one ELM-annotated SLiM instance and more than one annotated UniProt protein product.
All data sets are filtered for proteins of high similarity using UniRef90 (31) to limit bias introduced by homologous proteins with greater than 90% sequence identity.
Methods
The enrichment of functional units (SLiMs, phosphorylation sites, globular domains and functional elements [transmembrane domains and signal peptides]) within alternative sequences (AltSeqs) is assessed based on an approach outlined by Kriventseva et al. (6). This method aims to evaluate whether there is a preference for certain functional units to be altered between protein isoforms. This approach compares the expected number of instances—calculated with the assumption that there are no biases in the data set towards certain functional units being altered—with the observed number of instances altered between protein isoforms. AltSeqs are used in this approach, as they are continuous sections present in canonical protein sequences, as prescribed by UniProt, but missing in another protein isoform. AltSeqs, therefore, reflect the consequences of transcript changes at the protein level, for example, an AltSeq may represent two alternative exons that are always removed together, ensuring a whole globular domain is never only partially present.
The calculation of the expected number of occurrences (e) of functional units within AltSeqs uses a sliding window method. This approach requires that AltSeqs are randomly distributed within protein sequences. To test this assumption, the distribution of annotated UniProt AltSeqs is assessed. This analysis found no strong positional bias for AltSeqs [Supplementary Figure S1 and Kriventseva et al (6)]. A sliding window approach is, therefore, used to calculate the expected number of occurrences of functional units. For each AltSeq, a window of equal length to the AltSeq (Window) scans the AltSeq-containing protein progressing one amino acid at a time, counting the functional units (FUNCAS) overlapping (partial hits) or within (full hits) the window. The expected occurrences of partial/full domains, phosphorylation sites, transmembrane regions, signal peptides, PTMs and linear motifs are calculated using the following equation:
| (1) |
where FUNCAS = number of instances of a functional unit, j, in sliding windows; Window = number of sliding windows; AltSeqsCount = number of AltSeqs. A goodness of fit χ2 test is then used to compare the expected and observed proportions.
The expected number of occurrence of functional units within regions of intrinsic disorder [( )] is also assessed. A protein sequence is assessed for disorder using the IUPred algorithm (32), with the assumption that amino acids with IUPred scores over 0.4 are disordered (11,12). The following equation is used:
)] is also assessed. A protein sequence is assessed for disorder using the IUPred algorithm (32), with the assumption that amino acids with IUPred scores over 0.4 are disordered (11,12). The following equation is used:
| (2) |
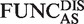 = number of functional units both in a disordered region and an AltSeqs; WindowDIS = sliding window count only including windows with an average IUPred score of over 0.4; AltSeqsCountDIS = number of AltSeqs in disordered regions (average IUPred score > 0.4). A goodness of fit χ2 test is then used to compare the expected and observed proportions.
= number of functional units both in a disordered region and an AltSeqs; WindowDIS = sliding window count only including windows with an average IUPred score of over 0.4; AltSeqsCountDIS = number of AltSeqs in disordered regions (average IUPred score > 0.4). A goodness of fit χ2 test is then used to compare the expected and observed proportions.
The assessment of the individual ELM classes for enrichment within AltSeqs by χ2 test is infeasible due to the limited number of instances in each class. An adaptation of the log-odds ratio calculation [LOG-odds domain (LOD) (5)] was, therefore, used to compare individual ELM classes with the observed occurrence of linear-motif removal, taking into account the number of instances in each ELM class:
| (3) |
IC = instance count, Pyy = observed probability of an instance in ELM class being in AltSeq; Pyx = observed probability of an instance in ELM class not being in AltSeq; Pxx = observed probability of an ELM instance in AltSeq, Pxy = Observed probability of an ELM instance not being in AltSeq.
Counts of recurring SLiMs or SLiM instances that have at least one other instance of the same ELM class in the same protein is calculated from the ELM resource’s annotated data. A recent survey of ELM identified that 34.9% of ELM-annotated instances were recurring (11). A goodness of fit χ2 test is used to assess whether the observed occurrences of recurring motifs within AltSeqs is present at a higher rate than expected (34.9%).
Structural disorder is assessed using the IDR predictor IUPred (32) with exons having an average score of greater than 0.4 considered as unstructured (11,12). The expected proportion of intrinsic disorder within an exon is calculated based on an analysis of the protein-coding exons annotated by EnsEMBL (33) found within the canonical UniProt human proteins. A goodness of fit χ2 test compares the intrinsic disorder of the average exon with the observed intrinsic disorder of the exons altered by alternative promoter usage or alternative splicing.
RESULTS
Alternative promoter exons are enriched in IDRs
AltSeqs produced by alternative splicing are enriched for IDRs (8); however, no investigation of protein-encoding AltSeqs specifically produced by alternative promoter usage has been undertaken. A comparison is therefore undertaken comparing the proportion of IDRs within the average human exon with the proportion of IDRs within exons removed or included by alternative promoter usage. We extract from the UniProt database (30), a non-redundant set of 188 altered splice variants derived from 124 genes produced solely by alternative promoter usage. The analysis of this data using the IUPred algorithm (scores > 0.4 considered disordered) identifies an enrichment of IDRs within exons altered by alternative promoter usage (χ2 P value: 0.033) (59 observed and 38 expected) (Figure 1). In addition to this, a significant under-representation of ordered regions is noted within those AltSeqs altered by alternative promoter usage (χ2 P value: 1.32 e−3) (61 observed and 102 expected). The exons removed or included by alternative splicing are also enriched for IDRs (χ2 P value: 2.20 e−16) as previously shown (8).
Figure 1.

A comparison of intrinsic disorder between exons. The proportion of exons predicted as intrinsically disordered, defined as residues that the IUPred algorithm predicted with a score above 0.4. Exons altered by alternative splicing and exons altered by alternative promoter usage were analysed for IDRs as compared with the average human exon. Error bars represent 90.0% error rate.
SLiMs are enriched in AltSeqs
The enrichment of IDRs within AltSeqs raises the question of whether known functional regions within IDRs occur at a higher or lower rate than expected within sequences altered between protein isoforms. The initial analysis of functional site enrichment within AltSeqs is undertaken using a data set of 1421 protein isoforms produced from 404 genes, which is limited to those genes with a protein isoform containing an annotated and experimentally validated SLiM instance from the ELM resource. As shown in Figure 2a, the proportion of AltSeqs (average length 112.3 residues, equivalent to 15.5% of average UniProt sequence length) containing a SLiM is at a higher frequency than expected (χ2 P value: 5.13 e−5) with 196 SLiMs (30.3% of SLiMs in proteins with alternative products or 12.1% of total ELM instances) observed in AltSeqs compared with the 123 expected. Phosphorylation sites are similarly enriched (χ2 P value: 5.76 e−4) with 61 more sites found in AltSeqs than the 128 expected. There is, however, a potential bias in this analysis, as IDRs are enriched in alternative exons (8,9) and SLiMs are enriched in IDRs (11,12). We, therefore, also assessed whether the aforementioned enrichment still occurs, when only regions predicted as disordered are investigated [IUPred (32) scores > 0.4 considered as an IDR]. In this case, SLiMs are the sole functional unit significantly enriched (χ2 P value: 2.40 e−4) (138 observed and 83 expected) (Figure 2b) suggesting a preference for SLiMs in AltSeqs. These results are validated using the independently annotated data from the Domino database of peptide-mediated interactions (21) consisting of 848 protein isoforms produced from 274 genes. Peptides, likely to contain SLiMs, are highly enriched within AltSeqs (χ2 P value: 4.74 e−5) (163 observed and 97 expected). This enrichment of SLiMs is again observed when only functional units within IDRs are investigated (χ2 P value: 5.71 e−3) (106 observed and 69 expected) (Supplementary Figure S3). For further assessment of functional site enrichment, additional instances of PTMs are extracted from the PhosphoSite Plus database (34). However, no enrichment is identified for these other PTMs in AltSeqs (Supplementary Table S1). This suggests that SLiMs represent a key regulatory element altered between protein isoforms.
Figure 2.

The distribution of functional units within AltSeqs. The observed and expected counts of AltSeqs disrupting an entire or partial SLiM, a phosphorylation site, an entire or partial globular domain, an entire or partial functional element (transmembrane domain or signal peptide) or no functional units from the annotated data set. The number of elements in each class is shown and their percentage in brackets. (A) The observed distribution of functional units within AltSeqs when compared with the expected distribution. (B) The observed distribution of functional units within AltSeqs when compared with the expected distribution within IDRs (regions with IUPred scores > 0.4). Both partial and entire transmembrane domains and signal peptides (functional elements) are combined, as their observed occurrences were very low.
The analysis does not show a bias towards a particular type of SLiM, for example, targeting motifs, to be in an AltSeq (Supplementary Figure S2). The observation that SLiMs are enriched within AltSeqs but no particular ELM type is significantly enriched raises the question, what type of SLiMs are present within these regions? We, therefore, assess the individual ELM functional classes for enrichment within alternative exons (Table 1). We identify a number of classes whose instances occur at a much higher frequency than expected in non-constitutive exons. The majority of these ELM classes bind to domains found within intracellular signal-transduction proteins (e.g. SH2 or PTB domains). However, the instances annotated in the ELM resource are limited in number, as only examples identified by low-throughput experimentation are included. To validate the observation that motif instances associated with domains in signal-transduction proteins being enriched in protein encoding alternative exons, motif instances identified as binding to PDZ (23), PTB (24), SH2 (13,25), SH3 (26) and WW (13,27) domains in HTP experiments and by specialist annotation are investigated further. As shown in Figure 3, the aforementioned enrichment of motifs binding to the SH2 (χ2 P value: 0.027) and PTB (χ2 P value: 0.033) domains is confirmed, as well as identifying that PDZ- (χ2 P value: 0.025) and WW-(χ2 P value: 0.092) domain-binding motifs have an increased likelihood of being removed or included between protein isoforms.
Table 1.
Preferential alteration of specific ELM classes
| ELM_ID | Regular expression | Binding domaina | No. removed | No. total | Percentage removed | LOD |
|---|---|---|---|---|---|---|
| LIG_SH2_STAT3 | (Y).Q | SH2 | 7 | 8 | 87.50 | 10.89 |
| LIG_SH2_STAT5 | (Y)[VLTFIC]. | SH2 | 7 | 12 | 58.33 | 7.95 |
| LIG_PTB_Apo_2 | [^P].NP.(Y) | PTB | 8 | 19 | 42.11 | 7.91 |
| MOD_TYR_ITSM | T.(Y).[IV] | SH2 | 6 | 11 | 54.55 | 6.55 |
| LIG_PTB_Phospho_1 | [^P].NP.[FY]. | PTB | 7 | 16 | 43.75 | 6.51 |
| LIG_SxIP_EBH_1 | [ST].[IL]P | EB1 | 5 | 9 | 55.56 | 5.52 |
| LIG_PDZ_Class_1 | [ST].[ACVILF]$ | PDZ | 6 | 15 | 40.00 | 5.11 |
| LIG_EVH1_2 | PP.F | WH1 | 4 | 8 | 50.00 | 4.13 |
| MOD_PKA_1 | [RK][RK].([ST])[^P]. | Pkinase | 7 | 23 | 30.43 | 3.62 |
| MOD_ProDKin_1 | ([ST])P. | Pkinase | 8 | 28 | 28.57 | 3.32 |
| TRG_ENDOCYTIC | Y.[LMVIF] | Adap_comp_sub | 3 | 7 | 42.86 | 2.74 |
The number of instances per an ELM class that occur in non-constitutive protein-encoding exons (removed), the total number of instances annotated and the log-odds ratio (LOD), Equation 3, for statistical significance as a function of the total counts of the domain (5) are presented. The regular expressions are annotated in the ELM resource. Only ELM classes with more than five annotated instances were assessed, and only those with a LOD score >2 are shown. $ = C terminal; (X) = residue X must be modified for binding (e.g. by phosphorylation); [^P] = proline residue not allowed; . = any amino acid; [XYZ] = either residue X, Y or Z is allowed at this position.
aNames based on annotated by the Pfam resource.
Figure 3.

A comparison of the occurrences of five highly studied binding motifs within alternative exons. The observed and expected occurrences of linear motifs identified in HTP experimental studies of proteins with known isoforms (30). The PDZ, PTB and SH2-binding sites are all enriched within AltSeqs (χ2, P < 0.05). The WW domain-binding motif is not enriched to a level of statistical significance but has 26 instances observed in AltSeqs compared with the 15.2 instances expected. The expected occurrence of binding motifs was calculated using Equation 1 except for the PDZ-binding motif, which was calculated based on the occurrence of AltSeqs at the C terminal. (*statistical enrichment and numbers = occurrences).
SLiMs tend to reoccur or have one (or multiple) other instances of the same ELM class in the same protein. This is highlighted by the fact that 34.9% of ELM-annotated SLiM instances are recurring (11). When we analyse the occurrence of these recurring motifs within alternative exons, we find that SLiMs known to reoccur multiple times in a protein sequence are significantly enriched within AltSeqs (50.7% are recurring: χ2 P value: 0.013) (107 observed and 73.6 expected). This suggests that the inclusion or removal of SLiM-containing exons may tune the multivalent cooperativity of an isoform’s interactions (35).
The removal or inclusion of complete globular domains also displays a weak statistical enrichment within AltSeqs (χ2 P value: 1.29 e−3) (204 observed and 144 expected) reflecting similar results by Kriventseva et al. (6). Conversely, splice variants with partial domains or domains that are partially encoded by an AltSeq are under-represented (χ2 P value < 1.59 e−3) (161 observed and 223 expected) (6,7). A similar finding is also observed for functional elements truncated by the removal or inclusion of AltSeqs (15 observed and 85 expected) (Figure 2a) (6).
SLiM prediction can aid understanding of protein isoforms
The diverse and often conflicting functions of the different protein products of a gene are frequently designated to one protein isoform. The use of bioinformatics to discriminate these differences, in particular by identifying isoform-specific SLiMs, may facilitate an understanding of the distinct properties of these splice variants, such as half-life, interaction partners and cellular localisation.
An apt example of this is p53, whose varied and often opposing functions have puzzled researchers for many years (36). The recent expansion in the number of known alternative protein products of this gene has given a tantalising opportunity to uncover the source of this functional diversity (37). A series of articles focusing on the transcriptional regulation of these splice isoforms [e.g. (37,38)] has enabled some of this diversity to be explored. In Figure 4, the alternative products of p53 are displayed along with their DNA-binding domains and SLiMs. The different phenotypes of p53 isoforms can often be attributed to the removal or inclusion of SLiM-containing exons. For example, the increased half-life of Δ40p53 (9.5 h compared with 5–20 min of full-length p53) (38) has been attributed to the loss of the MDM2-binding site (39), which marks full-length p53 for degradation by the attachment of ubiquitin (40). Similarly, the absence of the nuclear export signal in Δ40p53γ explains its exclusive nuclear localisation (in a similar manner to p53β and Δ133p53γ) (38). Other putative explanations of phenotypic observations include attributing the shorter half-life of p53β to the absence of the LIG_USP7_1 (41) and 14-3-3 (42) binding sites. Novel motifs can also arise by addition of an alternative exon, such as the putative KEN-box degron in p53γ, which could indicate a novel method of degradation for p53γ by the APC/C complex during anaphase. The expression and half-life of p53 is, therefore, carefully regulated by pre-and post-translational mechanisms that alter the availability of this protein’s interaction surfaces resulting in subtle but important phenotypic differences (37,38). Observations based on the interpretation of phenotypic data can help direct further experimentation, which may further elucidate the often-enigmatic differences between protein isoforms.
Figure 4.

Bioinformatics can identify functional differences between protein variants. (A) The exon sequence of the TP53 gene. The coloured (non-grey) exons are alternative exons that vary between protein isoforms. The yellow exons are absent in Δ40p53, the mauve exon is specific to p53β, the mint green exon is exclusive to p53γ and the orange exons are present only in p53α. (B) The four distinct isoforms of TP53 are shown with modular architecture annotated onto the full-length protein isoform (p53α) using ELM and Pfam, the exception being the KEN-box, which is predicted. The protein sequences of the TP53 isoforms are shown as a grey line, SLiMs in red, globular domains in blue and previously shown modular structures are opaque. Sequence diversity between the alternative protein products leads to changes in the SLiM content of the p53 alternative products, for example, p53γ loses a cyclin binding site and two USP7 binding sites, a nuclear export signal and a 14-3-3 binding site.
DISCUSSION
The importance of pre-translational variation within the cell for facilitating cell signalling and regulation is becoming increasingly apparent (43–45). The inclusion or removal of non-protein coding regions, for example, is known to influence mRNA stability, translational efficiency and mRNA localisation (46,47). In this article, we have investigated how the removal/inclusion of functional modules between protein isoforms can lead to functional diversity. In particular, we have focused on the inclusion/removal of SLiM-containing alternative exons known to create protein isoforms of differing functions (10). These differences include the targeting of protein splice variants to different sub-cellular locations [e.g. to the peroxisome rather than the mitochondria (48)], changes in interaction partners [e.g. PDZ SLiMs within membrane receptors (49)] or more dramatic changes such as altering a protein’s function from pro-apoptotic to anti-apoptotic (50).
In this article, we have confirmed previous observations that alternatively spliced exons are enriched for IDRs (8,9) as well as demonstrating a similar enrichment for IDRs in exons generated by alternative promoter usage (Figure 1). This observation prompted us to investigate the propensity of known functional protein modules to occur in regions altered by alternative splicing and/or by variable promoter usage. We identified an enrichment of SLiMs within AltSeqs indicating that the inclusion or removal of motif-containing exons is an important mechanism for modifying the functional properties of alternative protein products. In particular, exons containing SLiMs that bind to SH2 domains are commonly altered by pre-translational mechanisms (Table 1 and Figure 3). SH2-binding motifs are often present in the cytoplasmic tails of membrane receptors, and their inclusion or removal is, for example, known to affect the multivalent assembly of regulatory complexes important for signal propagation (51,52). Similarly, the inclusion or removal of PDZ motifs, also found enriched in AltSeqs, is known to create functional diversity. For example, in neurons, splice variants differing in their C-terminal PDZ motifs play specific roles in the regulation of neurotransmission, ion channel function and development (49).
The small footprint of linear motifs confers a number of advantages in terms of cell regulation and signalling (11,53,54). First, the limited number of residues in a SLiM that contribute to binding usually leads to a binding affinity for SLiM-mediated interactions in the micromolar range. Consequently, motif-mediated interactions are predominately both transient and reversible (14). This reliance on a limited number of amino acids means that SLiM-mediated interactions can be weakened (or strengthened) by PTMs, whose bulk and charge can disrupt (or enhance) this weak binding affinity. Similarly, the short length of linear motifs means these interaction modules can often occur multiple times in a single protein (11). This can facilitate mutually exclusive binding, when two motifs share a binding surface (for example, when they overlap) or promote high-avidity interactions, when motifs reoccur in separate positions along a protein sequence. These switching mechanisms act to regulate SLiM-mediated interaction. Alternative splicing and other pre-translational mechanisms can therefore alter the regulation of a protein by including or removing SLiM-containing exons. For example, altering the number of reoccurring motifs in a protein by exon removal/inclusion can change the avidity of SLiM-mediated interactions, tuning the sensitivity of signalling pathways in a temporal- and tissue-specific manner [e.g. (55)]. In this article, we have demonstrated that these reoccurring motifs are enriched in AltSeqs, suggesting that the inclusion/removal of reoccurring SLiMs is a mechanism commonly used by the cell. Similarly, an exon boundary intersecting these two overlapping SLiMs can facilitate the production of one isoform with an overlapping pair of motifs capable of acting as a regulatory switch and another isoform with just a single motif [e.g. (56,57)]. Linear motifs are, therefore, susceptible to a multiplicity of regulatory mechanisms that are important in regulating signalling within the cell. These regulatory features can be manipulated by the inclusion or removal of non-constitutive exons to create important but often subtle differences in the regulation and function of a protein.
The high false-positive rate of SLiM prediction (11) means the scope of this analysis is limited to the annotated SLiM data sets available from the ELM and Domino resources as well as data from HTP experimental studies. Despite this limitation, we have still been able to demonstrate a statistical enrichment of SLiMs within AltSeqs, suggesting an important role for motifs in the functional diversification and regulation of alternative protein products. An appreciation of how functional differences can arise between protein isoforms is key to our understanding of proteomic diversity. This is important as up to one-half of disease-causing mutations affect splicing (58) with several examples of the inclusion/exclusion of SLiM-containing exons producing disease-specific isoforms (59–61). An example of this is Hoyerall-Hreidarsson syndrome, a rare genetic disorder characterised by premature ageing, in which an aberrant splice variant of the Apollo gene is expressed that lacks a telomeric repeat-binding factor 2 (TRF2)-binding motif. This Apollo splice variant is unable to bind the TRF2 protein leading to telomeric dysfunction and cellular senescence (61). Approaches are being developed to target this type of aberrant splicing event by redirecting alternative splicing. The principal of this approach is to redirect the splicing of a transcript to promote the production of a favourable isoform in preference to the unfavourable splice variant (62). This could have therapeutic potential as demonstrated in Duchenne muscular dystrophy (63) and a melanoma model (64). An appreciation of the protein interaction modules most commonly altered between protein isoforms can help target these problems more precisely.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1, Supplementary Figures 1–3, Supplementary Methods, Supplementary Results and Supplementary Reference [65].
FUNDING
EMBL International PhD Program (EIPP) fellowship (R.J.W.) and an EMBL Interdisciplinary Postdoctoral (EIPOD) fellowship (N.E.D.). Funding for open access charge: EMBL.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank members of the Gibson Team, and in particular Kim Van Roey, for their critical reading of the manuscript.
REFERENCES
- 1.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davuluri RV, Suzuki Y, Sugano S, Plass C, Huang TH. The functional consequences of alternative promoter use in mammalian genomes. Trends Genet. 2008;24:167–177. doi: 10.1016/j.tig.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 3.Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E, Zhang K, Gao Y, Church GM. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 4.Stamm S, Ben-Ari S, Rafalska I, Tang Y, Zhang Z, Toiber D, Thanaraj TA, Soreq H. Function of alternative splicing. Gene. 2005;344:1–20. doi: 10.1016/j.gene.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 5.Resch A, Xing Y, Modrek B, Gorlick M, Riley R, Lee C. Assessing the impact of alternative splicing on domain interactions in the human proteome. J. Proteome. Res. 2004;3:76–83. doi: 10.1021/pr034064v. [DOI] [PubMed] [Google Scholar]
- 6.Kriventseva EV, Koch I, Apweiler R, Vingron M, Bork P, Gelfand MS, Sunyaev S. Increase of functional diversity by alternative splicing. Trends Genet. 2003;19:124–128. doi: 10.1016/S0168-9525(03)00023-4. [DOI] [PubMed] [Google Scholar]
- 7.Tress ML, Martelli PL, Frankish A, Reeves GA, Wesselink JJ, Yeats C, Olason PI, Albrecht M, Hegyi H, Giorgetti A, et al. The implications of alternative splicing in the ENCODE protein complement. Proc. Natl Acad. Sci. USA. 2007;104:5495–5500. doi: 10.1073/pnas.0700800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romero PR, Zaidi S, Fang YY, Uversky VN, Radivojac P, Oldfield CJ, Cortese MS, Sickmeier M, LeGall T, Obradovic Z, et al. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc. Natl Acad. Sci. USA. 2006;103:8390–8395. doi: 10.1073/pnas.0507916103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegyi H, Kalmar L, Horvath T, Tompa P. Verification of alternative splicing variants based on domain integrity, truncation length and intrinsic protein disorder. Nucleic Acids Res. 2011;39:1208–1219. doi: 10.1093/nar/gkq843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weatheritt RJ, Gibson TJ. Linear motifs: lost in (pre)translation. Trends Biochem. Sci. 2012;37:333–341. doi: 10.1016/j.tibs.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Davey NE, Van Roey K, Weatheritt RJ, Toedt G, Uyar B, Altenberg B, Budd A, Diella F, Dinkel H, Gibson TJ. Attributes of short linear motifs. Mol. Biosyst. 2012;8:268–281. doi: 10.1039/c1mb05231d. [DOI] [PubMed] [Google Scholar]
- 12.Fuxreiter M, Tompa P, Simon I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics. 2007;23:950–956. doi: 10.1093/bioinformatics/btm035. [DOI] [PubMed] [Google Scholar]
- 13.Dinkel H, Michael S, Weatheritt RJ, Davey NE, Van Roey K, Altenberg B, Toedt G, Uyar B, Seiler M, Budd A, et al. ELM—the database of eukaryotic linear motifs. Nucleic Acids Res. 2012;40:D242–D251. doi: 10.1093/nar/gkr1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diella F, Haslam N, Chica C, Budd A, Michael S, Brown NP, Trave G, Gibson TJ. Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front Biosci. 2008;13:6580–6603. doi: 10.2741/3175. [DOI] [PubMed] [Google Scholar]
- 15.Stein A, Aloy P. Contextual specificity in peptide-mediated protein interactions. PLoS One. 2008;3:e2524. doi: 10.1371/journal.pone.0002524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lo KW, Naisbitt S, Fan JS, Sheng M, Zhang M. The 8-kDa dynein light chain binds to its targets via a conserved (K/R)XTQT motif. J Biol Chem. 2001;276:14059–14066. doi: 10.1074/jbc.M010320200. [DOI] [PubMed] [Google Scholar]
- 17.Tompa P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 18.Chen FC, Wang SS, Chen CJ, Li WH, Chuang TJ. Alternatively and constitutively spliced exons are subject to different evolutionary forces. Mol. Biol. Evol. 2006;23:675–682. doi: 10.1093/molbev/msj081. [DOI] [PubMed] [Google Scholar]
- 19.Wagner W, Fodor E, Ginsburg A, Hammer JAr. The binding of DYNLL2 to myosin Va requires alternatively spliced exon B and stabilizes a portion of the myosin's coiled-coil domain. Biochemistry. 2006;45:11564–11577. doi: 10.1021/bi061142u. [DOI] [PubMed] [Google Scholar]
- 20.Nishioka K, Ohtsubo T, Oda H, Fujiwara T, Kang D, Sugimachi K, Nakabeppu Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell. 1999;10:1637–1652. doi: 10.1091/mbc.10.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ceol A, Chatr-aryamontri A, Santonico E, Sacco R, Castagnoli L, Cesareni G. DOMINO: a database of domain-peptide interactions. Nucleic Acids Res. 2007;35:D557–D560. doi: 10.1093/nar/gkl961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weatheritt RJ, Luck K, Petsalaki E, Davey NE, Gibson TJ. The identification of short linear motif-mediated interfaces within the human interactome. Bioinformatics. 2012;28:976–982. doi: 10.1093/bioinformatics/bts072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beuming T, Skrabanek L, Niv MY, Mukherjee P, Weinstein H. PDZBase: a protein-protein interaction database for PDZ-domains. Bioinformatics. 2005;21:827–828. doi: 10.1093/bioinformatics/bti098. [DOI] [PubMed] [Google Scholar]
- 24.Smith MJ, Hardy WR, Murphy JM, Jones N, Pawson T. Screening for PTB domain binding partners and ligand specificity using proteome-derived NPXY peptide arrays. Mol. Cell. Biol. 2006;26:8461–8474. doi: 10.1128/MCB.01491-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang H, Li L, Wu C, Schibli D, Colwill K, Ma S, Li C, Roy P, Ho K, Songyang Z, et al. Defining the specificity space of the human SRC homology 2 domain. Mol. Cell. Proteomics. 2008;7:768–784. doi: 10.1074/mcp.M700312-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Wu C, Ma MH, Brown KR, Geisler M, Li L, Tzeng E, Jia CY, Jurisica I, Li SS. Systematic identification of SH3 domain-mediated human protein-protein interactions by peptide array target screening. Proteomics. 2007;7:1775–1785. doi: 10.1002/pmic.200601006. [DOI] [PubMed] [Google Scholar]
- 27.Liou YC, Zhou XZ, Lu KP. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem. Sci. 2011;36:501–514. doi: 10.1016/j.tibs.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, Diella F. Phospho.ELM: a database of phosphorylation sites—update 2011. Nucleic Acids Res. 2011;39:D261–D267. doi: 10.1093/nar/gkq1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.UniProt Consortium. The universal protein resource (UniProt) in 2010. Nucleic Acids Res. 2010;38:D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li W, Jaroszewski L, Godzik A. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics. 2001;17:282–283. doi: 10.1093/bioinformatics/17.3.282. [DOI] [PubMed] [Google Scholar]
- 32.Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- 33.Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, et al. Ensembl 2012. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibson TJ. Cell regulation: determined to signal discrete cooperation. Trends Biochem. Sci. 2009;34:471–482. doi: 10.1016/j.tibs.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 36.Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harb. Perspect. Biol. 2010;2:a000893. doi: 10.1101/cshperspect.a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khoury MP, Bourdon JC. p53 Isoforms: an intracellular microprocessor? Genes Cancer. 2011;2:453–465. doi: 10.1177/1947601911408893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marcel V, Hainaut P. p53 isoforms—a conspiracy to kidnap p53 tumor suppressor activity? Cell. Mol. Life Sci. 2009;66:391–406. doi: 10.1007/s00018-008-8336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Courtois S, Verhaegh G, North S, Luciani MG, Lassus P, Hibner U, Oren M, Hainaut P. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene. 2002;21:6722–6728. doi: 10.1038/sj.onc.1205874. [DOI] [PubMed] [Google Scholar]
- 40.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 41.Sheng Y, Saridakis V, Sarkari F, Duan S, Wu T, Arrowsmith CH, Frappier L. Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat. Struct. Mol. Biol. 2006;13:285–291. doi: 10.1038/nsmb1067. [DOI] [PubMed] [Google Scholar]
- 42.Schumacher B, Mondry J, Thiel P, Weyand M, Ottmann C. Structure of the p53 C-terminus bound to 14-3-3: implications for stabilization of the p53 tetramer. FEBS Lett. 2010;584:1443–1448. doi: 10.1016/j.febslet.2010.02.065. [DOI] [PubMed] [Google Scholar]
- 43.Moroy T, Heyd F. The impact of alternative splicing in vivo: mouse models show the way. RNA. 2007;13:1155–1171. doi: 10.1261/rna.554607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore MJ, Wang Q, Kennedy CJ, Silver PA. An alternative splicing network links cell-cycle control to apoptosis. Cell. 2010;142:625–636. doi: 10.1016/j.cell.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tollervey JR, Wang Z, Hortobagyi T, Witten JT, Zarnack K, Kayikci M, Clark TA, Schweitzer AC, Rot G, Curk T, et al. Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res. 2011;21:1572–1582. doi: 10.1101/gr.122226.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiaruttini C, Sonego M, Baj G, Simonato M, Tongiorgi E. BDNF mRNA splice variants display activity-dependent targeting to distinct hippocampal laminae. Mol. Cell. Neurosci. 2008;37:11–19. doi: 10.1016/j.mcn.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 47.Hughes TA. Regulation of gene expression by alternative untranslated regions. Trends Genet. 2006;22:119–122. doi: 10.1016/j.tig.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Mubiru JN, Shen-Ong GL, Valente AJ, Troyer DA. Alternative spliced variants of the alpha-methylacyl-CoA racemase gene and their expression in prostate cancer. Gene. 2004;327:89–98. doi: 10.1016/j.gene.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 49.Sierralta J, Mendoza C. PDZ-containing proteins: alternative splicing as a source of functional diversity. Brain Res. Brain Res. Rev. 2004;47:105–115. doi: 10.1016/j.brainresrev.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 50.Scott M, Bonnefin P, Vieyra D, Boisvert FM, Young D, Bazett-Jones DP, Riabowol K. UV-induced binding of ING1 to PCNA regulates the induction of apoptosis. J. Cell. Sci. 2001;114:3455–3462. doi: 10.1242/jcs.114.19.3455. [DOI] [PubMed] [Google Scholar]
- 51.Gao Z, Monckton EA, Glubrecht DD, Logan C, Godbout R. The early isoform of disabled-1 functions independently of Reelin-mediated tyrosine phosphorylation in chick retina. Mol. Cell. Biol. 2010;30:4339–4353. doi: 10.1128/MCB.00545-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qazi AM, Tsai-Morris CH, Dufau ML. Ligand-independent homo- and heterodimerization of human prolactin receptor variants: inhibitory action of the short forms by heterodimerization. Mol. Endocrinol. 2006;20:1912–1923. doi: 10.1210/me.2005-0291. [DOI] [PubMed] [Google Scholar]
- 53.Akiva E, Friedlander G, Itzhaki Z, Margalit H. A dynamic view of domain-motif interactions. PLoS Comput. Biol. 2012;8:e1002341. doi: 10.1371/journal.pcbi.1002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Roey K, Gibson TJ, Davey NE. Motif switches: decision-making in cell regulation. Curr. Opin. Struct. Biol. 2012;22:378–385. doi: 10.1016/j.sbi.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 55.Mantovani F, Piazza S, Gostissa M, Strano S, Zacchi P, Mantovani R, Blandino G, Del Sal G. Pin1 links the activities of c-Abl and p300 in regulating p73 function. Mol. Cell. 2004;14:625–636. doi: 10.1016/j.molcel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 56.Lorenzo MJ, Gish GD, Houghton C, Stonehouse TJ, Pawson T, Ponder BA, Smith DP. RET alternate splicing influences the interaction of activated RET with the SH2 and PTB domains of Shc, and the SH2 domain of Grb2. Oncogene. 1997;14:763–771. doi: 10.1038/sj.onc.1200894. [DOI] [PubMed] [Google Scholar]
- 57.Wong A, Bogni S, Kotka P, de Graaff E, D'Agati V, Costantini F, Pachnis V. Phosphotyrosine 1062 is critical for the in vivo activity of the Ret9 receptor tyrosine kinase isoform. Mol. Cell. Biol. 2005;25:9661–9673. doi: 10.1128/MCB.25.21.9661-9673.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lopez-Bigas N, Audit B, Ouzounis C, Parra G, Guigo R. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 2005;579:1900–1903. doi: 10.1016/j.febslet.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 59.Guardavaccaro D, Frescas D, Dorrello NV, Peschiaroli A, Multani AS, Cardozo T, Lasorella A, Iavarone A, Chang S, Hernando E, et al. Control of chromosome stability by the beta-TrCP-REST-Mad2 axis. Nature. 2008;452:365–369. doi: 10.1038/nature06641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maretzky T, Le Gall SM, Worpenberg-Pietruk S, Eder J, Overall CM, Huang XY, Poghosyan Z, Edwards DR, Blobel CP. Src stimulates fibroblast growth factor receptor-2 shedding by an ADAM15 splice variant linked to breast cancer. Cancer Res. 2009;69:4573–4576. doi: 10.1158/0008-5472.CAN-08-4766. [DOI] [PubMed] [Google Scholar]
- 61.Touzot F, Callebaut I, Soulier J, Gaillard L, Azerrad C, Durandy A, Fischer A, de Villartay JP, Revy P. Function of Apollo (SNM1B) at telomere highlighted by a splice variant identified in a patient with Hoyeraal-Hreidarsson syndrome. Proc. Natl Acad. Sci. USA. 2010;107:10097–10102. doi: 10.1073/pnas.0914918107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zammarchi F, de Stanchina E, Bournazou E, Supakorndej T, Martires K, Riedel E, Corben AD, Bromberg JF, Cartegni L. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc. Natl Acad. Sci. USA. 2011;108:17779–17784. doi: 10.1073/pnas.1108482108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bauman JA, Li SD, Yang A, Huang L, Kole R. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 2010;38:8348–8356. doi: 10.1093/nar/gkq731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ, et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19:1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.