


Abstract
Using profiles of phylogenetic profiles (P-cubic) we compared the evolutionary dynamics of different kinds of functional associations. Ordered from most to least evolutionarily stable, these associations were genes in the same operons, genes whose products participate in the same biochemical pathway, genes coding for physically interacting proteins and genes in the same regulons. Regulons showed the most plastic functional interactions with evolutionary stabilities barely better than those of unrelated genes. Further regulon analyses showed that global regulators contain less evolutionarily stable associations than local regulators. Genes co-repressed by global regulators had a higher evolutionary conservation than genes co-activated by global regulators. However, the reverse was true for genes co-repressed and co-activated by local regulators. Of all the regulon-related associations, the relationship between regulators and their target genes showed the most evolutionary stability. Different negative data sets built to contrast against each of the analysed kinds of modules also differed in evolutionary conservation revealing further underlying genome organization. Applying P-cubic analyses to other genomes might help visualize genome organization, understand the evolutionary importance and plasticity of functional associations and compare the quality of data sets expected to reflect functional interactions, such as those coming from high-throughput experiments.
INTRODUCTION
The main idea behind phylogenetic profiles is that if the products of two genes have interdependent functions, both genes should be either present or absent within a given genome (1–3). Accordingly, previous work has shown that genes in the same operons, adjacent genes transcribed into a single messenger ribonucleic acid (4,5), tend to have more similar profiles than adjacent genes in different transcription units (6,7). However, the co-occurrence (co-occurring pairs of genes divided by the sum of co-occurring pairs plus genes that have lost their partners) of genes in operons of Escherichia coli can be as low as 0.2 in Archaea [updated data following (6)]. The loss of a functionally related partner could be due to events such as false negatives, where the tools available can no longer find an orthologous gene; non-orthologous gene displacement (8) and/or a particular function being unnecessary under a different environment. The loss of a gene partner might also reflect functional divergence, where the product of the remaining gene might be associated to a different cellular process perhaps in conjunction with other gene products. In other words, functionally related genes in one organism might not be functionally related in another.
As the co-occurrence analyses mentioned earlier might have shown that not all the functional associations of known operons are stable across organisms, genomic context tools, such as phylogenetic profiles, can be used to study and compare the evolutionary stability of other functional associations. In line with this idea, Snel and Huynen (9) presented a work using phylogenetic profiles to explore whether functional modules are also evolutionary modules. They analysed particular groups of genes within different kinds of functional gene modules. Herein, instead of studying such particular groups of genes (single operons and single regulons), we aimed to compare the evolutionary plasticity of different, experimentally determined types of functional associations (for instance, all genes known to be associated into operons compared against all genes in known regulons) using phylogenetic profiles. The objective was to compare the relative evolutionary stability of these kinds of associations. We have chosen functional modules of E. coli K12 because there are high-quality literature-derived databases of several types of experimentally determined modules in this model organism (Figure 1). The types of gene modules analysed were (i) operons (4,5), taken from RegulonDB (10,11); (ii) genes whose products participate in the same metabolic pathway, taken from EcoCyc (12,13); (iii) regulons, genes regulated by the same transcription factor (TF), also taken from RegulonDB and (iv) genes coding for physically interacting proteins (13–17).
Figure 1.
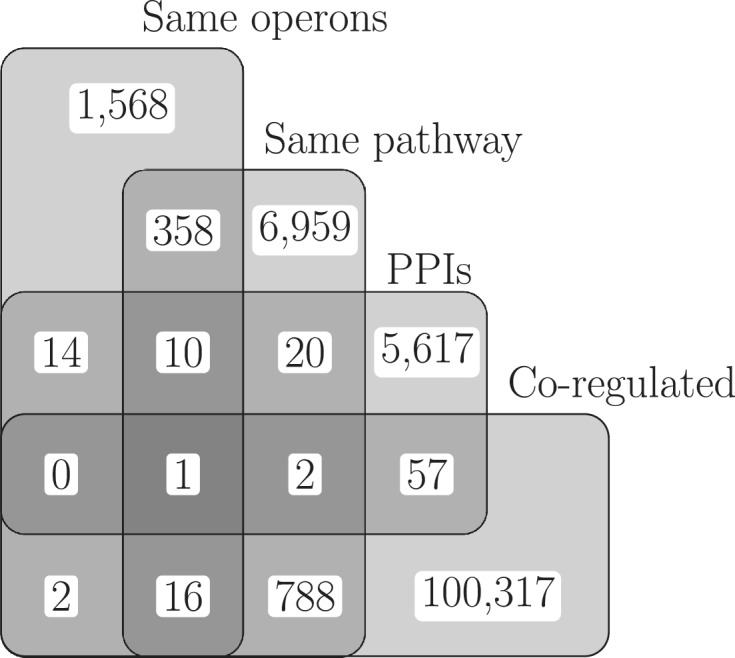
Venn diagram showing the overlaps between the four different kinds of functional modules from E. coli K12 substr. MG1655 analysed in this work. The few same-regulon genes overlapping with the same-operon data set are due to operons with internal promoters.
DATA AND METHODS
Phylogenetic profiles for each gene were represented as vectors where each item represented either the presence (number 1) or the absence (number 0) of an ortholog to the gene within a genome (18) (Figure 2). There are more elaborated vectors where the presences have been annotated with a number related to the score of the alignment of the gene and the corresponding ortholog (19). However, our preliminary results did not show a significant difference between such elaborated vectors and simpler binary vectors. Thus, we used here the binary (1/0) vectors to calculate mutual information scores for the phylogenetic profiles of all possible pairs of genes within the genome of E. coli K12 MG1655 using the formula described previously (19–21):
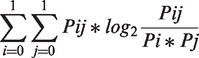 |
Figure 2.

Overview of the P-cubic method. Mutual information can be though of as a measure of how much two patterns coincide beyond what would be expected by chance. When the pattern for two proteins is almost the same, that is, the two proteins tend to co-occur across genomes, their mutual information is higher than when the patterns do not show co-occurrence. For example, despite the number of ‘1’ is approximately the same for genes A and C, their mutual information is low because their co-occurrences are more likely random. Genes whose products interact are expected to co-occur. However, this is not always the case, but the tendency is measurable as a higher proportion of co-occurring pairs than there would be among genes whose products are independent from each other (do not work together). A caveat of mutual information, however, is that if genes are abundant (or the opposite), then even though they might tend to co-occur, the patterns of co-occurrence might not result in high mutual information (genes E and F). As all gene pair sets have mutual information of 0 and better, all P-cubic curves start at ‘0’ [ln(1) = 0]. As the mutual information threshold increases, the proportion of gene pairs with that mutual information or better should decrease. More so for gene pairs that do not work together (less co-occurrence), than for genes whose products functionally interact (more co-occurrence).
Our working definition of orthology consisted of BLASTP reciprocal best hits and fusions as described elsewhere (22). We ran BLAST+ (23) to compare all the proteins annotated within ∼1300 prokaryotic genomes available at RefSeq (24,25) (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/) by May 2011. The E value cutoff was 1E−6, with a database size fixed at 5E8 (-dbsize 500 000 000), soft filtering of low information content sequences (-seg yes -soft_masking true) and a final Smith-Waterman alignment (-use_sw_tback). We also required coverage of at least 50% of any of the sequences in the alignment. The phylogenetic profiles were built using a non-redundant genome subset obtained as described elsewhere (26). Genomes smaller than 2.5 Mbp were not used for the analyses because obligate parasites and symbionts, which have severely reduced genomes, tend to lack TFs (27).
We compared data from four different kinds of modules of functionally related genes as follows: genes in the same operons, genes in the same biochemical pathways, genes in the same regulons and genes coding for physically interacting proteins (Figure 1).
To build data sets of pairs of genes within operons and of genes at transcription unit boundaries (TU borders), we used the current data set of transcription units of E. coli K12 substr MG1655 (28) found in RegulonDB (10,11) as explained previously (29,30). The current data sets contain 736 same-operon pairs and 567 TU borders. The operon data set included pairs of genes in the same operon even if they were not immediately adjacent to each other. We complemented TU borders with adjacent genes in different strands (divergently transcribed TU borders and convergently transcribed TU borders). This way the total number of operon pairs increased to 2536 and the total of TU borders increased to 1765.
To derive pairs of genes whose protein products participate in the same pathway, we used the EcoCyc database (12,13). As a contrasting data set, we used pairs of genes in different pathways constructed using pathways with no single gene in common. If the comparison of two pathways resulted in a single pair of genes found in the same-pathway set, the whole data set of different-pathway genes derived from such comparison was eliminated. The procedures resulted in 9524 same-pathway and 359 392 different-pathway pairs.
Physically interacting pairs (PPI) were genes coding for protein–protein interactions in E. coli as found by high-throughput methods (15–17), as well as manually curated interactions from low-throughput experiments, curated out of the database of interacting proteins (14) and the EcoCyc database (13,17). Negatives consisted of genes whose products are found in different compartments (17).
Genes in the same regulon were also derived from data in RegulonDB. As we wanted to explore the stability of the functional associations implied by the co-regulation due to the TF, we included only genes in different transcription units regulated by the same TF as same-regulon pairs. This way the data set remained exclusively composed of same-regulon pairs, rather than a combination of same-regulon and operon pairs. The difference with the TU borders mentioned earlier is the co-regulation by a common TF and that the transcription units can be anywhere in the genome. As some transcription units can be regulated by several TFs, we took care not to use any pair more than once. Genes in different regulons were built by comparing two regulons at a time. The compared regulons could not contain a single gene in common. Also, if the comparison of two regulons produced a pair of genes present in the same-regulon data set, the whole set of different-regulon pairs resulting from the comparison of the two regulons was eliminated. The procedure resulted in 92 376 same-regulon pairs and 140 259 different-regulon pairs. We did not consider genes transcribed by the same sigma factors as regulons. Note that the current data set of genomes contains several obligate symbionts and parasites. These organisms tend to display degraded genomes where the missing genes might have been lost because of the lack of selective pressure to keep them under this very particular lifestyle. A gene group particularly missing in obligate parasites is that of genes coding for TFs (27). Thus, the phylogenetic profiles for all the analyses presented, and the resulting P-cubic curves, did not include organisms with genomes smaller than 2.5 Mbp to avoid biases.
Figures in colour were produced in a colour-blind-friendly palette as suggested at: http://jfly.iam.u-tokyo.ac.jp/color/. A simple PERL module that we use to establish the palette for use in GNUPLOT and in LaTeX is offered ‘as is’ at: http://microbiome.wlu.ca/palette.
RESULTS AND DISCUSSION
Proof of concept: genes in the same operon are evolutionarily more stable than genes at TU borders
The contrast attainable in comparing profiles of phylogenetic profiles (P-cubic) is shown by using genes in the same operon, compared with genes at different transcription units (Figure 3a). Our measure of co-occurrence was mutual information (see ‘Data and Methods’ section). The phylogenetic profiles of functionally related genes should display higher mutual information than those of unrelated genes. Accordingly, the P-cubic analysis used to display and compare the evolutionary stability of different data sets consists on graphs showing the drop in the proportion of pairs of genes remaining as the mutual information threshold increases. Although a curve of direct proportion values could be used, taking the logarithm of these proportions helps to better compare the curves corresponding to each data set at higher mutual information thresholds.
Figure 3.

Profiles of phylogenetic-profile (P-cubic) are useful to compare the evolutionary stability of different gene sets. Sets of gene-pairs with the most evolutionarily stable functional interactions would have a higher proportion of pairs with high mutual information, thus their curves should drop less than those of unrelated genes. Accordingly, genes in the same operon (WO pairs), which are functionally related, show a higher P-cubic curve than genes at TU borders, which are not necessarily functionally related (a). Also in (a), as reported previously (31), convergently transcribed genes (cTU borders) are the least related of all adjacent genes in different TUs, followed by divergently transcribed TU borders (dTU borders) and adjacent TU borders in the same strand (TU borders). (b) Genes in the same biochemical pathway have an evolutionarily stable relationship when compared with genes in different pathways. However, the relationship is less stable than that among genes in the same operon. (c) Genes producing proteins that physically interact have a less stable functional relationship than genes in operons. The higher mutual information of verified protein–protein interactions shows that P-cubic analyses are also useful to verify the quality of large experimental data sets. (d) Genes in the same regulon have higher mutual information than genes in different regulons. However, the relationship seems to be subtle and so plastic throughout evolution that the P-cubic of genes in regulons is close to that of functionally unrelated genes. Transcriptional regulation might evolve very fast and be a major source of functional diversity and adaptation.
The P-cubic of operons drops slowly compared with those of the different sets of TU borders as the mutual information increases (Figure 3a). This is expected because operons are mainly formed of functionally related genes, and functionally related genes should have a higher tendency to co-occur than non-related genes. Different sets of TU borders, namely co-directional TU borders, divergently transcribed genes and convergently transcribed genes, also display differences in their co-occurrence (Figure 3a). Same-strand TU borders show the highest mutual information, meaning that a higher proportion of these gene pairs might be functionally related than those in the other TUB categories. The least related were the convergent TU borders. These results are in agreement with previous work showing that divergent TU borders have stronger tendencies towards conservation of gene order than convergent TU borders (31) and with work showing that some co-directional TU borders also have functional associations (31–33). Thus, the P-cubic reflects both the proportions of functionally related pairs of genes and the evolutionary stability of such associations. As most genes in operons are known, or expected, to have functional interactions, the main component of the operon curve should correspond to evolutionary stability of the functional association.
Genes in the same biochemical pathway are less evolutionarily stable than genes in the same operon
The next comparison consisted of genes in the same biochemical pathway against genes in different pathways. As expected again, genes in the same pathway show higher mutual information than those in different pathways (Figure 3b). However, same-pathway genes are not as evolutionarily stable as those in the same operon. Genes in the same operon are often thought to consist of genes whose products participate in the same biochemical pathways [see for instance (21,34)]. Accordingly, there is an overlap between the two data sets, operon pairs and same-pathway pairs, of 447 gene pairs. This number constitutes 17.6% of the operon pairs and 5.4% of the same-pathway pairs (Figure 1).
Genes in different pathways present a P-cubic curve that drops faster than the curve of overall TU borders (Figure 3b). Different-pathway genes are a large data set (359 392 pairs of genes) and, thus, make a cleaner and smoother negative sample than overall TU borders (1765 pairs). Thus, we decided to use the different-pathway set as a negative contrasting data set for the following analyses.
Genes coding for physically interacting proteins are less evolutionarily stable than genes in the same operon
To test whether genes coding for physically interacting proteins (PPI for protein–protein interactions) form an evolutionarily stable gene module, we compared their P-cubic with that of genes in the same operon. Only a small proportion of the proteins involved in PPIs are encoded by adjacent genes. Accordingly, only 28 of the manually curated low-throughput PPIs, and 47 of the 6047 total PPIs from Butland et al. (15) (21 of them among the 716 verified PPI pairs), are also in the operon data set. The data set of genes coding for manually curated PPIs shows the higher mutual information of all PPI sets, only slightly lower than operon pairs. The P-cubic comparison of operon pairs and PPI pairs shows that the functional association of genes in the same operon might be more stable throughout evolution than the functional association of genes coding for physically interacting proteins (Figure 3c). Also noteworthy, the data set of verified PPIs shows that this data set contains a higher proportion of evolutionarily stable pairs of genes than the non-verified data set. This result shows that P-cubic comparisons can also be used to compare the quality of different high-throughput experimental data sets. The negative PPI set, genes whose proteins are found in different cellular compartments show an interesting curve. A few pairs of genes show such high mutual information that they twist the curve, so that it does not drop as much as that for genes in different pathways. This makes sense given that proteins in different cellular compartments will not interact physically, but they still might have a functional association. Thus, similar to TU borders, the curve seems to reflect the presence of a proportion of functionally associated gene pairs.
Genes in the same regulon have the most evolutionarily plastic functional associations
The data set of genes in the same regulons contained very few pairs in common with the operons data set because we were interested in the association arising from the co-regulation brought about by the TF. The comparison of co-regulated (same regulon) genes against genes in different regulons shows a higher stability for the genes expected to be functionally associated (Figure 3d). However, same-regulon pairs display much lower mutual information than genes in the same operons and noticeably close to that of genes in different pathways. In other words, it would seem that pairs of genes in different pathways are almost as strongly associated as genes regulated by the same TF.
To better understand the evolutionary plasticity of the functional associations by co-regulation, we separated the regulon data into categories. We first separated the data into those involving global TFs and those involving local TFs as defined elsewhere (35). Each data set (overall, global and local) was separated into activation (positively co-regulated transcription units), repression (negatively co-regulated transcription units) and dual (dually co-regulated transcription units). The rationale being that if two transcription units are regulated in the same way, then it should be more probable for their gene products to have a stable functional relationship. Although the P-cubic of any of these data sets shows better evolutionary stability than that of genes in different regulons (Figure 4a), genes related by co-activation presented a higher P-cubic than genes related by co-repression. This makes sense because activation requires more information than repression, as for the latter it is enough for a protein to bind at an appropriate site impeding, for instance, the binding of a sigma factor. This has been the argument used to explain why repressors are the most abundant kind of TFs (36). However, co-activated genes still showed a lower P-cubic than any of the other functionally related groups analysed and very close to the P-cubic of TU borders (compare to Figure 3). In contrast to the results earlier, if we analyse regulons involving global regulators (Figure 4b), we find co-activated genes to be the least conserved, showing worse conservation than genes in different regulons. This result is contradictory given the rationale earlier that repression requires less information than activation. We suggest that the results are due to the dual nature of global regulators. As global TFs perform both activation and repression, they already have a way of interacting with sigma factors to provide activation and, thus, acting as repressors or as activators does not make too much of a difference. Accordingly, local TFs, most of which act as either repressors or activators, show higher conservation of co-activated gene pairs than of co-repressed pairs (Figure 4c). The reversal between co-activated and co-repressed P-cubic pairs seen when comparing overall regulons with global TFs is explained by the facts that genes co-activated by local TFs, which tend to be better conserved, constitute close to 26% of the overall co-activated pairs. Dually co-regulated genes show a somewhat higher P-cubic than co-repressed and co-activated pairs; only they contain no pairs with mutual information much higher than 0.4 bits in both the overall and the global TFs analyses and no pairs with mutual information higher than 0.2 bits in the local TFs analysis.
Figure 4.

P-cubic comparison of regulon subsets. Herein, we compared the P-cubic of positively co-regulated genes (activation), negatively co-regulated genes (repression) and the relationship between TFs and their TGs (TF–TG). (a) Overall, the most stable functional association corresponds to that between TFs and their TGs. Co-activated genes and dually regulated genes are next in stability with the lowest stability presented among co-repressed gene pairs. The relationships change when we analyse the subset of regulatory interactions by global regulators in (b). Although the TF–TG relationship shows the highest stability again, co-activated genes show a lower conservation than genes in different regulons, whereas dually co-regulated genes show the highest conservation. In the analyses involving local regulators (c), co-activated genes show more conservation than the TF–TG relationship. In agreement with previous results about the conservation of the TF–TG relationship among evolutionarily close Enterobacteria (38), we found that a higher proportion of repressor–TG relationships attain a higher mutual information than activator–TG relationships. However, even the most conserved interactions brought about by TFs remain close to those among overall TU borders shown in Figure 3b.
We also explored the relationship of the TF and their target genes (TGs). In agreement with a previous report that shows that the TFs and TGs seem to evolve independently (37), the P-cubic of the TF/TG interactions are close to that of overall TU pairs (compare the curves of TF–TG in Figure 4 with curves in Figure 3). However, except for regulons involving local TFs, it shows the highest P-cubic among the TF association groups analysed (Figure 4).
A previously published analysis found that the TF/TG association for positively regulated genes was less conserved among Enterobacteria than that of negatively regulated genes (38). To test whether we had a similar result using P-cubic, we separated our TF/TG pairs into activated, repressed and dually regulated (Figure 4d). In agreement with those previous results, the TF/TG P-cubic does not show activation TF/TG pairs with higher mutual information than 0.4 bits. Thus, our results confirm the previous finding. However, we note that despite this difference in TF/TG conservation, neither set seems to be more conserved than TU borders.
Given the results in this section, relationships arising from co-regulation are the most plastic of all the gene associations tested. Previous work has suggested that co-regulation is not well conserved in evolution [see for instance (37,39)], whereas other analyses have suggested high conservation [see for instance (40)]. Although more particular analyses are necessary, the results herein show that neither the association of genes by co-regulation nor the regulation of a gene by a particular TF is much more conserved than the relationship of genes with little evidence for a functional interaction. In other words, it would seem as if the evolution of operators, the DNA motif where a TF binds (5), is independent from the evolution of the genes they regulate. This suggestion is also in agreement with previous work suggesting that operators can evolve quickly (41).
CONCLUDING REMARKS
This work uses profiles of phylogenetic profiles (P-cubic) to compare the evolutionary stability of functional associations of different gene relationships, gaining insight into the structure and evolution of the genome. The results led us to conclude that conservation of interactions is not as ubiquitous as believed previously. Four aspects of gene relationships have been addressed in this work: genes in the same operon, genes in the same biochemical pathway, genes coding for physically interacting proteins and genes in the same regulons.
One of the expected results is that the P-cubic of gene pairs within operons would be more stable than otherwise adjacent genes; in other words, they would show more enduring relationships than the genes at the borders of transcription units. This particular result showing the general functional relationship of genes within an operon has been suggested in previous work (7,26,31). Thus, this first result also served as a confirmation of the concept.
Genes in different pathways show the lowest conservation of all negative sets, as those genes would rarely be expected to function as partners in the organism. Their lower co-occurrence compared with that of any TU borders set supports the idea of an underlying genome organization that keeps transcription units of related functions within a close network. Despite the fact that TU borders might be the proper contrast against operons, they might still contain some proportion of gene pairs with related functions. This should be expected given that previous works have shown that genes in the same biochemical pathways tend to be closer in the chromosome than would be expected by chance (42).
The analysis shows that pairs of genes coding for physically interacting proteins are not as conserved as operon pairs, this can mainly be attributed to the protein redundancy within the cell. Losing the physical interaction in a protein interactome does not mark a loss of functional interaction, just an evolutionary replacement of a protein with one that has a higher efficiency or more commonly the shuffling in amino acid composition through evolutionary time (43).
The finding with the most wide-ranging implications is the counterintuitive result stemming from a comparison of genes within a regulon, as opposed to gene pairs outside regulons and gene pairs in the other three gene modules tested. When the different regulon pairs and same regulon pairs are compared, the same-regulon pairs show a greater conservation over increasing mutual information than genes in different regulons. This is to be expected, genes within the same regulons are more likely to be part of the same functional module compared with genes regulated differently. However, same-regulon pairs are only slightly more conserved than pairs in different regulons, and both are overall less evolutionarily stable than any of the other gene modules. Although it has been previously postulated that regulons show plasticity (37,39), the relationship has not been compared with other gene modules. From this analysis (Figure 3d) it is clear that regulon conservation is only slightly higher than that of baseline results from gene pairs in different pathways. This suggests that regulons are the gene modules that evolve the most readily and offer more information on the evolution of prokaryotic organisms than previously thought (44). Comparing the plasticity of regulatory interactions with that of acquisition of genes by horizontal gene transfer, and gene loss, is not possible by this method. It is noteworthy, however, that many genomic islands, such as pathogenicity ones, contain TFs (45) and that a high proportion of genes coding for regulatory proteins in E. coli K12 might come from horizontal gene transfer (46), pointing to a possible relationship between regulatory plasticity and the plasticity of gene content, as the quick evolution of transcriptional regulation might allow these islands to quickly become part of the network of functional interactions of their hosts. Such relationship could be worth exploring in future research.
Individual regulatory changes have been implicated in large evolutionary changes of both eukaryotes (47,48) and prokaryotes (37,39), but it has not been clear to what extent this line of evidence extends due to the inherent difficulty of comparing many individual regulatory systems between species. This work presents a broad view over a large data set of gene pairs in regulons to clearly show that their evolutionary plasticity is great enough to account for an important part of variation at the species level in prokaryotes. One can thus suggest that the regulatory networks of all life forms are governed by the same principle (49), which offers insights deeply into evolutionary theory.
FUNDING
Discovery grant from Natural Sciences and Engineering Research Council of Canada (to G.M.-H.). Funding for open access charge: Natural Sciences and Engineering Research Council of Canada.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors acknowledge an equipment grant from WLU, Gary Molenkamp from SHARCNET for computer cluster assistance and SHARCNET for computer cluster usage. They also thank the Laboratory of Andrew Emili of the University of Toronto for providing data sets of physically interacting proteins, as well as J. Javier Díaz-Mejía, also of the University of Toronto, for the carefully curated data sets of low-throughput PPI data sets and the negative data sets consisting of proteins working in different compartments.
REFERENCES
- 1.Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 2.Gaasterland T, Ragan MA. Microbial genescapes: phyletic and functional patterns of ORF distribution among prokaryotes. Microb. Comp. Genomics. 1998;3:199–217. doi: 10.1089/omi.1.1998.3.199. [DOI] [PubMed] [Google Scholar]
- 3.Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl Acad. Sci. USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacob F, Perrin D, Sanchez C, Monod J. [Operon: a group of genes with the expression coordinated by an operator.] C. R. Hebd. Seances. Acad. Sci. 1960;250:1727–1729. [PubMed] [Google Scholar]
- 5.Jacob F, Perrin D, Sanchez C, Monod J, Edelstein S. [The operon: a group of genes with expression coordinated by an operator. C. R. Acad. Sci. Paris 250 (1960) 1727–1729] Comptes. Rendus. Biol. 2005;328:514–520. doi: 10.1016/j.crvi.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Moreno-Hagelsieb G, Trevino V, Perez-Rueda E, Smith TF, Collado-Vides J. Transcription unit conservation in the three domains of life: a perspective from Escherichia coli. Trends Genet. 2001;17:175–177. doi: 10.1016/s0168-9525(01)02241-7. [DOI] [PubMed] [Google Scholar]
- 7.Moreno-Hagelsieb G, Collado-Vides J. Operon conservation from the point of view of Escherichia coli, and inference of functional interdependence of gene products from genome context. In Silico Biol. 2002;2:87–95. [PubMed] [Google Scholar]
- 8.Koonin EV, Mushegian AR, Bork P. Non-orthologous gene displacement. Trends Genet. 1996;12:334–336. [PubMed] [Google Scholar]
- 9.Snel B, Huynen MA. Quantifying modularity in the evolution of biomolecular systems. Genome Res. 2004;14:391–397. doi: 10.1101/gr.1969504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huerta AM, Salgado H, Thieffry D, Collado-Vides J. RegulonDB: a database on transcriptional regulation in Escherichia coli. Nucleic Acids Res. 1998;26:55–59. doi: 10.1093/nar/26.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gama-Castro S, Salgado H, Peralta-Gil M, Santos-Zavaleta A, Muniz-Rascado L, Solano-Lira H, Jimenez-Jacinto V, Weiss V, Garcia-Sotelo JS, Lopez-Fuentes A, et al. RegulonDB version 7.0: transcriptional regulation of Escherichia coli K-12 integrated within genetic sensory response units (Gensor Units) Nucleic Acids Res. 2011;39:D98–D105. doi: 10.1093/nar/gkq1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karp PD, Riley M, Paley SM, Pelligrini-Toole A. EcoCyc: an encyclopedia of Escherichia coli genes and metabolism. Nucleic Acids Res. 1996;24:32–39. doi: 10.1093/nar/24.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keseler IM, Collado-Vides J, Santos-Zavaleta A, Peralta-Gil M, Gama-Castro S, Muniz-Rascado L, Bonavides-Martinez C, Paley S, Krummenacker M, Altman T, et al. EcoCyc: a comprehensive database of Escherichia coli biology. Nucleic Acids Res. 2011;39:D583–D590. doi: 10.1093/nar/gkq1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–D451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butland G, Peregrin-Alvarez JM, Li J, Yang W, Yang X, Canadien V, Starostine A, Richards D, Beattie B, Krogan N, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- 16.Arifuzzaman M, Maeda M, Itoh A, Nishikata K, Takita C, Saito R, Ara T, Nakahigashi K, Huang HC, Hirai A, et al. Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome Res. 2006;16:686–691. doi: 10.1101/gr.4527806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu P, Janga SC, Babu M, Diaz-Mejia JJ, Butland G, Yang W, Pogoutse O, Guo X, Phanse S, Wong P, et al. Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol. 2009;7:e96. doi: 10.1371/journal.pbio.1000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl Acad. Sci. USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Date SV, Marcotte EM. Discovery of uncharacterized cellular systems by genome-wide analysis of functional linkages. Nat. Biotechnol. 2003;21:1055–1062. doi: 10.1038/nbt861. [DOI] [PubMed] [Google Scholar]
- 20.Huynen M, Snel B, Lathe W, III, Bork P. Predicting protein function by genomic context: quantitative evaluation and qualitative inferences. Genome Res. 2000;10:1204–1210. doi: 10.1101/gr.10.8.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng Y, Roberts RJ, Kasif S. Genomic functional annotation using co-evolution profiles of gene clusters. Genome Biol. 2002;3:61–69. doi: 10.1186/gb-2002-3-11-research0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moreno-Hagelsieb G, Latimer K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics. 2008;24:319–324. doi: 10.1093/bioinformatics/btm585. [DOI] [PubMed] [Google Scholar]
- 23.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maglott DR, Katz KS, Sicotte H, Pruitt KD. NCBI's LocusLink and RefSeq. Nucleic Acids Res. 2000;28:126–128. doi: 10.1093/nar/28.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreno-Hagelsieb G, Janga SC. Operons and the effect of genome redundancy in deciphering functional relationships using phylogenetic profiles. Proteins. 2008;70:344–352. doi: 10.1002/prot.21564. [DOI] [PubMed] [Google Scholar]
- 27.Ranea JA, Buchan DW, Thornton JM, Orengo CA. Evolution of protein superfamilies and bacterial genome size. J. Mol. Biol. 2004;336:871–887. doi: 10.1016/j.jmb.2003.12.044. [DOI] [PubMed] [Google Scholar]
- 28.Blattner FR, Plunkett G, III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 29.Salgado H, Moreno-Hagelsieb G, Smith TF, Collado-Vides J. Operons in Escherichia coli: genomic analyses and predictions. Proc. Natl Acad. Sci. USA. 2000;97:6652–6657. doi: 10.1073/pnas.110147297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moreno-Hagelsieb G, Collado-Vides J. A powerful non-homology method for the prediction of operons in prokaryotes. Bioinformatics. 2002;18(Suppl. 1):S329–S336. doi: 10.1093/bioinformatics/18.suppl_1.s329. [DOI] [PubMed] [Google Scholar]
- 31.Korbel JO, Jensen LJ, von Mering C, Bork P. Analysis of genomic context: prediction of functional associations from conserved bidirectionally transcribed gene pairs. Nat. Biotechnol. 2004;22:911–917. doi: 10.1038/nbt988. [DOI] [PubMed] [Google Scholar]
- 32.Ermolaeva MD, White O, Salzberg SL. Prediction of operons in microbial genomes. Nucleic Acids Res. 2001;29:1216–1221. doi: 10.1093/nar/29.5.1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janga SC, Moreno-Hagelsieb G. Conservation of adjacency as evidence of paralogous operons. Nucleic Acids Res. 2004;32:5392–5397. doi: 10.1093/nar/gkh882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Romero PR, Karp PD. Using functional and organizational information to improve genome-wide computational prediction of transcription units on pathway-genome databases. Bioinformatics. 2004;20:709–717. doi: 10.1093/bioinformatics/btg471. [DOI] [PubMed] [Google Scholar]
- 35.Martinez-Antonio A, Collado-Vides J. Identifying global regulators in transcriptional regulatory networks in bacteria. Curr. Opin. Microbiol. 2003;6:482–489. doi: 10.1016/j.mib.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 36.Perez-Rueda E, Gralla JD, Collado-Vides J. Genomic position analyses and the transcription machinery. J. Mol. Biol. 1998;275:165–170. doi: 10.1006/jmbi.1997.1465. [DOI] [PubMed] [Google Scholar]
- 37.Lozada-Chavez I, Janga SC, Collado-Vides J. Bacterial regulatory networks are extremely flexible in evolution. Nucleic Acids Res. 2006;34:3434–3445. doi: 10.1093/nar/gkl423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hershberg R, Margalit H. Co-evolution of transcription factors and their targets depends on mode of regulation. Genome Biol. 2006;7:R62. doi: 10.1186/gb-2006-7-7-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Teichmann SA, Babu MM. Conservation of gene co-regulation in prokaryotes and eukaryotes. Trends Biotechnol. 2002;20:407–410; discussion 410. doi: 10.1016/s0167-7799(02)02032-2. [DOI] [PubMed] [Google Scholar]
- 40.Snel B, van Noort V, Huynen MA. Gene co-regulation is highly conserved in the evolution of eukaryotes and prokaryotes. Nucleic Acids Res. 2004;32:4725–4731. doi: 10.1093/nar/gkh815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider TD. Evolution of biological information. Nucleic Acids Res. 2000;28:2794–2799. doi: 10.1093/nar/28.14.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yellaboina S, Goyal K, Mande SC. Inferring genome-wide functional linkages in E. coli by combining improved genome context methods: comparison with high-throughput experimental data. Genome Res. 2007;17:527–535. doi: 10.1101/gr.5900607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Dam TJ, Snel B. Protein complex evolution does not involve extensive network rewiring. PLoS Comput. Biol. 2008;4:e1000132. doi: 10.1371/journal.pcbi.1000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wray GA. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 2007;8:206–216. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt H, Hensel M. Pathogenicity islands in bacterial pathogenesis. Clin. Microbiol. Rev. 2004;17:14–56. doi: 10.1128/CMR.17.1.14-56.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Price MN, Dehal PS, Arkin AP. Horizontal gene transfer and the evolution of transcriptional regulation in Escherichia coli. Genome Biol. 2008;9:R4. doi: 10.1186/gb-2008-9-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hahn MW, Rockman MV, Soranzo N, Goldstein DB, Wray GA. Population genetic and phylogenetic evidence for positive selection on regulatory mutations at the factor VII locus in humans. Genetics. 2004;167:867–877. doi: 10.1534/genetics.103.025726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cretekos CJ, Wang Y, Green ED, Martin JF, Rasweiler JJT, Behringer RR. Regulatory divergence modifies limb length between mammals. Genes Dev. 2008;22:141–151. doi: 10.1101/gad.1620408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hinman VF, Davidson EH. Evolutionary plasticity of developmental gene regulatory network architecture. Proc. Natl Acad. Sci. USA. 2007;104:19404–19409. doi: 10.1073/pnas.0709994104. [DOI] [PMC free article] [PubMed] [Google Scholar]