

Abstract
The p16INK4A (hereafter p16) tumor suppressor is encoded by the INK4A/ARF locus which is among the most commonly dysregulated sequences in human cancer. By inhibiting cyclin-dependent kinases, p16 activates the G1-S checkpoint, and this response is often considered to be critical for establishing a senescence-like growth arrest. Not all studies support a universal role for p16 in senescence. Single-cell analysis of noncancerous human fibroblast cultures undergoing senescence as a function of culture age (replicative senescence) has revealed that p16 is not expressed in the majority (>90%) of cells that exhibit features of senescence (e.g., flattened and enlarged morphology coupled with senescence-associated β-galactosidase expression), ruling out a requirement for p16 in this process. In addition, ionizing radiation triggers premature senescence in human cancer cell lines that do not express p16. These observations are made with cells that express wild-type p53, a key mediator of the DNA damage response. In this paper, we examine the growing evidence suggesting a negative regulatory relationship between p16 and p53 and discuss recent reports that implicate a role for p16 in replicative senescence and ionizing radiation-induced premature senescence in human cells that lack wild-type p53 function.
1. Introduction
Normal somatic human cells in culture undergo a finite number of divisions before entering a state of irreversible growth arrest termed ‘‘replicative senescence” [1]. This phenotype is characterized by the acquisition of flattened and enlarged cell morphology, presence of β-galactosidase activity at suboptimal conditions (i.e., pH 6), and absence of cell division in metabolically active cells. Replicative senescence is triggered by erosion and dysfunction of telomeres and is mediated by multibranched signaling processes [2, 3]. Exposure of certain immortalized cell types (e.g., p53-proficient human solid tumor-derived cell lines), as well as “young” (early-passage) normal human cells (e.g., skin fibroblasts) to DNA-damaging agents can also trigger a state of sustained growth arrest resembling senescence. The DNA damage-triggered response is commonly called “stress-induced premature senescence” (SIPS). Unlike replicative senescence, SIPS is independent of telomere length or function [3].
Bypassing replicative senescence is a prerequisite step in immortalization and malignant transformation [4], and escape from SIPS can lead to the emergence of highly metastatic and therapy-resistant cells [5, 6]. Accordingly, a great deal of research has been directed towards understanding the molecular basis for different forms of senescence in an attempt to identify novel targets for the treatment of pre-neoplastic lesions and malignant disease.
Ectopic expression of numerous cancer-associated cell-cycle genes (e.g., p21WAF1, p16INK4A, p27KIP1, p15INK4B, pRB, and CHK2) in human cells has been reported to trigger senescence (reviewed in [7]). In the absence of artificial gene manipulation, upregulation of the cyclin-dependent kinase (CDK) inhibitors p16INK4A and p21WAF1 (hereafter called p16 and p21, resp.) has also been consistently reported to be associated with senescence [8–13]. While the pivotal role of p21 in orchestrating replicative senescence and DNA damage-induced SIPS has been well established [6], attempts to elucidate a role for p16 in these processes have led to inconsistent outcomes, with some reports providing strong evidence for p16-driven senescence (e.g., in human fibroblasts [8] and melanocytes [13] undergoing telomere-directed senescence), and other reports demonstrating the induction of senescence in the absence of p16 (e.g., in human fibroblasts [2] and endothelial cells [14], also undergoing telomere-directed senescence).
Although p16 has been extensively characterized for its ability to decelerate cell progression from G1 to S phase, it has emerged as a multifunctional protein capable of forming a negative regulatory loop with p53, a key mediator of the DNA damage response. In addition, recent work with noncancerous human fibroblast strains and solid tumor-derived cell lines with differing p53 status has implicated the involvement of p16 in a redundant pathway for senescence, triggering this response only in the absence of wild-type p53 activity. Here, we will consider the evidence for these properties of p16.
2. Regulation of p16 Expression
The human INK4/ARF locus is located on chromosome 9p21 and generates p16 and at least two other transcriptional variants, p14ARF (alternative reading frame) and p12 [15, 16]. Regulation of this locus is complex, involving several tumor-relevant and/or stress signaling pathways [17]. The p38 mitogen-activated protein kinase (MAPK) pathway mediates p16INK4A induction, the RNA binding protein AUF1 negatively regulates p16INK4A mRNA stability [17], and the T box proteins (e.g., Tbx2) [18], the polycomb group proteins (e.g., BMI-1) [19, 20], histone deacetylases [21, 22], and the transcription regulators E2F1 and c-MYC [21, 23] repress p16INK4A expression.
In 2005, Jacobs and de Lange proposed that the p53 tumor suppressor might also contribute to p16 regulation [17]. This notion was based on the observation that upregulation of p16 following DNA damage was unexpectedly delayed, occurring after the initial increase of p53 and its subsequent decline to background levels. That p16 is a target of p53-mediated repression is now well documented. Hernández-Vargas et al. [24], for example, reported that p53 transcriptionally activates the helix-loop-helix transcriptional regulator protein Id1, a well-known repressor of p16INK4A [25, 26]. Additionally, p53 is known to downregulate p16 through Id1-independent mechanisms [27].
3. Multiple Functions of p16
The p16 protein was discovered in the early 1990s, and was extensively studied for its ability to influence cell progression from G1 to S phase. p16 was shown to inhibit the kinase activities of the cyclin D-dependent kinases CDK4 [28] and CDK6 [29]. As cyclin D levels rise in G1, cyclin D binds the constitutively expressed CDK4 and CDK6 molecules. The resultant cyclin/CDK complexes phosphorylate pRB, leading to the release of active E2F that mediates transcriptional activation of a variety of proteins necessary for G1 to S progression and DNA replication, including cyclin E, cyclin A, and thymidine kinase [30]. Inhibition of pRB phosphorylation and E2F release in turn lead to inhibition of G1-S progression.
A number of additional biochemical and biological functions have since been documented for p16 (Figure 1). Numerous reports published in the late 1990s implicated a role for p16 in regulating angiogenesis [31], tumor invasion [32], cell spreading [33], and other fundamental cellular processes [34–37]. In part, p16 was shown to elicit such pleiotropic effects by modulating the expression or function of distinct target molecules, such as transcriptional downregulation of genes that encode vascular endothelial growth factor (VEGF) [31], matrix metalloproteinase 2 (MMP-2) [32], nuclear factor κB (NF-κB) [38], and pRB [39].
Figure 1.
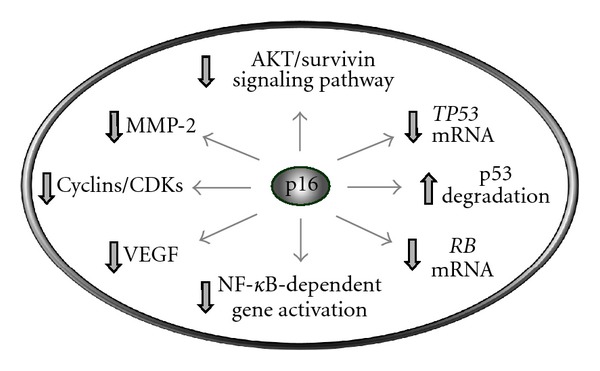
Multiple functions of the p16 tumor suppressor. This protein inhibits the kinase activities of CDK4 and CDK6 that mediate pRB phosphorylation [28–30], promotes MDM2-dependent degradation of p53 [40], downregulates AKT/survivin signaling [41], and represses the transcription of several genes including RB [39], TP53 [40], VEGF (vascular endothelial growth factor) [13], MMP-2 (matrix metalloproteinase 2) [32], and NF-κB [38].
More recently, p16 was shown to negatively or positively regulate apoptotic cell death depending on the stimuli. Thus, p16 protected cells from undergoing apoptosis after DNA damage by downregulating the intrinsic-mitochondrial pathway [43], whereas it promoted the detachment-triggered apoptosis (a process called anoikis; Greek word for homeless) by downregulating AKT/survivin signaling [41].
In addition, a reciprocal relationship was demonstrated between p16 and p53, a key regulator of apoptosis. Huschtscha et al. [40], for example, reported that p16 regulates p53 expression by both decreasing TP53 transcription and increasing Mdm2-mediated p53 degradation. Functioning at the hub of the DNA damage surveillance network, p53 regulates many DNA-damage-triggered responses including transcription, DNA repair, cell-cycle checkpoints, apoptosis, and SIPS (reviewed in [6]). A reciprocal relationship between p16 and p53 was documented not only with cultured human and murine cells [27, 40], but also with human tumor xenografts [44] and with a transgenic mouse model that carries the entire human p16INK4A locus [45]. As extensively discussed by Rayess et al. [23], these and related studies established a critical role for p16 in cell-fate determination following genotoxic stress when p53 is inactivated. Thus, genotoxic stress (e.g., DNA damage, oncogenic RAS expression) triggers the increased generation of reactive oxygen species that activate the MAPK pathway, leading to MAPK-mediated p16 expression and p16-mediated responses (e.g., SIPS) in p53-deficient cells.
In short, p16 is a multifunctional tumor suppressor capable of forming a negative regulatory loop with p53 and influencing the expression of a large number of cancer-associated genes both directly (e.g., RB, TP53) and indirectly by inhibiting the transcription regulators NF-κB and p53.
4. p16 Expression in Human Fibroblasts Undergoing Replicative Senescence
As mentioned earlier, evaluating the roles of p21 and p16 in different forms of senescence has been the subject of intensive research since their discovery in the 1990s. Initial studies suggested a sequential involvement of these proteins in replicative senescence of diploid human fibroblasts, with p21 activating cell-cycle arrest at the early stage of senescence, and p16 being crucial to maintain the senescent cell-cycle arrest [46, 47]. p16 was proposed to elicit growth arrest by inhibiting pRB phosphorylation, which results in sequestration and inhibition of the E2F family of transcription factors [1]. It was subsequently demonstrated that pRB is downregulated in cells undergoing senescence and becomes barely detectable in “late” senescent cells [47], suggesting that the long-term maintenance of the senescence phenotype can occur in the absence of pRB. Brookes et al. [48] observed considerable variability in the basal levels and kinetics of p16 accumulation in different human fibroblast strains. The levels of p16 increased with population doublings in two of the four normal fibroblast strains tested, with the other two stains showing little or no increase in p16 at late passages. These authors further demonstrated that p16-deficient human fibroblast strains are arrested at late passages and exhibit features of replicative senescence, and concluded that p16 might not be essential for the termination of fibroblast life span.
In 2007, two comprehensive review papers were published on the role of p16 in replicative senescence, with opposing conclusions. Thus, Cánepa et al. [49] suggest that p16 is a key regulator of replicative senescence and identified p16 as a molecular marker of this process, whereas Zhang [3] suggests that p16 is not related to replicative senescence mediated by telomere shortening, although the global expression of this CDK inhibitor might be increased in senescent cells. Examining the basis for such conflicting ideas is beyond the scope of the current paper. In what follows we will mainly focus on our own findings with human fibroblasts with different genetic backgrounds.
Most studies addressing the role of p16 in telomere-directed senescence relied on the evaluation of global p16 protein levels. However, such measurements can be misleading because not all proteins are uniformly expressed among cells within a putatively “clonal” population. Indeed, Herbig et al. [2] employed single-cell evaluation techniques and demonstrated that p16 protein levels can be heterogeneous among cells within a given senescent culture, with the majority of cells exhibiting undetectable levels of p16, and only a small proportion containing extremely high levels. We have reported similar observations with human normal and ataxia telangiectasia (AT) fibroblast strains that express wild-type p53 [42]. Normal and AT fibroblast cultures entered the state of senescence after approximately 70 and 50 population doublings, respectively. The majority (>90%) of cells undergoing replicative senescence exhibited strong nuclear accumulation of p21, but did not express p16 [42].
A different scenario was apparent in Li-Fraumeni syndrome (LFS) fibroblast strains. The LFS strains studied by us are heterozygous for TP53 mutations at either codon 254 (strains 2675T and 2674T) or codon 234 (strain 2800T). Such mutations result in either compromise (codon 254) or absence (codon 234) of p53-dependent transcription, as evident from the ability of the cells to upregulate p21WAF1 mRNA and p21 protein in response to DNA damage [50, 51]. Given that the p53-p21 pathway is a key mediator of senescence [6], it was of interest to determine the fate of LFS cells as a function of culture age. Vaziri et al. [52] reported that strains 2674T and 2675T lose the wild-type TP53 allele at late passages and (surprisingly) undergo replicative senescence. We demonstrated that all three LFS stains undergo replicative senescence after ~80 (2800T), ~90 (2675T), and ~100 (2674T) population doublings and that such cells fail to express p21 but express very high levels of p16 [42]. Early-passage cultures of these LFS strains do not express p21 or p16.
Collectively, these results led us to propose the model presented in Figure 2, in which p16 functions in a redundant pathway of replicative senescence in human fibroblasts, triggering this process only in the absence of wild-type p53 activity. This model is consistent with the aforementioned recent discoveries demonstrating a negative interrelationship between p16 and p53.
Figure 2.
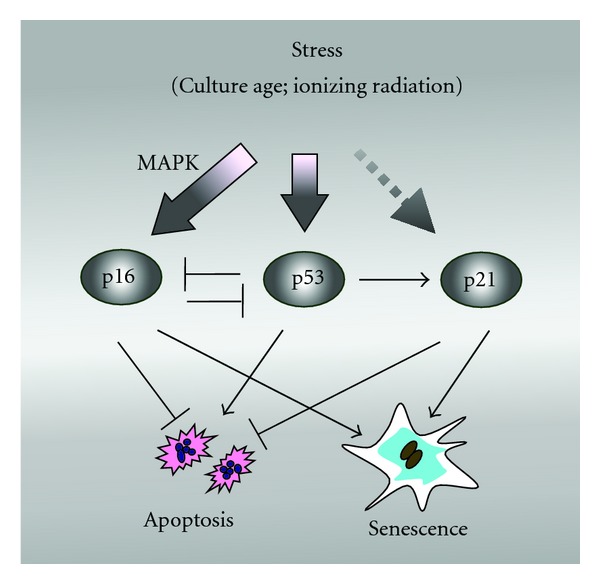
Model illustrating the involvement of the p16, p53, and p21 tumor suppressors in senescence of human fibroblast cultures [42]. In p53-proficient (normal) fibroblasts, telomerase shortening (e.g., as a function of culture age) or exposure to DNA-damaging agents results in activation of p53, which represses p16 and transcriptionally activates p21. The latter protein suppresses apoptosis and triggers senescence. On the other hand, p53-deficient (Li-Fraumeni syndrome) fibroblasts respond to stress by upregulating p16 which suppresses apoptosis and triggers senescence.
5. p16 Expression in Human Fibroblasts Undergoing SIPS
In our previous work, ionizing radiation exposure triggered extensive SIPS but marginal (if any) apoptosis in early-passage cultures of p53-proficient (normal and AT) and p53-deficient (LFS) fibroblasts [42]. The proportion of cells undergoing SIPS after irradiation correlated with the proportion of cells expressing p21 but not p16 in p53-proficient cultures, and with the proportion of cells expressing p16 but not p21 in p53-deficient (strain 2800T) cultures. These observations are consistent with the properties of p21 [6] and p16 (see above), both of which are known to downregulate the intrinsic-mitochondrial pathway of apoptosis and to induce senescence. It is important to note that a small proportion (<5%) of cells within cultures of normal and AT strains did express p16 before radiation exposure which remained virtually unchanged after irradiation.
In an earlier work, we also determined the relationship between SIPS and expression of p21 and p16 in normal human fibroblasts exposed to ultraviolet light (UV). UV exposure triggered extensive SIPS which was associated with sustained nuclear accumulation of p21 [53]. Normal fibroblasts did not express p16 before and after exposure to UV [53].
These observations provide further support for our model (Figure 2) in which p16 and p53/p21 function in non-overlapping pathways of senescence.
6. p16 Expression in Human Cancer Cells Undergoing SIPS
In 1994, several reports demonstrated that the majority (~85%) of human cancer cell lines do not express p16 due to deletion, mutation, or silencing of the INK4A locus [54–56]. This discovery led to the notion that such cancer cells might not undergo growth arrest through the process of senescence. In 1999, however, Chang et al. [57] reported the induction of SIPS in p16-null and p53 wild-type human cancer cells (e.g., HT1080 fibrosarcoma) after exposure to different genotoxic agents, including ionizing radiation. Numerous reports have since demonstrated the induction of SIPS by ionizing radiation and chemotherapeutic agents in different solid tumor-derived cell lines that express wild-type p53. Among the cell lines studied by us, HCT116, A172, and SKNSH showed a response to ionizing radiation similar to normal human fibroblasts in terms of clonogenic survival, SIPS, and p21 expression [58]. As seen with normal fibroblasts, these cancer cell lines did not express p16 before and at different times (between 24 and 96 h) after ionizing radiation exposure (unpublished observation cited in [58]). Lack of p16 expression in HCT116 cells is consistent with the presence of a frameshift mutation in one allele of the INK4A gene, and hypermethylation of the promoter of the other allele [59]. Like HCT116, the widely used cancer cell lines A549 and MCF7 that undergo SIPS after DNA damage express wild-type p53 but do not express p16 [60–62].
These observations, together with our findings with human normal and LFS fibroblasts suggesting the involvement of p16 in a redundant pathway of senescence [42], prompted Wang and associates [62] to test whether this model is also applicable to solid tumor-derived cells. Ionizing radiation exposure triggered SIPS not only in a p53-wild-type and p16-deficient cell line (A549 lung carcinoma), but also in two p16-proficient and p53-mutated cell lines (ABC-1 adenocarcinoma and HCC44 lung carcinoma). Immunofluorescence analysis revealed that the induction of SIPS in these p16-proficient and -deficient cancer cell lines was associated with nuclear accumulation of p16 and p21, respectively. Consistent with our findings with LFS fibroblasts [42], the induction of p16 in ABC-1 and HCC44 cells was observed within days following 8 Gy irradiation [62].
We extended these studies to the mutant p53-expressing cell lines MDA-MB-435s, SKMEL-28, and MDD2; the MCF7 cell line was also evaluated as a control. Both MDA-MB-435s and SKMEL-28 cell lines lack wild-type p53 activity due to TP53 mutations, but do express p16 [63–65]. The MDD2 cell line is a variant derived from MCF7 by transfection with a dominant negative mutant of p53 [66]. Unlike MCF7, MDD2 cells lack wild-type p53 activity [66, 67]. The results for seven days after irradiation are presented in Figures 3 and 4. As expected, ionizing radiation (8 Gy) triggered SIPS in p53 wild-type cells (MCF7), which correlated with expression of p21 but not of p16. Irradiation of mutant p53-expressing cell lines also triggered SIPS and, surprisingly, this response was associated with induction of p21 but not of p16.
Figure 3.
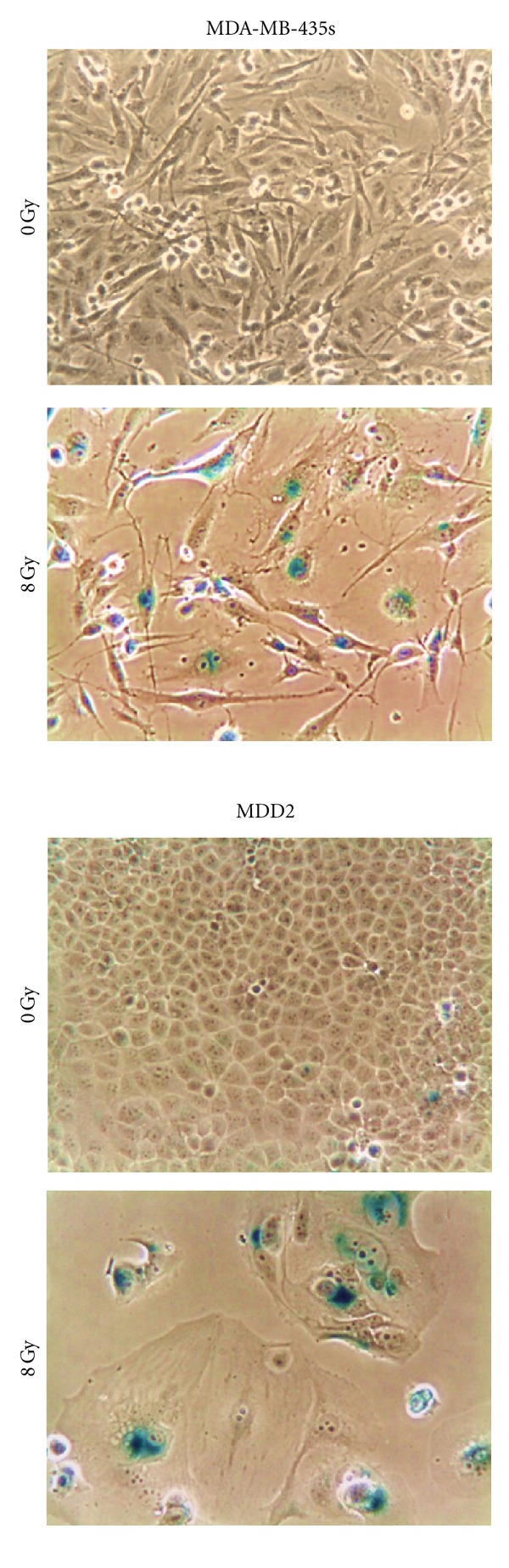
Phase-contrast photomicrographs showing SIPS in breast cancer cell lines that express mutant p53. Cells were exposed to 60Co γ-radiation (8 Gy) or sham-irradiated (0 Gy), incubated for seven days, and evaluated for features of SIPS (flattened and enlarged cellular morphology and positive (blue) staining in the senescence-associated β-galactosidase assay).
Figure 4.
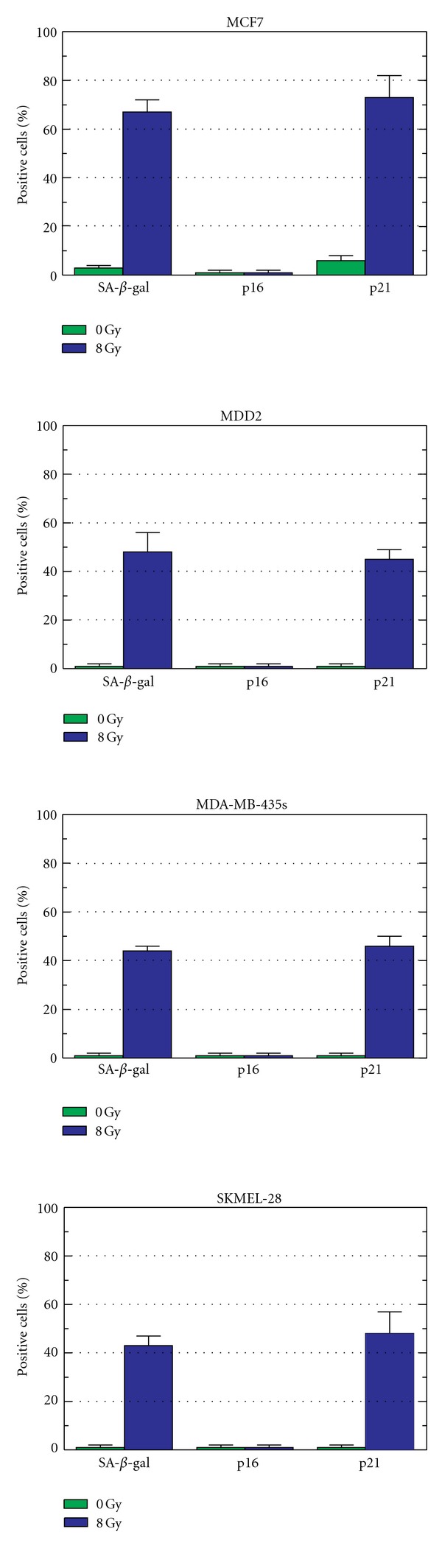
Relationship between the proportion of senescence-associated β-galactosidase (SA-β-gal)-positive cells, p16-expressing cells, and p21-expressing cells before and seven days after exposure to γ-radiation (8 Gy). Bars, SE.
In short, the results discussed above clearly demonstrate that DNA damage can trigger SIPS in human cancer cell lines expressing wild-type or mutant p53, and that this response is associated with nuclear accumulation of p21 in the majority of cases, and with induction of p16 in some cases. Further studies are warranted to determine the basis for the nuclear accumulation of p21 at late times (7 days) after irradiation in some cancer cell lines that express mutant p53 and to elucidate the reason why some p53-mutated cell lines (e.g., MDA-MB-453 s and SKMEL-24, but not ABC-1 and HCC44) exhibit the delayed nuclear accumulation of p21 after irradiation but not of p16. It is noteworthy that p21 functions as a repressor in p53-mediated downregulation of genes such as BCL-2, MCL-1, survivin, and MDR-1 (reviewed in [6]). As a working model, we propose that p21 might also be responsible for repressing p16, which might explain why some p16-proficient cell lines that exhibit nuclear accumulation of p21 after DNA damage do not concomitantly exhibit nuclear accumulation of p16.
7. Conclusion
Since their discovery in the 1990s, the CDK inhibitors p21 [6] and p16 (Figures 1 and 2) have been shown to exhibit a variety of biochemical and biological functions, most of which are independent of their major influence on pRB-regulated G1-S progression. Herein, we have highlighted the growing diversity of p16 functions, examined recent studies that implicate a role for p16 in a redundant pathway of senescence that operates in cells lacking wild-type p53, and reported new data demonstrating the complexity of DNA damage-induced SIPS in human solid tumor-derived cell lines expressing mutant p53.
At first glance, the findings discussed above may appear contradictory to several reports which suggested p16-directed senescence induced by different stimuli in p53 wild-type (e.g., normal) human cells, including SIPS triggered by DNA-damaging agents (e.g., bleomycin) (reviewed in [3]). However, it is important to note that the majority of studies that did not support a role for p16 in SIPS used single-cell measurements at 7 days (this report) or shorter times [42, 53, 62] after exposure, whereas most studies that did support a relationship between p16 expression and SIPS used global protein measurements and observed significant p16 induction at much longer times (e.g., 30 days) after introduction of DNA damage (e.g., [68]). Thus, it is reasonable to conclude that p16 might be dispensable for activation and relatively short-term (several days) maintenance of SIPS in p53 wild-type cells.
An open question remains: is p16 required for the long-term maintenance of the SIPS response in cells expressing wild-type p53? Addressing this question is of particular importance in the context of cancer therapy in view of a large body of recent evidence demonstrating that a proportion of cells undergoing SIPS after DNA damage can eventually escape this response, giving rise to aneuploid offspring exhibiting highly metastatic and therapy-resistant properties (reviewed in [5–7]).
Acknowledgments
This work was supported by the Canadian Breast Cancer Foundation-Prairies/NWT region and the Alberta Cancer Foundation donor designated grant courtesy of Mr. Wayne Stanton.
References
- 1.Huschtscha LI, Reddel RR. p16INK4a and the control of cellular proliferative life span. Carcinogenesis. 1999;20(6):921–926. doi: 10.1093/carcin/20.6.921. [DOI] [PubMed] [Google Scholar]
- 2.Herbig U, Jobling WA, Chen BPC, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a . Molecular Cell. 2004;14(4):501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 3.Zhang H. Molecular signaling and genetic pathways of senescence: its role in tumorigenesis and aging. Journal of Cellular Physiology. 2007;210(3):567–574. doi: 10.1002/jcp.20919. [DOI] [PubMed] [Google Scholar]
- 4.Vergel M, Carnero A. Bypassing cellular senescence by genetic screening tools. Clinical and Translational Oncology. 2010;12(6):410–417. doi: 10.1007/s12094-010-0528-2. [DOI] [PubMed] [Google Scholar]
- 5.Rajaraman R, Guernsey DL, Rajaraman MM, Rajaraman SR. Stem cells, senescence, neosis and self-renewal in cancer. Cell Biology International. 2005;29(12):1084–1097. doi: 10.1016/j.cellbi.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Mirzayans R, Andrais B, Scott A, Murray D. New insights into p53 signaling and cancer-cell response to DNA damage: implications for cancer therapy. Journal of Biomedicine and Biotechnology. 2012;2012:16 pages. doi: 10.1155/2012/170325.170325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mirzayans R, Murray D. Cellular senescence: implications for cancer therapy. In: Garvey RB, editor. New Research on Cell Aging. New York, NY, USA: Nova Science; 2007. pp. 1–64. [Google Scholar]
- 8.Beauséjour CM, Krtolica A, Galimi F, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. The EMBO Journal. 2003;22(16):4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roninson IB. Tumor cell senescence in cancer treatment. Cancer Research. 2003;63(11):2705–2715. [PubMed] [Google Scholar]
- 10.Itahana K, Zou Y, Itahana Y, et al. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Molecular and Cellular Biology. 2003;23(1):389–401. doi: 10.1128/MCB.23.1.389-401.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Huang X, Halicka D, et al. Contribution of p16INK4a and p21CIP1 pathways to induction of premature senescence of human endothelial cells: permissive role of p53. American Journal of Physiology. 2006;290(4):H1575–H1586. doi: 10.1152/ajpheart.00364.2005. [DOI] [PubMed] [Google Scholar]
- 12.Kunieda T, Minamino T, Nishi JI, et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation. 2006;114(9):953–960. doi: 10.1161/CIRCULATIONAHA.106.626606. [DOI] [PubMed] [Google Scholar]
- 13.Sviderskaya EV, Gray-Schopfer VC, Hill SP, et al. p16/cyclin-dependent kinase inhibitor 2A deficiency in human melanocyte senescence, apoptosis, and immortalization: possible implications for melanoma progression. Journal of the National Cancer Institute. 2003;95(10):723–732. doi: 10.1093/jnci/95.10.723. [DOI] [PubMed] [Google Scholar]
- 14.Freedman DA, Folkman J. CDK2 translational down-regulation during endothelial senescence. Experimental Cell Research. 2005;307(1):118–130. doi: 10.1016/j.yexcr.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 15.Robertson KD, Jones PA. Tissue-specific alternative splicing in the human INK4a/ARF cell cycle regulatory locus. Oncogene. 1999;18(26):3810–3820. doi: 10.1038/sj.onc.1202737. [DOI] [PubMed] [Google Scholar]
- 16.Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochimica et Biophysica Acta. 2002;1602(1):73–87. doi: 10.1016/s0304-419x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- 17.Jacobs JJL, de Lange T. p16INK4a as a second effector of the telomere damage pathway. Cell Cycle. 2005;4(10):1364–1368. doi: 10.4161/cc.4.10.2104. [DOI] [PubMed] [Google Scholar]
- 18.Jacobs JJL, Keblusek P, Robanus-Maandag E, et al. Senescence bypass screen identifies TBX2, which represses Cdkn2a (p19ARF) and is amplified in a subset of human breast cancers. Nature Genetics. 2000;26(3):291–299. doi: 10.1038/81583. [DOI] [PubMed] [Google Scholar]
- 19.Gil J, Bernard D, Martínez D, Beach D. Polycomb CBX7 has a unifying role in cellular lifespan. Nature Cell Biology. 2004;6(1):67–72. doi: 10.1038/ncb1077. [DOI] [PubMed] [Google Scholar]
- 20.Jacobs JL, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and polycombgroup gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397(6715):164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 21.Jung JW, Lee S, Seo MS, et al. Histone deacetylase controls adult stem cell aging by balancing the expression of polycomb genes and jumonji domain containing 3. Cellular and Molecular Life Sciences. 2010;67(7):1165–1176. doi: 10.1007/s00018-009-0242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng Y, Wang X, Xu L, et al. The transcription factor ZBP-89 suppresses p16 expression through a histone modification mechanism to affect cell senescence. FEBS Journal. 2009;276(15):4197–4206. doi: 10.1111/j.1742-4658.2009.07128.x. [DOI] [PubMed] [Google Scholar]
- 23.Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. International Journal of Cancer. 2012;130(8):1715–1725. doi: 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hernández-Vargas H, Ballestar E, Carmona-Saez P, et al. Transcriptional profiling of MCF7 breast cancer cells in response to 5-fluorouracil: relationship with cell cycle changes and apoptosis, and identification of novel targets of p53. International Journal of Cancer. 2006;119(5):1164–1175. doi: 10.1002/ijc.21938. [DOI] [PubMed] [Google Scholar]
- 25.Alani RM, Young AZ, Shifflett CB. Id1 regulation of cellular senescence through transcriptional repression of p16/Ink4a. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(14):7812–7816. doi: 10.1073/pnas.141235398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polsky D, Young AZ, Busam KJ, Alani RM. The transcriptional repressor of p16/Ink4a, Id1, is up-regulated in early melanomas. Cancer Research. 2001;61(16):6008–6011. [PubMed] [Google Scholar]
- 27.Leong WF, Chau JFL, Li B. p53 deficiency leads to compensatory up-regulation of p16INK4a . Molecular Cancer Research. 2009;7(3):354–360. doi: 10.1158/1541-7786.MCR-08-0373. [DOI] [PubMed] [Google Scholar]
- 28.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366(6456):704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 29.Parry D, Bates S, Mann DJ, Peters G. Lack of cyclin D-Cdk complexes in Rb-negative cells correlates with high levels of p16INK4/MTS1 tumour suppressor gene product. The EMBO Journal. 1995;14(3):503–511. doi: 10.1002/j.1460-2075.1995.tb07026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2(2):103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 31.Harada H, Nakagavva K, Iwata S, et al. Restoration of wild-type p16 down-regulates vascular endothelial growth factor expression and inhibits angiogenesis in human gliomas. Cancer Research. 1999;59(15):3783–3789. [PubMed] [Google Scholar]
- 32.Castellano M, Pollock PM, Walters MK, et al. CDKN2A/p16 is inactivated in most melanoma cell lines. Cancer Research. 1997;57(21):4868–4875. [PubMed] [Google Scholar]
- 33.Fåhraeus R, Lane DP. The p16INK4a tumour suppressor protein inhibits α v β 3 integrin-mediated cell spreading on vitronectin by blocking PKC-dependent localization of α v β 3 to focal contacts. The EMBO Journal. 1999;18(8):2106–2118. doi: 10.1093/emboj/18.8.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vojta PJ, Barrett JC. Genetic analysis of cellular senescence. Biochimica et Biophysica Acta. 1995;1242(1):29–41. doi: 10.1016/0304-419x(95)00002-w. [DOI] [PubMed] [Google Scholar]
- 35.Sandig V, Brand K, Herwig S, Lukas J, Bartek J, Strauss M. Adenovirally transferred p16INK4/CDKN2 and p53 genes cooperate to induce apoptotic tumor cell death. Nature Medicine. 1997;3(3):313–319. doi: 10.1038/nm0397-313. [DOI] [PubMed] [Google Scholar]
- 36.Naruse I, Heike Y, Hama S, Mori M, Saijo N. High concentrations of recombinant adenovirus expressing p16INK4a gene induces apoptosis in lung cancer cell lines. Anticancer Research. 1998;18(6 A):4275–4282. [PubMed] [Google Scholar]
- 37.Plath T, Detjen K, Welzel M, et al. A novel function for the tumor suppressor p16INK4a: induction of anoikis via upregulation of the α 5 β 1 fibronectin receptor. Journal of Cell Biology. 2000;150(6):1467–1477. doi: 10.1083/jcb.150.6.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolff B, Naumann M. INK4 cell cycle inhibitors direct transcriptional inactivation of NF-κB. Oncogene. 1999;18(16):2663–2666. doi: 10.1038/sj.onc.1202617. [DOI] [PubMed] [Google Scholar]
- 39.Fang X, Jin X, Xu HJ, et al. Expression of p16 induces transcriptional downregulation of the RB gene. Oncogene. 1998;16(1):1–8. doi: 10.1038/sj.onc.1201525. [DOI] [PubMed] [Google Scholar]
- 40.Huschtscha LI, Moore JD, Noble JR, et al. Normal human mammary epithelial cells proliferate rapidly in the presence of elevated levels of the tumor suppressors p53 and p21WAF1/CIP1 . Journal of Cell Science. 2009;122(16):2989–2995. doi: 10.1242/jcs.044107. [DOI] [PubMed] [Google Scholar]
- 41.Hu H, Li Z, Chen J, et al. P16 reactivation induces anoikis and exhibits antitumour potency by downregulating Akt/survivin signalling in hepatocellular carcinoma cells. Gut. 2011;60(5):710–721. doi: 10.1136/gut.2010.220020. [DOI] [PubMed] [Google Scholar]
- 42.Mirzayans R, Andrais B, Scott A, Paterson MC, Murray D. Single-cell analysis of p16INK4a and p21WAF1 expression suggests distinct mechanisms of senescence in normal human and Li-Fraumeni syndrome fibroblasts. Journal of Cellular Physiology. 2010;223(1):57–67. doi: 10.1002/jcp.22002. [DOI] [PubMed] [Google Scholar]
- 43.Al-Mohanna MA, Manogaran PS, Al-Mukhalafi Z, Al-Hussein KA, Aboussekhra A. The tumor suppressor p16INK4a gene is a regulator of apoptosis induced by ultraviolet light and cisplatin. Oncogene. 2004;23(1):201–212. doi: 10.1038/sj.onc.1206927. [DOI] [PubMed] [Google Scholar]
- 44.Zhang D, Shimizu T, Araki N, et al. Aurora A overexpression induces cellular senescence in mammary gland hyperplastic tumors developed in p53-deficient mice. Oncogene. 2008;27(31):4305–4314. doi: 10.1038/onc.2008.76. [DOI] [PubMed] [Google Scholar]
- 45.Yamakoshi K, Takahashi A, Hirota F, et al. Real-time in vivo imaging of p16INK4a reveals cross talk with p53. Journal of Cell Biology. 2009;186(3):393–407. doi: 10.1083/jcb.200904105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16INK4a in replicative senescence of normal human fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(24):13742–13747. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stein GH, Drullinger LF, Soulard A, Dulić V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Molecular and Cellular Biology. 1999;19(3):2109–2117. doi: 10.1128/mcb.19.3.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brookes S, Rowe J, Gutierrez Del Arroyo A, Bond J, Peters G. Contribution of p16INK4a to replicative senescence of human fibroblasts. Experimental Cell Research. 2004;298(2):549–559. doi: 10.1016/j.yexcr.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 49.Cánepa ET, Scassa ME, Ceruti JM, et al. INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life. 2007;59(7):419–426. doi: 10.1080/15216540701488358. [DOI] [PubMed] [Google Scholar]
- 50.Barley RDC, Enns L, Paterson MC, Mirzayans R. Aberrant p21WAF1-dependent growth arrest as the possible mechanism of abnormal resistance to ultraviolet light cytotoxicity in Li-Fraumeni syndrome fibroblast strains heterozygous for TP53 mutations. Oncogene. 1998;17(5):533–543. doi: 10.1038/sj.onc.1202271. [DOI] [PubMed] [Google Scholar]
- 51.Mirzayans R, Severin D, Murray D. Relationship between DNA double-strand break rejoining and cell survival after exposure to ionizing radiation in human fibroblast strains with differing ATM/p53 status: implications for evaluation of clinical radiosensitivity. International Journal of Radiation Oncology Biology Physics. 2006;66(5):1498–1505. doi: 10.1016/j.ijrobp.2006.08.064. [DOI] [PubMed] [Google Scholar]
- 52.Vaziri H, West MD, Allsopp RC, et al. ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. The EMBO Journal. 1997;16(19):6018–6033. doi: 10.1093/emboj/16.19.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mirzayans R, Scott A, Andrais B, Pollock S, Murray D. Ultraviolet light exposure triggers nuclear accumulation of p21WAF1 and accelerated senescence in human normal and nucleotide excision repair-deficient fibroblast strains. Journal of Cellular Physiology. 2008;215(1):55–67. doi: 10.1002/jcp.21284. [DOI] [PubMed] [Google Scholar]
- 54.Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368(6473):753–756. doi: 10.1038/368753a0. [DOI] [PubMed] [Google Scholar]
- 55.Kamb A, Gruis NA, Weaver-Feldhaus J, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264(5157):436–440. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- 56.Spruck CH, 3rd, Gonzalez-Zulueta M, Shibata A, et al. p16 gene in uncultured tumours. Nature. 1994;370(6486):183–184. doi: 10.1038/370183a0. [DOI] [PubMed] [Google Scholar]
- 57.Chang BD, Broude EV, Dokmanovic M, et al. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Research. 1999;59(15):3761–3767. [PubMed] [Google Scholar]
- 58.Mirzayans R, Scott A, Cameron M, Murray D. Induction of accelerated senescence by γ radiation in human solid tumor-derived cell lines expressing wild-type TP53 . Radiation Research. 2005;163(1):53–62. doi: 10.1667/rr3280. [DOI] [PubMed] [Google Scholar]
- 59.Myöhänen SK, Baylin SB, Herman JG. Hypermethylation can selectively silence individual p16INK4A alleles in neoplasia. Cancer Research. 1998;58(4):591–593. [PubMed] [Google Scholar]
- 60.Zhang W, Zhu J, Bai J, et al. Comparison of the inhibitory effects of three transcriptional variants of CDKN2A in human lung cancer cell line A549. Journal of Experimental and Clinical Cancer Research. 2010;29(1, article 74) doi: 10.1186/1756-9966-29-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jones KR, Elmore LW, Jackson-Cook C, et al. p53-dependent accelerated senescence induced by ionizing radiation in breast tumour cells. International Journal of Radiation Biology. 2005;81(6):445–458. doi: 10.1080/09553000500168549. [DOI] [PubMed] [Google Scholar]
- 62.Wang M, Morsbach F, Sander D, et al. EGF receptor inhibition radiosensitizes NSCLC cells by inducing senescence in cells sustaining DNA double-strand breaks. Cancer Research. 2011;71(19):6261–6269. doi: 10.1158/0008-5472.CAN-11-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reed JA, Loganzo F, Shea CR, et al. Loss of expression of the p16/cyclin-dependent kinase inhibitor 2 tumor suppressor gene in melanocytic lesions correlates with invasive stage of tumor progression. Cancer Research. 1995;55(13):2713–2718. [PubMed] [Google Scholar]
- 64.Abd Elmageed ZY, Gaur RL, Williams M, et al. Characterization of coordinated immediate responses by p16INK4a and p53 pathways in UVB-irradiated human skin cells. The Journal of Investigative Dermatology. 2009;129(1):175–183. doi: 10.1038/jid.2008.208. [DOI] [PubMed] [Google Scholar]
- 65.Hollestelle A, Nagel JHA, Smid M, et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Research and Treatment. 2010;121(1):53–64. doi: 10.1007/s10549-009-0460-8. [DOI] [PubMed] [Google Scholar]
- 66.Galmarini CM, Falette N, Tabone E, et al. Inactivation of wild-type p53 by a dominant negative mutant renders MCF-7 cells resistant to tubulin-binding agent cytotoxicity. British Journal of Cancer. 2001;85(6):902–908. doi: 10.1054/bjoc.2001.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ostrakhovitch EA, Olsson PE, Von Hofsten J, Cherian MG. P53 mediated regulation of metallothionein transcription in breast cancer cells. Journal of Cellular Biochemistry. 2007;102(6):1571–1583. doi: 10.1002/jcb.21381. [DOI] [PubMed] [Google Scholar]
- 68.Robles SJ, Adami GR. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene. 1998;16(9):1113–1123. doi: 10.1038/sj.onc.1201862. [DOI] [PubMed] [Google Scholar]