

Abstract
Treacher Collins syndrome is a genetic disorder resulting in congenital craniofacial malformation. Patients typically present with downslanting palpebral fissures, lower eyelid colobomas, microtia, and malar and mandibular hypoplasia. This autosomal dominant disorder has a variable degree of phenotypic expression, and patients have no associated developmental delay or neurologic disease. Care for these patients requires a multidisciplinary team from birth through adulthood. Proper planning, counseling and surgical techniques are essential for optimizing patient outcomes. Here the authors review the features, genetics, and treatment of Treacher Collins syndrome.
Keywords: Treacher Collins syndrome, Franceschetti-Klein syndrome, mandibulofacial dysostosis, microtia, microsomia, mandibular distraction, malar hypoplasia, facial clefting
Treacher Collins syndrome (TCS) is a rare genetic disorder of facial dysmorphism that affects structures of the first and second pharyngeal arches. Patients have normal intelligence, but often face social challenges throughout life because of their physical appearance. Here we will discuss the clinical features, etiology, current scientific understanding, and treatment protocols for this complex craniofacial syndrome.
History
TCS was first described in 1889 as a congenital neonatal deformity with the colobomata of the lower eyelids by George Andreas Berry.1 In 1900, an ophthalmologist named Edward Treacher Collins presented two patients with these ocular and periorbital sequelae at a meeting in London and subsequently published one of earliest case reports describing his findings.2 In the 1940s, Adolphe Franceschetti of Switzerland further characterized the disorder and published extensive reviews with his colleague David Klein. They further characterized the facial features and coined the description, mandibulofacial dysostosis.3 Today, there are several eponyms for this syndrome as it is known in the United States and United Kingdom as Treacher Collins syndrome, in Europe as Franceschetti-Klein syndrome, and otherwise as mandibulofacial dysostosis.1
Embryology/Genetics
TCS is an autosomal dominant disorder with a high degree of penetrance but variable phenotypic expression. The clinical features are a result of a loss of function mutation of the TCOF1 gene on chromosome 5.4 There have been multiple exons identified within the gene and there are different splicing patterns resulting in several variants of the mutant gene. So far, over 120 mutations have been identified; however, combined analysis of the variants and clinical features has not demonstrated a clear relationship between genotype and phenotype. Spontaneous mutations can occur as well as the inherited form; however, there is no gender predilection, and mutations can be spliced, nonsense, or deletion variants.4 All of the mutations result in insertion of a premature termination codon.
The TCOF1 protein is named Treacle, which functions in the ribosome biogenesis pathway. Treacle is part of the rRNA preprocessing complex and methylation. It is thought that the loss of function mutation results in haploinsufficiency.5
Treacle has been implicated in craniofacial development in mouse studies as the maximum TCOF1 expression was found in the neuroepithelium prior to fusion and in the embryonic frontonasal process and branchial arches. Additional studies have shown that though TCOF1 mutations do not affect neural crest cell migration, there are decreased neural crest cells overall in heterozygote mouse studies.5
Clinical Features
The clinical characteristics of TCS are well described and distinctive. The majority of pathology affects structures arising from the first and second pharyngeal arches. Unlike some other cleft and craniofacial syndromes, TCS is generally a bilateral disease. However, despite the bilateral distribution, the skeletal and soft tissue structures are rarely symmetrical.6 These structures should be evaluated systematically and in concert with the appropriate consultants.
Ocular and Periorbital
TCS is associated with prominent ocular and periorbital findings (Fig. 1). Classically, these patients have skeletal dysmorphism of the orbits, with malar hypoplasia and ellipsoid shape. Relatively normal nasal and maxillary prominences may result in an enophthalmic appearance. Downslanting “antimongoloid” palpebral fissures are universally described.
Figure 1.

Characteristic findings of Treacher Collins syndrome include downward slanting palpebral fissures, lower eyelid colobomas, midface and zygomatic hypoplasia, microtia, and mandibular microretrognathia. Patients can be fitted with a removable bone-assisted hearing aid to help speech and language development prior to definitive implantation after ear reconstruction.
The soft tissues surrounding the eye are typically hypoplastic with thin lower lid skin, and attenuated or absent orbicularis oculi muscles and Meibomian glands. Lid notching, or colobomas, has been reported as well as periorbital dermoids. The lid margin can have significant laxity and absence of lashes, particularly in the medial third of the lower lid, which is a pathognomonic finding. Lacrimal duct atresia and absence of puncta are also reported.
The eyes themselves can be affected with vision loss (33%), strabismus (37%), congenital cataracts, and even occasional microphthalmia or anophthalmia.7 This is usually due to amblyopia from corneal scarring, strabismus, and significant refractive errors. Despite these abnormalities, vision is generally normal in TCS because the retina does not develop from the affected branchial arches and the majority of patients retain at least one eye with normal vision.
Auricular
The external ear is notably deformed in TCS patients. Patients usually present with bilateral microtia or anotia of varying severity. Any auricular remnants are usually malpositioned. This is accompanied by stenosis or atresia of the external auditory meatus. The tympanic membrane is usually misshapen as well. The middle ear may be deformed or absent entirely. The inner ear is usually morphologically normal; however, from a functional standpoint, the pathologic ossicular chain results in a conductive hearing loss.8 This can be a maximum conductive hearing loss of up to 60 dB; however, the degree of deficit can vary.
Zygoma and Malar Region
Midfacial structures are distinctive in TCS patients. Malar hypoplasia is a cardinal feature. Posnick et al described the deficient zygoma anthropometrically using linear measurements of axial computed tomography (CT) scans.9 They were able to demonstrate statistically smaller interzygomatic arch distance and zygomatic arch length in TCS patients. These findings confirmed the clinical impression of decreased midfacial width.
The TCS zygoma has been volumetrically evaluated demonstrating marked deficiency, but a spectrum of dysmorphology and intrapatient asymmetry (Wong, unpublished data, 2012). This analysis stratified the severity of zygomatic hypoplasia into a classification system (Fig. 2).
Figure 2.

The Treacher Collins syndrome zygoma can be variable in morphology and volume. Classification of this deformity is based on the presence of all (type I), some (type II), or none (type III) of the normal components and buttresses.
Maxilla and Mandible
The TCS maxillomandibular skeleton also has characteristic features. Overall, the facial profile in TCS is dramatically convex due to the pronounced retrognathia (Fig. 3). In addition to being retruded, the mandible is often malformed altogether. The mandibular angle can be significantly underdeveloped and may be totally absent. There can be high antegonial notching and decreased height of the lower third of the face.10 Cephalometric studies demonstrate that TCS patients have decreased sella-nasion-B point (SNB) angles and generally normal sella-nasion-A point (SNA). These features are further accentuated because of decreased posterior facial height and clockwise rotation of the occlusal plane.11,12 The most severe forms of TCS may have significantly deficient proximal mandible or even lacking the ramus/condyle unit altogether.
Figure 3.
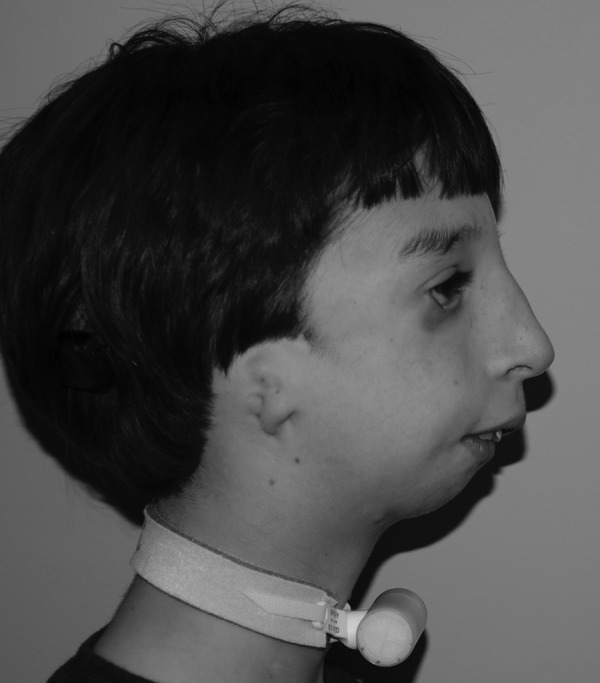
Treacher Collins syndrome patients have a convex facial profile with a “birdlike” appearance due to pronounced retrognathia and a relatively normal nasal projection.
Three-dimensional CT analysis has shown a significant decrement in mandibular volume and shape. In particular, volumetric studies have demonstrated that the mandibular hypoplasia is most severe proximally—the condyle as the most hypoplastic structure followed by the ramus then the body.6 This not only implies that growth from the condylar process is greatly affected, but also that the normal articulation at the temporomandibular joint (TMJ) and zygomatic relationship is severely affected (Fig. 4). The deficient mandible results in a high incidence of TMJ dysfunction and ankylosis in TCS patients.
Figure 4.
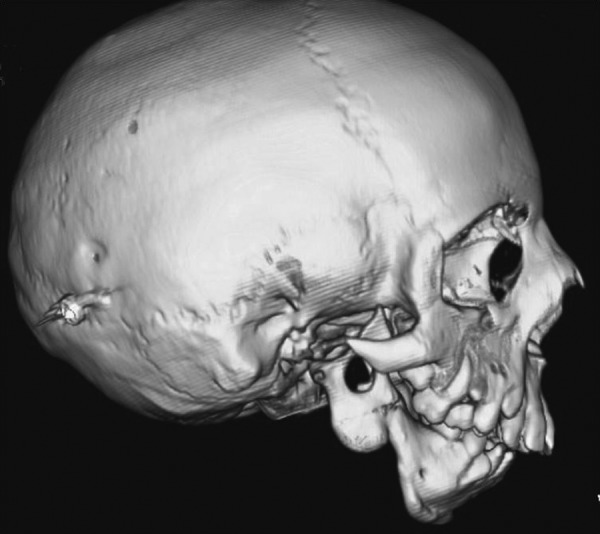
The mandible and temporomandibular joint can be severely affected in Treacher Collins syndrome. Condylar malformation can be the most severely affected portion of the mandible and the normal ramus-angle-body relationship can be totally absent.
Other Findings
Additional abnormal structures that are occasionally found in TCS include choanal atresia, complete or submucous cleft palate, absent parotid glands, cervical spine malformation, cryptorchidism, extremity malformation, renal anomalies, and congenital heart disease; however, these are not hallmark features and are not always consistent findings in affected patients. The nose is often described as “beaked”; however, anthropometric studies of TCS patients' noses show relatively normal nose measurements and the hypoplasia of the surrounding tissue is the main contributor to the abnormal facial balance.13
Screening and Diagnosis
TCS can be detected using prenatal screening ultrasound.14,15 Usually, it is difficult to get an adequate view of facial structures until after 30 weeks. In addition, traditional two-dimentional imaging is limited and may not be sufficient to assess the fetal profile. Three-dimensional sonographic imaging has been shown to detect these subtle features including downslanting palpebral fissures, micrognathia, and low-set ears/microtia.16 Polyhydramnios is seen as well. Once suspected, other testing can be done to confirm the diagnosis. Amniocentesis may be performed to identify the mutation and rule out other facial dysostoses such as Goldenhar or Nager syndromes, which can have similar appearance on ultrasound.17,18 High-risk families with or without ultrasound findings should be referred for genetic counseling.
Neonatal Management
Early priorities should focus on airway management, feeding, and growth. Patients may require continuous apnea or oxygen-saturation monitoring. A formal sleep study can document the severity and type of apnea. Targeted assessment is also required to determine the cause of airway obstruction as multiple sites may be involved such as the nasopharynx (choanal atresia), oropharynx/hypopharynx (glossoptosis/micrognathia), or laryngopharynx (laryngomalacia). Intervention is based on the severity and site. In mild cases without significant anatomic constriction, conservative management (suctioning or nasopharyngeal trumpet) and positioning (side/prone) may be sufficient. If the obstruction is more severe, an early tongue–lip adhesion or mandibular advancement may be required. In the most severe cases, intubation or tracheostomy may be necessary.
Other urgent considerations include corneal scarring, feeding, and growth. Depending on the degree of eye exposure, early tarsorrhaphy may be necessary to prevent corneal scarring, ulceration, and blindness. Parenteral feeding should commence as soon as possible to aid in growth and development. Parents should be counseled on feeding techniques and prevention of aspiration. In cases with overt or submucous cleft palate, specific feeders may also be needed.
Once the child is stable, feeding, breathing without significant obstruction, and has adequate corneal protection, the remaining dysmorphology can be addressed using a staged, coordinated approach. Here we will discuss surgical considerations by anatomic structure and in order of repair.
Surgical Treatment
Preoperative Planning
Preoperative planning and evaluation should begin as early as possible. A multidisciplinary craniofacial team approach is critical to coordinate oral, ocular, dental, pediatric, and craniofacial care. Patients will require preoperative imaging usually as CT scans for planning, measurement, and implant fabrication. Newer computer-aided design/computer-aided manufacturing (CAD/CAM) technology allows preoperative simulation—either virtually or by model—to assess developing tooth buds, design osteotomies, hardware placement, and tongue position. TCS patients typically will need multiple surgical procedures and staged treatment may continue throughout the patient's childhood and adolescence.
Mandible
The characteristic microretrognathic TCS mandible is a priority for surgical evaluation because it can be markedly hypoplastic and can lead to glossoptosis and upper airway obstruction. In addition, the pharynx has been noted to be abnormally hypoplastic and narrow.19 Historically, many of these patients required tracheostomy that was maintained until they were grown enough for conventional mandibular advancement. However, newer techniques in distraction osteogenesis enable neonatal mandible advancement to relieve airway obstruction.20,21 Possible downsides to neonatal distraction include difficulties with hardware fixation due to poor bone quality and stock. Also, absence of a functional TMJ and/or an aplastic condyle may preclude distraction and tracheostomy may still be required.
If the airway is stable, mandibular distraction can be delayed until early childhood. Mandibular distraction planning can be challenging because the TCS mandible may be uniplanar without a true mandibular angle. To correct this phenotype, two vectors of distraction must be applied—one to recreate mandibular height (ramus) and one to recreate mandibular length (body).22 Recent strategies to simplify this process include the use of curvilinear distraction devices (Fig. 5).23,24 The endpoint of distraction is to ideally bring the mandible to an overcorrected, class III position to help overcome its deficient growth potential, and to obviate probable relapse.25
Figure 5.

Distraction osteogenesis of the Treacher Collins syndrome mandible usually necessitates two vectors of distraction: Inferior bony length and anterior projection are needed. Newer curvilinear distraction devices aim to correct this multivector deficiency.
Distraction may be delayed until later childhood or until permanent dentition erupts26; however, many centers advocate early distraction. Despite efforts to correct the skeletal abnormalities in infancy, there is a possibility of relapse toward the original deformity. This can lead to latent airway obstruction as the patient grows but the mandible remains retruded. The growth potential of these hypoplastic bones is thought to be limited as well; therefore, repeat distraction is anticipated. The primary goals of this sequence is to maintain airway and engineer more bone stock to make future sagittal split/mandibular osteotomies less complicated, and minimize the potential need for bone graft and the degree of definitive orthognathic advancement. Although distraction opponents may argue that orthognathic surgery should be performed initially, most surgeons express concern about further hindering any residual growth potential and prefer to delay two-jaw correction until the mature skeleton is established.
Oral Surgery/Dentition
Malocclusion is present in nearly all TCS patients. The more severe pathology often shows a steep occlusal plane, an anterior open bite, and aberrant dentition. The shortened mandible in combination with normal anterior maxillary height results in a bird-like appearance with a prominent nose and midface with a retruded lower third of the face.
Although some distraction advocates argue that lowering the mandibular plane will allow descent and growth of the posterior maxilla, usually this compensation, if any, is not enough to close a residual posterior open bite. Once skeletal maturity is reached, bimaxillary surgery may be performed. LeFort I with bilateral sagittal split osteotomy is typically performed in late adolescence to level the occlusal plane. Different osteotomy variations have been described, occasionally utilizing intervening or onlay bone grafts; however, regardless of technique, the goal of orthognathic repair is to restore a class I occlusion, level occlusal plane, and restore normal facial projection.27
Dentition can also be abnormal with missing, malrotated, or malpositioned teeth.28 Oral hygiene can be problematic and high plaque buildup has been shown to be significant in TCS patients.29 Orthodontics can help establish improved bite and intercuspation. However, extraction and implants may be necessary in some cases as repeated surgical intervention combined with poor bone stock subject the tooth roots to potential devascularization and injury.30
Palatoplasty
Cleft palate is a commonly reported finding in TCS and is present in approximately one-third of cases. Once the airway is secure and stable, palatoplasty can be planned. In addition to speech, palate repair is important for feeding, growth, and development. Ideally, palatoplasty is timed as early as the first year of life. However, unlike other cleft palate patients, TCS cleft palates may be more challenging because of a high arch, smaller oropharynx, limited interincisal opening, and thin, atrophic soft tissues. Bresnick et al noted an increased risk for fistula formation in TCS patients compared with other syndromic cleft palate patients (50% vs 8.7%) after palate repair.31 They propose that the vascularity of the mucoperiosteum in TCS is limited; therefore, minimal flap undermining and elevation should be done when performing palatoplasty in TCS.
Ear Reconstruction
Bilateral microtia is one of the most prominent features of TCS and is present to some degree in 85% of patients.32 This deformity can be absent or mild, or the patient can have complete anotia without any auricular remnant. Because of the ear's prominent location, predisposition to trauma, delicate skin envelope, and complex anatomy, reconstructive techniques have become more intricate.33 As implant technology has improved, prosthetic reconstruction has become an area of controversy. Although a simpler to reconstruct and more precisely machined anatomic reproduction can be made on a prosthesis, opponents cite the high rate of implant failure, infection, and extrusion as reasons to avoid foreign bodies. Although historically silastic implants have been used, modern materials have focused on porous polyethylene (MedPor) products, which are advertised for their inertness and resistance to extrusion.
Still the majority of plastic surgeons argue that the best reconstruction is autologous tissue. Modern techniques derived from Tanzer, Brent, Nagata, and Firmin have greatly improved reconstructive outcomes.34,35,36,37 All of these techniques require a staged approach. Ear reconstruction typically begins as early as age 535; however, many authors report improved results when reconstruction is delayed until age 10.33,35,36,37 In the first stage, the costal cartilage is harvested and molded into an auricular framework. A template is usually created if there is a normal ear or from a family member. The costal cartilage is harvested from the sixth to ninth ribs, leaving the visceral perichondrium intact. Authors vary whether they use the ipsilateral or contralateral cartilage, but agree that minimizing the donor site morbidity is important to reduce chest wall deformity and growth limitations. The framework is then carved from a base block with elevations for helical rim, superior and inferior crura, tragus, and antitragus. Some surgeons support the framework with additional base projection, whereas others bank this cartilage for elevation at a later stage (Fig. 6).
Figure 6.

Auricular reconstruction is performed with autogenous costal cartilage grafts, fabrication of a framework, and staged implant and elevation.
Care is taken to preserve any auricular remnants, which are usually atrophic and malpositioned lobular segments. The skin envelope is elevated and the remnant is rotated/debulked33 while the framework is inset. Care should be taken when earlier TMJ or mandibular surgery is performed so as to protect and not disrupt the periauricular skin that is ultimately incorporated into microtia repair.
The second stage is usually done 6 months after the framework is well healed. The banked rib cartilage is harvested and an incision is made posteriorly along the skin pocket. The cartilage is fabricated into a tapered buttress, recreating the convexity of the conchal bowl, supporting the framework against the skull and creating the postauricular sulcus. This is covered with a fascial flap and split thickness skin graft. In the Brent technique, the second stage is only focused on lobule transposition, conchal excavation and occasionally tragal construction.35 Brent believes this leaves a more natural lobule allowing the patient to wear earrings. However, it delays the framework elevation necessitating a third stage.
Bone-Assisted Hearing Aid
As mentioned, TCS patients have aberrant internal ear structures and the ossicles do not function properly. This results in a conductive hearing loss in the majority of patients. Most centers advocate for definitive ear reconstruction to be complete before bone-assisted hearing aid (BAHA) implantation; however, delayed hearing can cause significant language and social delays in children. Therefore, many patients wear removable BAHA headbands (Softband) that allow early auditory rehabilitation (Fig. 1).
Once definitive BAHA placement is planned, it is usually positioned 4 to 5 cm superoposterior to the external auditory meatus.38 The abutment hardware is drilled directly into the hardware, but care must be taken as there is a high degree of irregularity and weak bone stock in the TCS calvarium. Osseointegration is essential to device function.
Eyelid Correction
Other than neonatal tarsorrhaphy to protect corneal exposure, most periorbital correction is delayed. Some surgeons prefer to stage coloboma correction after zygoma and orbit reconstruction because the extent of lower lid skin deficit can be best assessed after skeletal correction. Depending on the dimensions of the deformity, orbicularis transposition, skin grafting, Z-plasty, and/or canthopexy can be performed.39
Midface/Soft Tissue Resuspension
Malar reconstruction is often undertaken to correct the absent or hypoplastic zygomas. Historically, bone grafting to the zygomas was delayed until late childhood. Prior techniques involved split rib grafts to augment the inferior orbital rim and malar eminences. This was complicated by a high rate of bony resorption and regression.39 Many centers now use nonvascularized split calvarial bone grafts, which seem to persist with less degree of resorption.40 Alloplastic implants have also been used such as MedPor or customized CAD/CAM malar implants; however, as with all prostheses, concern for infection, migration, and extrusion exists. Tissue engineering and zygomatic cell in growth constructs may be an additional option to reconstruct the craniofacial skeleton, including the zygoma.41,42
Despite skeletal correction, TCS patients have atrophic skin, fat, and facial muscles so many patients have significant residual deformity. Over the past several decades, vascularized soft tissue reconstruction has been used to correct this problem. Microvascular free flaps have been described by Saadeh et al and have demonstrated long-term stability.43,44
More recently, alternative methods for soft tissue augmentation have been proposed.45 Fat grafting has become a popular technique for its technical ease and low donor site morbidity. Furthermore, adipose-derived stem cells have been hypothesized to help establish graft survival, overlying tissue revascularization and surrounding tissue regeneration. Although the physiology of fat graft survival has yet to be clearly elucidated, the technique has grown in popularity and is a useful biologic filler.
Treatment Timeline
A treatment timeline is presented in Fig. 7 for surgical considerations and staging throughout the patient's childhood.
Figure 7.

A treatment timeline is presented for surgical considerations and staging throughout the patient's childhood.
Summary
Treacher Collins syndrome is a complex congenital disorder with a variable degree of craniofacial deformity. Most patients do not have neurodevelopmental impairment; thus, reconstructive treatment is important for their social and psychological development. It is important to anticipate staged treatment throughout childhood and beginning in early neonatal life. Airway assessment is the priority, followed by oropharyngeal repair, and midface reconstruction. Auricular fabrication and BAHA placement should be done in late childhood. Lastly, orthodontic and dental correction should be undertaken once definitive skeletal repair is finalized. This complex sequence of treatment should be undertaken at dedicated craniofacial centers for the best chance of a successful outcome.
References
- 1.Scully C, Langdon J, Evans J. Marathon of eponyms: 20 Treacher Collins syndrome. Oral Dis. 2011;17(6):619–620. doi: 10.1111/j.1601-0825.2009.01552.x. [DOI] [PubMed] [Google Scholar]
- 2.Collins E T. Cases with symmetrical congenital notches in the outer part of each lid and defective development of the malar bones. Trans Ophthalmol Soc U K. 1900;20:190–192. [Google Scholar]
- 3.Franceschetti A, Klein D. The mandibulofacial dysostosis; a new hereditary syndrome. Acta Ophthalmol (Copenh) 1949;27(2):143–224. [PubMed] [Google Scholar]
- 4.Dixon M J. Treacher Collins syndrome. J Med Genet. 1995;32(10):806–808. doi: 10.1136/jmg.32.10.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon J, Trainor P, Dixon M J. Treacher Collins syndrome. Orthod Craniofac Res. 2007;10(2):88–95. doi: 10.1111/j.1601-6343.2007.00388.x. [DOI] [PubMed] [Google Scholar]
- 6.Terner J S, Travieso R, Chang C, Bartlett S P, Steinbacher D M. An analysis of mandibular volume in Treacher Collins syndrome. Plast Reconstr Surg. 2012;129(4):751e–753e. doi: 10.1097/PRS.0b013e318245e903. [DOI] [PubMed] [Google Scholar]
- 7.Hertle R W, Ziylan S, Katowitz J A. Ophthalmic features and visual prognosis in the Treacher-Collins syndrome. Br J Ophthalmol. 1993;77(10):642–645. doi: 10.1136/bjo.77.10.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fernandez A O, Ronis M L. The Treacher-Collins Syndrome. Arch Otolaryngol. 1964;80:505–520. doi: 10.1001/archotol.1964.00750040519006. [DOI] [PubMed] [Google Scholar]
- 9.Posnick J C, al-Qattan M M, Moffat S M, Armstrong D. Cranio-orbito-zygomatic measurements from standard CT scans in unoperated Treacher Collins syndrome patients: comparison with normal controls. Cleft Palate Craniofac J. 1995;32(1):20–24. doi: 10.1597/1545-1569_1995_032_0020_cmfscs_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 10.Chong D K, Murray D J, Britto J A, Tompson B, Forrest C R, Phillips J H. A cephalometric analysis of maxillary and mandibular parameters in Treacher Collins syndrome. Plast Reconstr Surg. 2008;121(3):77e–84e. doi: 10.1097/01.prs.0000299379.64906.2e. [DOI] [PubMed] [Google Scholar]
- 11.Steinbacher D M, Bartlett S P. Relation of the mandibular body and ramus in Treacher Collins syndrome. J Craniofac Surg. 2011;22(1):302–305. doi: 10.1097/SCS.0b013e3181f7df87. [DOI] [PubMed] [Google Scholar]
- 12.Posnick J C, Tiwana P S, Costello B J. Treacher Collins syndrome: comprehensive evaluation and treatment. Oral Maxillofac Surg Clin North Am. 2004;16(4):503–523. doi: 10.1016/j.coms.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 13.Farkas L G, Posnick J C. Detailed morphometry of the nose in patients with Treacher Collins syndrome. Ann Plast Surg. 1989;22(3):211–219. doi: 10.1097/00000637-198903000-00008. [DOI] [PubMed] [Google Scholar]
- 14.Cohen J, Ghezzi F, Gonçalves L, Fuentes J D, Paulyson K J, Sherer D M. Prenatal sonographic diagnosis of Treacher Collins syndrome: a case and review of the literature. Am J Perinatol. 1995;12(6):416–419. doi: 10.1055/s-2007-994511. [DOI] [PubMed] [Google Scholar]
- 15.Ochi H, Matsubara K, Ito M, Kusanagi Y. Prenatal sonographic diagnosis of Treacher Collins syndrome. Obstet Gynecol. 1998;91(5 Pt 2):862. doi: 10.1016/s0029-7844(97)00676-5. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka Y, Kanenishi K, Tanaka H, Yanagihara T, Hata T. Antenatal three-dimensional sonographic features of Treacher Collins syndrome. Ultrasound Obstet Gynecol. 2002;19(4):414–415. doi: 10.1046/j.1469-0705.2002.00533_2.x. [DOI] [PubMed] [Google Scholar]
- 17.Hsu T Y, Hsu J J, Chang S Y, Chang M S. Prenatal three-dimensional sonographic images associated with Treacher Collins syndrome. Ultrasound Obstet Gynecol. 2002;19(4):413–422. doi: 10.1046/j.1469-0705.2002.00533_1.x. [DOI] [PubMed] [Google Scholar]
- 18.Ellis P E Dawson M Dixon M J Mutation testing in Treacher Collins syndrome J Orthod 2002294293–297., discussion 278 [DOI] [PubMed] [Google Scholar]
- 19.Shprintzen R J, Croft C, Berkman M D, Rakoff S J. Pharyngeal hypoplasia in Treacher Collins syndrome. Arch Otolaryngol. 1979;105(3):127–131. doi: 10.1001/archotol.1979.00790150017005. [DOI] [PubMed] [Google Scholar]
- 20.Miloro M. Mandibular distraction osteogenesis for pediatric airway management. J Oral Maxillofac Surg. 2010;68(7):1512–1523. doi: 10.1016/j.joms.2009.09.099. [DOI] [PubMed] [Google Scholar]
- 21.Steinbacher D M, Kaban L B, Troulis M J. Mandibular advancement by distraction osteogenesis for tracheostomy-dependent children with severe micrognathia. J Oral Maxillofac Surg. 2005;63(8):1072–1079. doi: 10.1016/j.joms.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 22.Singh D J, Glick P H, Bartlett S P. Mandibular deformities: single-vector distraction techniques for a multivector problem. J Craniofac Surg. 2009;20(5):1468–1472. doi: 10.1097/SCS.0b013e3181b09ab2. [DOI] [PubMed] [Google Scholar]
- 23.Miller J J, Schendel S A. Invited discussion: surgical treatment of Treacher Collins syndrome. Ann Plast Surg. 2006;56(5):555–556. doi: 10.1097/01.sap.0000205823.77358.47. [DOI] [PubMed] [Google Scholar]
- 24.Kaban L B, Seldin E B, Kikinis R, Yeshwant K, Padwa B L, Troulis M J. Clinical application of curvilinear distraction osteogenesis for correction of mandibular deformities. J Oral Maxillofac Surg. 2009;67(5):996–1008. doi: 10.1016/j.joms.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shetye P R, Grayson B H, Mackool R J, McCarthy J G. Long-term stability and growth following unilateral mandibular distraction in growing children with craniofacial microsomia. Plast Reconstr Surg. 2006;118(4):985–995. doi: 10.1097/01.prs.0000232830.12603.eb. [DOI] [PubMed] [Google Scholar]
- 26.Kobus K, Wójcicki P. Surgical treatment of Treacher Collins syndrome. Ann Plast Surg. 2006;56(5):549–554. doi: 10.1097/01.sap.0000196580.56569.d2. [DOI] [PubMed] [Google Scholar]
- 27.Obwegeser H L. Variations of a standard approach for correction of the bird-face deformity. J Craniomaxillofac Surg. 1988;16(6):247–265. doi: 10.1016/s1010-5182(88)80059-3. [DOI] [PubMed] [Google Scholar]
- 28.da Silva Dalben G, Costa B, Gomide M R. Prevalence of dental anomalies, ectopic eruption and associated oral malformations in subjects with Treacher Collins syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101(5):588–592. doi: 10.1016/j.tripleo.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 29.da Silva Dalben G Teixeira das Neves L Ribeiro Gomide M Oral health status of children with Treacher Collins syndrome Spec Care Dentist 200626271–75., quiz 85–87 [DOI] [PubMed] [Google Scholar]
- 30.da Silva Freitas R, Tolazzi A R, Alonso N, Cruz G A, Busato L. Evaluation of molar teeth and buds in patients submitted to mandible distraction: long-term results. Plast Reconstr Surg. 2008;121(4):1335–1342. doi: 10.1097/01.prs.0000304444.43168.fa. [DOI] [PubMed] [Google Scholar]
- 31.Bresnick S, Walker J, Clarke-Sheehan N, Reinisch J. Increased fistula risk following palatoplasty in Treacher Collins syndrome. Cleft Palate Craniofac J. 2003;40(3):280–283. doi: 10.1597/1545-1569_2003_040_0280_ifrfpi_2.0.co_2. [DOI] [PubMed] [Google Scholar]
- 32.Jahrsdoerfer R A, Jacobson J T. Treacher Collins syndrome: otologic and auditory management. J Am Acad Audiol. 1995;6(1):93–102. [PubMed] [Google Scholar]
- 33.Bauer B S. Reconstruction of microtia. Plast Reconstr Surg. 2009;124(1, Suppl):14e–26e. doi: 10.1097/PRS.0b013e3181aa0e79. [DOI] [PubMed] [Google Scholar]
- 34.Tanzer R C. Total reconstruction of the auricle. The evolution of a plan of treatment. Plast Reconstr Surg. 1971;47(6):523–533. doi: 10.1097/00006534-197106000-00001. [DOI] [PubMed] [Google Scholar]
- 35.Brent B Microtia repair with rib cartilage grafts: a review of personal experience with 1000 cases Clin Plast Surg 2002292257–271., vii vii [DOI] [PubMed] [Google Scholar]
- 36.Firmin F. Ear reconstruction in cases of typical microtia. Personal experience based on 352 microtic ear corrections. Scand J Plast Reconstr Surg Hand Surg. 1998;32(1):35–47. doi: 10.1080/02844319850158930. [DOI] [PubMed] [Google Scholar]
- 37.Nagata S Total auricular reconstruction with a three-dimensional costal cartilage framework Ann Chir Plast Esthet 1995404371–399., discussion 400–403 [PubMed] [Google Scholar]
- 38.Marsella P, Scorpecci A, Pacifico C, Tieri L. Bone-anchored hearing aid (Baha) in patients with Treacher Collins syndrome: tips and pitfalls. Int J Pediatr Otorhinolaryngol. 2011;75(10):1308–1312. doi: 10.1016/j.ijporl.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 39.Goodrich J T, Hall C D. New York, NY: Thieme Medical Publishers; 1995. Craniofacial Anomalies: Growth and Development from a Surgical Perspective; p. 213. [Google Scholar]
- 40.Posnick J C, Ruiz R L. Treacher Collins syndrome: current evaluation, treatment, and future directions. Cleft Palate Craniofac J. 2000;37(5):434. doi: 10.1597/1545-1569(2000)037<0434:TCSCET>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 41.Taylor J A. Bilateral orbitozygomatic reconstruction with tissue-engineered bone. J Craniofac Surg. 2010;21(5):1612–1614. doi: 10.1097/SCS.0b013e3181edc829. [DOI] [PubMed] [Google Scholar]
- 42.Abukawa H, Shin M, Williams W B, Vacanti J P, Kaban L B, Troulis M J. Reconstruction of mandibular defects with autologous tissue-engineered bone. J Oral Maxillofac Surg. 2004;62(5):601–606. doi: 10.1016/j.joms.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 43.Jones N F. The contribution of microsurgical reconstruction to craniofacial surgery. World J Surg. 1989;13(4):454–464. doi: 10.1007/BF01660757. [DOI] [PubMed] [Google Scholar]
- 44.Saadeh P B, Chang C C, Warren S M, Reavey P, McCarthy J G, Siebert J W. Microsurgical correction of facial contour deformities in patients with craniofacial malformations: a 15-year experience. Plast Reconstr Surg. 2008;121(6):368e–378e. doi: 10.1097/PRS.0b013e3181707194. [DOI] [PubMed] [Google Scholar]
- 45.Coleman S R. Facial augmentation with structural fat grafting. Clin Plast Surg. 2006;33(4):567–577. doi: 10.1016/j.cps.2006.09.002. [DOI] [PubMed] [Google Scholar]