

Abstract
Voltage-gated sodium channels initiate action potentials in nerve, muscle and other excitable cells. The sodium current that initiates the nerve action potential was discovered by Hodgkin and Huxley using the voltage clamp technique in their landmark series of papers in The Journal of Physiology in 1952. They described sodium selectivity, voltage-dependent activation and fast inactivation, and they developed a quantitative model for action potential generation that has endured for many decades. This article gives an overview of the legacy that has evolved from their work, including development of conceptual models of sodium channel function, discovery of the sodium channel protein, analysis of its structure and function, determination of its structure at high resolution, definition of the mechanism and structural basis for drug block, and exploration of the role of the sodium channel as a target for disease mutations. Structural models for sodium selectivity and conductance, voltage-dependent activation, fast inactivation and drug block are discussed. A perspective for the future envisions new advances in understanding the structural basis for sodium channel function, the role of sodium channels in disease and the opportunity for discovery of novel therapeutics.

William A. Catterall received a BA in Chemistry from Brown University in 1968, a PhD in Physiological Chemistry from Johns Hopkins in 1972, and postdoctoral training in neurobiology and molecular pharmacology as a Muscular Dystrophy Association Fellow with Dr Marshall Nirenberg at the National Institutes of Health from 1972 to 1974. Following three years as a staff scientist at NIH, he joined the University of Washington in 1977 as Associate Professor of Pharmacology, became Professor in 1981 and Chair in 1984. He discovered the voltage-gated sodium and calcium channel proteins, which initiate electrical and chemical signalling in excitable cells, and his work has contributed much to understanding their structure, function, regulation and molecular pharmacology. He is a member of several science academies, including the National Academy of Sciences and the Institute of Medicine (in the USA), and is a Foreign Member of The Royal Society. He has received numerous awards, including the Gairdner International Award of Canada in 2010.
Introduction
The modern era of research on electrical signalling in nerve, muscle and other excitable cells began in 1952 with the publication of a series of four seminal papers by Hodgkin and Huxley on analysis of the action potential of the squid giant axon using the voltage clamp procedure (Hodgkin & Huxley, 1952a,b,c,d). Their work showed that electrical signals in nerves are initiated by voltage-dependent activation of sodium current that carries Na+ inward. The sodium current then inactivates within 1–2 ms, and electrical signalling is terminated by activation of the voltage-gated potassium current, which carries K+ outward and re-establishes the original balance of electrical charges across the membrane. Their research revealed the voltage-gated sodium current for the first time and began studies of this key electrical signalling mechanism. This article gives an overview of the legacy of Hodgkin and Huxley as exemplified in current studies of the structure, function and pathophysiology of sodium channels.
Following Hodgkin and Huxley, Hille and Armstrong introduced the idea that the sodium and potassium currents are conducted by specific ion channels in the 1960s, and many investigators used the voltage clamp technique to further define the functional properties of sodium channels and develop conceptual models for their function through the 1960s and 1970s. Hille analysed the ion selectivity, saturation and block of sodium permeation and developed a detailed model of the ion selectivity filter of the sodium channel and its function in sodium selectivity (Hille, 1971, 1972, 1975a). The four-barrier, three-site model envisaged partial dehydration of Na+ through interaction with a high-field-strength site containing a carboxyl side chain at the extracellular end of the pore followed by rehydration in the lumen of the pore and escape into the intracellular milieu. He also established that local anaesthetics and related drugs that act on sodium channels bind to a receptor site in the pore of the channel, which can be accessed either through the open activation gate at the intracellular end of the pore or, for small hydrophobic drugs, through a membrane access pathway (Hille, 1977). Armstrong mapped the shape of the intracellular mouth of the pore using substituted alkyl ammonium potassium channel blockers added inside the cell (Armstrong, 1971). Armstrong and Bezanilla used high-resolution electrophysiological recording methods to detect the transmembrane movement of the gating charges (Armstrong & Bezanilla, 1973), the ‘electrically charged particles’ associated with sodium channels that respond to changes in electrical field and drive the rapid activation of the channels as predicted by Hodgkin & Huxley (1952d). Armstrong also made key insights into the process of fast sodium channel inactivation, showing that it is mediated by protein components on the intracellular surface of the sodium channel that were hypothesized to fold into the pore and block it during inactivation (Armstrong et al. 1973). Together, the studies of Hille, Armstrong and others in the 1970s established enduring conceptual models of sodium channel function (reviewed in Hille, 2001), but there was no information on the actual structure of ion channel proteins at that time.
Discovery of the sodium channel protein
Through the 1970s, work in several laboratories demonstrated that neurotoxins act on multiple receptor sites to modify the ion conductance and voltage-dependent gating of sodium channels (reviewed in Catterall, 1980). In 1980, scorpion toxins were used to identify the protein subunits of the sodium channel by photoaffinity labelling (Beneski & Catterall, 1980), revealing large α subunits of 260 kDa and smaller β subunits of 30–40 kDa (Fig. 1A). After solubilization and purification, brain sodium channels were found to contain an α subunit with a non-covalently associated β1 subunit and a disulfide-linked β2 subunit (Fig. 1A and B; Hartshorne & Catterall, 1981, 1984; Hartshorne et al. 1982), and this purified complex was sufficient to reconstitute voltage-gated sodium channel function with the correct pharmacology, single channel conductance and voltage sensitivity after insertion into phospholipid vesicles and bilayers (Fig. 1A; Talvenheimo et al. 1982; Tamkun et al. 1984; Hartshorne et al. 1985). Sodium channel α subunits were also purified from eel electroplax (Agnew et al. 1980), and a complex of α and β subunits was purified from skeletal muscle (Barchi, 1983). Together, these studies provided the first identification and functional reconstitution of a voltage-gated ion channel protein and established the principle that these ion channels are composed of primary pore-forming subunits in association with auxiliary subunits.
Figure 1. Subunit structure of voltage-gated sodium channels.
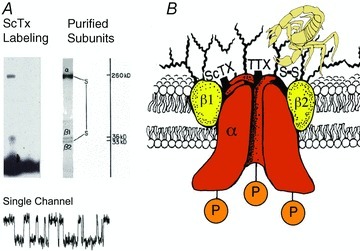
A, SDS polyacrylamide gel electrophoresis patterns illustrating the α and β subunits of the brain sodium channels. Left, α and β subunits covalently labelled with 125I-labelled scorpion toxin (Beneski & Catterall, 1980). Lane 1, specific labelling; lane 2, non-specific labelling. Right, sodium channel purified from rat brain showing the α, β1 and β2 subunits and their molecular weights (Hartshorne et al. 1982). As illustrated, the α and β2 subunits are linked by a disulfide bond. Tetrodotoxin and scorpion toxins bind to the α subunits of sodium channels as indicated and were used a molecular tags to identify and purify the sodium channel protein (Beneski & Catterall, 1980; Hartshorne et al. 1982; Hartshorne & Catterall, 1984). Inset, single channel currents conducted by a single purified sodium channel incorporated into a planar bilayer (Hartshorne et al. 1985). B, drawing of the subunit structure of the brain sodium channel based on biochemical data.
Primary structures of sodium channel subunits
Cloning and sequencing cDNA encoding the α subunits of sodium channels established their primary structures and showed that mRNA encoding the α subunit is sufficient for expression of functional sodium channels (Noda et al. 1984, 1986; Goldin et al. 1986). Sodium channel α subunits are composed of approximately 2000 amino acid residues organized in four homologous domains, which each contains six transmembrane segments (Fig. 2). Determination of the primary structure of the sodium channel α subunit was a major technical achievement, as it was the largest protein whose amino acid sequence had been determined by cDNA cloning and sequencing at that time. Later biochemical analyses and cDNA cloning showed that sodium channel β subunits are composed of an N-terminal extracellular immunoglobulin-like fold, a single transmembrane segment, and a short intracellular segment (Fig. 2; Isom et al. 1992, 1995). These subunits are thought to form heterodimeric and heterotrimeric complexes composed of a single α subunit and one or two β subunits in excitable cell membranes, and co-expression of β subunits modulates the kinetics and voltage dependence of sodium channel activation and inactivation.
Figure 2. The primary structures of the subunits of the voltage-gated sodium channels.
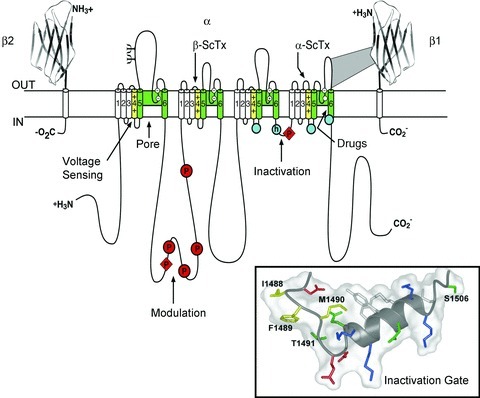
Cylinders represent probable α-helical segments. Bold lines represent the polypeptide chains of each subunit with length approximately proportional to the number of amino acid residues in the brain sodium channel subtypes. The extracellular domains of the β1 and β2 subunits are shown as immunoglobulin-like folds. Y, sites of probable N-linked glycosylation; P in red circles, sites of demonstrated protein phosphorylation by PKA (circles) and PKC (diamonds); green, pore-lining segments; white circles, the outer (EEEE) and inner (DEKA) rings of amino residues that form the ion selectivity filter and the tetrodotoxin binding site; yellow, S4 voltage sensors; h in blue circle, inactivation particle in the inactivation gate loop; blue circles, sites implicated in forming the inactivation gate receptor. Sites of binding of α- and β-scorpion toxins and a site of interaction between α and β1 subunits are also shown. Tetrodotoxin is a specific blocker of the pore of sodium channels (Hille, 1975b), whereas the α- and β-scorpion toxins block fast inactivation and enhance activation, respectively, and thereby generate persistent sodium current that causes depolarization block of nerve conduction (Catterall et al. 2007). Tetrodotoxin has been used as a tool to probe the pore of the sodium channel, whereas the scorpion toxins have been valuable as probes of voltage sensor function. Inset, structure of the inactivation gate in solution determined by NMR (Rohl et al. 1999).
Diversity of sodium channel subunits
Sodium channel α subunits are encoded by 10 genes, which are expressed in different excitable tissues (Table 1; Goldin, 2001). NaV1.1, 1.2, 1.3 and 1.6 are the primary sodium channels in the central nervous system. NaV1.7, 1.8 and 1.9 are the primary sodium channels in the peripheral nervous system. NaV1.4 is the primary sodium channel in skeletal muscle, whereas NaV1.5 is primary in heart. Most of these sodium channels also have significant levels of expression outside of their primary tissues. The 10th sodium channel protein is not voltage-gated and is involved in salt sensing (Watanabe et al. 2000). Two additional sodium channel β subunits have been identified by genomic analyses and cDNA cloning to give a small family of four NaVβ subunits in total (Morgan et al. 2000; Yu et al. 2003). β1 and β3 are associated non-covalently with α subunits and resemble each other most closely in amino acid sequence, whereas β2 and β4 form disulfide bonds with α subunits and also resemble each other closely. The structure of Navβ subunits resembles the family of cell adhesion molecules (Isom et al. 1995), and increasing evidence supports their role in localization and immobilization of sodium channels in specific locations in excitable cells (Brackenbury & Isom, 2011). NaVβ subunits are promiscuous in their associations with α subunits, and no evidence for strict specificity has emerged to allow prediction of functional associations in specific tissues or cell types.
Table 1.
Mammalian sodium channel α subunits
| Type | Gene symbol | Chromosomal location | Primary tissues |
|---|---|---|---|
| Nav1.1 | SCN1A | Mouse 2 Human 2q24 | CNS neurons |
| Nav1.2 | SCN2A | Mouse 2 Human 2q23–24 | CNS neurons |
| Nav1.3 | SCN3A | Mouse 2 Human 2q24 | CNS neurons |
| Nav1.4 | SCN4A | Mouse 11 Human 17q23–25 | SkM |
| Nav1.5 | SCN5A | Mouse 9 Human 3p21 | Uninnervated SkM, heart |
| Nav1.6 | SCN8A | Mouse 15 Human 12q13 | CNS neurons |
| Nav1.7 | SCN9A | Mouse 2 Human 2q24 | PNS neurons |
| Nav1.8 | SCN10A | Mouse 9 Human 3p22–24 | DRG neurons |
| Nav1.9 | SCN11A | Mouse 9 Human 3p21–24 | DRG neurons |
| Nax | SCN7A SCN6A | Mouse 2 Human 2q21–23 | uterus, astrocytes, hypothalamus |
The ion channel protein superfamily
Analysis of the human genome revealed that there are 143 ion channel proteins whose pore-forming segments are related to sodium channels, and they are associated with at least 10 distinct families of auxiliary subunits (Yu & Catterall, 2004). The voltage-gated ion channels and their molecular relatives are one of the largest superfamilies of signalling proteins and one of the most prominent targets for drugs used in the therapy of human diseases. The voltage-gated sodium channels were the founders of this large superfamily in terms of discovery of their function by Hodgkin and Huxley and the later discovery of the sodium channel protein itself. Surprisingly, the sodium channel family is ancient in evolution. The bacterial sodium channel NaChBac and several prokaryotic relatives are composed of homotetramers of a single subunit whose structure resembles one of the domains of a vertebrate sodium channel (Ren et al. 2001; Koishi et al. 2004). It is likely that these bacterial sodium channels are the evolutionary ancestors of the larger, four-domain sodium channels in eukaryotes.
Sodium channel structure at atomic resolution
Sodium channel architecture has recently been revealed in three-dimensions by determination of the crystal structure of the bacterial sodium channel NavAb at high resolution (2.7 Å) (Fig. 3; Payandeh et al. 2011). This remarkable structure has revealed a wealth of new information about the structural basis for sodium selectivity and conductance, the mechanism of block of the channel by therapeutically important drugs, and the mechanism of voltage-dependent gating. As viewed from the top, NavAb has a central pore surrounded by four pore-forming modules composed of S5 and S6 segments and the intervening pore loop (Fig. 3A, blue). Four voltage-sensing modules composed of S1–S4 segments are symmetrically associated with the outer rim of the pore module (Fig. 3A and B). The transmembrane architecture of NavAb shows that the adjacent subunits have swapped their functional domains such that each voltage-sensing module is most closely associated with the pore-forming module of its neighbour (Fig. 3C, warmer colors), similar to voltage-gated potassium channels (Long et al. 2007). It is likely that this domain-swapped arrangement enforces concerted gating of the four subunits or domains of sodium and potassium channels.
Figure 3. Structure of NavAb.
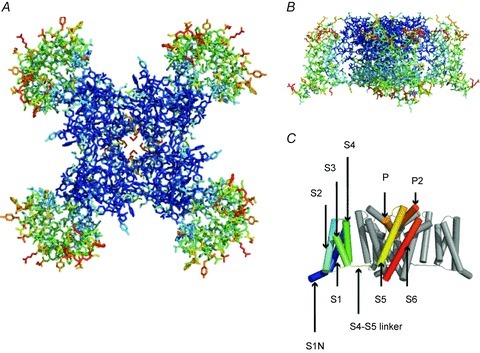
A, top view of NavAb channels coloured according to crystallographic temperature factors of the main chain (blue < 50 Å2 to red > 150 Å2). B, side view of NavAb. C, structural elements in NavAb. The structural components of one subunit are highlighted (1–6, transmembrane segments S1–S6).
The pore of sodium channels
The region of the sodium channel that forms the outer end of the pore was first determined by identifying the amino acid residues that form the binding site of the pore-blocking toxin tetrodotoxin in the short α-helical segments between S5 and S6 (Noda et al. 1989; Terlau et al. 1991). Mutations of the same amino acid residues in the pore loop were shown to control ion selectivity (Heinemann et al. 1992). This initial view of pore structure of sodium channels has been illuminated at the atomic level by the structure of the NavAb channel (Payandeh et al. 2011). This crystal structure reveals an overall pore architecture with a large external vestibule, a narrow ion selectivity filter containing the amino acid residues shown to determine ion selectivity, a large central cavity that is lined by the S6 segments and is water filled, and an intracellular activation gate formed at the crossing of the S6 segments at the intracellular surface of the membrane (Fig. 4A; Payandeh et al. 2011). The activation gate is tightly closed in the NavAb structure (Fig. 4B). This general pore architecture reveals the structural basis for gated access of blocking ions and drugs to the lumen of the pore observed in classical studies of ion selectivity and pore block (Armstrong, 1971; Hille, 1975a, 1977).
Figure 4. NavAb pore and selectivity filter.
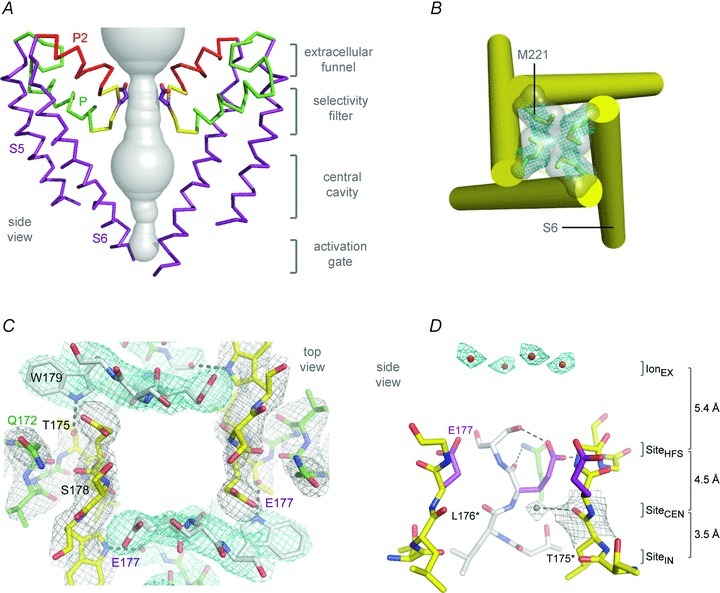
A, architecture of the NavAb pore. Glu177 side-chains, purple; pore volume, grey. B, the closed activation gate at the intracellular end of the pore illustrating the close interaction of Met221 residues in closing the pore. C, top view of the ion selectivity filter. Symmetry-related molecules are coloured white and yellow; P-helix residues are coloured green. Hydrogen bonds between Thr175 and Trp179 are indicated by grey dashes. Electron-densities from Fo–Fc omit maps are contoured at 4.0 σ (blue and grey) and subtle differences can be appreciated (small arrows). D, side view of the selectivity filter. Glu177 (purple) interactions with Gln172, Ser178 and the backbone of Ser180 are shown in the far subunit. Fo–Fc omit map, 4.75 σ (blue); putative cations or water molecules (red spheres, IonEX). Electron-density around Leu176 (grey; Fo–Fc omit map at 1.75 σ) and a putative water molecule is shown (grey sphere). Na+-coordination sites: SiteHFS, SiteCEN and SiteIN.
Ion selectivity and conductance
Although the overall pore architecture of sodium and potassium channels is similar, the structures of their ion selectivity filters and their mechanisms of ion selectivity and conductance are completely different. Potassium channels select K+ by direct interaction with a series of four ion coordination sites formed by the backbone carbonyls of the amino acid residues that comprise the ion selectivity filter (Zhou et al. 2001). No water molecules intervene between K+ and its interacting backbone carbonyls in the ion selectivity filter of potassium channels (Zhou et al. 2001). In contrast, the NavAb ion selectivity filter has a high-field-strength site at its extracellular end (Fig. 4C), formed by the side chains of four glutamate residues (Payandeh et al. 2011), which are highly conserved and are key determinants of ion selectivity in vertebrate sodium and calcium channels (Heinemann et al. 1992). Considering its dimensions of approximately 4.6 Å square, Na+ with two planar waters of hydration could fit in this high-field-strength site. This outer site is followed by two ion coordination sites formed by backbone carbonyls (Fig. 4D). These two carbonyl sites are perfectly designed to bind Na+ with four planar waters of hydration but would be much too large to bind Na+ directly. In fact, the NavAb selectivity filter is large enough to fit the entire potassium channel ion selectivity filter inside it (Payandeh et al. 2011). Thus, the chemistry of Na+ selectivity and conductance is opposite to that of K+: negatively charged residues interact with Na+ to remove most (but not all) of its waters of hydration, and Na+ is conducted as a hydrated ion interacting with the pore through its inner shell of bound waters. This structure of the ion selectivity filter of NavAb is remarkably similar to the four-barrier, three-site model of ion selectivity, which predicted an outer high-field-strength site that would partially dehydrate the permeating ion and two inner sites that would conduct and rehydrate the permeant Na+ ion (Hille, 1975a). This congruence of theory and structure gives clear insight into the chemistry and biophysics of sodium permeation.
Drug receptor sites in sodium channels
Sodium channels are blocked by drugs used clinically as local anaesthetics, antiarrhythmics and antiepileptics. Site-directed mutagenesis studies of sodium channels revealed the receptor site for local anaesthetics and related drugs, which is formed by amino acid residues in the S6 segments in domains I, III and IV (Ragsdale et al. 1994, 1996; Wang et al. 1998; Yarov-Yarovoy et al. 2001, 2002). These drugs bind to a common receptor site in the pore of sodium channels and impede ion permeation. The structure of NavAb places this drug receptor site in three-dimensional context (Payandeh et al. 2011). The amino acid residues that form the receptor sites for sodium channel blockers line the inner surface of the S6 segments and create a three-dimensional drug receptor site whose occupancy would block the pore (Fig. 5). Access to this receptor site by large or hydrophilic drugs would require opening of the intracellular activation gate, which is tightly closed in our structure. This tight closure of the activation gate provides a structural basis for use-dependent block of sodium channels by local anaesthetics and related drugs (Hille, 1977), as they would bind much more rapidly when the channel is frequently opened. Remarkably, as also predicted by the modulated receptor hypothesis (Hille, 1977), fenestrations lead from the lipid phase of the membrane sideways into the drug receptor site, providing a specific hydrophobic access pathway for drug binding in the resting state of the channel (Fig. 5, pore portals; Payandeh et al. 2011). Access to the drug binding site in NavAb channels is controlled by the side chain of a single amino acid residue, Phe203 (Fig. 5; Payandeh et al. 2011), which is homologous to amino acid residues identified in previous structure–function studies that control drug access and egress from the local anaesthetic receptor site in mammalian cardiac and brain sodium channels (Ragsdale et al. 1994; Qu et al. 1995).
Figure 5. Membrane access to the central cavity in NavAb.
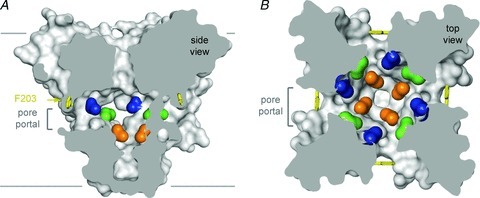
A, side-view through the pore module illustrating fenestrations (portals) and hydrophobic access to central cavity. Phe203 side-chains, yellow sticks. Surface representations of NavAb residues aligning with those implicated in drug binding and block; Thr206, blue; Met209, green; Val213, orange. Membrane boundaries, grey lines. Electron-density from an Fo–Fc omit map is contoured at 2.0 σ. B, top-view sectioned below the selectivity filter, coloured as in A.
Fast inactivation
As first described by Hodgkin & Huxley (1952c), sodium channels in metazoans open in response to depolarization and then inactivate within 1–2 ms. This fast inactivation process is required for repetitive firing of action potentials in neural circuits and for control of excitability in nerve and muscle cells. Studies with site-directed anti-peptide antibodies showed that the short intracellular loop connecting homologous domains III and IV of the sodium channel α subunit is responsible for fast inactivation (Fig. 2; Vassilev et al. 1988). This fast inactivation gate serves as an intracellular blocking particle that folds into the channel structure and blocks the pore during inactivation (Vassilev et al. 1988). Binding of a site-directed antibody to this peptide segment results in slowed entry of single channels into the inactivated state (Vassilev et al. 1989). Cutting this loop by expression of the sodium channel in two pieces also greatly slows inactivation (Stuhmer et al. 1989). The key amino acid motif IFM is required to maintain closure of the inactivation gate (West et al. 1992), and peptides containing this inactivation gate sequence motif can restore fast inactivation to mutant sodium channels (Eaholtz et al. 1994). The inactivation gate bends at a key pair of glycine residues to fold into the intracellular mouth of the pore, bind and block sodium conductance as a hinged lid (Kellenberger et al. 1996, 1997a,b). Analysis of the structure of the inactivation gate by NMR showed that it contains a rigid α-helix preceded by two loops of protein that array the IFM motif and a neighbouring Thr residue on their surface for interaction with and block of the open pore of the channel (Fig. 2, inset; Rohl et al. 1999). The fast inactivation gate is not present in homotetrameric bacterial sodium channels, so further structural analysis must await determination of the three-dimensional structure of a metazoan sodium channel.
Voltage sensing and voltage-dependent activation
Voltage-dependent activation of sodium channels was first demonstrated by Hodgkin and Huxley, and they predicted that the steep voltage dependence of sodium channel activation would require movement of three ‘electrically charged particles’ across the cell membrane through the full extent of the transmembrane electric field (Hodgkin & Huxley, 1952d). The predicted transmembrane movement of these gating charges was detected as a small capacitative gating current in high-resolution voltage clamp studies of the squid giant axon (Armstrong & Bezanilla, 1973), fulfilling a major tenet of the Hodgkin–Huxley model for channel function. The S4 transmembrane segments of sodium channels, which contain four to eight repeated motifs of a positively charged amino acid residue (usually arginine) followed by two hydrophobic residues, was proposed to carry the gating charges of sodium channels in the sliding helix or helical screw model of voltage sensing (Catterall, 1986a,b; Guy & Seetharamulu, 1986; Yarov-Yarovoy et al. 2006). The S4 segment is proposed to be in a transmembrane position in both resting and activated states; the gating charges are stabilized in their transmembrane position by forming ion pairs with neighbouring negatively charged residues; and their outward movement is catalysed by exchange of these ion pair partners (Catterall, 1986a,b; Guy & Seetharamulu, 1986; Yarov-Yarovoy et al. 2006). Extensive studies of sodium channels now provide strong support for all of the elements of this model (reviewed in Bezanilla, this volume, and Catterall, 2010).
The transmembrane position of the S4 segment in sodium channels has been confirmed by mapping the receptor sites for scorpion toxins in detail and showing that these toxins bind to the outer end of the S3–S4 loop of the voltage sensors in both resting and activated states, thereby establishing that the S4 segment remains in a transmembrane position in both of these states (Catterall, 1979; Rogers et al. 1996; Cestèle et al. 1998, 2006; Wang et al. 2011; Zhang et al. 2011). Mutation of the arginine residues in the S4 segment of sodium channels reduces the steepness of voltage-dependent gating, consistent with the idea that these residues serve as gating charges (Stuhmer et al. 1989; Kontis et al. 1997). Covalent labelling and voltage clamp fluorescence studies show that the S4 segments of sodium channels move outward and rotate upon membrane depolarization and transport the gating charges from an inner water-accessible vestibule to an outer water-accessible vestibule (Yang & Horn, 1995; Yang et al. 1996; Chanda & Bezanilla, 2002). Molecular modelling of sodium channel voltage sensors with rigid scorpion toxins bound has yielded high-resolution models of the voltage sensor in its resting and activated states (Wang et al. 2011; Zhang et al. 2011), which also illustrate the outward movement of the S4 segment and exchange of ion pair partners in the transition from resting to activated states. Based on the X-ray crystal structures of the KV1.2 and NavAb channels with activated voltage sensors, ab initio molecular modelling using the Rosetta algorithm provides a detailed structural model of the resting states of the voltage sensor and charts the sequence of conformational changes and the gating charge interactions with negative charges and hydrophilic groups in the voltage sensor during activation of the channel (Fig. 6; Yarov-Yarovoy et al. 2006, 2011). These structural models show that the S4 segment and its gating charges move through a narrow gating pore that focuses the transmembrane electric field to a distance of approximately 5 Å normal to the membrane (hydrophobic constriction site, HCS; Fig. 6) and allows the gating charges to move from an intracellular aqueous vestibule to an extracellular aqueous vestibule with a short transit through the channel protein (see Bezanilla review, this volume). This mechanism of outward movement of the gating charges during the activation process has been confirmed by extensive disulfide-locking studies of the ion-pair interactions in sodium channels predicted from the sliding-helix model of gating (DeCaen et al. 2008, 2009, 2011; Yarov-Yarovoy et al. 2011). This model is also consistent with metal ion and sulfhydryl crosslinking studies of Shaker potassium channels (Campos et al. 2007; Broomand & Elinder, 2008; Lin et al. 2011). Together, these studies define the detailed mechanism of voltage sensing and voltage-dependent activation of the voltage sensor of sodium channels through a series of resting and activated states at the atomic level (Fig. 6; Yarov-Yarovoy et al. 2011).
Figure 6. Model of conformational changes in the voltage sensor during gating.
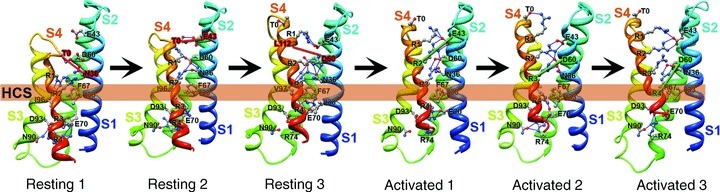
Transmembrane view of the ribbon representation of Rosetta models of three resting and three activated states of the VSD of NaChBac. Segments S1 through S4 coloured individually and labelled. Side chain atoms of the gating charge carrying arginines in S4 (coloured dark blue), negatively charged residues in S1, S2 and S3 segments (coloured red), polar residues in S1, S3 and S4 (coloured purple), and key hydrophobic residues in S1, S2 and S3 (coloured grey) are shown in sphere representation and labelled. The HCS is highlighted by the orange bar.
Neuromodulation of sodium channel function
Although the primary mechanism of regulation of sodium channel activity is voltage-dependent gating on the millisecond time scale, the brain sodium channels NaV1.1 and NaV1.2 are also regulated by dopamine, acetylcholine, serotonin and other neurotransmitters that act through G-protein coupled receptors and activate protein kinase A and C (reviewed in Cantrell & Catterall, 2001). These kinases phosphorylate a series of sites in the intracellular loop connecting domains I and II and act synergistically to reduce sodium channel activity (Fig. 2; Cantrell & Catterall, 2001). Their effects are mediated by enhancement of the intrinsic slow inactivation process of sodium channels, which serves to reduce sodium channel activity during trains of action potentials (Carr et al. 2003; Chen et al. 2006). Protein kinase C also phosphorylates a site in the inactivation gate and subtly slows fast inactivation (Cantrell & Catterall, 2001).
In contrast to brain sodium channels, the activity of NaV1.8 channels that are specifically expressed in sensory neurons is enhanced by activation of PGE2 receptors and phosphorylation by PKA and PKC through analogous phosphorylation sites that regulate brain sodium channels, even though the direction of regulation is opposite (Fitzgerald et al. 1999; Vijayaragavan et al. 2004). This form of regulation may contribute to up-regulation of sodium channel activity in hyperalgesic states. The NaV1.7 and NaV1.8 channels are also regulated by the p38 mitogen-activated protein kinase and by ERKI/II protein kinases (Hudmon et al. 2008; Stamboulian et al. 2010), offering additional mechanisms through which sodium channel function could be regulated in pain states.
Pathophysiology of sodium channel mutations
A remarkable and unexpected facet of sodium channel biology that has evolved from the discovery of sodium currents by Hodgkin and Huxley and the subsequent understanding of sodium channel genes, structure and function has been the discovery of a large number of genetic diseases of sodium channels, including inherited forms of periodic paralysis, cardiac arrhythmia, epilepsy and chronic pain (Lehman-Horn & Jurkat-Rott, 1999; Table 2). In most cases, these are genetically dominant diseases in which the mutations cause a gain-of-function effect at both the molecular and cellular levels. In skeletal muscle, mutations in NaV1.4 channels increase channel activity by impairing fast and/or slow inactivation in paramyotonia congenita and hyperkalaemic periodic paralysis (Jurkat-Rott & Lehmann-Horn, 2006; Venance et al. 2006). In the heart, mutations in NaV1.5 channels cause long QT syndrome by impairing sodium channel inactivation (Keating & Sanguinetti, 2001). In sensory neurons, mutations in NaV1.7 channels cause erythromelalgia by shifting the voltage dependence of activation to more negative membrane potentials and cause paroxysmal extreme pain disorder by impairing sodium channel inactivation (Dib-Hajj et al. 2007; and see review by Waxman in this volume). In the rare recessive pain disorder congenital indifference to pain, loss-of-function mutations in both alleles of the gene encoding NaV1.7 channels cause complete loss of pain sensation (Cox et al. 2006).
Table 2.
Major inherited diseases of sodium channels
| Disease | Gene | Channel |
|---|---|---|
| Generalized epilepsy with febrile seizures plus | SCN1A | Nav1.1 |
| Dravet syndrome (severe myoclonic epilepsy in infancy) | SCN1A | NaV1.1 |
| Benign neonatal convulsions | SCN1A | NaV1.1 |
| Familial hemiplegic migraine type III | SCN1A | NaV1.1 |
| Benign familial neonatal–infantile seizures | SCN2A | NaV1.2 |
| Hypokalaemic periodic paralysis type II | SCN4A | Nav1.4 |
| K+-sensitive normokalaemic periodic paralysis | SCN4A | Nav1.4 |
| Hyperkalaemic periodic paralysis | SCN4A | Nav1.4 |
| Paramyotonia congenita | SCN4A | NaV1.4 |
| Long QT syndrome type III | SCN5A | Nav1.5 |
| Brugada syndrome | SCN5A | NaV1.5 |
| Erythromelalgia | SCN9A | NaV1.7 |
| Paroxysmal extreme pain disorder | SCN9A | NaV1.7 |
| Congenital indifference to pain | SCN9A | NaV1.7 |
In three of the autosomal dominant ion channelopathies, unexpected new mechanisms of increased cellular excitability have emerged from recent structure–function studies. In Brugada syndrome, loss-of-function mutations of NaV1.5 channels create inhomogeneity of conduction across the ventricular wall and generate arrhythmias (Terrenoire et al. 2007). In Dravet syndrome, and possibly also in generalized epilepsy with febrile seizures plus, loss-of-function mutations in NaV1.1 channels selectively impair the excitability of GABAergic inhibitory neurons and thereby create hyperexcitability in neural circuits and cause epilepsy (Catterall et al. 2010). In hypokalaemic periodic paralysis, mutations of the gating charges in the voltage sensor of NaV1.4 channels cause an ionic leak through the gating pore, resulting in excess Na+ influx through the leaky voltage sensor, accumulation of intracellular Na+, depolarization and conduction block that lead to episodic paralysis (Sokolov et al. 2007, 2008; Struyk & Cannon, 2007). As these novel mechanisms of ion channelopathies illustrate, there is much more to do in analysing the full extent of the pathophysiological implications of sodium channel dysfunction.
Conclusion
The legacy of the discoveries by Hodgkin and Huxley is rich indeed. The sodium current they discovered is now well understood in terms of its voltage-dependent gating and ion selectivity. The sodium channel protein has been discovered and characterized in biochemical and molecular detail, even to atomic resolution. Structural and functional studies from many laboratories have combined synergistically to give a clear view of the molecular basis for sodium channel function. The ion selectivity filter is defined as a rigid high-field-strength site of approximately 4.6 Å square formed by carboxyl side chains of glutamates followed by two wider ion coordination sites formed by backbone carbonyls, and the structural basis for selectivity and conductance of Na+ as a hydrated cation has been uncovered. Voltage-dependent activation is understood in terms of the outward movement of gating charges, the ‘electrically charged particles’ of Hodgkin and Huxley, which are located in the S4 segments of the voltage sensors, are stabilized in their transmembrane position by interactions with negative charges and hydrophilic moieties in neighbouring transmembrane segments, and move outward upon depolarization by exchange of ion pair partners that catalyse their voyage through the narrow gating pore and across the membrane permeability barrier. The mechanism of coupling of voltage sensor activation to pore opening remains tentative, but it is likely to yield to structural biology soon. The mechanism of fast inactivation has also been well defined, with clear structural and functional evidence for closure of an intracellular fast inactivation gate formed by the intracellular loop connecting domains III and IV of metazoan sodium channels, which acts as a hinged lid and closes the intracellular mouth of the open pore. The role of sodium channels as a crucial drug target has been revealed, the complex mechanism of use-dependent block has been explained by the modulated receptor hypothesis, and both the receptor site for drug block and the structural basis for control of drug access to the receptor site have been defined at atomic resolution. Finally, the sodium channel has been revealed as a target of disease mutations causing a wide range of ion channelopathies, and the molecular understanding of these diseases has led to important advances in their therapy. Even with all of this progress, one can look to the future with anticipation of new discoveries of the structural basis of sodium channel gating and ion conductance as well as new developments in sodium channel pathophysiology, pharmacology, and therapeutics. The use of new structural information on drug binding to sodium channels promises to advance development of drugs that would target the individual sodium channel subtypes that are specifically expressed in brain, sensory neurons and heart, and thereby lead to a new generation of drugs for more safe and effective treatment of epilepsy, chronic and neuropathic pain, and cardiac arrhythmia.
References
- Agnew WS, Moore AC, Levinson SR, Raftery MA. Identification of a large molecular weight peptide associated with a tetrodotoxin binding proteins from the electroplax of Electrophorus electricus. Biochem Biophys Res Commun. 1980;92:860–866. doi: 10.1016/0006-291x(80)90782-2. [DOI] [PubMed] [Google Scholar]
- Armstrong CM. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axon. J Gen Physiol. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F. Currents related to movement of the gating particles of the sodium channels. Nature. 1973;242:459–461. doi: 10.1038/242459a0. [DOI] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F, Rojas E. Destruction of sodium conductance inactivation in squid axons perfused with pronase. J Gen Physiol. 1973;62:375–391. doi: 10.1085/jgp.62.4.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barchi RL. Protein components of the purified sodium channel from rat skeletal sarcolemma. J Neurochem. 1983;36:1377–1385. doi: 10.1111/j.1471-4159.1983.tb13580.x. [DOI] [PubMed] [Google Scholar]
- Beneski DA, Catterall WA. Covalent labeling of protein components of the sodium channel with a photoactivable derivative of scorpion toxin. Proc Natl Acad Sci U S A. 1980;77:639–643. doi: 10.1073/pnas.77.1.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury WJ, Isom LL. Na channel beta subunits: overachievers of the ion channel family. Front Pharmacol. 2011;2:53. doi: 10.3389/fphar.2011.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broomand A, Elinder F. Large-scale movement within the voltage-sensor paddle of a potassium channel-support for a helical-screw motion. Neuron. 2008;59:770–777. doi: 10.1016/j.neuron.2008.07.008. [DOI] [PubMed] [Google Scholar]
- Campos FV, Chanda B, Roux B, Bezanilla F. Two atomic constraints unambiguously position the S4 segment relative to S1 and S2 segments in the closed state of Shaker K channel. Proc Natl Acad Sci U S A. 2007;104:7904–7909. doi: 10.1073/pnas.0702638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell AR, Catterall WA. Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci. 2001;2:397–407. doi: 10.1038/35077553. [DOI] [PubMed] [Google Scholar]
- Carr D, Day M, Cantrell AR, Scheuer T, Catterall WA, Surmeier DJ. G-protein coupled receptors modulate sodium channel gating by promoting entry into a slow inactivated state a novel form of activity-dependent plasticity. Neuron. 2003;39:793–806. doi: 10.1016/s0896-6273(03)00531-2. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Binding of scorpion toxin to receptor sites associated with sodium channels in frog muscle. Correlation of voltage-dependent binding with activation. J Gen Physiol. 1979;74:375–391. doi: 10.1085/jgp.74.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Neurotoxins that act on voltage-sensitive sodium channels in excitable membranes. Annu Rev Pharmacol Toxicol. 1980;20:15–43. doi: 10.1146/annurev.pa.20.040180.000311. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Molecular properties of voltage-sensitive sodium channels. Annu Rev Biochem. 1986a;55:953–985. doi: 10.1146/annurev.bi.55.070186.004513. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Voltage-dependent gating of sodium channels: correlating structure and function. Trends Neurosci. 1986b;9:7–10. [Google Scholar]
- Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67:915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Cestele S, Yarov-Yarovoy V, Yu FH, Konoki K, Scheuer T. Voltage-gated ion channels and gating modifier toxins. Toxicon. 2007;49:124–141. doi: 10.1016/j.toxicon.2006.09.022. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Kalume F, Oakley JC. NaVV1.1 channels and epilepsy. J Physiol. 2010;588:1849–1859. doi: 10.1113/jphysiol.2010.187484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestèle S, Qu Y, Rogers JC, Rochat H, Scheuer T, Catterall WA. Voltage sensor-trapping: Enhanced activation of sodium channels by β-scorpion toxin bound to the S3-S4 loop in domain II. Neuron. 1998;21:919–931. doi: 10.1016/s0896-6273(00)80606-6. [DOI] [PubMed] [Google Scholar]
- Cestele S, Yarov-Yarovoy V, Qu Y, Sampieri F, Scheuer T, Catterall WA. Structure and function of the voltage sensor of sodium channels probed by a beta-scorpion toxin. J Biol Chem. 2006;281:21332–21344. doi: 10.1074/jbc.M603814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B, Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol. 2002;120:629–645. doi: 10.1085/jgp.20028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yu FH, Surmeier DJ, Scheuer T, Catterall WA. Neuromodulation of Na+ channel slow inactivation via cAMP-dependent protein kinase and protein kinase C. Neuron. 2006;49:409–420. doi: 10.1016/j.neuron.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Scheuer T, Catterall WA. Gating charge interactions with the S1 segment during activation of a Na+ channel voltage sensor. Proc Natl Acad Sci U S A. 2011;108:18825–18830. doi: 10.1073/pnas.1116449108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Sharp EM, Scheuer T, Catterall WA. Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proc Natl Acad Sci U S A. 2009;106:22498–22503. doi: 10.1073/pnas.0912307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Zhao Y, Scheuer T, Catterall WA. Disulfide locking a sodium channel voltage sensor reveals ion pair formation during activation. Proc Natl Acad Sci U S A. 2008;105:15142–15147. doi: 10.1073/pnas.0806486105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. From genes to pain: Nav1.7 and human pain disorders. Trends Neurosci. 2007;30:555–563. doi: 10.1016/j.tins.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Eaholtz G, Scheuer T, Catterall WA. Restoration of inactivation and block of open sodium channels by an inactivation gate peptide. Neuron. 1994;12:1041–1048. doi: 10.1016/0896-6273(94)90312-3. [DOI] [PubMed] [Google Scholar]
- Fitzgerald EM, Okuse K, Wood JN, Dolphin AC, Moss SJ. cAMP-dependent phosphorylation of the tetrodotoxin-resistant voltage-dependent sodium channel SNS. J Physiol. 1999;516:433–446. doi: 10.1111/j.1469-7793.1999.0433v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL. Resurgence of sodium channel research. Annu Rev Physiol. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- Goldin AL, Snutch T, Lubbert H, Dowsett A, Marshall J, Auld V, Downey W, Fritz LC, Lester HA, Dunn R, Catterall WA, Davidson N. Messenger RNA coding for only the α subunit of the rat brain Na channel is sufficient for expression of functional channels in Xenopus oocytes. Proc Natl Acad Sci U S A. 1986;83:7503–7507. doi: 10.1073/pnas.83.19.7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy HR, Seetharamulu P. Molecular model of the action potential sodium channel. Proc Natl Acad Sci U S A. 1986;508:508–512. doi: 10.1073/pnas.83.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorne RP, Catterall WA. Purification of the saxitoxin receptor of the sodium channel from rat brain. Proc Natl Acad Sci U S A. 1981;78:4620–4624. doi: 10.1073/pnas.78.7.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorne RP, Catterall WA. The sodium channel from rat brain. Purification and subunit composition. J Biol Chem. 1984;259:1667–1675. [PubMed] [Google Scholar]
- Hartshorne RP, Keller BU, Talvenheimo JA, Catterall WA, Montal M. Functional reconstitution of the purified brain sodium channel in planar lipid bilayers. Proc Natl Acad Sci U S A. 1985;82:240–244. doi: 10.1073/pnas.82.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorne RP, Messner DJ, Coppersmith JC, Catterall WA. The saxitoxin receptor of the sodium channel from rat brain Evidence for two nonidentical beta subunits. J Biol Chem. 1982;257:13888–13891. [PubMed] [Google Scholar]
- Heinemann SH, Terlau H, Stühmer W, Imoto K, Numa S. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature. 1992;356:441–443. doi: 10.1038/356441a0. [DOI] [PubMed] [Google Scholar]
- Hille B. The permeability of the sodium channel to organic cations in myelinated nerve. J Gen Physiol. 1971;59:599–619. doi: 10.1085/jgp.58.6.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. The permeability of the sodium channel to metal cations in myelinated nerve. J Gen Physiol. 1972;59:637–658. doi: 10.1085/jgp.59.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ionic selectivity, saturation, and block in sodium channels. A four-barrier model. J Gen Physiol. 1975a;66:535–560. doi: 10.1085/jgp.66.5.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. The receptor for tetrodotoxin and saxitoxin: a structural hypothesis. Biophys J. 1975b;15:615–619. doi: 10.1016/S0006-3495(75)85842-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. 3rd ed. Sunderland, MA: Sinauer Associates Inc; 2001. [Google Scholar]
- Hodgkin AL, Huxley AF. The components of membrane conductance in the giant axon of Loligo. J Physiol. 1952a;116:473–496. doi: 10.1113/jphysiol.1952.sp004718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J Physiol. 1952b;116:449–472. doi: 10.1113/jphysiol.1952.sp004717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J Physiol. 1952c;116:497–506. doi: 10.1113/jphysiol.1952.sp004719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952d;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Choi JS, Tyrrell L, Black JA, Rush AM, Waxman SG, Dib-Hajj SD. Phosphorylation of sodium channel Nav1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J Neurosci. 2008;28:3190–3201. doi: 10.1523/JNEUROSCI.4403-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Patton DE, Reber BFX, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA. Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- Isom LL, Ragsdale DS, De Jongh KS, Westenbroek RE, Reber BFX, Scheuer T, Catterall WA. Structure and function of the beta-2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM-motif. Cell. 1995;83:433–442. doi: 10.1016/0092-8674(95)90121-3. [DOI] [PubMed] [Google Scholar]
- Jurkat-Rott K, Lehmann-Horn F. Paroxysmal muscle weakness: the familial periodic paralyses. J Neurol. 2006;253:1391–1398. doi: 10.1007/s00415-006-0339-0. [DOI] [PubMed] [Google Scholar]
- Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- Kellenberger S, Scheuer T, Catterall WA. Movement of the Na+ channel inactivation gate during inactivation. J Biol Chem. 1996;271:30971–30979. doi: 10.1074/jbc.271.48.30971. [DOI] [PubMed] [Google Scholar]
- Kellenberger S, West JW, Catterall WA, Scheuer T. Molecular analysis of potential hinge residues in the inactivation gate of brain type IIA Na+ channels. J Gen Physiol. 1997a;19:607–617. doi: 10.1085/jgp.109.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger S, West JW, Scheuer T, Catterall WA. Molecular analysis of the putative inactivation particle in the inactivation gate of brain type IIA Na+ channels. J Gen Physiol. 1997b;109:589–605. doi: 10.1085/jgp.109.5.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koishi R, Xu H, Ren D, Navarro B, Spiller BW, Shi Q, Clapham DE. A superfamily of voltage-gated sodium channels in bacteria. J Biol Chem. 2004;279:9532–9538. doi: 10.1074/jbc.M313100200. [DOI] [PubMed] [Google Scholar]
- Kontis KJ, Rounaghi A, Goldin AL. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. J Gen Physiol. 1997;110:391–401. doi: 10.1085/jgp.110.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiol Rev. 1999;79:1317–1372. doi: 10.1152/physrev.1999.79.4.1317. [DOI] [PubMed] [Google Scholar]
- Lin MC, Hsieh JY, Mock AF, Papazian DM. R1 in the Shaker S4 occupies the gating charge transfer center in the resting state. J Gen Physiol. 2011;138:155–163. doi: 10.1085/jgp.201110642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- Morgan K, Stevens EB, Shah B, Cox PJ, Dixon AK, Lee K, Pinnock RD, Hughes J, Richardson PJ, Mizuguchi K, Jackson AP. β3: An additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc Natl Acad Sci U S A. 2000;97:2308–2313. doi: 10.1073/pnas.030362197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Ikeda T, Suzuki T, Takeshima H, Takahashi T, Kuno M, Numa S. Expression of functional sodium channels from cloned cDNA. Nature. 1986;322:826–828. doi: 10.1038/322826a0. [DOI] [PubMed] [Google Scholar]
- Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N, Kangawa K, Matsuo H, Raftery M, Hirose T, Inayama S, Hayashida H, Miyata T, Numa S. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312:121–127. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- Noda M, Suzuki H, Numa S, Stuhmer W. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS letters. 1989;259:213–216. doi: 10.1016/0014-5793(89)81531-5. [DOI] [PubMed] [Google Scholar]
- Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475:353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Rogers J, Tanada T, Scheuer T, Catterall WA. Molecular determinants of drug access to the receptor site for antiarrhythmic drugs in the cardiac Na+ channel. Proc Natl Acad Sci U S A. 1995;270:25696–25701. doi: 10.1073/pnas.92.25.11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale DR, McPhee JC, Scheuer T, Catterall WA. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc Natl Acad Sci U S A. 1996;93:9270–9275. doi: 10.1073/pnas.93.17.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of sodium channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- Ren D, Navarro B, Xu H, Yue L, Shi Q, Clapham DE. A prokaryotic voltage-gated sodium channel. Science. 2001;294:2372–2375. doi: 10.1126/science.1065635. [DOI] [PubMed] [Google Scholar]
- Rogers JC, Qu Y, Tanada TN, Scheuer T, Catterall WA. Molecular determinants of high affinity binding of α-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel α subunit. J Biol Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- Rohl CA, Boeckman FA, Baker C, Scheuer T, Catterall WA, Klevit RE. Solution structure of the sodium channel inactivation gate. Biochemistry. 1999;38:855–861. doi: 10.1021/bi9823380. [DOI] [PubMed] [Google Scholar]
- Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature. 2007;446:76–78. doi: 10.1038/nature05598. [DOI] [PubMed] [Google Scholar]
- Sokolov S, Scheuer T, Catterall WA. Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc Natl Acad Sci U S A. 2008;105:19980–19985. doi: 10.1073/pnas.0810562105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamboulian S, Choi JS, Ahn HS, Chang YW, Tyrrell L, Black JA, Waxman SG, Dib-Hajj SD. ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel Nav1.7 and alters its gating properties. J Neurosci. 2010;30:1637–1647. doi: 10.1523/JNEUROSCI.4872-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struyk AF, Cannon SC. A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J Gen Physiol. 2007;130:11–20. doi: 10.1085/jgp.200709755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhmer W, Conti F, Suzuki H, Wang X, Noda M, Yahadi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Talvenheimo JA, Tamkun MM, Catterall WA. Reconstitution of neurotoxin-stimulated sodium transport by the voltage-sensitive sodium channel purified from rat brain. J Biol Chem. 1982;257:11 868–11 871. [PubMed] [Google Scholar]
- Tamkun MM, Talvenheimo JA, Catterall WA. The sodium channel from rat brain. Reconstitution of neurotoxin-activated ion flux and scorpion toxin binding from purified components. J Biol Chem. 1984;259:1676–1688. [PubMed] [Google Scholar]
- Terlau H, Heinemann SH, Stühmer W, Pusch M, Conti F, Imoto K, Numa S. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 1991;293:93–96. doi: 10.1016/0014-5793(91)81159-6. [DOI] [PubMed] [Google Scholar]
- Terrenoire C, Simhaee D, Kass RS. Role of sodium channels in propagation in heart muscle: how subtle genetic alterations result in major arrhythmic disorders. J Cardiovasc Electrophysiol. 2007;18:900–905. doi: 10.1111/j.1540-8167.2007.00838.x. [DOI] [PubMed] [Google Scholar]
- Vassilev P, Scheuer T, Catterall WA. Inhibition of inactivation of single sodium channels by a site-directed antibody. Proc Natl Acad Sci U S A. 1989;86:8147–8151. doi: 10.1073/pnas.86.20.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- Venance SL, Cannon SC, Fialho D, Fontaine B, Hanna MG, Ptacek LJ, Tristani-Firouzi M, Tawil R, Griggs RC. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. 2006;129:8–17. doi: 10.1093/brain/awh639. [DOI] [PubMed] [Google Scholar]
- Vijayaragavan K, Boutjdir M, Chahine M. Modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by protein kinase A and protein kinase C. J Neurophysiol. 2004;91:1556–1569. doi: 10.1152/jn.00676.2003. [DOI] [PubMed] [Google Scholar]
- Wang GK, Quan C, Wang S. A common local anesthetic receptor for benzocaine and etidocaine in voltage-gated mu1 Na+ channels. Pflugers Arch. 1998;435:293–302. doi: 10.1007/s004240050515. [DOI] [PubMed] [Google Scholar]
- Wang J, Yarov-Yarovoy V, Kahn R, Gordon D, Gurevitz M, Scheuer T, Catterall WA. Mapping the receptor site for α-scorpion toxins on a Na+ channel voltage sensor. Proc Natl Acad Sci U S A. 2011;108:15426–15431. doi: 10.1073/pnas.1112320108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe E, Fujikawa A, Matsunaga H, Yasoshima Y, Sako N, Yamamoto T, Saegusa C, Noda M. Nav2/NaG channel is involved in control of salt-intake behavior in the CNS. J Neurosci. 2000;20:7743–7751. doi: 10.1523/JNEUROSCI.20-20-07743.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proc Natl Acad Sci U S A. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, George AL, Jr, Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- Yang N, Horn R. Evidence for voltage-dependent S4 movement in sodium channel. Neuron. 1995;15:213–218. doi: 10.1016/0896-6273(95)90078-0. [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Baker D, Catterall WA. Voltage sensor conformations in the open and closed states in ROSETTA structural models of K+ channels. Proc Natl Acad Sci U S A. 2006;103:7292–7297. doi: 10.1073/pnas.0602350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Brown J, Sharp E, Clare JJ, Scheuer T, Catterall WA. Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na+ channel a subunit. J Biol Chem. 2001;276:20–27. doi: 10.1074/jbc.M006992200. [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Decaen PG, Westenbroek RE, Pan CY, Scheuer T, Baker D, Catterall WA. Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci U S A. 2011;109:E93–102. doi: 10.1073/pnas.1118434109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, McPhee JC, Idsvoog D, Pate C, Scheuer T, Catterall WA. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the sodium channel α subunit in voltage-dependent gating and drug block. J Biol Chem. 2002;277:35393–35401. doi: 10.1074/jbc.M206126200. [DOI] [PubMed] [Google Scholar]
- Yu FH, Catterall WA. The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE. 2004;2004:re15. doi: 10.1126/stke.2532004re15. [DOI] [PubMed] [Google Scholar]
- Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, Scheuer T, Curtis R. Sodium channel β-4, a new disulfide-linked auxiliary subunit with similarity to β-2. J Neurosci. 2003;23:7577–7585. doi: 10.1523/JNEUROSCI.23-20-07577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JZ, Yarov-Yarovoy V, Scheuer T, Karbat I, Cohen L, Gordon D, Gurevitz M, Catterall WA. Structure-function map of the receptor site for β-scorpion toxins in domain II of voltage-gated sodium channels. J Biol Chem. 2011;286:33641–33651. doi: 10.1074/jbc.M111.282509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a potassium channel-Fab complex at 2.0 A resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]