

Abstract
M1476I, a French Canadian founder mutation of Na+ channel Nav1.4, causes potassium-aggravated myotonia, with cold-induced myotonia as the most distinctive clinical feature. Mexiletine, a class 1B local anaesthetic, relieves the myotonic symptoms of patients carrying the M1476I mutation. We used the patch-clamp method to investigate the functional characteristics of this mutation by heterologous expression in tsA201 cells. The M1476I mutation caused an increased persistent Na+ current, a 2- to 3-fold slower fast inactivation, a 6.4 mV depolarizing shift in the midpoint of steady-state inactivation, and an accelerated recovery from fast inactivation compared to the wild-type (WT) channel. Cooling slowed the kinetics of both channel types and increased the amplitude of the persistent current in M1476I channels. Mexiletine suppressed the persistent Na+ current generated by the M1476I mutation and blocked both WT and M1476I channels in a use-dependent manner. The inactivation-deficient M1476I channels were less susceptible to mexiletine during repetitive pulses. The decreased use-dependent block of M1476I channels might have resulted from the slower onset of mexiletine block, and/or the faster recovery from mexiletine block, given that the affinity of mexiletine for the inactivated state of the WT and mutant channels was similar. Increased extracellular concentrations of potassium had no effect on either M1476I or WT currents. These results indicated that cooling can augment the disruption of the voltage dependence of fast inactivation by M1476I channels. The therapeutic efficacy of mexiletine in M1476I carriers may be partly due to the open-channel block targeting the persistent Na+ currents generated by M1476I channels.
Key points
Na+ channels are pores present at the surface of every muscle cell; the initiation of muscle contraction requires the opening of a large number of Na+ channels.
Nav1.4 channels are encoded by the SCN4A gene and represent over 90% of Na+ channels in adult skeletal muscle cells; the M1476I mutation of Nav1.4 causes potassium-aggravated myotonia in a French Canadian population of the Saguenay-Lac-Saint-Jean region of Quebec.
Individuals carrying this mutation exhibit typical features ranging from asymptomatic myotonic discharges on electromyography to severe diffuse myotonia, as well as unusual cold-induced, painful myotonia.
Our study provides a detailed characterization of the underlying biophysical defect of the M1476I mutation, including an increased persistent Na+ current, a disruption of fast inactivation and an accelerated recovery from inactivation; cooling further enhances the abnormalities of fast inactivation of the mutant channels.
Our data suggest that mexiletine could be used as a therapeutic for patients carrying this mutation.
Introduction
Hodgkin and Huxley, have not only recognized the importance of the sodium channel conductance for the rising phase of AP in neurons in the 1950's, but also developed algorithms to describe the ionic basis of nerve excitability (Hodgkin et al., 1952; Hodgkin & Huxley, 1952). Nav1.4 channels, which are encoded by the SCN4A gene, comprise over 90% of voltage-gated Na+ channels in postnatal skeletal muscle. Like other Na+ channel subtypes, Nav1.4 channels are transmembrane glycoprotein complexes composed of a pore-forming α subunit and one or more regulatory β subunits. The α subunit is a large protein with four internally homologous domains (DI–IV), each containing six α-helical transmembrane segments (S1–S6) (Catterall, 2000; Chahine et al. 2005). These Nav1.4 channels are located in the sarcolemma and T-tubular membranes, with a high density near the endplate of the muscle cell.
Disturbances in the function of Nav1.4 channels cause autosomal dominant Na+ channelopathies, including hypokalaemic periodic paralysis (HypoPP), hyperkalaemic periodic paralysis (HyperPP), paramyotonia congenita (PMC), potassium-aggravated myotonia (PAM, or sodium channel myotonia), and congenital myasthenic syndrome (CMS) (Jurkat-Rott et al. 2010). Myotonia is the predominant feature of PMC and PAM (Ebers et al. 1991; Lehmann-Horn & Rudel, 1996). More than 10 SCN4A mutations have been identified in association with PAM. The three most common mutations, Gly1306Ala/Val/Glu, occur at the same residue of the DIII–DIV linker (Hayward et al. 1996), which acts as a ‘hinged lid’ to block the permeation pathway after activation to inactivate the Na+ channel (Yu & Catterall, 2003). Other PAM mutations such as V445M (Rosenfeld et al. 1997), S804F (McClatchey et al. 1992) and V1589M (Heine et al. 1993) are situated in the S5 and S6 segments of four domains of the Nav1.4 channel. These regions form part of the inner vestibule of the central pore to which the inactivation gate binds. PAM mutations thus produce a pattern of Nav1.4 channel dysfunction that includes disruption of entry into fast inactivation, an increase in the persistent Na+ current, and accelerated recovery from fast inactivation (Mitrovic et al. 1995; Hayward et al. 1996; Lerche et al. 1996; Richmond et al. 1997; Green et al. 1998). PAM is exacerbated by K+ ingestion (e.g. fruit juices) or strenuous work. Myotonia of this phenotype does not worsen significantly after exposure to cold and is not associated with attacks of weakness. Among PAM mutations, only V1589M has been reported to cause cold-aggravated myotonia (Heine et al. 1993). The severity of PAM often fluctuates temporally and ranges from mild to severe episodes of muscle stiffness.
M1476I, a new dominant missense SCN4A mutation in exon 24, was found in a French Canadian population of the Saguenay–Lac-Saint-Jean region of Quebec. This mutation (substitution of methionine for isoleucine) is located in the cytoplasmic S4–S5 loop of DIV. Individuals carrying this mutation exhibit a PAM phenotype with typical features ranging from asymptomatic myotonic discharges on electromyography (EMG) to severe diffuse myotonia, as well as unusual cold-induced (41% of patients), painful myotonia (18% of patients). Mexiletine is an effective treatment for M1476I carriers, especially compared to other class 1B antiarrhythmic agents and local anaesthetics (Rossignol et al. 2007). To elucidate the mechanism underlying the M1476I phenotype, we utilized a whole-cell patch-clamp technique to study tsA201 cells expressing WT or M1476I channels. The mutation exhibited biophysical defects similar to other PAM-causing mutations, in particular impaired fast inactivation and accelerated recovery from inactivation. To better understand the unusual cold sensitivity of patients carrying the M1476I mutation, we compared the biophysical properties of the mutant channels at different temperatures (10°C, 18°C and 23°C). We also studied the effect of higher extracellular K+ concentrations and the action of mexiletine on M1476I channels.
Methods
Na+ channel mutagenesis
Mutant human Nav1.4/M1476I was generated using QuickChange site-directed mutagenesis kits according to the manufacturer's instructions (Stratagene, La Jolla, CA, USA). Oligonucleotide primers containing the corresponding M1476I mutation were synthesized using following sequences:
5′-ctg ctg ttc gcc ctc ata atg tcg ctg cct gcc-3′ (forward primer) and
5′-ggc agg cag cga cat tat gag ggc gaa cag cag-3′ (reverse primer).
The mutated site is underlined. Mutant and WT Nav1.4 channels were inserted in the pcDNA1 plasmid, amplified in E. coli DH5α, and purified using Qiagen columns (Qiagen Inc., Chatsworth, CA, USA).
Gene transfection and cell culture
TsA201 cells are derived from HEK 293 cells by stable transfection with SV40 large T antigen (Margolskee et al. 1993). The cells were grown using standard tissue culture conditions (5% CO2, 37°C) in high glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal bovine serum (FBS) (10%), l-glutamine (2 mm), penicillin (100 U ml−1), and streptomycin (10 mg ml−1) (Gibco BRL Life Technologies, Burlington, ON, Canada). The cells were transiently transfected with wild-type (WT) or mutant Nav1.4 cDNA and the human β1 subunit by calcium phosphate precipitation as previous described (Zhao et al. 2007). The β1 subunit and CD8 were constructed in the piRES vector (piERS/CD8/β1) (Invitrogen Corp., Carlsbad, CA, USA). After preincubation with CD8 antibody-coated beads (Dynabeads M-450 CD8-a) prior to recording, cells expressing the piRES/CD8/β1 bicistronic vector were decorated with CD8 beads. The transfected cells were identified under a light microscope for patch-clamp analyses (Margolskee et al. 1993).
Whole-cell voltage-clamp recordings
Macroscopic Na+ currents from transfected tA201 cells were recorded using the whole-cell configuration of the patch-clamp technique. A correction for the liquid junction potential between the patch pipette and bath solutions (−7 mV) was applied to command pulses before seal formation. Unless indicated otherwise, Na+ current recordings were initiated after an equilibration period of 10 min once the whole-cell configuration was established. Command pulses were generated and currents were recorded using pCLAMP software 10.2 and an Axopatch 200B amplifier with a CV 203BU headstage (Molecular Devices, Sunnyvale, CA, USA). Patch electrodes were fashioned from borosilicate glass (Corning 8161) and coated with silicone elastomer (Sylgard, Dow-Corning, Midland, MI, USA) to minimize stray capacitance. Current recordings were made using low-resistance electrodes (<1 MΩ), and the series resistance was compensated at values ≥80% to minimize voltage-clamp errors. Whole-cell currents were filtered at 5 kHz, digitized at 10 kHz, and stored on a microcomputer equipped with an AD converter (Axon Digidata 1440A, Molecular Devices, Sunnyvale, CA, USA). The current signal was low-pass filtered at 5 kHz and digitalized at a sampling rate of 100 μs during acquisition. Na+ currents were recorded at 10°C, 18°C and 23°C (room temperature). The temperature was controlled using a water bath equipped with a bipolar temperature controller (Model TC-202, Medical Systems Corp., Greenvale, NY, USA) and was measured as close as possible to the cells using a bath sensor (Thermistor BSC-T3, Harvard Apparatus, Holliston, MA, USA). Except where indicated, currents were recorded at 23°C.
Peak currents were measured during a current–voltage protocol. Na+ current densities (pA pF−1) were obtained by dividing the peak current by the cell capacitance. Average I–V curves were obtained by plotting normalized peak currents versus the voltage. For the construction of activation curves, the Na+ conductance (GNa) was calculated from the peak current (INa) using the following equation: GNa = INa/(V–ENa), where V is the test potential and ENa is the reversal potential. The normalized GNa was plotted against the test potential. Steady state activation was fitted to a Boltzmann equation of the following form:
where V1/2 is the voltage at which channels are half-maximally activated and kv is the slope factor. For the construction of inactivation curves, the peak current (I) was normalized relative to the maximal value (Imax) and was plotted against the conditioning pulse potential. Steady-state activation and inactivation curves were fitted to a Boltzmann equation of the following form:
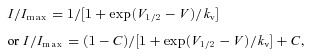 |
where V1/2 is the voltage at which channels are half-maximal inactivated, kv is the slope factor, and C is the non-zero pedestal. Time constants of fast inactivation were assessed by fitting a single exponential curve to the current decay. For the recovery from inactivation or mexiletine block, the test pulse peak current (Itest) was normalized to the corresponding prepulse current (Icont). Itest/Icont was plotted against the interpulse interval and was fitted to a single or double exponential function with a single time constant (τ), or two time constants (τfast and τslow for recovery from fast inactivation, τ1 and τ2 for recovery from mexiletine block).
The overlap between steady-state inactivation and activation has been called the window current. Na+ channels are not inactivated by the mechanism of steady-state inactivation and are available for opening over this window of voltages where the two curves overlap. Using the V1/2 and kv values of voltage-dependent activation and inactivation, the probability of a Na+ channel being within the window was calculated using the following equation:
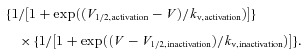 |
The probability of a Na+ channel being a mixture of persistent and window currents was calculated using the following equation:
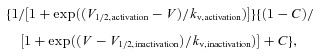 |
where C is a constant representing the fraction of the persistent Na+ current.
Solutions and reagents
For whole-cell patch-clamp recordings of tsA201 cells, the pipette solution contained (in mm): 35 NaCl, 105 CsF, 10 EGTA and 10 Cs-Hepes. The pH was adjusted to 7.4 using 1 n CsOH. The standard extracellular solution generally contained (in mm) 150 NaCl, 2 KCl, 1.5 CaCl2, 1 MgCl2, 10 glucose and 10 Na-Hepes (pH 7.4). The pH was adjusted to 7.4 with 1 n NaOH. To evaluate the effect of variable extracellular concentrations of K+ on the gating properties of WT and M1476I channels, the concentration of KCl in the extracellular solution was increased to 4, 6, or 9 mm. To maintain constant external osmolarity, the concentrations of NaCl were correspondingly decreased to 148, 146, or 143 mm. Stock solutions of 10 mm mexiletine and 10 μm TTX were prepared and were diluted in bath solution. The effects of these drugs were recorded 10 min after application to allow for drug equilibration.
Data analysis and statistics
Data were analysed using a combination of pCLAMP software 10.2 (Molecular Devices), Microsoft Excel, and SigmaPlot for Windows version 11.0 (SPSS, Chicago, IL, USA). Data were expressed as means ± SEM. Statistical significance was determined by comparing control and mutant groups using Student's unpaired t test, and the level of statistical significance was set at P < 0.05.
Results
M1476I mutation induces a persistent Na+ current
A common feature of most naturally occurring Nav1.4 mutations causing PAM is a persistent or non-inactivating Na+ current. To verify the presence of the non-inactivating component in M1476I channels, we measured whole-cell Na+ currents from tsA201 cells transiently expressing WT or M1476I mutant Nav1.4 channels. Representative Na+ currents are shown in Fig. 1A. Compared to WT channels, only a small fraction of M1476I channels were resistant to inactivation (persistent current) during prolonged depolarization (>20 ms). Cells expressing WT and M1476I channels were studied by first constructing normalized peak current versus voltage (I–V) curves (Fig. 1B). Both channel types activated around −70 mV and peaked around −30 mV. On average, we observed no difference in peak current densities for cells expressing WT (523.9 ± 49.2 pA pF−1; n = 21) or M1476I channels (567.1 ± 42.5 pA pF−1; n = 35). The ratio of the persistent current was determined by comparing the current that persisted during the last 20 ms of depolarization to the peak transient current recorded in the same cell. This ratio gave an estimate of the fraction of channels that failed to inactivate. The peak persistent current measured at a −30 mV test potential was 1.5% of the peak current, as shown in the inset of Fig. 1B.
Figure 1. Whole-cell properties of WT and M1476I channels.
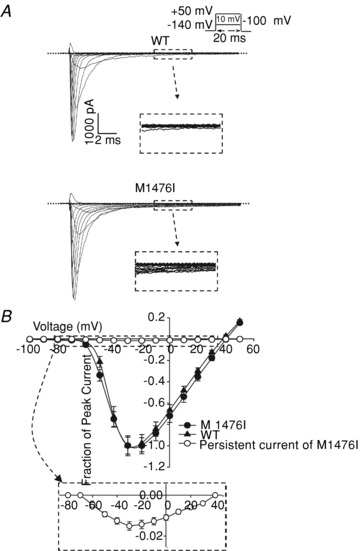
A, representative whole-cell traces recorded from tsA201 cells expressing either WT (top) or M1476I (bottom) Na+ channels. Currents were elicited from a holding potential of –140 mV and were depolarized to potentials ranging from –100 to 50 mV in 10 mV increments lasting 20 ms for each step (protocol shown in the inset above the WT trace). Insets under the WT and M1476I traces show a zoom-in of the persistent current of both channel types. The dashed line represents zero current. B, normalized current/voltage relationships of the WT (▴, n = 21), M1476I (•, n = 35) channels, and a persistent current of the M1476I (⊚, n = 28) channel. The transient WT and M1476I currents were normalized to their peak currents. The persistent currents were calculated as the mean amplitude of the last 20 ms of the test pulse and are expressed as a percentage of the peak transient current recorded from the same cell. The inset shows a zoom-in of the persistent current–voltage relationship of M1476I channels.
M1476I channels exhibit a defective steady-state inactivation
The kinetics of fast inactivation were assessed by fitting the decay of Na+ whole-cell currents elicited at voltages of –50 to +30 mV with a single exponential equation. A defect in the fast inactivation of M1476I channels was seen as a 2- to 3-fold slower time constant than that of WT channels (Fig. 2A) (P < 0.05 or P < 0.01).
Figure 2. Gating properties of WT and M1476I channels and the enlarged window current induced by the M1476I mutation.
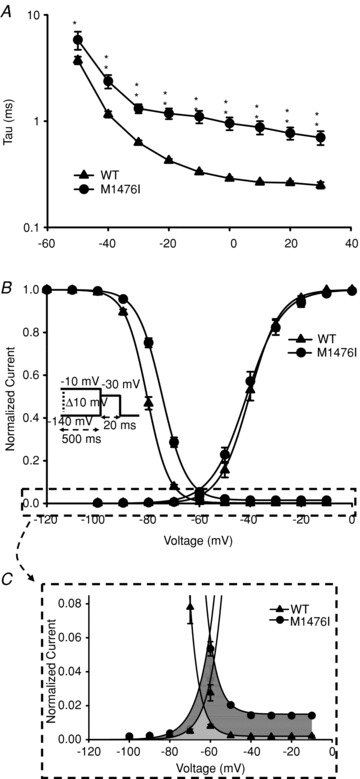
A, voltage dependence of inactivation time constants of the WT (▴, n = 14) and M1476I (•, n = 12) currents. The time course of the current decay elicited at depolarized voltages was best fitted to a single exponential function, and the resulting time constants were plotted versus voltage. B, voltage dependence of activation and inactivation of WT (▴) and M1476I (•) Na+ currents fitted with a Boltzmann equation. Activation curves were generated using the same protocol as shown in Fig. 1A. Steady-state inactivation was determined using a 20 ms test pulse to –30 mV after a 500 ms prepulse to potentials ranging from –140 mV to –10 mV (see inset under inactivation curves for the protocol). The V1/2 and kv values of the activation and inactivation curves are shown in Table 1. C shows a zoom-in of the enlarged window current (the sum of the dark grey and of the grey areas) induced by the M1476I mutation, that is the enlarged overlapping area between the voltage-dependent activation and inactivation curves. The grey area shows the window current induced by WT channels.
The I–V curves shown in Fig. 1B were converted to conductance versus voltage (G–V) curves and were fitted using the Boltzmann function described in Methods. The voltage dependence of activation was unaffected by the M1476I mutation (WT: V1/2 = –40.3 ± 1.2 mV, kv = –5.0 ± 0.2 mV, n = 21; M1476I: V1/2 = –41.7 ± 1.2 mV, kv = –5.2 ± 0.1 mV, n = 35; P > 0.05; Fig. 2B and Table 1). Fast inactivation is a process by which Na+ channels switch to a non-conducting state milliseconds after the onset of depolarization. This process contributes to the repolarization of the action potential. Figure 2B shows that the M1476I mutation caused a 6.4 mV shift in the midpoint (V1/2) of steady-state inactivation in a depolarizing direction (WT: V1/2 = –80.6 ± 0.5 mV, kv = 4.2 ± 0.1 mV, n = 23; M1476I: V1/2 = –74.2 ± 0.5 mV, kv = 4.5 ± 0.04 mV, n = 35; P < 0.01). Unlike WT currents, the fast inactivation of mutant currents was incomplete, and the fraction of non-inactivated (residual) M1476I currents was approximately 1.5% (Fig. 2C). This finding was consistent with the observed persistent Na+ current shown in Fig. 1A and B.
Table 1.
Effects of temperature and extracellular K+ on Nav1.4 WT and M1476I currents
| Recovery from inactivation | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Activation | Inactivation | τ (ms) with 500 ms conditioning pulse | |||||||||
| [K+]o | V1/2 (mV) | Kv (mV) | V1/2 (mV) | Kv (mV) | τ (ms) with 40 ms conditioning pulse | τ/τfast (ms) | τslow (ms) | % of τfast | % of τslow | ||
| 10°C | 2 mm | M1476I | −40.3 ± 1.9 (n = 10) | −6.6 ± 0.4 (n = 10) | −85.6 ± 1.6† (n = 9) | 5.1 ± 0.2 (n = 9) | 2.16 ± 0.4† (n = 10) | 2.18 ± 0.2 (n = 9) | |||
| WT | −40.2 ± 1.8 (n = 8) | −6.2 ± 0.5 (n = 8) | −91.5 ± 1.4 (n = 8) | 4.8 ± 0.1 (n = 8) | 3.8 ± 0.2 (n = 8) | 4.2 ± 0.4 (n = 6) | 139 ± 30 (n = 6) | 95 ± 0.6 (n = 6) | 4.7 ± 0.7 (n = 6) | ||
| 18°C | 2 mm | M1476I | −40.9 ± 1.1 (n = 12) | −5.7 ± 0.3 (n = 12) | −79.6 ± 0.9† (n = 12) | 4.7 ± 0.1* (n = 12) | 0.78 ± 0.04† (n = 12) | 0.9 ± 0.05 (n = 12) | |||
| WT | −39.3 ± 1.0 (n = 9) | −5.8 ± 0.4 (n = 9) | −84.1 ± 0.7 (n = 9) | 4.4 ± 0.1 (n = 9) | 1.8 ± 0.1 (n = 8) | 2.0 ± 0.1 (n = 7) | 111.5 ± 17.3 (n = 7) | 93.4 ± 1.1 (n = 7) | 8.1 ± 0.3 (n = 7) | ||
| 23°C | 2 mm | M1476I | −41.7 ± 1.2 (n = 35) | −5.2 ± 0.1 (n = 35) | −74.2 ± 0.5† (n = 35) | 4.5 ± 0.04† (n = 35) | 0.51 ± 0.03† (n = 27) | 0.65 ± 0.03† (n = 26) | 65.4 ± 3.6 (n = 26) | 90.72 ± 0.5 (n = 26) | 8.8 ± 0.5 (n = 26) |
| WT | −40.3 ± 1.2 (n = 21) | −5.0 ± 0.2 (n = 21) | −80.6 ± 0.5 (n = 21) | 4.2 ± 0.1 (n = 21) | 0.86 ± 0.02 (n = 11) | 1.1 ± 0.1 (n = 11) | 71.7 ± 5.5 (n = 11) | 89.4 ± 0.8 (n = 11) | 10.0 ± 0.6 (n = 11) | ||
| 23°C | 4 mm | M1476I | −42.8 ± 1.4 (n = 7) | −4.7 ± 0.2 (n = 7) | −76.8 ± 1.3† (n = 7) | 4.1 ± 0.1 (n = 7) | 0.54 ± 0.01† (n = 7) | 0.59 ± 0.2† (n = 7) | 76.9 ± 4.8 (n = 7) | 90.8 ± 1.1 (n = 7) | 8.5 ± 1.1 (n = 7) |
| WT | −41.9 ± 1.4 (n = 7) | −5.0 ± 0.7 (n = 7) | −81.5 ± 1.5 (n = 5) | 4.2 ± 0.2 (n = 5) | 0.97 ± 0.03 (n = 5) | 1.1 ± 0.07 (n = 5) | 79.6 ± 4.7 (n = 5) | 87.4 ± 0.6 (n = 5) | 11.7 ± 0.6 (n = 5) | ||
| 23°C | 6 mm | M1476I | −43.6 ± 1.4 (n = 7) | −4.6 ± 0.3 (n = 7) | 77.1 ± 1.0† (n = 7) | 4.1 ± 0.1 (n = 7) | 0.59 ± 0.03† (n = 6) | 0.62 ± 0.04† (n = 6) | 76.1 ± 4.9 (n = 6) | 88.7 ± 1.3 (n = 6) | 10.8 ± 1.1 (n = 6) |
| WT | −42.9 ± 1.0 (n = 6) | −4.8 ± 0.2 (n = 6) | −82.3 ± 1.0 (n = 6) | 4.3 ± 0.2 (n = 6) | 1.2 ± 0.1 (n = 6) | 1.3 ± 0.1 (n = 6) | 79.7 ± 4.3 (n = 6) | 89.3 ± 0.6 (n = 6) | 10.2 ± 0.5 (n = 6) | ||
| 23°C | 9 mm | M1476I | −42.1 ± 1.5 (n = 12) | −4.7 ± 0.3 (n = 12) | −75.6 ± 1.0† (n = 10) | 4.5 ± 0.1 (n = 10) | 0.57 ± 0.03† (n = 7) | 0.68 ± 0.04† (n = 8) | 73.5 ± 5.1 (n = 8) | 87.5 ± 1.2 (n = 8) | 11.6 ± 1.1 (n = 8) |
| WT | −40.3 ± 0.9 (n = 9) | −5.2 ± 0.2 (n = 9) | −82.0 ± 1.6 (n = 9) | 4.3 ± 0.1 (n = 9) | 1.1 ± 0.1 (n = 7) | 1.3 ± 0.1 (n = 7) | 75.9 ± 3.1 (n = 7) | 88.2 ± 1.2 (n = 7) | 11.3 ± 1.2 (n = 7) | ||
P < 0.5,
P < 0.01, data was significantly different for mutant channels when compared to WT.
M1476I increased the Na+ window current
The window current results from the overlapping area of the activation and inactivation curves that identifies a voltage range (window) in which Na+ channels are not inactivated by the mechanism of steady-state inactivation and are available for opening by depolarization (Cummins et al. 1993). In the present study, the shift of the inactivation curve of M1476I channels along the voltage axis resulted in an increased overlap between steady-state activation and steady-state inactivation. This increased window current, plus the residual Na+ current during steady-state inactivation, predicted that a larger fraction of M1476I channels would be available to open in the voltage range of –80 to –10 mV (Fig. 2B). Figure 3A shows a plot of the probability of Na+ channels opening at the window or at more depolarized voltages calculated from the V1/2 and kv values of the fitted activation and inactivation curves. The resulting probability was a biphasic function of voltage, with a combination of two components: a relatively small component with a peak near –70 mV and a larger component at depolarized voltages greater than –50 mV. This biphasic function could be separated into a window component and a persistent component, as shown in Fig. 3B and C. The window component was defined by the overlap of the Na+ channel activation and inactivation curves. Compared to the WT, the M1476I mutation shifted the peak toward a more depolarized voltage (WT: –74 mV vs. M1476I: –66 mV) and enlarged the window, as indicated by a 6-fold increase in the probability of channel opening at the peak current (WT: 0.02%vs. M1476I: 0.13%). To characterize a ‘pure’ persistent component, we subtracted the window component shown in Fig. 3B from the biphasic function shown in Fig. 3A. The persistent component occurred over a more depolarized range of voltages (>–50 mV). For WT channels, the persistent component increased over the range of voltages where Na+ channels activate and reached a peak probability of 0.19% at voltages more depolarized than –30 mV. The M1476I mutation increased the peak probability of channel opening 8-fold. The increased window and persistent current caused by the mutation would tend to depolarize skeletal muscle cells in PAM patients.
Figure 3. Effects of the M1476I mutation on the window current.
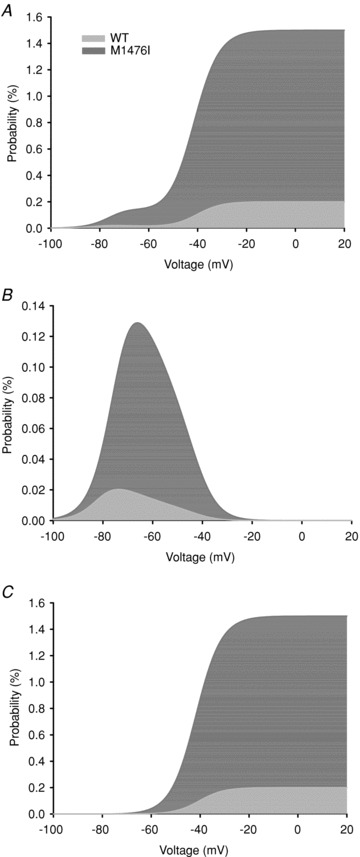
The overlap of the activation and inactivation curves defines a range of voltages or window where Na+ channels are non-inactivated and are available for activation. A depolarizing shift of the inactivation curve caused by the M1476I mutation increased the overlap, resulting in an increased window current. A shows the probability of a M1476I channel being non-inactivated and available for activation at depolarized voltages calculated using the equation described in Methods. This probability is a biphasic function of voltage, with a peak at around –70 mV and at depolarized voltages greater than –50 mV. The contribution of the window component plotted in B was calculated using the equation described in Methods. The M1476I mutation enhanced the peak opening probability of the channel being in the window by 6-fold and shifted the peak in a more depolarizing direction. C is the contribution of the persistent component calculated by subtracting the window component shown in B from A. The M1476I mutation increased the amplitude of the persistent current by 7-fold but did not alter the onset of the persistent component.
M1476I accelerated recovery from fast inactivation
Recovery from fast inactivation determines the time required for inactivated Na+ channels to move back to the active state. It thus limits the maximal firing rate and permits repetitive firing of muscle cells. Recovery from fast inactivation was assessed using a double-pulse protocol (see inset of Fig. 4). Cells were held at –140 mV, depolarized by a 40 ms or 500 ms conditioning pulse to –30 mV, and then repolarized to the recovery potential for increasing durations. With a 40 ms conditioning pulse, a single-exponential function was sufficient for fitting WT and M1476I currents, yielding only a single time constant (τ). In contrast, a double exponential equation was required to obtain an accurate fit with a 500 ms conditioning pulse and yielded two time constants (τfast and τslow). The τfast had a relatively large weight (∼90%) compared with the τslow (∼10%). We thus used τfast to compare WT and M1476I channels. The time constants of M1476I channels under these two conditioning pulses were significantly lower (τ or τfast) (Fig. 4 and Table 1), indicating that the M1476I mutation accelerated recovery from inactivation, as is the case for other PAM and PC-causing mutations (Chahine et al. 1994; Mitrovic et al. 1994, 1995).
Figure 4. Recovery from inactivation of WT and M1476I Na+ channels.
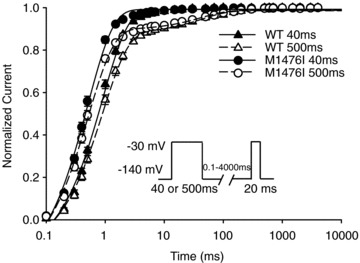
From a holding potential of –140 mV, the cells were depolarized to –30 mV for 40 ms (WT: ▴, n = 11; M1476I: •, n = 27) or 500 ms (WT: ▵, n = 11; M1476I: ⊚, n = 26) to inactivate all the Na+ channels and then repolarized to various recovery potentials. The time course of recovery from inactivation with a 40 ms conditioning pulse was well fitted by a single exponential equation, yielding a single time constant (τ), whereas a double exponential equation was required to obtain a good fit with a 500 ms conditioning pulse, yielding two time constants (τfast and τslow). The values of the time constants are listed in Table 1.
Elevating extracellular K+ concentrations had no effect on either M1476I or WT currents
In PAM patients, myotonia attacks are precipitated by elevated serum K+ concentrations (Jurkat-Rott et al. 2010). We thus examined the effects of different extracellular K+ concentrations on the expression levels and gating properties of WT and M1476I channels. Elevating extracellular K+ concentrations from 2 mm to 4 mm, 6 mm, or 9 mm had no detectable effect on the peak current amplitudes of WT or M1476I currents. In addition, the ratio of persistent current, voltage dependence of activation and fast inactivation, and the time course of recovery from fast inactivation were similar at various concentrations of extracellular K+ (Table 1).
Effects of temperature
One of the most distinctive clinical features of patients carrying the M1476I mutation is the exacerbation of muscle stiffness by cold, while it is not a cardinal symptom of PAM (Lehmann-Horn & Jurkat-Rott, 2007). We thus investigated the effect of cooling on the biophysical properties of WT and M1476I channels. Compared to current traces recorded at 23°C (Fig. 1A), lowering the temperature to 10°C or 18°C slowed the kinetics of both WT and M1476I channels (Fig. 6A). As soon as the whole-cell configuration was established, WT and M1476I channels both exhibited a significant increase in the persistent Na+ current at lower temperatures, which was more marked with the mutant channels and reached a maximum at 10°C (data not shown). Interestingly, persistent Na+ currents ‘ran down’ over time. For example, for M1476I channels at 10°C, the persistent current measured at a –30 mV test potential 1 min after the whole-cell configuration was established was 13.6 ± 1.1% (n = 12), then dropped to 6.7 ± 1.1% (n = 11) after 5 min, and remained stable at around 3.5% after 10 min (10 min: 3.7 ± 0.6%, n = 11; 20 min: 3.1 ± 0.3%, n = 8). The same significant alterations in persistent currents were also observed at 18°C, but were less prominent than at 10°C (Fig. 5A).
Figure 6. Effects of Na+ channel blockers on persistent Na+ currents.
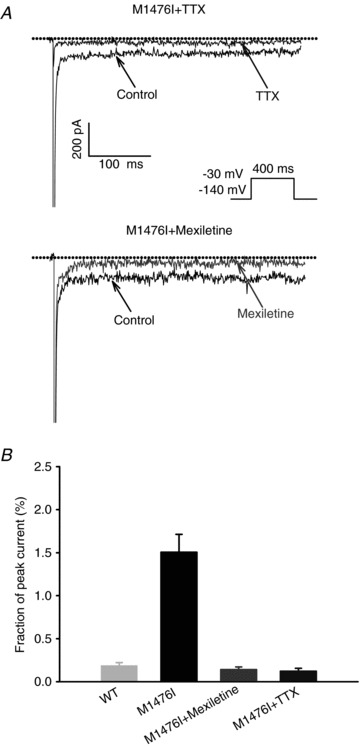
A, effects of 50 nm TTX (top), or 100 μm mexiletine (bottom) on persistent Na+ currents. The dashed line represents zero current. The persistent Na+ currents were induced by a depolarizing step to –30 mV for 400 ms from a holding potential of –140 mV (see protocol in inset). The top current in A represents the blocking effect of TTX, and the top current in B represents the blocking effect of mexiletine. B, bar graph showing the fraction of persistent Na+ currents before and after the administration of Na+ channel blockers.
Figure 5. Effects of temperature on WT and M1476I channels.
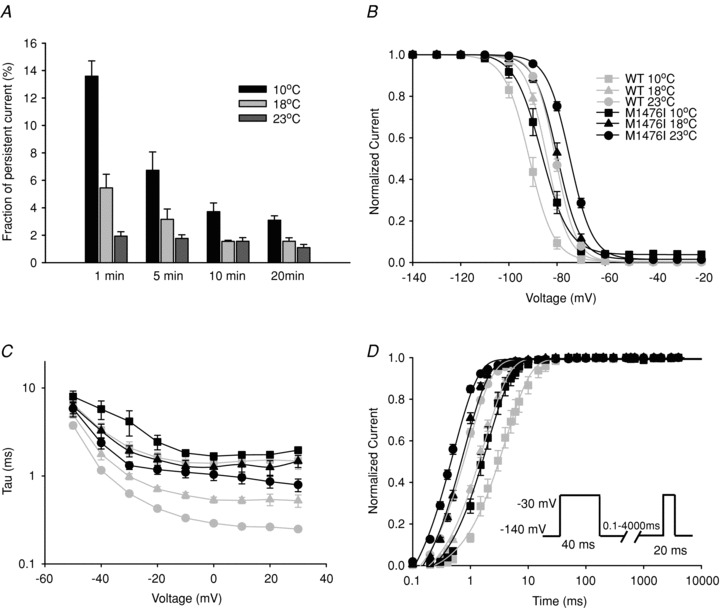
A, histogram showing the amplitudes of the persistent currents of M1476I channels recorded at different time points (1 min, 5 min, 10 min and 20 min) after the whole-cell configuration was established at 10°C, 18°C and 23°C. The persistent currents of the M1476I channel appeared to run down and then become stable 10 min after the whole-cell configuration was established. The effects of cooling on steady-state inactivation, the decay kinetics of the Na+ current, and the recovery from inactivation are shown in B, C, and D. The functional properties at room temperature (23°C) (WT: ○; M1476I: •) are also shown in order to compare the changes caused by lowering the temperature to 10°C (WT: □; M1476I: ▪) or 18°C (WT: ▵; M1476I: ▴). The voltage dependence of steady-state fast inactivation of WT and M1476I channels at different temperatures was recorded using the protocol in Fig. 2B. V1/2 and kv inactivation values are listed in Table 1. C, voltage-dependent time constants of inactivation of WT and M1476I channels at 10°C, 18°C and 23°C. The time course of the current decay elicited at depolarized voltages was best fitted to a single exponential function, and the resulting time constants were plotted versus voltage. D, recovery from inactivation with a 40 ms conditioning pulse at 10°C, 18°C and 23°C. The protocol is shown in the inset, and values of time constants are listed in Table 1.
At room temperature, M1476I channels activated at the same negative potentials as WT channels, as reflected in the absence of a shift in V1/2 of steady-state activation (Fig. 2B). Similarly, no significant shift was observed in the V1/2 of activation for M1476I channels compared to WT channels at 10°C or 18°C, suggesting that the opening of channels was unaffected by the cooling or the M1476I mutation (Table 1).
The effects of cooling on two main characteristics of fast inactivation were also investigated, that is the Voltage-dependence of steady-state inactivation and the time constant of inactivation. The V1/2 of steady-state inactivation was shifted in a similar manner for both WT and M1476I channels in a hyperpolarized direction by cooling from 23°C to 18°C and 10°C: for WT from –80.6 ± 0.5 mV (23°C, n = 23) to –84.1 ± 0.7 mV (18°C, n = 9) and –91.5 ± 1.4 mV (10°C, n = 8), and for M1476I from –74.2 ± 0.5 mV (23°C, n = 35) to –79.6 ± 0.9 mV (18°C, n = 12) and –85.6 ± 1.6 mV (10°C, n = 9) (Fig. 5B and Table 1). Cooling thus caused a similar hyperpolarizing shift in steady-state inactivation for both channel types. The inactivation time constants were significantly slower for both channel types when the recording solution was cooled (Fig. 5C). At a test potential of –30 mV, for example, WT channels inactivated with time constants of 0.6 ± 0.03 ms (23°C, n = 14), 1.0 ± 0.1 ms (18°C, n = 11), and 2.1 ± 0.4 ms (10°C, n = 8), while M1476I channels inactivated with time constants of 1.3 ± 0.1 ms (23°C, n = 12), 1.9 ± 0.3 ms (18°C, n = 16), and 4.2 ± 1.3 ms (10°C, n = 11).
Figure 5D shows a plot of the time course of recovery from inactivation for WT and M1476I channels at 10°C, 18°C and 23°C with a 40 ms conditioning pulse. Both channel types exhibited slower recovery kinetics at lower temperatures. Time constants of recovery from inactivation at 10°C were significantly larger than those at 18°C, while the time constants at 18°C were larger than those at 23°C. Similar alterations were also observed for recovery from inactivation with a 500 ms conditioning pulse at 10°C, 18°C and 23°C (Table 1).
Effects of mexiletine
As mentioned above, the persistent current and the rapid recovery from inactivation caused by the M1476I mutation appeared to underlie the PAM phenotype. Mexiletine showed promising potential for the treatment of patients carrying this mutation. We were interested in the therapeutic mechanism of mexiletine. We first assessed the ability of mexiletine to reduce the amplitude of the persistent current exhibited by M1476I channels, which may be the basis for its therapeutic efficacy (Wang et al. 1997). The persistent current recorded from M1476I channels was significantly inhibited after a 100 μm mexiletine treatment for 10 min (Fig. 6, bottom panel). In general, the persistent current was reduced by approximately 90% (Fig. 6B). At the same time, the block of the peak current in M1476I channels was minimal, unlike the total block of the current caused by a 10 min treatment with 50 nm TTX (Fig. 6A, top panel).
Many local anaesthetics and antiarrhythmic drugs act by preferentially binding to the inactivated state of Na+ channels, which is characterized by the use-dependent block elicited by repetitive stimulation (Hille, 2001). We measured the use-dependent block of WT and M1476I currents by mexiletine with a series of 50 depolarizing pulses to –30 mV at different frequencies (from 2 to 50 Hz) for 10 ms. Peak currents were measured, normalized to the amplitude of the first pulse, and plotted against the pulse number. In the absence of mexiletine, there was no decrease in the current amplitude for the two channel types at frequencies up to 50 Hz. In the presence of mexiletine, the use-dependent block increased with higher frequencies. The steady-state level of block was significantly lower for M1476I channels than for WT channels (Fig. 7B). Figure 7A shows that the use-dependent block by mexiletine reached a steady-state level after a few depolarizing pulses at 50 Hz. The peak current amplitude of the last pulse normalized to the first pulse (P50/P1) indicated a use-dependent block of 49.6 ± 1.9% for WT (n = 13) and 55.2 ± 1.6% for M1476I (n = 14). We next investigated whether the reduced use-dependent block in M1476I channels was due to: (1) a lower affinity of mexiletine for the inactivated state; (2) a faster recovery from mexiletine block; or (3) the slower onset of mexiletine block.
Figure 7. Use-dependent inhibition by mexiletine.
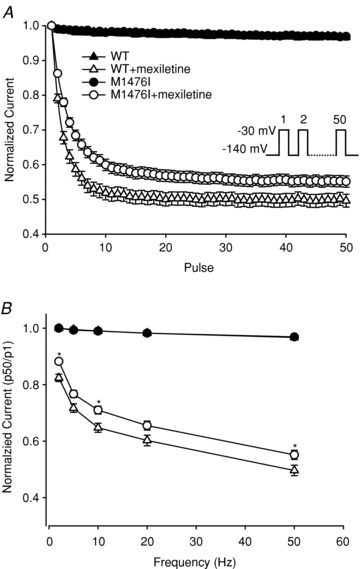
A, to record the use-dependent inhibition of WT and M1476I channels by 100 μm mexiletine, a 50-pulse train was applied at –30 mV for 10 ms from a holding potential of –140 mV at 50 Hz. Each normalized peak current was plotted against the pulse number. B, relative amplitudes of the 50th sweep (P50/P1) with and without mexiletine at different pulse frequencies (2–50 Hz). Before the mexiletine treatment, there were no differences in normalized currents between WT (▴, n = 11) and M1476I (•, n = 19) channels. After the mexiletine treatment, the normalized currents of both WT (▵, n = 12) and M1476I (○, n = 16) were inhibited. However, the steady-state level of inhibition was lower for M1476I at different frequency stimuli ranging from 2 Hz to 50 Hz (*P < 0.05 compared to WT+mexiletine).
The affinity of mexiletine for the inactivated state was estimated by observing the effect of mexiletine on steady-state inactivation. Figure 8A is a composite figure showing the voltage dependence of steady-state activation and inactivation of WT and M1476I channels expressed in tsA201 cells. Adding 100 μm mexiletine did not affect the peak current density (data not shown), but significantly shifted the voltage-dependent activation and inactivation curves of both WT and M1476I channels in a hyperpolarizing direction. The extent of shifts in the activation and inactivation curves induced by mexiletine was similar for WT and M1476I channels, indicating that the M1476I mutation does not interfere with the affinity of mexiletine for the inactivation state.
Figure 8. Effects of mexiletine on the gating properties of WT and M1476I channels.
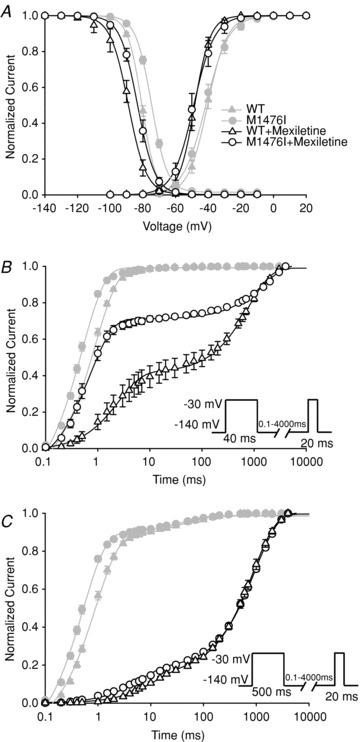
The properties in the absence of 100 μm mexiletine (WT: ▵; M1476I: ○) are also shown in order to compare the changes induced by mexiletine (WT: ▵; M1476I: ⊚). A, voltage dependence of activation and inactivation of WT and M1476I channels in the absence or presence of 100 μm mexiletine. Currents were evoked using the same protocol as in Fig. 1A (activation) and Fig. 2B (inactivation). The grey curves representing the activation and inactivation curves of WT and M1476I channels in the absence of mexiletine are the same as those shown in Fig. 2B in order to compare the blocking effect of mexiletine. Recovery from inactivation and mexiletine block was assessed using standard double-pulse protocols with 40 ms (B) or 500 ms conditioning pulses (C). Protocols are given in the insets under the recovery vs. time curves of B and C. The black smooth curves were fitted to a double exponential equation with two time constants (τ1 and τ2). The grey curves shown in B and C represent the recovery from inactivation in the absence of mexiletine in order to compare the blocking effect of this drug. The V1/2 and kv values of steady-state activation and inactivation, and the values of time constants of recovery from inactivation and mexiletine block are listed in Table 2.
In terms of recovery from inactivated-state block after a treatment with 100 μm mexiletine, double exponentials were required to describe the recovery time courses and yielded two time constants (τ1 and τ2). With a brief conditioning pulse (40 ms) at –30 mV, the fast time constants (τ1) remained similar to those (τ in Table 1) measured in the absence of mexiletine. With this conditioning pulse, the recovery from fast inactivation had fully developed, and the contribution of slow inactivation was minimal (Fig. 4). The additional slow time constant (τ2) thus represented the recovery from mexiletine block. Compared to WT channels, M1476I channels exhibited both accelerated recovery from mexiletine block and lower levels of mexiletine block. The level of block was approximately 60% for WT channels and only about 30% for M1476I channels in the same conditions (Fig. 8B and Table 2). With a 500 ms conditioning pulse at –30 mV, the level of mexiletine block was similar for both WT and M1476I channels (75%). The fast time constants (τ1) had a relatively small weight (<18%), corresponding to a mixture of the fast and slow components of recovery from inactivation (τfast and τslow), as shown in Fig. 4. The slow time constant (τ2) showing the recovery from mexiletine block was not significantly different between WT and mutant channels with a 500 ms conditioning pulse (Fig. 8C and Table 2). With shorter conditioning pulse durations (i.e. 40 ms), M1476I channels had less use-dependent block (i.e. showed greater recovery within 20 ms) compared to WT channels. The use-dependent block was similar for both channels types with longer conditioning pulse (i.e. 500 ms), suggesting a slower onset of mexiletine block for M1476I channels compared to WT channels. Since the affinity of mexiletine for the inactivated state was the same for WT and M1476I channels (Fig. 5A), the decreased use-dependent block (Fig. 7A and B) of M1476I channels may result from the slower onset of mexiletine block, and/or the faster recovery from mexiletine block of mutant channels.
Table 2.
Effects of mexiletine on Nav1.4 WT and M1476I currents
| Recovery from mexiletine block | ||||||||
|---|---|---|---|---|---|---|---|---|
| Activation | Steady-state inactivation | 40 ms conditioning pulse | 500 ms conditioning pulse | |||||
| V1/2 (mV) (n) | kv (mV) (n) | V1/2 (mV) (n) | kv (mV) (n) | τ1 (ms) Fraction τ1 (%) (n) | τ2 (ms) Fraction τ2 (%) (n) | τ1 (ms) Fraction τ1 (%) (n) | τ2 (ms) Fraction τ2 (%) (n) | |
| M1476I | −48.7 ± 1.4 (10) | −4.8 ± 0.2 (10) | −83.1 ± 1.3† (15) | 4.5 ± 0.1* (15) | 0.75 ± 0.01† | 530.8 ± 54.4* | 6.93 ± 0.7† | 867.38 ± 45.1 |
| 35.9 ± 2.3 | 71.0 ± 1.5 | 18.4 ± 0.7 | 82.4 ± 0.9 | |||||
| (12) | (12) | (10) | (10) | |||||
| WT | −48.0 ± 1.3 (9) | −4.6 ± 0.2 (9) | −88.6 ± 0.8 (14) | 4.2 ± 0.1 (14) | 3.5 ± 0.4 | 700.8 ± 51.2 | 11.4 ± 1.0 | 963.8 ± 37.7 |
| 41.1 ± 3.7 | 61.2 ± 3.5 | 15.1 ± 1.0 | 84.9 ± 1.2 | |||||
| (10) | (10) | (10) | (10) | |||||
P < 0.5,
P < 0.01, data were significantly different for mutant channels when compared to WT.
Discussion
Genotype–phenotype relationships
M1476I currents measured in tsA201 cells revealed an increased persistent current, a slower current decay, a more positive midpoint voltage of fast inactivation, and an accelerated recovery from fast inactivation. A depolarizing shift in steady-state inactivation produces an enhanced overlap between activation and inactivation curves, which leads to an increased window current. The accelerated recovery from fast inactivation in M1476I channels indicated that they had a shorter refractory period after the action potential than WT channels. These gain-of-function defects of M1476I channels observed in our study were consistent with those of other PAM mutations (Mitrovic et al. 1995; Hayward et al. 1996; Lerche et al. 1996; Richmond et al. 1997; Green et al. 1998). The functional disruption of M1476I channels enhances the excitability of the cell membrane, which increases the movement of Na+ ions into skeletal muscle cells. This extra Na+ influx is enough to change the membrane potential and trigger prolonged muscle contractions, which underlie episodes of myotonia characteristic of PAM (Cannon, 1996).
Both the PAM mutation M1476I (Rossignol et al. 2007) and the adjacent PMC mutation F1473S (Fleischhauer et al. 1998) reside in the S4–5 cytoplasmic loop of domain IV, a docking site of the inactivation gate according to the ‘hinged lid’ mechanism (Goldin, 2003). These two mutations induce an increase in the number of non-inactivating Na+ channels and a depolarizing shift in the fast inactivation, indicating that the DIV/S4–5 cytoplasmic loop is involved in the inactivation gating of Na+ channels.
The varying extents of depolarization caused by defective inactivation may explain how these mutations produce different clinical phenotypes. The F1473S mutation exhibits a more pronounced disruption of fast inactivation than the M1476I mutation (M1476I: 1.6% persistent current, 6.4 mV rightward shift of fast inactivaton vs. F1473S: 1.9% persistent current, 18 mV rightward shift of inactivation). A slight depolarization caused by PAM mutations (e.g. M1476I in our study) results in a long-lasting hyperexcitability of the membrane, which causes muscle stiffness, an intermediate depolarization caused by PMC mutations (e.g. F1473S) results in muscle stiffness followed by weakness (Fleischhauer et al. 1998), and a strong depolarization induced by HyperPP mutations (e.g. T698M) results in membrane hypoexcitability and paralysis (Cummins & Sigworth, 1996).
Role of extracellular K+ in PAM
Myotonia is provoked or aggravated by K+ loading in patients with PAM mutations (Heine et al. 1993; Ricker et al. 1994; Kubota et al. 2009). While a K+-loading test was not performed during the diagnosis of patients carrying the M1476I mutation (Rossignol et al. 2007), we examined the effects of elevated extracellular K+ concentrations on M1476I channels to determine whether the biophysical defects of this mutation are also K+ sensitive. Increasing extracellular K+ from 2 mm to 4 mm, 6 mm, or 9 mm had no effect on the gating properties of either WT or M1476I channels. Over 10 Nav1.4 mutations associated with PAM have been reported to date, and muscle stiffness is aggravated by K+ in patients with these mutations. Of these mutations, only G1306E has been reported to be sensitive to the elevation of extracellular K+ concentrations, with a 4-fold increase in extracellular K+ from 4 mm to 16 mm causing a 10–20% slowing of current decay and an increase in the amplitude of the persistent current (Hayward et al. 1996). However, a study by Mitrovic et al. (1995) using the same mutation and the same expression system failed to detect any effect of high extracellular K+ concentrations on WT and M1476I channels. The mechanisms by which elevated serum K+ concentrations trigger myotonia attacks remain to be determined. High extracellular K+ concentrations (7–9 mm) have been shown to enhance depolarization in intact muscle fibres by decreasing the resting membrane potential from –80 to –63 mV (Lehmann-Horn et al. 1987). The depolarized voltage induced by the elevation in extracellular K+ concentrations in T-tubules incorporates the non-inactivated component exhibited by mutant Nav1.4 channels and may trigger repetitive after-discharges following driven action potentials. After-discharges cause stiffness or delayed relaxation of muscles in patients with myotonia. This hypothesis is supported by mathematical models of muscle cells. For example, S804F is a PAM mutation that causes fast inactivation abnormalities. A model of muscle cell containing this mutation exhibits enhanced excitability but does not display after-discharges unless the extracellular K+ concentration is raised to 5.5 mm (Hayward et al. 1996; Green et al. 1998; Cannon, 2000).
Cold-induced myotonia
Typically, myotonia in patients carrying the M1476I mutation is aggravated when the patients are exposed to cold temperatures. However, these patients exhibit a PAM phenotype, which is not very sensitive to cold (Lehmann-Horn & Jurkat-Rott, 2007). How then does this Na+ channel mutation cause a peculiar phenotype of PAM with unusual sensitivity to cold? Cold-induced stiffness and subsequent weakness are in fact characteristic features of PMC. PMC mutations lead to biophysical defects that are clearly pathogenic at low temperatures (Jurkat-Rott et al. 2010). Experimental cooling of mutant PMC channels results in a slowing of the kinetics, a hyperpolarizing shift in the steady-state inactivation, and an increase in the amplitude of persistent currents (Lerche et al. 1996; Fleischhauer et al. 1998; Carle et al. 2009). The same changes in fast inactivation and persistent currents were also observed with our cold-sensitive M1476I mutation as well as with other PAM mutations that are not sensitive to cold (Hayward et al. 1996). These changes are thus not sufficient to explain why myotonias in M1476I carriers are aggravated by cold. One possible mechanism might be that the both the slowing of fast inactivation and the increased persistent current have to reach a certain threshold to trigger myotonia at low temperatures (Hayward et al. 1996; Lerche et al. 1996). In the present study, cooling slowed the kinetics for both WT and M1476I currents (Fig. 5C). However, M1476I channels displayed a slower inactivation than WT channels at all temperatures investigated, as is the case for other PAM-causing mutations (Mitrovic et al. 1994; Hayward et al. 1996; Green et al. 1998). While cooling markedly increased the persistent Na+ current of the M1476I channel, it ‘run down’ over time, reaching 13.6 to 3.1% at the lowest temperature (10°C) 1–20 min after the whole-cell configuration was established. This ‘run down’ of the persistent current has also been reported for some Nav1.4 mutations associated with the HyperPP phenotype such as I1495F and T698M (Cummins & Sigworth, 1996; Bendahhou et al. 1999). A persistent current up to 2% is sufficient to elicit repetitive spikes in muscles (myotonia) in the computer model of Cannon et al. whereas a state of depolarization block (paralysis) occurs when the amplitude of the persistent current exceeds a certain level, as in HyperPP mutations (Cannon et al. 1993). Based on this model, the large persistent current observed in our study at 23°C (1.1–1.9%) was close to the ‘borderline’ of myotonia.
Other temperature-dependent processes, such as the activity of the Na+,K+-ATPase, can also affect membrane excitability and trigger myotonia. Cooling decreased the activity of the Na+,K+-ATPase, resulting in a decreased Na+ ion efflux (accumulation of Na+ ions inside the muscle fibres) and a decreased K+ ion influx. Cooling from 37 to 13°C induced up to 20 mV depolarization in rat skeletal myotubes (Dlouha et al. 1980; Brodie et al. 1987). In addition, compared with PMC and PAM mutations, cooling induces different defects of Na+ channels gating in Nav1.4 mutations associated with a paralytic phenotype. For example, the P1158S mutation responsible for cold-induced paralysis and myotonia is located in the DIII/S4–5 cytoplasmic loop (Sugiura et al. 2003). Lowering the temperature from 37°C to 25°C induced a hyperpolarizing shift in activation and a depolarizing shift in slow inactivation, but fast inactivation was not substantially affected. The destabilization of slow inactivation in combination with a defective activation of P1158S channels is expected to increase the risk of prolonged membrane depolarization and leads to loss of excitability with paralysis (Webb & Cannon, 2008).
Effects of mexiletine
The electrophysiological basis of myotonia is repetitive action potentials following voluntary contraction. Multiple defects in channel gating caused by SCN4A mutations have been identified as important mechanisms. The therapeutic effect of antimyotonic drugs such as mexiletine is commonly attributed to their well-known use-dependent block of Na+ channels that prevents repetitive firing of action potentials (Lehmann-Horn & Jurkat-Rott, 1999). In the present study, mexiletine induced a pronounced use-dependent block in both WT and M1476I channels, albeit to a lesser extent in the mutant channels. Mexiletine stabilizes inactivation by causing a hyperpolarizing shift in the steady-state inactivation curves, which is similar for both WT and M1476I channels, suggesting that the M1476I mutation does not substantially alter the affinity of mexiletine in the inactivated state. Thus, the decreased use-dependent block of M1476I channels may due to two mechanisms: a slower onset of mexiletine block (Fig. 8B and C), and/or a fast recovery from mexiletine block (Fig. 8B). The M1476I mutation may accelerate the recovery by facilitating the untrapping of mexiletine from the non-inactivating mutant, as previously described for flecainide in Nav1.5 channels (Ramos & O'Leary, 2004).
Since the use-dependent phenomenon is non-specific for most class 1B local anaesthetics (Lehmann-Horn & Jurkat-Rott, 1999), it is unclear why mexiletine is more efficacious than similar drugs for treating M1476I carriers (Rossignol et al. 2007). One possible explanation is that mexiletine has a 20-fold higher affinity for the open state than for the inactivated state of Nav1.4 channels, with an IC50 of 3.3 μm for the open state vs. 67.8 μm for the inactivated state. At therapeutic concentrations (2.8–11 μm), mexiletine preferentially blocks persistent late open channels (Wang et al. 2004). This open-channel block of mexiletine by selectively targeting persistent Na+ currents may be the therapeutic basis of antiarrhythmic drugs (Wang et al. 1997; Huang et al. 2011) and may also play a dominant role in preventing the repetitive firing of action potentials that cause muscle stiffness. Further studies are needed to determine why mexiletine is more efficacious in treating patients carrying this mutation.
Altogether, the present study indicated that cold-induced myotonia, a phenotypic peculiarity of the M1476I mutation of Nav1.4 channels, may be explained by a defective inactivation that increases Na+ influx, triggers depolarizations of muscle fibres, and induces membrane hyperexcitability and muscle stiffness. Cooling further enhances the abnormalities of fast inactivation of the mutant channels. The clinical efficacy of mexiletine in treating M1476I carriers may be partly due to the open-channel block targeting the persistent Na+ currents generated by the M1476I mutation, with the exception of the use-dependent block.
Acknowledgments
This work was supported by grants from the Heart and Stroke Foundation of Quebec and Canadian Institutes of Health Research Grant MT-13181. The authors wish to thank Valerie Pouliot for her technical assistance.
Glossary
Abbreviations
- PAM
potassium-aggravated myotonia
- PMC
paramyotonia congenita
Author contributions
J.Z. performed the experiments involving the biophysical characterization, analyzed the data, and helped to prepare the manuscript. N.D. and J.P. were at the origin the clinical aspects of the project, recruited family members, genotyped the index patient and his family. M.C. designed the project and experiments, performed some current measurements, analyzed and interpreted the data, and drafted the manuscript.
The work was done at Le Centre de recherche en neurosciences, Institut universitaire en santé mentale de Québec, 2601 Chemin de la Canardière, Quebec, QC, G1J 2G3, Canada
References
- Bendahhou S, Cummins TR, Tawil R, Waxman SG, Ptacek LJ. Activation and inactivation of the voltage-gated sodium channel: role of segment S5 revealed by a novel hyperkalaemic periodic paralysis mutation. J Neurosci. 1999;19:4762–4771. doi: 10.1523/JNEUROSCI.19-12-04762.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie C, Bak A, Shainberg A, Sampson SR. Role of Na-K ATPase in regulation of resting membrane potential of cultured rat skeletal myotubes. J Cell Physiol. 1987;130:191–198. doi: 10.1002/jcp.1041300204. [DOI] [PubMed] [Google Scholar]
- Cannon SC. Spectrum of sodium channel disturbances in the nondystrophic myotonias and periodic paralyses. Kidney Int. 2000;57:772–779. doi: 10.1046/j.1523-1755.2000.00914.x. [DOI] [PubMed] [Google Scholar]
- Cannon SC. Ion-channel defects and aberrant excitability in myotonia and periodic paralysis. Trends Neurosci. 1996;19:3–10. doi: 10.1016/0166-2236(96)81859-5. [DOI] [PubMed] [Google Scholar]
- Cannon SC, Brown RH, Jr, Corey DP. Theoretical reconstruction of myotonia and paralysis caused by incomplete inactivation of sodium channels [see comments] Biophys J. 1993;65:270–288. doi: 10.1016/S0006-3495(93)81045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carle T, Fournier E, Sternberg D, Fontaine B, Tabti N. Cold-induced disruption of Na+ channel slow inactivation underlies paralysis in highly thermosensitive paramyotonia. J Physiol. 2009;587:1705–1714. doi: 10.1113/jphysiol.2008.165787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Chahine M, George AL, Jr, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Chahine M, Ziane R, Vijayaragavan K, Okamura Y. Regulation of Nav channels in sensory neurons. Trends Pharmacol Sci. 2005;26:496–502. doi: 10.1016/j.tips.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Sigworth FJ. Impaired slow inactivation in mutant sodium channels. Biophys J. 1996;71:227–236. doi: 10.1016/S0006-3495(96)79219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Zhou J, Sigworth FJ, Ukomadu C, Stephan M, Ptácek LJ, Agnew WS. Functional consequences of a Na+ channel mutation causing hyperkalemic periodic paralysis. Neuron. 1993;10:667–678. doi: 10.1016/0896-6273(93)90168-q. [DOI] [PubMed] [Google Scholar]
- Dlouha H, Donselaar Y, Teisinger J, Vyskocil F. Effect of temperature and ouabain on th Na+–K+ activated membrane ATPase and electrogenic ionic pump of the golden hamster and mouse diaphragm. Physiol Bohemoslov. 1980;29:543–552. [PubMed] [Google Scholar]
- Ebers GC, George AL, Barchi RL, Ting-Passador SS, Kallen RG, Lathrop GM, Beckmann JS, Hahn AF, Brown WF, Campbell RD, et al. Paramyotonia congenita and hyperkalemic periodic paralysis are linked to the adult muscle sodium channel gene. Ann Neurol. 1991;30:810–816. doi: 10.1002/ana.410300610. [DOI] [PubMed] [Google Scholar]
- Fleischhauer R, Mitrovic N, Deymeer F, Lehmann-Horn F, Lerche H. Effects of temperature and mexiletine on the F1473S Na+ channel mutation causing paramyotonia congenita. Pflugers Arch. 1998;436:757–765. doi: 10.1007/s004240050699. [DOI] [PubMed] [Google Scholar]
- Goldin AL. Mechanisms of sodium channel inactivation. Curr Opin Neurobiol. 2003;13:284–290. doi: 10.1016/s0959-4388(03)00065-5. [DOI] [PubMed] [Google Scholar]
- Green DS, George AL, Jr, Cannon SC. Human sodium channel gating defects caused by missense mutations in S6 segments associated with myotonia: S804F and V1293I. J Physiol. 1998;510:685–694. doi: 10.1111/j.1469-7793.1998.685bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward LJ, Brown RH, Jr, Cannon SC. Inactivation defects caused by myotonia-associated mutations in the sodium channel III-IV linker. J Gen Physiol. 1996;107:559–576. doi: 10.1085/jgp.107.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine R, Pika U, Lehmann-Horn F. A novel SCN4A mutation causing myotonia aggravated by cold and potassium. Hum Mol Genet. 1993;2:1349–1353. doi: 10.1093/hmg/2.9.1349. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. 3rd edn. Sunderland, MA: Sinauer Associates, Inc; 2001. [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF, Katz B. Measurement of current-voltage relations in the membrane of the giant axon of Loligo. J Physiol. 1952;116:424–448. doi: 10.1113/jphysiol.1952.sp004716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Priori SG, Napolitano C, O'Leary ME, Chahine M. Y1767C, a novel SCN5A mutation, induces a persistent Na+ current and potentiates ranolazine inhibition of Nav1.5 channels. Am J Physiol Heart Circ Physiol. 2011;300:H288–H299. doi: 10.1152/ajpheart.00539.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkat-Rott K, Holzherr B, Fauler M, Lehmann-Horn F. Sodium channelopathies of skeletal muscle result from gain or loss of function. Pflugers Arch. 2010;460:239–248. doi: 10.1007/s00424-010-0814-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T, Kinoshita M, Sasaki R, Aoike F, Takahashi MP, Sakoda S, Hirose K. New mutation of the Na channel in the severe form of potassium-aggravated myotonia. Muscle Nerve. 2009;39:666–673. doi: 10.1002/mus.21155. [DOI] [PubMed] [Google Scholar]
- Lehmann-Horn F, Jurkat-Rott K. Myotonic disorders. Handb Clin Neurol. 2007;86:61–76. doi: 10.1016/S0072-9752(07)86003-9. [DOI] [PubMed] [Google Scholar]
- Lehmann-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiol Rev. 1999;79:1317–1372. doi: 10.1152/physrev.1999.79.4.1317. [DOI] [PubMed] [Google Scholar]
- Lehmann-Horn F, Kuther G, Ricker K, Grafe P, Ballanyi K, Rudel R. Adynamia episodica hereditaria with myotonia: a non-inactivating sodium current and the effect of extracellular pH. Muscle Nerve. 1987;10:363–374. doi: 10.1002/mus.880100414. [DOI] [PubMed] [Google Scholar]
- Lehmann-Horn F, Rudel R. Channelopathies: the nondystrophic myotonias and periodic paralyses. Semin Pediatr Neurol. 1996;3:122–139. doi: 10.1016/s1071-9091(96)80041-6. [DOI] [PubMed] [Google Scholar]
- Lerche H, Mitrovic N, Dubowitz V, Lehmann-Horn F. Paramyotonia congenita: the R1448P Na+ channel mutation in adult human skeletal muscle. Ann Neurol. 1996;39:599–608. doi: 10.1002/ana.410390509. [DOI] [PubMed] [Google Scholar]
- Margolskee RF, McHendry-Rinde B, Horn R. Panning transfected cells for electrophysiological studies. Biotechniques. 1993;15:906–911. [PubMed] [Google Scholar]
- McClatchey AI, McKenna-Yasek D, Cros D, Worthen HG, Kuncl RW, DeSilva SM, Cornblath DR, Gusella JF, Brown RH., Jr Novel mutations in families with unusual and variable disorders of the skeletal muscle sodium channel. Nat Genet. 1992;2:148–152. doi: 10.1038/ng1092-148. [DOI] [PubMed] [Google Scholar]
- Mitrovic N, George AL, Jr, Heine R, Wagner S, Pika U, Hartlaub U, Zhou M, Lerche H, Fahlke C, Lehmann-Horn F. K+-aggravated myotonia: destabilization of the inactivated state of the human muscle Na+ channel by the V1589M mutation. J Physiol. 1994;478:395–402. doi: 10.1113/jphysiol.1994.sp020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic N, George AL, Jr, Lerche H, Wagner S, Fahlke C, Lehmann-Horn F. Different effects on gating of three myotonia-causing mutations in the inactivation gate of the human muscle sodium channel. J Physiol. 1995;487:107–114. doi: 10.1113/jphysiol.1995.sp020864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos E, O'Leary ME. State-dependent tapping of flecainide in the cardiac sodium channel. J Physiol. 2004;560:37–49. doi: 10.1113/jphysiol.2004.065003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, VanDeCarr D, Featherstone DE, George AL, Jr, Ruben PC. Defective fast inactivation recovery and deactivation account for sodium channel myotonia in the I1160V mutant. Biophys J. 1997;73:1896–1903. doi: 10.1016/S0006-3495(97)78220-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricker K, Moxley RT, III, Heine R, Lehmann-Horn F. Myotonia fluctuans. A third type of muscle sodium channel disease. Arch Neurol. 1994;51:1095–1102. doi: 10.1001/archneur.1994.00540230033009. [DOI] [PubMed] [Google Scholar]
- Rosenfeld J, Sloan-Brown K, George AL., Jr A novel muscle sodium channel mutation causes painful congenital myotonia. Ann Neurol. 1997;42:811–814. doi: 10.1002/ana.410420520. [DOI] [PubMed] [Google Scholar]
- Rossignol E, Mathieu J, Thiffault I, Tetreault M, Dicaire MJ, Chrestian N, Dupre N, Puymirat J, Brais B. A novel founder SCN4A mutation causes painful cold-induced myotonia in French-Canadians. Neurology. 2007;69:1937–1941. doi: 10.1212/01.wnl.0000290831.08585.2c. [DOI] [PubMed] [Google Scholar]
- Sugiura Y, Makita N, Li L, Noble PJ, Kimura J, Kumagai Y, Soeda T, Yamamoto T. Cold induces shifts of voltage dependence in mutant SCN4A, causing hypokalemic periodic paralysis. Neurology. 2003;61:914–918. doi: 10.1212/01.wnl.0000086820.54065.a0. [DOI] [PubMed] [Google Scholar]
- Wang DW, Yazawa K, Makita N, George AL, Jr, Bennett PB. Pharmacological targeting of long QT mutant sodium channels. J Clin Invest. 1997;99:1714–1720. doi: 10.1172/JCI119335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GK, Russell C, Wang SY. Mexiletine block of wild-type and inactivation-deficient human skeletal muscle hNav1.4 Na+ channels. J Physiol. 2004;554:621–633. doi: 10.1113/jphysiol.2003.054973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb J, Cannon SC. Cold-induced defects of sodium channel gating in atypical periodic paralysis plus myotonia. Neurology. 2008;70:755–761. doi: 10.1212/01.wnl.0000265397.70057.d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, Catterall WA. Overview of the voltage-gated sodium channel family. Genome Biol. 2003;4:207. doi: 10.1186/gb-2003-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ziane R, Chatelier A, O'Leary ME, Chahine M. Lidocaine promotes the trafficking and functional expression of Nav1.8 sodium channels in mammalian cells. J Neurophysiol. 2007;98:467–477. doi: 10.1152/jn.00117.2007. [DOI] [PubMed] [Google Scholar]