

Abstract
Successful oncolytic virus treatment of malignant glioblastoma multiforme depends on widespread tumor-specific lytic virus replication and escape from mitigating innate immune responses to infection. Here we characterize a new HSV vector, JD0G, that is deleted for ICP0 and the joint sequences separating the unique long and short elements of the viral genome. We observed that JD0G replication was enhanced in certain glioblastoma cell lines compared to HEL cells, suggesting that a vector backbone deleted for ICP0 may be useful for treatment of glioblastoma. The innate immune response to virus infection can potentially impede oncolytic vector replication in human tumors. Indoleamine-2,3-dioxygenase (IDO) is expressed in response to interferon γ (IFNγ) and has been linked to both antiviral functions and to the immune escape of tumor cells. We observed that IFNγ treatment of human glioblastoma cells induced the expression of IDO and that this expression was quelled by infection with both wild-type and JD0G viruses. The role of IDO in inhibiting virus replication and the connection of this protein to the escape of tumor cells from immune surveillance suggest that IDO downregulation by HSV infection may enhance the oncolytic activity of vectors such as JD0G.
1. Introduction
Glioblastoma multiforme (GBM) is the most common type of primary brain tumor with an incidence rate of approximately 3 cases per 100,000 people per year in the United States (2004-2005, http://www.cbtrus.org/). The current treatment modality for GBM typically involves surgery to remove the tumor, followed by radiotherapy and adjuvant chemotherapy with temozolomide [1]. The infiltrative nature of the tumor makes complete surgical removal difficult, and tumor recurrence is common at the tumor margin. The median survival for patients with GBM is generally less than two years despite treatment. Clearly, new and effective therapies are needed.
Oncolytic vectors (OVs), viruses that have been designed to undergo lytic replication specifically in tumor cells, provide a novel therapeutic approach to treatment of GBM. Herpes simplex virus-based oncolytic vectors (oHSV) have been used in early-phase clinical trials for a variety of tumors including recurrent GBM. Recent trials using the genetically engineered HSV strains G207, HSV1716, OncoVEXGM-CSF and NV1020 have been encouraging, demonstrating that these viruses are relatively nontoxic for normal tissue and showing instances of promising patient responses [2–6]. However, despite their antitumor potential, the overall effectiveness of these vectors has been limited. This observation warrants the exploration of other mutant vector backbones that may improve tumor cell-dependent vector replication.
The engineering of an oHSV vector relies upon the deletion of viral genes that are needed to efficiently complete the virus life cycle in normal cells, but that are complemented by genetic alterations that commonly occur in tumor cells. The ICP0 protein is an attractive target for the design of an oncolytic vector. ICP0 is a nonessential, immediate early gene product that performs a wide range of functions important for virus replication such as counteracting the IFN response [7–9], maintaining the viral genome in a transcriptionally active state [10–12], and targeting specific cellular proteins for degradation by the proteasome, including proteins important in modulating DNA repair pathways, apoptosis and cell-cycle progression [13–19]. Importantly, mutants lacking ICP0 protein are impaired for growth at low multiplicity of infection (MOI) in most cell lines [20], while vigorous replication has been observed in tumor cell lines such as U2OS osteosarcoma cells [21, 22]. It has recently been demonstrated that other tumor cell lines enhance the replication of ICP0-deficient vectors, and an HSV-1 mutant lacking ICP0 protein expression was shown to increase survival in a mouse breast adenocarcinoma model [23, 24]. The tumor cell-specific replication of ICP0-deficient HSV suggests that this mutant backbone may represent a new class of oHSV vector useful for treatment of GBM.
The innate immune response to virus infection can potentially inhibit virus replication and spread. For example, natural killer (NK) cells recruited to the site of HSV infection release interferon γ (IFNγ) a cytokine that triggers multiple downstream pathways that can inhibit virus replication [25]. The production of IFNγ has been shown to inhibit replication of certain oHSV in vitro and virus gene expression in vivo [25, 26]. Local production of IFNγ induces the synthesis of indoleamine-2,3-dioxygenase (IDO), an enzyme that catalyzes the degradation of the essential amino-acid tryptophan producing kynurenine and multiple downstream metabolites. The consequent depletion of tryptophan from the cell and its surroundings impedes virus replication [27]. Furthermore, kynurenine along with other catabolites has been linked to the escape of tumor cells from immune surveillance [28–30]. Thus, IDO may present obstacles to both OV replication and immune clearance of tumor cells.
In this report, we characterized an ICP0-deficient oHSV vector, JD0G [31], for its ability to replicate in glioblastoma cells in the absence and presence of IFNγ. We show that JD0G virus replication was enhanced in glioblastoma cell lines compared to HEL cells, suggesting that the former complement the loss of ICP0 protein. Both wild-type KOS virus and JD0G were moderately sensitive to IFNγ at low MOI in glioblastoma cells, an effect that was dramatically reduced at higher MOI. Moreover, while glioblastoma cell lines produced IDO protein when stimulated with IFNγ, IDO induction had a minimal effect on virus replication. This observation may be related to our finding that IFNγ-induced IDO levels were substantially reduced in glioma cells infected with KOS or JD0G
2. Results
2.1. JD0G Structure
To create JD0G, the internal repeat sequences “joint” were deleted from the strain KOS HSV-1 genome, a modification that removes one copy each of the gamma 34.5 gene and the immediate early genes encoding ICP0 and ICP4. The removal of the joint enhances vector stability by eliminating internal genome rearrangements that occur during UL/US isomerization [38]. The joint deletion removes a region spanning positions from 116,982 to 132,605 of the HSV genome (nucleotide positions based on NC_001806). The remaining copy of ICP0 was then deleted, and an eGFP expression construct driven by the human cytomegalovirus (hCMV) major immediate early promoter was inserted in its place. The structure of the mutant virus was confirmed by PCR, sequencing, and Southern blot analysis. Figure 1 shows the structure of the JD0G genome compared to that of wild-type HSV-1 KOS.
Figure 1.
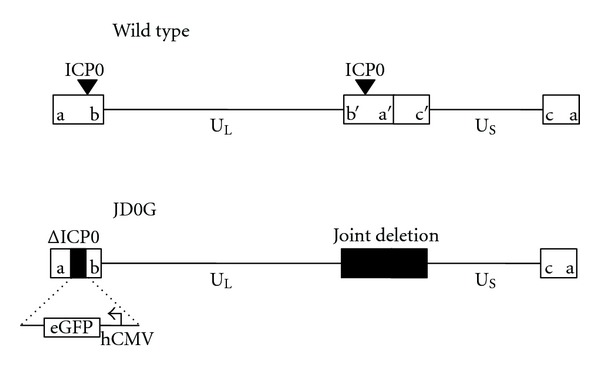
Structure of the JD0G virus compared to the wild-type HSV-1 virus. Schematics depicting the genome structure of wild-type HSV-1 (top) and the JD0G virus (bottom). The unique long (UL) and unique short (US) portions of the viral genome are indicated with solid lines, and the repeat regions of the genome (ab/b′a′, a′c′/ca) are shown as open boxes. The locations of the two ICP0 loci are designated with inverted triangles. Black boxes represent deleted regions of the JD0G genome. The joint deletion removes a region spanning positions from 116,982 to 132,605 of the HSV genome (nucleotide positions based on NC_001806). The terminal ICP0-deletion (ΔICP0) replaces the ICP0 coding sequence with an hCMV:eGFP expression cassette.
2.2. Glioblastoma Cell Lines Support Replication of the JD0G Virus
Successful oncolytic virus treatment of GBM requires efficient, tumor-specific replication, and lysis of the tumor cell. We compared JD0G replication with that of wild-type KOS virus and another oncolytic vector, MGH2, a derivative of the well-studied vector G207 [32]. MGH2 lacks expression of γ34.5 and produces a nonfunctional ICP6 protein fused with GFP from the U L 39 locus. In addition, MGH2 carries two genes that encode prodrug activating enzymes, CYP2B1 (cytochrome p450), and shiCE (secreted human intestinal carboxylesterase). ICP0-deficient viruses are impaired for replication at low MOI in cells such as human embryonic lung fibroblasts (HEL cells), while certain tumor cell lines have been shown to allow efficient replication [22]. To assess the ability of human glioblastoma cell lines to support virus replication, HEL, SNB19, and U251 glioblastoma cells were infected at an MOI of 0.1 (based on U2OS titers), supernatants were collected at 24 hours postinfection (hpi) and virus yields were measured by titration on U2OS cells. Infections with KOS consistently generated slightly higher virus yields on HEL cells than on the two glioma cell lines (Figure 2(a)). In contrast, infection of the glioma cells with JD0G virus produced titers that were on average 10-fold higher than those obtained from HEL cells (Figure 2(a)). A similar comparison of MGH2 titers across the cell lines demonstrated that while MGH2 was able to replicate in HEL cells, its replication was substantially more impaired than that of JD0G on the two glioblastoma cell lines (Figure 2(a)). These results suggest that both glioblastoma cell lines provided an enhanced environment for JD0G growth, but not for MGH2, and that these tumor cells partially complement the ICP0 deficit.
Figure 2.
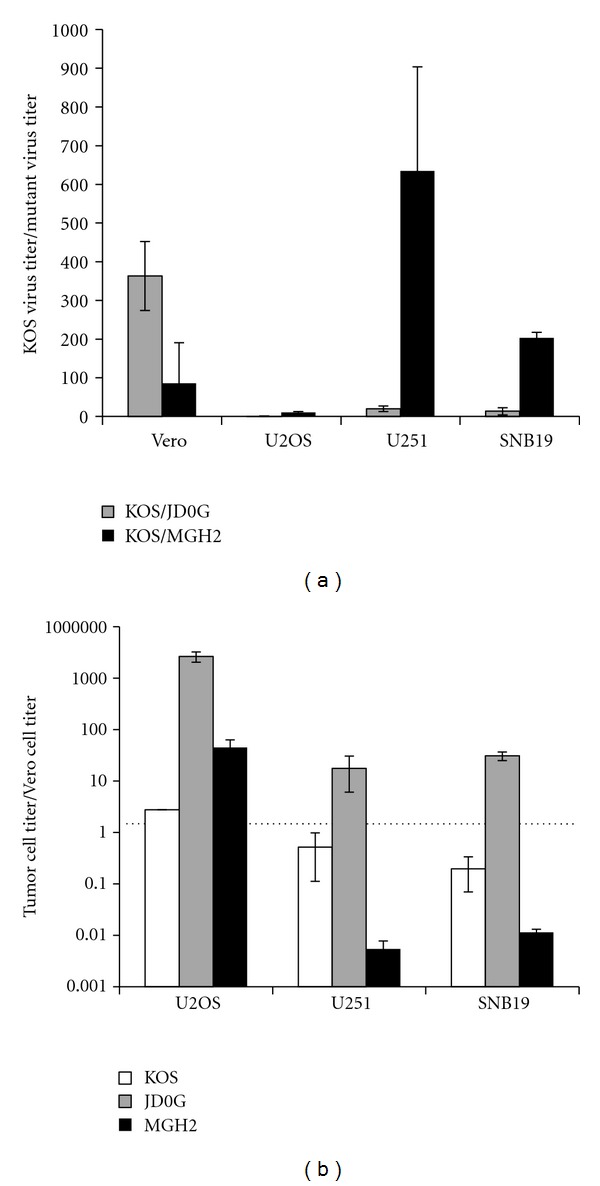
JD0G infection of glioblastoma cells results in efficient virus replication and tumor-selective cell death. (a) The replication of JD0G was evaluated in comparison to KOS and MGH2. HEL, U251, and SNB19 cell lines were infected in parallel with KOS, MGH2, or JD0G at an MOI of 0.1 based on U2OS titers. Supernatants were collected at 24 hpi, and the amount of virus produced was assessed by titration on U2OS cells. The data represent the average of three independent infections, and error bars represent the standard deviation. For all three viruses the differences in virus yield observed between HEL cells and either glioma cell line are statistically significant as determined by the student t-test (P values less than 0.03). (b) Tumor-specific cell death was assessed by counting the number of surviving cells following infection with either JD0G or MGH2 virus. HEL, U251 or SNB19 cells were infected at an MOI of 0.1, and Trypan Blue staining was performed to count the number of viable cells 48 h postinfection. The data represent the average of three independent infections, and error bars represent the standard deviation. All differences are statistically significant (P values less than 0.01).
We then examined the ability of the JD0G and MGH2 oncolytic viruses to cause tumor-specific cell death. HEL, SNB19, and U251 cells were infected at an MOI of 0.1, and the surviving cells were counted at 48 hpi (Figure 2(b)). Compared to mock-infected HEL cells, JD0G infection reduced the number of HEL cells by less than 2-fold. In contrast, the number of glioma cells that survived infection with JD0G was reduced by 10-fold or greater. MGH2 infection did not exhibit similar tumor-specific cell-killing activity, causing more cell death in the HEL cell line than in either glioma cell line. Thus, the JD0G oncolytic vector both exhibited a better tumor-selective replication profile than MGH2 and demonstrated clear glioma-specific cell death.
2.3. IFNγ Treatment Inhibits Virus Replication in an MOI-Dependent Manner
IFNγ presents a potential impediment to virus replication in tumor cells in vivo. To evaluate the sensitivity of KOS and JD0G to type II IFN, SNB19 and U251 cells were treated with IFNγ for 24 h and infected with either KOS or JD0G at an MOI of 0.1 or 10. Virus yields at 24 hpi were assessed by titration on U2OS cells. The results (Figures 3(a) and 3(b)) showed that (i) growth of KOS and JD0G was inhibited by IFNγ at low MOI on both cell lines; (ii) JD0G was somewhat more sensitive than KOS; (iii) both viruses at low MOI were hypersensitive to IFNγ on U251 cells; and (iv) high-MOI infection minimized IFNγ sensitivity. These results suggested that ICP0 deficiency has a moderate (≤5-fold) effect on IFNγ sensitivity at low MOI, a condition that likely models in vivo infection more realistically than high-MOI infection of cultured cells. In addition, the data indicated that both the antiviral response to IFNγ and the contribution of ICP0 to viral escape from this response at high MOI vary between glioblastoma lines.
Figure 3.
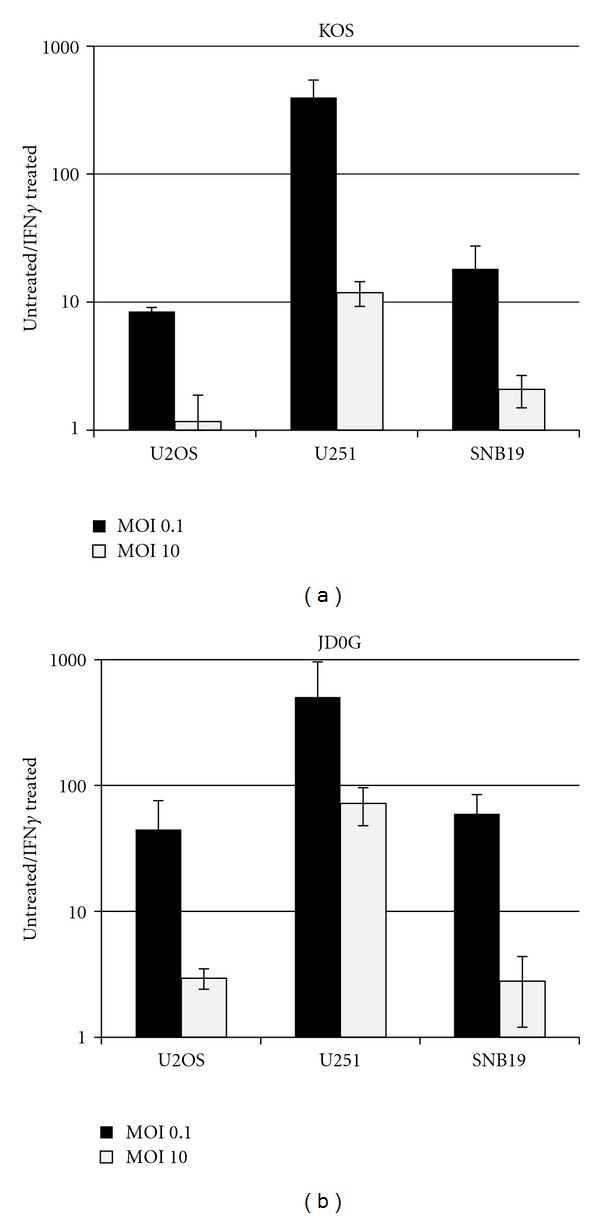
IFNγ treatment of glioblastoma cells results in a decrease in virus yield in an MOI-dependent manner. U251 and SNB19 cells were pretreated or mock treated with IFNγ (500 U/mL) 24 h prior to infection with either KOS or JD0G at an MOI of 0.1 (a) or 10 (b), and the supernatants were titered at 24 hpi. The data present the average of two independent infections, and the error bars represent the standard deviation; these data are representative of replicate experiments.
2.4. IFNγ-Induced IDO Protein Has a Minimal Effect on Virus Replication
Local production of IFNγ induces the synthesis of indoleamine-2,3-dioxygenase (IDO), an enzyme that catalyzes degradation of the essential amino acid tryptophan producing kynurenine. IDO production in response to IFNγ has been linked to the inhibition of HSV replication in certain cell lines in vitro [27]. In order to determine if the glioblastoma cells used in this study produce active IDO protein either constitutively or in response to IFNγ, IDO-enzymatic activity was assessed by measuring the level of kynurenine in the supernatants of IFNγ-treated cells. Kynurenine levels increased in a dose-dependent manner in response to increasing levels of IFNγ (Figure 4(a)). These data illustrated that active IDO protein was produced in response to IFNγ treatment of glioblastoma cells.
Figure 4.
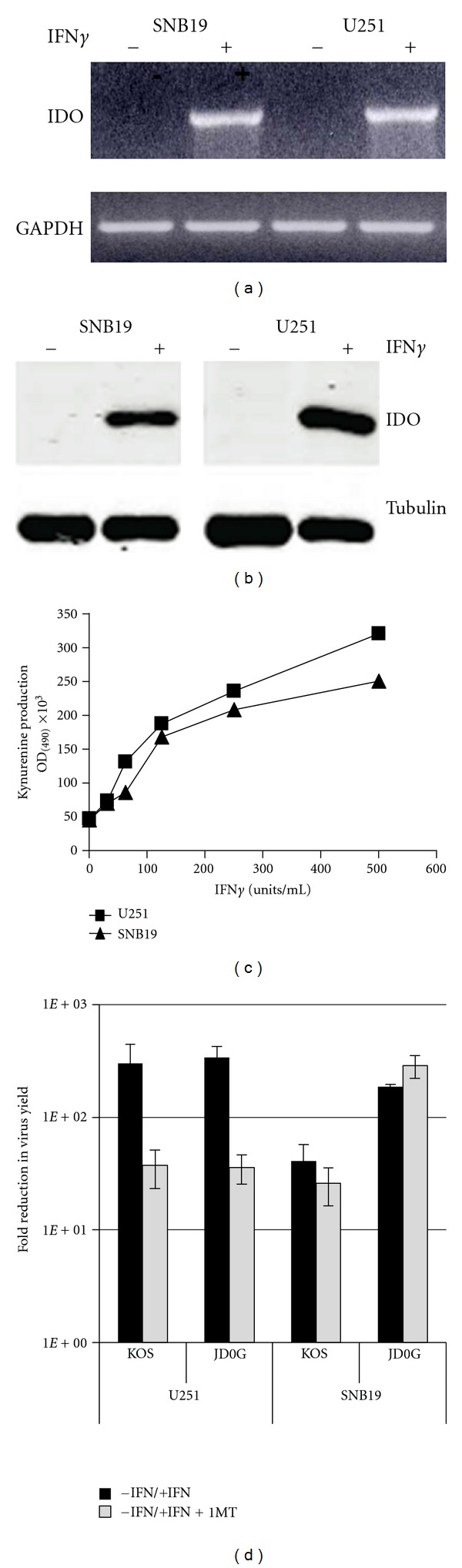
IFNγ-induced IDO protein has a minimal effect on virus replication. (a) SNB19 or U251 cells were pretreated with varying concentrations of IFNγ (0–500 U/mL) in media supplemented with 100 μg/mL of L tryptophan. After 3 days of incubation, IDO activity was measured by determining kynurenine content in the cell supernatants. (b) U251 or SNB19 cell lines were pretreated or mock treated with IFNγ (500 U/mL) with or without 1-MT (300 μM) 24 h prior to infection with JD0G or KOS virus, and the supernatants were titered at 24 hpi. The data represent the average of two independent infections, and the error bars represent the standard deviation; these data are representative of replicate experiments.
We used the small molecule inhibitor of IDO activity, 1-methyl-tryptophan (1-MT), to assess the role of IDO in the observed reduction in virus yields from glioblastoma cells treated with IFNγ prior to low-MOI infection (Figure 4(b)). In U251 cells, IFNγ decreased both KOS and JD0G yields over 100-fold, and the addition of 1-MT consistently reduced this loss to approximately 15-fold; 1-MT alone had no effect on virus growth (data not shown). In contrast, the presence of 1-MT did not significantly alter the reduced yields of KOS and JD0G on IFNγ-treated SNB19 cells. These data suggested that IDO contributes to the IFNγ hypersensitivity of the two viruses on U251 cells, but not to their lower sensitivity on SNB19 cells. In both instances, however, the main effect of IFNγ appeared to be mediated via a different pathway.
2.5. Virus Infection Reduces the Amount of IDO mRNA and Protein in IFNγ-Treated Cells
Although active IDO protein was produced in response to IFNγ in both U251 and SNB19 cells, the 1-MT experiments described above indicated that IDO activity did not significantly influence virus replication in at least one of these lines, SNB19. This may suggest that HSV-1 can counteract the IDO pathway to allow virus replication. The level of IDO protein present in U251 and SNB19 cells was measured following IFNγ treatment and virus infection (Figure 5(a)). Untreated cells and cells infected with virus alone did not show detectable levels of IDO protein at 12 and 24 hpi (Figure 5(a)). Cells treated with IFNγ produced IDO protein in both cell lines. However, cells treated with IFNγ and then infected with either KOS or JD0G virus showed a substantial reduction in the amount of IDO protein (Figures 5(a) and 5(b)). At 24 hpi, less than 20% of the IDO protein remained in SNB19 cells and less than 30% in U251 cells (Figure 5(b)). These data, representative of 3 independent experiments, suggested that infection of cells with HSV-1 can lead to downregulation of IDO protein.
Figure 5.
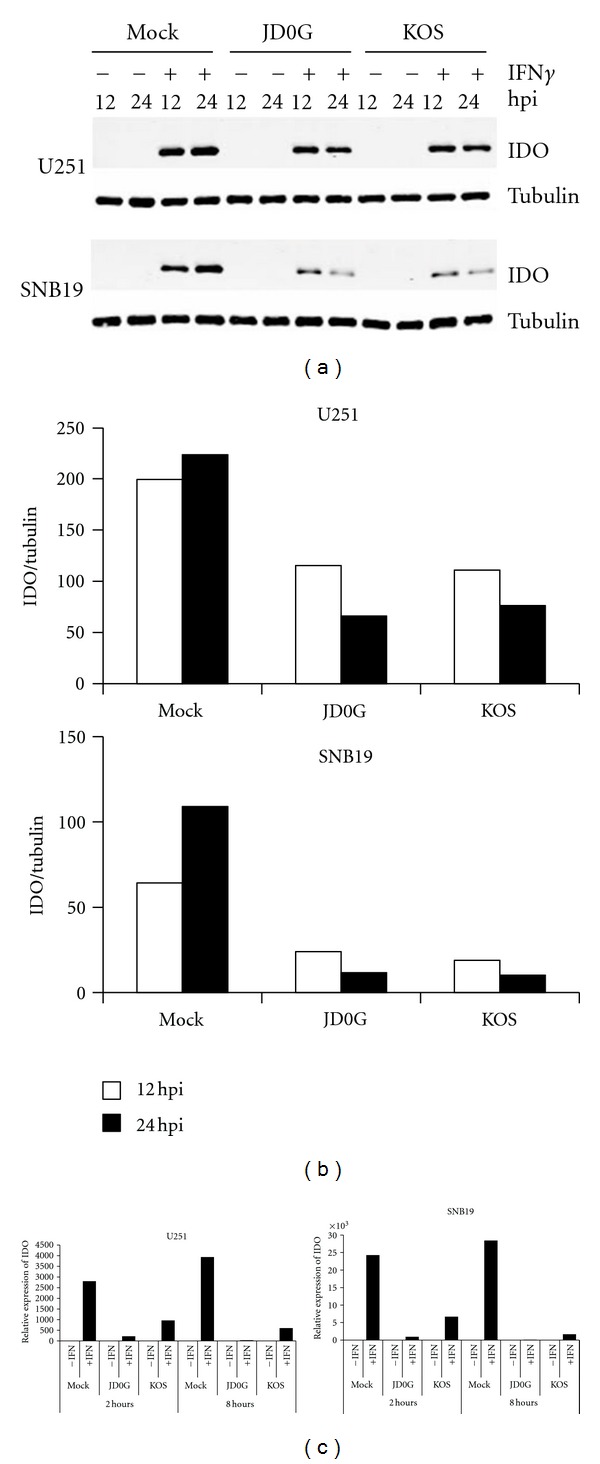
Reduction in IFNγ-induced IDO expression following virus infection of glioblastoma cells. (a) The effect of virus infection on IFNγ-induced IDO protein levels in SNB19 cells (top) and U251 cells (bottom) was visualized by Western blot. Cells were treated with IFNγ or mock treated for 24 h prior to infection with either JD0G or KOS virus. At 12 and 24 hpi, cell lysates were analyzed by Western blot with antibodies against IDO protein (upper panels) or α-tubulin (lower panels). (b) IDO protein levels were quantified using the Odyssey Infrared Imaging System, and the relative amount of IDO protein (IDO/α-tubulin) in each sample is shown. Data are representative of three experiments using different time points. (c) The effect of virus infection on IDO mRNA levels in SNB19 and U251 cells was assessed by quantitative RT-PCR. Cells were treated with IFNγ or mock treated for 24 h prior to infection with either JD0G or KOS virus, and total cellular RNA was collected at 2 and 8 hpi. IDO mRNA levels for each sample were normalized to GAPDH levels and the fold change in IDO expression for each sample is shown relative to the untreated, mock-infected control.
Downregulation of IDO in response to virus infection can occur either at the protein or mRNA level. We therefore measured IDO mRNA expression by quantitative RT-PCR at 2 and 8 hours following virus infection of IFNγ-treated cells. The results (Figure 5(c)) demonstrated that IDO mRNA levels were reduced as early as 2 hours postinfection with either KOS or JD0G viruses when compared to mock-treated controls. Together these data suggest that infection of glioblastoma cells with HSV results in reduced expression of both IDO mRNA and protein, thereby minimizing the inhibitory effects of this protein on virus replication.
3. Discussion
The development of attenuated viruses that are adapted for preferential replication in solid tumors is an attractive approach to treatment of malignancies where more standard therapies are either ineffective or difficult to apply. HSV-1-oncolytic vectors used in early-phase clinical trials for the treatment of GBM have shown some success without serious side effects [33, 34]. Tumor specificity can be achieved by deleting viral genes that permit mutant virus replication in tumor cells while profoundly impairing virus replication in normal host cells [1]. The vector prototype is G207, that is, deleted for γ34.5 and produces a nonfunctional ICP6-LacZ fusion protein [35]. ICP6 encodes the large subunit of the viral ribonucleotide reductase, a protein that permits virus growth in nondividing cells by maintaining the nucleotide pool, while γ34.5 counteracts the virus-induced activation of the PKR pathway. The more advanced oncolytic vector examined in this study, MGH2, was derived from G207 by replacing lacZ with eGFP at the ICP6 locus and inserting two antitumor genes, CYP2B1 (encoding cytochrome p450) and shiCE (encoding secreted human intestinal carboxylesterase) [32]. These enzymes activate the anticancer drugs cyclophosphamide and irinotecan, respectively, both of which are potent tumor toxic products. In one study, MGH2 showed oncolytic activity in vivo only with the addition of cyclophosphamide and irinotecan [32], indicating that the MGH2 vector backbone alone does not function as an effective OV. Some evidence suggests that vector replication in certain tumor cells may require γ34.5 activity and that oHSV is susceptible to innate immune responses, potentially limiting the effectiveness of this and other oHSV vectors [36]. We therefore sought to examine other mutant backbones that may overcome these limitations.
In this study we characterize an oHSV vector (JD0G) that is deleted for ICP0 and the joint elements of the viral genome. HSV-1 mutants defective for the production of ICP0 protein provide an attractive alternative to the MGH2 vector backbone. ICP0-deficient vectors are impaired for growth at low MOI in most cell lines, while replication preferentially occurs in certain tumor cell lines [22, 23, 37]. Moreover, Hummel and colleagues have reported that an HSV-1 double mutant lacking VP16 and ICP0 protein expression replicates efficiently in a variety of tumor cells derived from prostate, lung, colon, and mammary carcinomas with evidence of oncolytic activity in animal models [24]. Using the JD0G backbone, we sought to determine whether the unique interaction of an ICP0-deleted vector with tumor cells could be exploited to develop novel oncolytic viruses suitable for treatment of brain tumors. Our findings showed that JD0G replication was significantly enhanced in the U251 and SNB19 glioblastoma cell lines compared to HEL cells, while MGH2 replication was highly impaired, showing 2-3 logs lower yields on the glioblastoma lines than JD0G. These findings point to the possibility that genetic changes related to glioblastoma development relieve the need for ICP0 expression and permit JD0G replication, while the mutations in MGH2 are detrimental to vector growth and not complemented in these lines. Furthermore, the difference in replication between these two vectors was most pronounced at low multiplicity (data not shown), a condition that may be more relevant to circumstances in vivo than high-MOI infections in cell culture. An oHSV vector that can replicate efficiently from a small number of initial infectious particles is most likely to be effective in tumors.
It is likely that the tumor-specific replication of the JD0G vector can be ascribed to the ICP0 deficiency and not to the removal of the joint components of the viral genome. The HSV genome consists of two unique elements (UL and US) that are each flanked by repeated sequences (ab/b′a′; a′c′/ca) (Figure 1). This arrangement creates two terminal repeat regions that together contain one copy of the genes for ICP34.5, ICP4, ICP0, and the latency-associated transcript (LAT), and an internal repeat element (joint) that contains a second copy of each gene. Thus, the joint deletion generated in the construction of JD0G removes a single copy of each gene, reducing but not eliminating gene expression. This modification alone has a minimal effect on virus replication (unpublished observations, [38]). The benefit of this deletion is that it eliminates the possibility of genome isomerization events that typically occur between the repeated elements of the viral genome and generate four possible HSV isomers. Hence, the JD0G genome has enhanced stability over the wild-type KOS genome.
Following inoculation of oHSV in vivo, IFNγ can be produced by resident microglia, recruited macrophages, dendritic and NK cells at the growing tumor site [25, 39]. In response to IFNγ, dendritic cells and tumor cells may produce IDO, whose enzymatic activity leads to the degradation of tryptophan and the production of toxic catabolites such as kynurenine and quinolinate [40]. Additionally, tumor cells including glioblastoma cells have been reported to express IDO without IFNγ induction [41]. Tryptophan depletion can impede the production of viral proteins and, together with kynurenine, can induce effector T-cell anergy [30]. Therefore, IFNγ can negatively affect both oncolytic virus replication and tumor-specific immunity at several levels. Although IFNγ-mediated inhibition of oHSV replication may be overcome in part by the administration of immunosuppressive drugs such as cyclophosphamide, a drawback to this approach is that it will also inhibit the induction of tumor-specific immunity.
In view of these considerations, and to explore the tumor-related changes that may potentially influence the innate immune response to virus infection, KOS and JD0G viral replication in glioblastoma cells were tested for their sensitivity to IFNγ treatment. Our data demonstrate that both viruses were sensitive to IFNγ at low MOI, but the ability of IFNγ to control virus replication was largely lost at elevated MOI. However, we observed marked differences between the glioblastoma cell lines in both the IFNγ sensitivity of virus replication and the involvement of IDO activity in the inhibition of virus replication. SNB19 cells produced lower levels of IDO protein than U251 cells, and virus replication in SNB19 cells was less sensitive to IFNγ treatment. In the IFNγ-hypersensitive U251 cells, both KOS and JD0G were found to downregulate, but not eliminate, IDO mRNA and protein, and the IDO inhibitor 1-MT was able to improve the efficiency of virus replication. In contrast, both KOS and JD0G infection nearly eliminated IDO expression in IFNγ-treated SNB19 cells, and 1-MT did not improve virus replication. Together, these results indicate that IDO expression above a certain level can inhibit virus replication in glioblastoma cells, but also that the majority of IFNγ-induced inhibition of virus replication occurs via a different pathway. The limited role of IDO in controlling virus replication may be attributable to the virus-mediated downregulation of IDO observed on infection with both KOS and JD0G. This downregulation may not only allow for vector replication in the presence of IFNγ, but also minimize adverse effects on antitumor immunity. Thus, we propose that vector-induced IDO downregulation may be an important aspect of the therapeutic potential of oHSV vectors.
4. Methods
4.1. Cell Lines and Viruses
Human glioblastoma SNB19 and U251 [42], osteosarcoma U2OS (ATCC, Manassas, VA, USA), embryonic lung HEL (ATCCs), and monkey kidney Vero cells (ATCC) were cultured by standard methods. HEL cells were maintained in RPMI supplemented with 10% fetal bovine serum (FBS). All other cells were maintained in Dulbecco's Modified Eagle Medium (Atlanta Biologicals, Norcross, GA, USA) supplemented with 10% FBS at 37°C in 5% CO2. The wild-type HSV-1 (KOS) virus stock was prepared on Vero cells.
The JD0G mutant HSV-1 virus was derived by deletion of ICP0 by mixed infection/transfection of the Joint Deleted vector with plasmid 28E3. In order to obtain 28E.3 we initially deleted the fragment StuI/HpaI from the plasmid 28.1 (which consists of the Dra I fragment of psg28 [43], and a Bgl II linker was inserted into this site. Plasmid 28E3 was created by cloning a Bgl II fragment containing a HCMV eGFP expression construct from pEGFPN-1 (Clontech, Palo Alto, CA, USA) into the Bgl II site of 28B.
JD0G virus stocks were prepared on U2OS cells. The MGH2 virus was kindly provided by Antonio Chiocca (Ohio State University). For comparison purposes, MOIs were based throughout on viral titers determined on U2OS cells.
4.2. Virus Growth
1 × 105 cells were seeded in 24-well dishes prior to infection. Confluent monolayers of cells were infected at an MOI of 0.1 in serum-free DMEM (Atlanta Biologicals) for 2 h. Supernatants were collected at 24 hpi, and viral titers were determined by serial dilution on U2OS cells. Plaques were visualized by crystal violet staining.
4.3. IFNγ Treatment
7 × 104 cells were seeded in 24-well dishes and pretreated or mock treated for 24 h with 500 U/mL of recombinant human IFNγ (Cell Sciences, Canton, MA, USA). The IDO inhibitor 1-methyl-L-tryptophan (Sigma, St. Louis, MO, USA) was used at a final concentration of 300 μM. Pretreated cells were infected at the appropriate MOI in serum-free DMEM for 2 h, virus was removed, and the cells were fed with fresh media plus IFNγ. Supernatants were collected at 24 hpi and viral titers were determined by serial dilution on U2OS cells. Plaques were visualized by crystal violet staining.
4.4. Quantitative RT-PCR
3 × 105 cells were seeded in 6-well dishes and pretreated or mock treated for 20 h with 500 U/mL of IFNγ. Pretreated cells were infected at an MOI of 20 in serum-free DMEM with either KOS or JD0G for 2 h, virus was removed, and fresh media with IFNγ was added to the wells. Cells were harvested at the indicated times postinfection (either 2 or 8 h), and total RNA was extracted using the RNeasy kit (QIAGEN, Valencia, CA, USA). 2 μg of total RNA was used to synthesize the first cDNA strand using Superscript First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA). Quantitative PCR was performed as previously described [44]. Average C T values for IDO were normalized with average C T values for GAPDH, and data were plotted as the fold change relative to untreated, mock-infected controls using the 2−ΔΔCT method [45]. The following primers and probes were obtained from PE Applied Biosystems (Foster City, CA, USA): IDO Fwd, GGTCATGGAGATGTCCGTAA; IDO Rev, ACCAATAGAGAGACCAGGAAGAA; IDO Probe, 6FAM-CTGTTCCTTACTGCCAACTCTCCAAGAAACTG-Tamra; GAPDH-FWD, CCCCACACACATGCACTTACC; GAPDH-REV, CCTACTCCCAGGGCTTTGATT; GAPDH-Claw-Probe, 6FAM-AAAGAGCTAGGAAGGACAGGCAACTTGGC-TAMRA.
4.5. Kynurenine Assay
3 × 104 cells were pretreated with varying concentrations of IFNγ (0–500 U/mL) in media supplemented with 100 μg/mL L-tryptophan. After 3 days of incubation, supernatants were harvested for determination of kynurenine content. Samples of 160 μL were incubated with 10 μL of 30% TCA for 30 min at 50°C and centrifuged at 3,000 rpm for 20 min at room temperature. 100 μL of each sample was reacted with an equal volume of Ehrlich reagent (Sigma) at room temperature for 10 min, and the OD490 measured using a microplate reader.
4.6. Western Blot
3 × 105 cells were seeded in 6-well dishes and pretreated or mock treated for 20 h with 500 U/mL of IFNγ. Pretreated cells were infected at an MOI of 20 in serum-free DMEM with either KOS or JD0G for 2 h, virus was removed, and fresh media with IFNγ was added to the wells. Cells were harvested at the indicated times postinfection (either 12, or 24 h), resuspended in 200 μL lysis buffer (10 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA pH 8, 0.1% Triton-X, 1 mM DTT, 1 mM PMSF, 1 mM NaVO4, and 0.2% Protease Inhibitor Cocktail [Sigma]), and incubated on ice for 30 min. 5 μg of total protein was resolved on an 8% SDS-PAGE gel and transferred to Protran 0.45 μm nitrocellulose membrane (Whatman, Piscataway, NJ, USA). The membrane was blocked for 1 h at room temperature in Tween-20-free Odyssey blocking buffer (LI-Cor Biosciences, Lincoln, NE, USA) and incubated overnight at 4°C with a 1 : 5,000 dilution of ALX-210-429 rabbit anti-IDO antibody (Alexis Biochemicals, Lausanne, Switzerland) and a 1 : 20,000 dilution of mouse anti-α-tubulin antibody (T6074, Sigma) in 1x PBS/0.05% Tween-20. The membrane was washed with 1x PBS/0.05% Tween-20, incubated for 1 h at room temperature with goat anti-mouse IRDye800CW-green and goat anti-rabbit IRDye680-red secondary antibodies (both from LI-Cor), and washed with 1x PBS/0.05% Tween-20. The membrane was scanned with Odyssey Infrared Imaging System (LI-Cor) and analyzed with Odyssey 3.0 software as specified by the manufacturer.
4.7. Cell Killing
1 × 106 cells were seeded in 6-well dishes and infected 24 hours later at an MOI of 0.1 in serum-free DMEM (Atlanta Biologicals). Media was replaced with fresh serum-containing media after 1.5 h and 48 h postinfection viable cells were stained with Trypan Blue and counted.
Conflict of Interests
The authors declare no conflict of interests.
Acknowledgments
The authors thank David Klinkenberg and Qiwei Han for technical assistance and David Clawson and Bill Goins for qPCR analysis. This research was supported by NIH Grants NS040923 and CA119298 to J. C. Glorioso, and by a grant from the Copeland Foundation to P. Grandi. B. Reinhart was supported by a fellowship from the NIH Training Program in Molecular Microbial Persistence and Pathogenesis.
References
- 1.Grandi P, Peruzzi P, Reinhart B, Cohen JB, Chiocca EA, Glorioso JC. Design and application of oncolytic HSV vectors for glioblastoma therapy. Expert Review of Neurotherapeutics. 2009;9(4):505–517. doi: 10.1586/ern.09.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu JC, Booth MJ, Tripuraneni G, et al. A novel HSV-1 virus, JS1/34.5-/47—purges contaminating breast cancer cells from bone marrow. Clinical Cancer Research. 2006;12(22):6853–6862. doi: 10.1158/1078-0432.CCR-06-1228. [DOI] [PubMed] [Google Scholar]
- 3.Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant G207 for the treatment of malignant glioma: results of a phase I trial. Gene Therapy. 2000;7(10):867–874. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 4.Papanastassiou V, Rampling R, Fraser M, et al. The potential for efficacy of the modified (ICP 34.5-) herpes simplex virus HSV1716 following intratumoural injection into malignant glioma: a proof of principle study. Gene Therapy. 2002;9(6):398–406. doi: 10.1038/sj.gt.3301664. [DOI] [PubMed] [Google Scholar]
- 5.Rampling R, Cruickshank G, Papanastassiou V, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Therapy. 2000;7(10):859–866. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 6.Senzer NN, Kaufman HL, Amatruda T, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. Journal of Clinical Oncology. 2009;27(34):5763–5771. doi: 10.1200/JCO.2009.24.3675. [DOI] [PubMed] [Google Scholar]
- 7.Everett RD, Orr A. Herpes simplex virus type 1 regulatory protein ICP0 aids infection in cells with a preinduced interferon response but does not impede interferon-induced gene induction. Journal of Virology. 2009;83(10):4978–4983. doi: 10.1128/JVI.02595-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halford WP, Weisend C, Grace J, et al. ICP0 antagonizes Stat 1-dependent repression of herpes simplex virus: implications for the regulation of viral latency. Virology Journal. 2006;3, article 44 doi: 10.1186/1743-422X-3-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. Journal of Experimental Medicine. 1999;189(4):663–672. doi: 10.1084/jem.189.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu H, Roizman B. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(43):17134–17139. doi: 10.1073/pnas.0707266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai W, Schaffer PA. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. Journal of Virology. 1992;66(5):2904–2915. doi: 10.1128/jvi.66.5.2904-2915.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Silverstein S. Herpes simplex viruses with mutations in the gene encoding ICP0 are defective in gene expression. Journal of Virology. 1992;66(5):2916–2927. doi: 10.1128/jvi.66.5.2916-2927.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boutell C, Everett R, Hilliard J, Schaffer P, Orr A, Davido D. Herpes simplex virus type 1 ICP0 phosphorylation mutants impair the E3 ubiquitin ligase activity of ICP0 in a cell type-dependent manner. Journal of Virology. 2008;82(21):10647–10656. doi: 10.1128/JVI.01063-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boutell C, Orr A, Everett RD. PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. Journal of Virology. 2003;77(16):8686–8694. doi: 10.1128/JVI.77.16.8686-8694.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boutell C, Sadis S, Everett RD. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. Journal of Virology. 2002;76(2):841–850. doi: 10.1128/JVI.76.2.841-850.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chelbi-Alix MK, De Thé H. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene. 1999;18(4):935–941. doi: 10.1038/sj.onc.1202366. [DOI] [PubMed] [Google Scholar]
- 17.Everett RD. Herpes simplex virus type 1 regulatory protein ICP0 does not protect cyclins D1 and D3 from degradation during infection. Journal of Virology. 2004;78(18):9599–9604. doi: 10.1128/JVI.78.18.9599-9604.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin R, Noyce RS, Collins SE, Everett RD, Mossman KL. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. Journal of Virology. 2004;78(4):1675–1684. doi: 10.1128/JVI.78.4.1675-1684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lomonte P, Sullivan KF, Everett RD. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. Journal of Biological Chemistry. 2001;276(8):5829–5835. doi: 10.1074/jbc.M008547200. [DOI] [PubMed] [Google Scholar]
- 20.Stow ND, Stow EC. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. Journal of General Virology. 1986;67(12):2571–2585. doi: 10.1099/0022-1317-67-12-2571. [DOI] [PubMed] [Google Scholar]
- 21.Sacks WR, Schaffer PA. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. Journal of Virology. 1987;61(3):829–839. doi: 10.1128/jvi.61.3.829-839.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao F, Schaffer PA. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. Journal of Virology. 1995;69(10):6249–6258. doi: 10.1128/jvi.69.10.6249-6258.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sobol PT, Hummel JL, Rodrigues RM, Mossman KL. PML has a predictive role in tumor cell permissiveness to interferon-sensitive oncolytic viruses. Gene Therapy. 2009;16(9):1077–1087. doi: 10.1038/gt.2009.68. [DOI] [PubMed] [Google Scholar]
- 24.Hummel JL, Safroneeva E, Mossman KL. The role of ICP0-Null HSV-1 and interferon signaling defects in the effective treatment of breast adenocarcinoma. Molecular Therapy. 2005;12(6):1101–1110. doi: 10.1016/j.ymthe.2005.07.533. [DOI] [PubMed] [Google Scholar]
- 25.Fulci G, Breymann L, Gianni D, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(34):12873–12878. doi: 10.1073/pnas.0605496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedman A, Tian JP, Fulci G, Chiocca EA, Wang J. Glioma virotherapy: effects of innate immune suppression and increased viral replication capacity. Cancer Research. 2006;66(4):2314–2319. doi: 10.1158/0008-5472.CAN-05-2661. [DOI] [PubMed] [Google Scholar]
- 27.Adams O, Besken K, Oberdörfer C, MacKenzie CR, Rüßing D, Däubener W. Inhibition of human herpes simplex virus type 2 by interferon γ and tumor necrosis factor α is mediated by indoleamine 2,3-dioxygenase. Microbes and Infection. 2004;6(9):806–812. doi: 10.1016/j.micinf.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. Journal of Clinical Investigation. 2007;117(5):1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zamanakou M, Germenis AE, Karanikas V. Tumor immune escape mediated by indoleamine 2,3-dioxygenase. Immunology Letters. 2007;111(2):69–75. doi: 10.1016/j.imlet.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 30.Katz JB, Muller AJ, Prendergast GC. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunological Reviews. 2008;222(1):206–221. doi: 10.1111/j.1600-065X.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- 31.Hong CS, Fellows W, Niranjan A, et al. Ectopic matrix metalloproteinase-9 expression in human brain tumor cells enhances oncolytic HSV vector infection. Gene Therapy. 2010;17(10):1200–1205. doi: 10.1038/gt.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tyminski E, LeRoy S, Terada K, et al. Brain tumor oncolysis with replication-conditional herpes simplex virus type 1 expressing the prodrug-activating genes, CYP2B1 and secreted human intestinal carboxylesterase, in combination with cyclophosphamide and irinotecan. Cancer Research. 2005;65(15):6850–6857. doi: 10.1158/0008-5472.CAN-05-0154. [DOI] [PubMed] [Google Scholar]
- 33.Markert JM, Liechty PG, Wang W, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Molecular Therapy. 2009;17(1):199–207. doi: 10.1038/mt.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Markert JM, Parker JN, Buchsbaum DJ, Grizzle WE, Gillespie GY, Whitley RJ. Oncolytic HSV-1 for the treatment of brain tumours. Herpes. 2006;13(3):66–71. [PubMed] [Google Scholar]
- 35.Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nature Medicine. 1995;1(9):938–943. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- 36.Mossman KL, Smiley JR. Herpes simplex virus ICP0 and ICP34.5 counteract distinct interferon-induced barriers to virus replication. Journal of Virology. 2002;76(4):1995–1998. doi: 10.1128/JVI.76.4.1995-1998.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sacks WR, Schaffer PA. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. Journal of Virology. 1987;61(3):829–839. doi: 10.1128/jvi.61.3.829-839.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jenkins FJ, Roizman B. Herpes simplex virus 1 recombinants with noninverting genomes frozen in different isomeric arrangements are capable of independent replication. Journal of Virology. 1986;59(2):494–499. doi: 10.1128/jvi.59.2.494-499.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prestwich RJ, Errington F, Diaz RM, et al. The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Human gene therapy. 2009;20(10):1119–1132. doi: 10.1089/hum.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyazaki T, Moritake K, Yamada K, et al. Indoleamine 2,3-dioxygenase as a new target for malignant glioma therapy: laboratory investigation. Journal of Neurosurgery. 2009;111(2):230–237. doi: 10.3171/2008.10.JNS081141. [DOI] [PubMed] [Google Scholar]
- 41.Guillemin GJ, Cullen KM, Lim CK, et al. Characterization of the kynurenine pathway in human neurons. Journal of Neuroscience. 2007;27(47):12884–12892. doi: 10.1523/JNEUROSCI.4101-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okano F, Storkus WJ, Chambers WH, Pollack IF, Okada H. Identification of a novel HLA-A∗ 0201-restricted, cytotoxic T lymphocyte epitope in a human glioma-associated antigen, interleukin 13 receptor alpha2 chain. Clinical Cancer Research. 2002;8:2851–2855. [PubMed] [Google Scholar]
- 43.Alvira MR, Goins WF, Cohen JB, Glorioso JC. Genetic studies exposing the splicing events involved in herpes simplex virus type 1 latency-associated transcript production during lytic and latent infection. Journal of Virology. 1999;73(5):3866–3876. doi: 10.1128/jvi.73.5.3866-3876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsvitov M, Frampton AR, Shah WA, et al. Characterization of soluble glycoprotein D-mediated herpes simplex virus type 1 infection. Virology. 2007;360(2):477–491. doi: 10.1016/j.virol.2006.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]